

Uncovering the Secrets of Prostate Cancer’s Radiotherapy Resistance: Advances in Mechanism Research
 , ,
, ,
Abstract
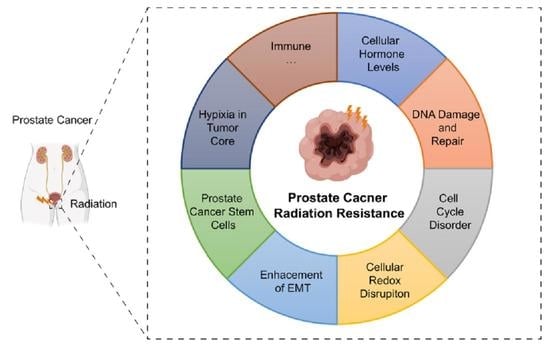
1. Introduction
2. Impact of Cellular Hormone Levels on PCa
3. Maintaining DNA Repair Deficiency and Disorders of the DNA Damage Repair System
4. Cell Cycle Disorder
5. Disruption of Cellular Redox Homeostasis
6. Enhance of Epithelial–Mesenchymal Transitions (EMT)
7. The Existence of Prostate Cancer Stem Cells (PCSCs) in Foci
8. Hypoxia in Tumor Core
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ferraro, S.; Biganzoli, D.; Rossi, R.S.; Palmisano, F.; Bussetti, M.; Verzotti, E.; Gregori, A.; Bianchi, F.; Maggioni, M.; Ceriotti, F.; et al. Individual risk prediction of high grade prostate cancer based on the combination between total prostate-specific antigen (PSA) and free to total PSA ratio. Clin. Chem. Lab. Med. 2023, 61, 1327–1334. [Google Scholar] [CrossRef]
- Bagshaw, M.A. External Radiation Therapy of Carcinoma of the Prostate. Cancer 1980, 45 (Suppl. 7), 1912–1921. [Google Scholar] [CrossRef]
- Ray, G.R.; Cassady, J.R.; Bagshaw, M.A. Definitive radiation therapy of carcinoma of the prostate. A report on 15 years of experience. 1973. J. Urol. 2002, 167, 990–998. [Google Scholar] [CrossRef]
- Kishan, A.U.; Cook, R.R.; Ciezki, J.P.; Ross, A.E.; Pomerantz, M.M.; Nguyen, P.L.; Shaikh, T.; Tran, P.T.; Sandler, K.A.; Stock, R.G.; et al. Radical Prostatectomy, External Beam Radiotherapy, or External Beam Radiotherapy with Brachytherapy Boost and Disease Progression and Mortality in Patients with Gleason Score 9–10 Prostate Cancer. JAMA 2018, 319, 896–905. [Google Scholar] [CrossRef]
- Deek, M.P.; Yu, C.; Phillips, R.; Song, D.Y.; Deville, C.; Greco, S.; DeWeese, T.L.; Antonarakis, E.S.; Markowski, M.; Paller, C.; et al. Radiation Therapy in the Definitive Management of Oligometastatic Prostate Cancer: The Johns Hopkins Experience. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 948–956. [Google Scholar] [CrossRef]
- Huggins, C. Studies on prostatic cancer: 1. The effect of castration, of estrogen and of androgen injection on serum phoshatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1. [Google Scholar] [CrossRef]
- Huggins, C.; Stevens, R.R.; Hodges, C.V. Studies on prostate cancer. II. The effect of castration on advanced carcinoma of the prostate gland. Arch. Surg. 1941, 43, 209–223. [Google Scholar] [CrossRef]
- Barfeld, S.J.; Itkonen, H.M.; Urbanucci, A.; Mills, I.G. Androgen-regulated metabolism and biosynthesis in prostate cancer. Endocr. Relat. Cancer 2014, 21, T57–T66. [Google Scholar] [CrossRef]
- Jamroze, A.; Chatta, G.; Tang, D.G. Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Lett. 2021, 518, 1–9. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Li, Y.; Yang, R.; Henzler, C.M.; Ho, Y.; Passow, C.; Auch, B.; Carreira, S.; Nava Rodrigues, D.; Bertan, C.; Hwang, T.H.; et al. Diverse AR Gene Rearrangements Mediate Resistance to Androgen Receptor Inhibitors in Metastatic Prostate Cancer. Clin. Cancer Res. 2020, 26, 1965–1976. [Google Scholar] [CrossRef]
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- Young, A.; Berry, R.; Holloway, A.F.; Blackburn, N.B.; Dickinson, J.L.; Skala, M.; Phillips, J.L.; Brettingham-Moore, K.H. RNA-seq profiling of a radiation resistant and radiation sensitive prostate cancer cell line highlights opposing regulation of DNA repair and targets for radiosensitization. BMC Cancer 2014, 14, 808. [Google Scholar] [CrossRef]
- Al-Ubaidi, F.L.; Schultz, N.; Loseva, O.; Egevad, L.; Granfors, T.; Helleday, T. Castration therapy results in decreased Ku70 levels in prostate cancer. Clin. Cancer Res. 2013, 19, 1547–1556. [Google Scholar] [CrossRef]
- Liu, R.F.; Li, J.; Zhang, J.; Bai, P.D.; Yang, Y.F.; Li, W.; Wu, Z.; Zheng, J.X. Crosstalk between apoptosis and autophagy in prostate epithelial cells under androgen deprivation. Exp. Ther. Med. 2018, 15, 2263–2268. [Google Scholar] [CrossRef]
- Pollack, A.; Salem, N.; Ashoori, F.; Hachem, P.; Sangha, M.; von Eschenbach, A.C.; Meistrich, M.L. Lack of prostate cancer radiosensitization by androgen deprivation. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 1002–1007. [Google Scholar] [CrossRef]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef]
