

High-Accuracy ncRNA Function Prediction via Deep Learning Using Global and Local Sequence Information


Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset
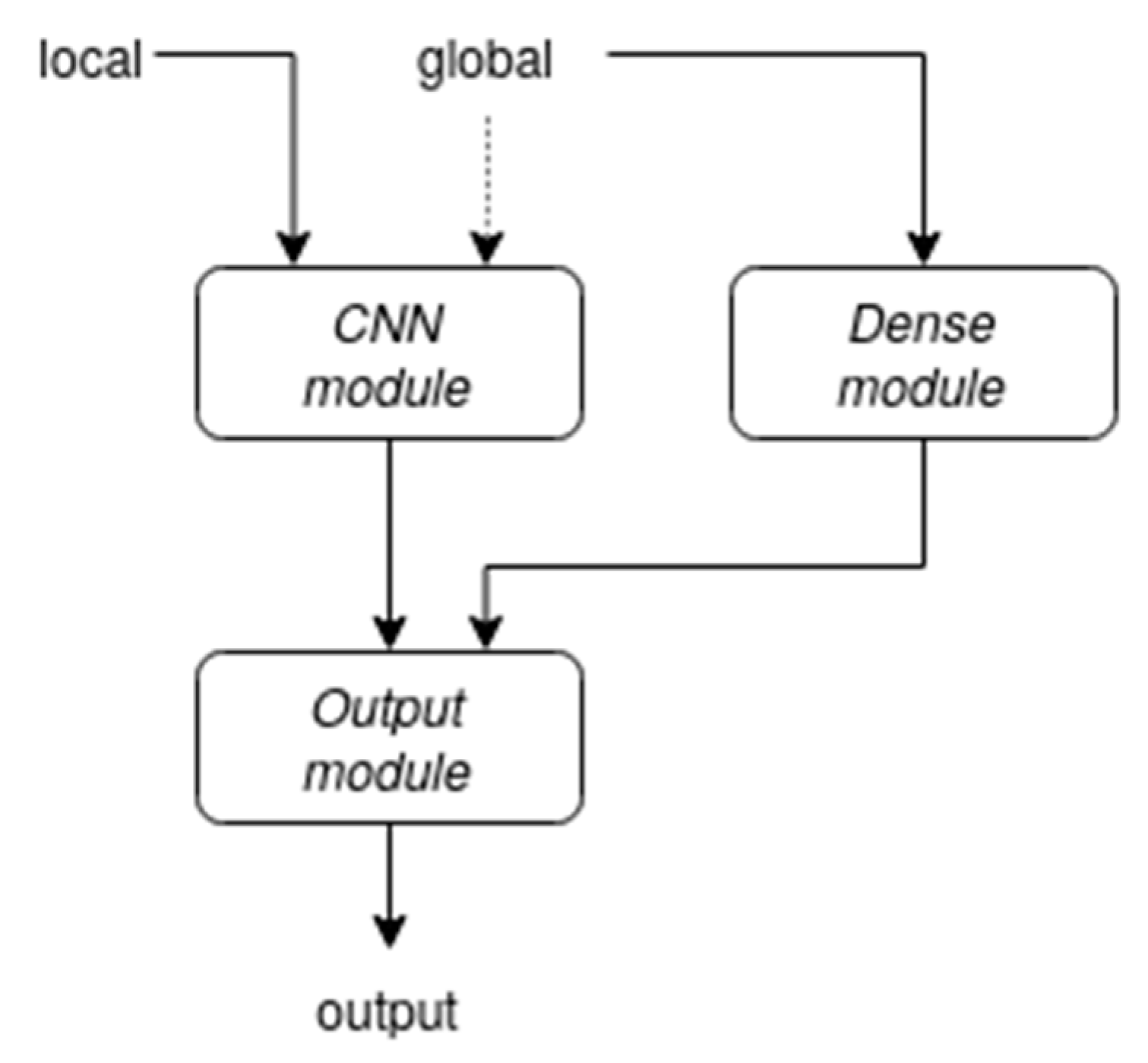
2.2. Architecture
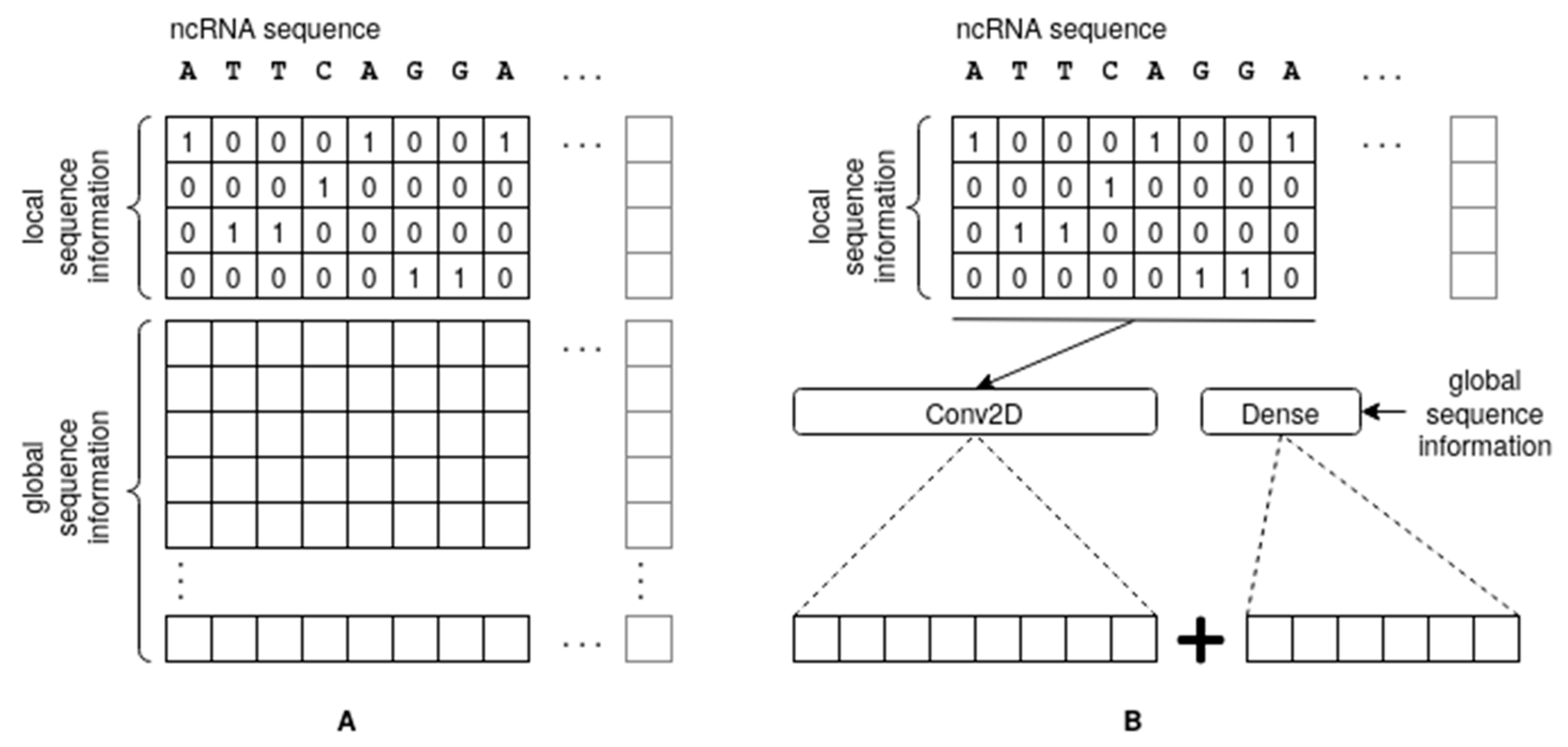
2.3. Global Information Representation
- K-mer statistics using small values of k;
- Class propensities derived from medium-length pattern occurrences; and
- Instance-based class propensities.
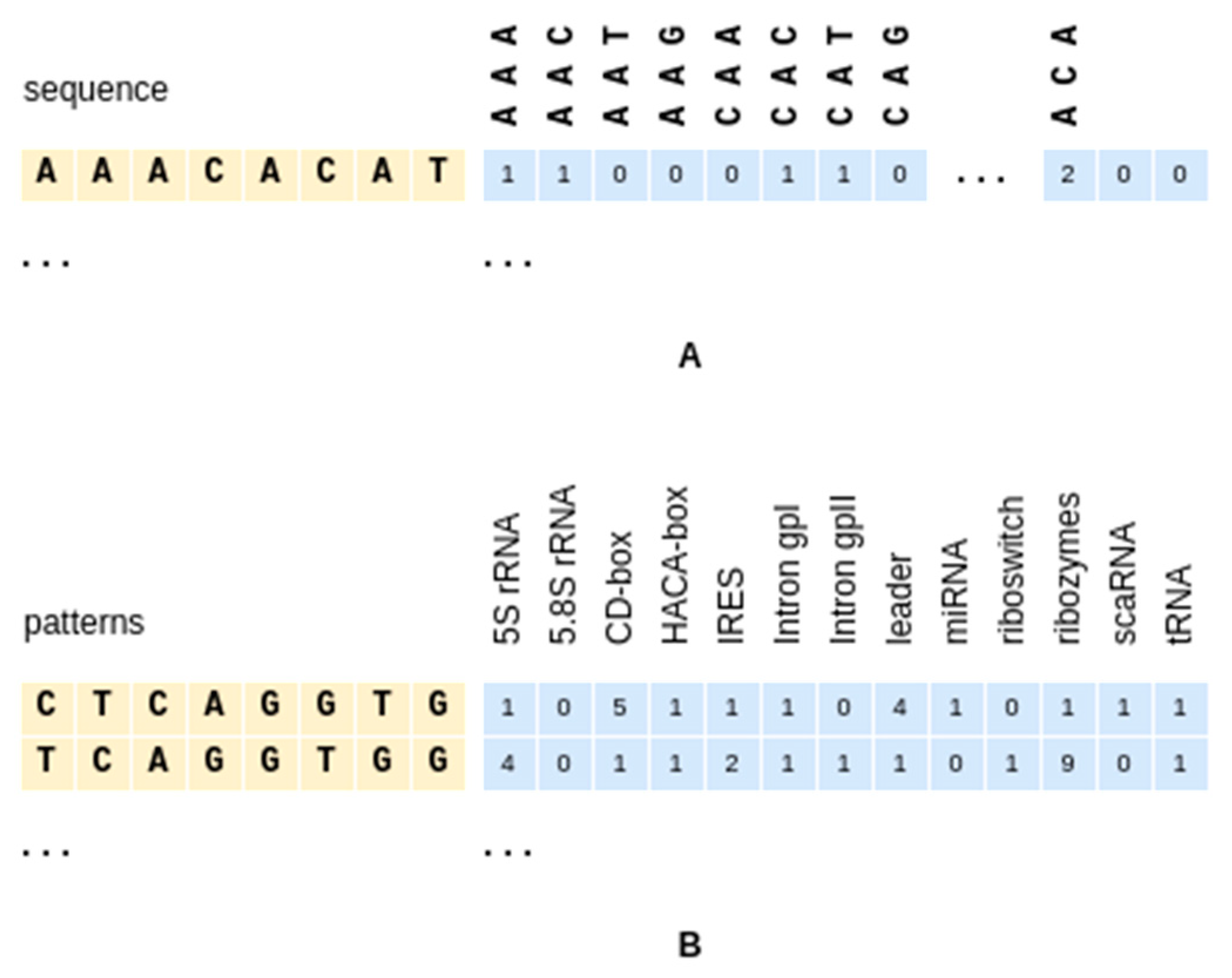
2.3.1. Small K-mers
2.3.2. Sequence Patterns
2.3.3. Instance-Based Class Propensities
2.4. Neural Network Configuration
3. Results
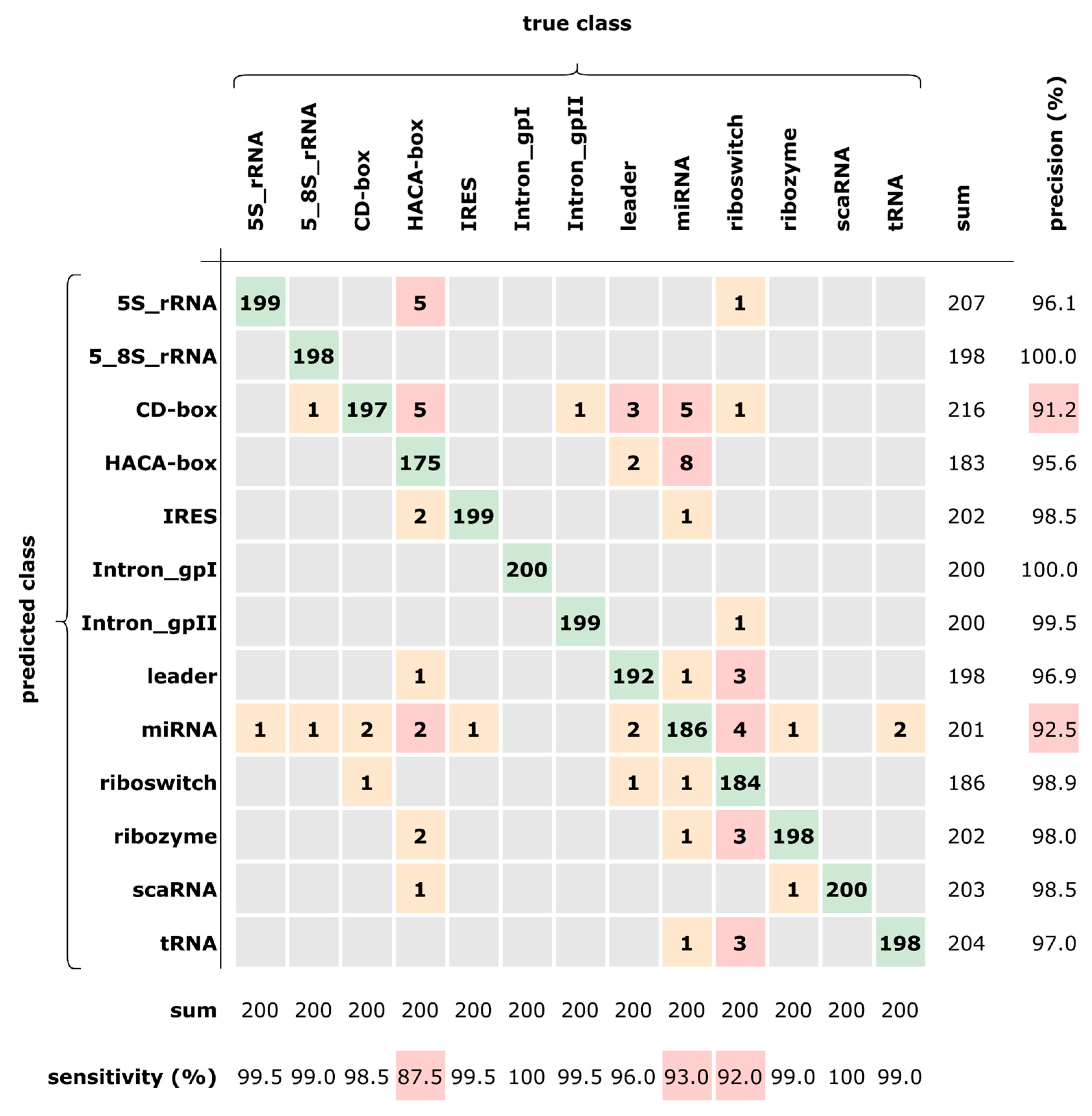
3.1. Impact of Introducing Global Information
3.2. Comparison with Other Methods
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wery, M.; Kwapisz, M.; Morillon, A. Noncoding RNAs in gene regulation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 728–738. [Google Scholar] [CrossRef]
- Romero-Barrios, N.; Legascue, M.F.; Benhamed, M.; Ariel, F.; Crespi, M. Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 2018, 46, 2169–2184. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Lin, H. Noncoding RNAs in the regulation of DNA replication. Trends Biochem. Sci. 2014, 39, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Lekka, E.; Hall, J. Noncoding RNAs in disease. FEBS Lett. 2018, 592, 2884–2900. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 924–933. [Google Scholar] [CrossRef]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Tian, H.; Zhou, C.; Yang, J.; Li, J.; Gong, Z. Long and short noncoding RNAs in lung cancer precision medicine: Opportunities and challenges. Tumor Biol. 2017, 39, 1010428317697578. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.S.; Whitty, E.; Yoo, B.; Moore, A.; Sempere, L.F.; Medarova, Z. Clinical Applications of Short Non-Coding RNA-Based Therapies in the Era of Precision Medicine. Cancers 2022, 14, 1588. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef]
- Amaral, P.P.; Clark, M.B.; Gascoigne, D.K.; Dinger, M.E.; Mattick, J.S. lncRNAdb: A reference database for long noncoding RNAs. Nucleic Acids Res. 2011, 39, D146–D151. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Yang, J.-H.; Shao, P.; Zhou, H.; Chen, Y.-Q.; Qu, L.-H. deepBase: A database for deeply annotating and mining deep sequencing data. Nucleic Acids Res. 2010, 38, D123–D130. [Google Scholar] [CrossRef]
- Szymanski, M.; Erdmann, V.A.; Barciszewski, J. Noncoding RNAs database (ncRNAdb). Nucleic Acids Res. 2007, 35, D162–D164. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Chantsalnyam, T.; Lim, D.Y.; Tayara, H.; Chong, K.T. ncRDeep: Non-coding RNA classification with convolutional neural network. Comput. Biol. Chem. 2020, 88, 107364. [Google Scholar] [CrossRef] [PubMed]
- Noviello, T.M.R.; Ceccarelli, F.; Ceccarelli, M.; Cerulo, L. Deep learning predicts short non-coding RNA functions from only raw sequence data. PLoS Comput. Biol. 2020, 16, e1008415. [Google Scholar] [CrossRef]
- Childs, L.; Nikoloski, Z.; May, P.; Walther, D. Identification and classification of ncRNA molecules using graph properties. Nucleic Acids Res. 2009, 37, e66. [Google Scholar] [CrossRef]
- Fiannaca, A.; La Rosa, M.; La Paglia, L.; Rizzo, R.; Urso, A. nRC: Non-coding RNA Classifier based on structural features. BioData Min. 2017, 10, 1–18. [Google Scholar] [CrossRef]
- Sato, K.; Kato, Y.; Hamada, M.; Akutsu, T.; Asai, K. IPknot: Fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 2011, 27, i85–i93. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Navarin, N.; Costa, F. An efficient graph kernel method for non-coding RNA functional prediction. Bioinformatics 2017, 33, 2642–2650. [Google Scholar] [CrossRef]
- Rossi, E.; Monti, F.; Bronstein, M.; Liò, P. NcRNA Classification with Graph Convolutional Networks. arXiv 2019, arXiv:1905.06515. [Google Scholar]
- Liu, X.-Q.; Li, B.-X.; Zeng, G.-R.; Liu, Q.-Y.; Ai, D.-M. Prediction of Long Non-Coding RNAs Based on Deep Learning. Genes 2019, 10, 273. [Google Scholar] [CrossRef]
- Khalil, K.; Eldash, O.; Kumar, A.; Bayoumi, M. Economic LSTM Approach for Recurrent Neural Networks. IEEE Trans. Circuits Syst. II: Express Briefs 2019, 66, 1885–1889. [Google Scholar] [CrossRef]
- Pascanu, R.; Mikolov, T.; Bengio, Y. On the Difficulty of Training Recurrent Neural Networks. arXiv 2013, arXiv:1211.5063. [Google Scholar]
- Dash, T.; Chitlangia, S.; Ahuja, A.; Srinivasan, A. A review of some techniques for inclusion of domain-knowledge into deep neural networks. Sci. Rep. 2022, 12, 1040. [Google Scholar] [CrossRef] [PubMed]
- Muralidhar, N.; Islam, M.R.; Marwah, M.; Karpatne, A.; Ramakrishnan, N. Incorporating Prior Domain Knowledge into Deep Neural Networks. In Proceedings of the IEEE International Conference on Big Data, Seattle, WA, USA, 10–13 December 2018; pp. 36–45. [Google Scholar] [CrossRef]
- Cannon, J.W.; Thurston, W.P. Group invariant Peano curves. Geom. Topol. 2007, 11, 1315–1355. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ncRNA Class | Description | Length |
|---|---|---|
| miRNA | microRNA, small single-stranded non-coding ribonucleic acid (ncRNA) mainly involved in RNA silencing and gene expression post-transcriptional regulation. | 10 ± 41 |
| 5S rRNA | 5S ribosomal RNA plays a role in the stabilization of the ribosome, of which it is one of the components | 119 ± 9 |
| 5.8S rRNA | 5.8S ribosomal RNA, similar to the 5S rRNA, is a component of the ribosome in eukaryotes; it plays a role in protein translation | 153 ±15 |
| ribozymes | Ribozymes (ribonucleic acid enzymes) have enzymatic behavior in catalyzing specific biochemical reactions | 260 ± 158 |
| CD-box | C/D box guide snoRNA, a subclass of small nucleolar RNAs (snoRNAs) involved in the methylation of RNA molecules | 106 ± 42 |
| HACA-box | H/ACA box snoRNA, a subclass of small nucleolar RNAs (snoRNAs) involved in the pseudouridylation of RNA molecules | 140 ± 35 |
| scaRNA | Small Cajal body-specific RNA plays a guiding role in the methylation and pseudouridylation of RNA polymerase II transcribed spliceosomal RNA | 174 ± 76 |
| tRNA | Transfer RNA plays a role in translation, binds the ribosome, and transfers a specific amino acid of a growing polypeptide chain | 78 ± 13 |
| Intron gpI | Group I Intron is a type of ribozyme able to extract itself from another RNA molecule; it has catalytic activity and is involved in intron splicing | 342 ± 99 |
| Intron gpII | Group II intron is similar to Group I but uses a different type of splicing reaction | 96 ± 26 |
| IRES | Internal Ribosome Entry Site is an RNA involved in protein synthesis | 235 ± 103 |
| leader | Leader RNA is a term that refers to the region of a messenger RNA that precedes the starting codon and has an important role in the regulation of translation of a transcript | 125 ± 30 |
| riboswitch | Riboswitch is a region of an mRNA molecule that regulates the corresponding protein encoding through the action of a specific binding ligand | 142 ± 50 |
| Type of Global Information | Step75 | F1 Score (%) | Accuracy (%) |
|---|---|---|---|
| Local-only | 1433 | 85.96 | 86.03 |
| KM | 18 | 91.55 | 91.64 |
| SP | 681 | 89.97 | 90.10 |
| IB | 461 | 95.46 | 95.49 |
| KM + SP | 14 | 94.11 | 94.22 |
| KM + IB | 14 | 96.87 | 96.90 |
| SP + IB | 27 | 95.75 | 95.80 |
| KM + SP + IB | 9 | 97.11 | 97.20 |
| Method | F1 Score (%) | Accuracy (%) |
|---|---|---|
| EDeN (*) | 65 | 67 |
| nRC | 81.81 | 81.66 |
| RNAGCN | 85.73 | 85.61 |
| CNN improved (*) | 96 | 96 |
| This work | 97.11 | 97.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orro, A.; Trombetti, G.A. High-Accuracy ncRNA Function Prediction via Deep Learning Using Global and Local Sequence Information. Biomedicines 2023, 11, 1631. https://doi.org/10.3390/biomedicines11061631
Orro A, Trombetti GA. High-Accuracy ncRNA Function Prediction via Deep Learning Using Global and Local Sequence Information. Biomedicines. 2023; 11(6):1631. https://doi.org/10.3390/biomedicines11061631
Chicago/Turabian StyleOrro, Alessandro, and Gabriele A. Trombetti. 2023. "High-Accuracy ncRNA Function Prediction via Deep Learning Using Global and Local Sequence Information" Biomedicines 11, no. 6: 1631. https://doi.org/10.3390/biomedicines11061631
APA StyleOrro, A., & Trombetti, G. A. (2023). High-Accuracy ncRNA Function Prediction via Deep Learning Using Global and Local Sequence Information. Biomedicines, 11(6), 1631. https://doi.org/10.3390/biomedicines11061631