- Spratt, D.E.; Evans, M.J.; Davis, B.J.; Doran, M.G.; Lee, M.X.; Shah, N.; Wongvipat, J.; Carnazza, K.E.; Klee, G.G.; Polkinghorn, W.; et al. Androgen Receptor Upregulation Mediates Radioresistance after Ionizing Radiation. Cancer Res. 2015, 75, 4688–4696. [Google Scholar] [CrossRef]
- Ciccarese, C.; Santoni, M.; Brunelli, M.; Buti, S.; Modena, A.; Nabissi, M.; Artibani, W.; Martignoni, G.; Montironi, R.; Tortora, G.; et al. AR-V7 and prostate cancer: The watershed for treatment selection? Cancer Treat. Rev. 2016, 43, 27–35. [Google Scholar] [CrossRef]
- Desai, K.; McManus, J.M.; Sharifi, N. Hormonal Therapy for Prostate Cancer. Endocr. Rev. 2021, 42, 354–373. [Google Scholar] [CrossRef]
- Luo, H.; Liu, Y.; Li, Y.; Zhang, C.; Yu, B.; Shao, C. Androgen receptor splicing variant 7 (ARv7) promotes DNA damage response in prostate cancer cells. FASEB J. 2022, 36, e22495. [Google Scholar] [CrossRef]
- Yin, Y.; Li, R.; Xu, K.; Ding, S.; Li, J.; Baek, G.; Ramanand, S.G.; Ding, S.; Liu, Z.; Gao, Y.; et al. Androgen Receptor Variants Mediate DNA Repair after Prostate Cancer Irradiation. Cancer Res. 2017, 77, 4745–4754. [Google Scholar] [CrossRef]
- Kornberg, Z.; Chou, J.; Feng, F.Y.; Ryan, C.J. Prostate cancer in the era of “Omic” medicine: Recognizing the importance of DNA damage repair pathways. Ann. Transl. Med. 2018, 6, 161. [Google Scholar] [CrossRef]
- Hillman, G.G.; Singh-Gupta, V. Soy isoflavones sensitize cancer cells to radiotherapy. Free Radic. Biol. Med. 2011, 51, 289–298. [Google Scholar] [CrossRef]
- Goode, E.L.; Ulrich, C.M.; Potter, J.D. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1513–1530. [Google Scholar]
- Chan, J.M.; Gann, P.H.; Giovannucci, E.L. Role of diet in prostate cancer development and progression. J. Clin. Oncol. 2005, 23, 8152–8160. [Google Scholar] [CrossRef]
- Fachal, L.; Gómez-Caamaño, A.; Peleteiro, P.; Carballo, A.; Calvo-Crespo, P.; Sánchez-García, M.; Lobato-Busto, R.; Carracedo, A.; Vega, A. Association of a XRCC3 polymorphism and rectum mean dose with the risk of acute radio-induced gastrointestinal toxicity in prostate cancer patients. Radiother. Oncol. 2012, 105, 321–328. [Google Scholar] [CrossRef]
- Lockett, K.L.; Snowhite, I.V.; Hu, J.J. Nucleotide-excision repair and prostate cancer risk. Cancer Lett. 2005, 220, 125–135. [Google Scholar] [CrossRef]
- Zou, Y.F.; Tao, J.H.; Ye, Q.L.; Pan, H.F.; Pan, F.M.; Su, H.; Ye, D.Q. Association of XPC gene polymorphisms with susceptibility to prostate cancer: Evidence from 3936 subjects. Genet. Test. Mol. Biomark. 2013, 17, 926–931. [Google Scholar] [CrossRef]
- Hartwig, A. Cadmium and cancer. Met. Ions Life Sci. 2013, 11, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Damaraju, S.; Murray, D.; Dufour, J.; Carandang, D.; Myrehaug, S.; Fallone, G.; Field, C.; Greiner, R.; Hanson, J.; Cass, C.E.; et al. Association of DNA repair and steroid metabolism gene polymorphisms with clinical late toxicity in patients treated with conformal radiotherapy for prostate cancer. Clin. Cancer Res. 2006, 12, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Rajput, M.; Mishra, D.; Kumar, K.; Singh, R.P. Silibinin Radiosensitizes EGF Receptor-knockdown Prostate Cancer Cells by Attenuating DNA Repair Pathways. J. Cancer Prev. 2022, 27, 170–181. [Google Scholar] [CrossRef]
- Hasegawa, T.; Someya, M.; Hori, M.; Tsuchiya, T.; Fukushima, Y.; Matsumoto, Y.; Sakata, K.I. Prediction of Results of Radiotherapy with Ku70 Expression and an Artificial Neural Network. In Vivo 2020, 34, 2865–2872. [Google Scholar] [CrossRef] [PubMed]
- Hoey, C.; Ray, J.; Jeon, J.; Huang, X.; Taeb, S.; Ylanko, J.; Andrews, D.W.; Boutros, P.C.; Liu, S.K. miRNA-106a and prostate cancer radioresistance: A novel role for LITAF in ATM regulation. Mol. Oncol. 2018, 12, 1324–1341. [Google Scholar] [CrossRef]
- Xie, X.; Xu, Z.; Wang, C.; Fang, C.; Zhao, J.; Xu, L.; Qian, X.; Dai, J.; Sun, F.; Xu, D.; et al. Tip60 is associated with resistance to X-ray irradiation in prostate cancer. FEBS Open Bio 2018, 8, 271–278. [Google Scholar] [CrossRef]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Zhao, H.; Luoto, K.R.; Lundin, C.; Coackley, C.; Chan, N.; Joshua, A.M.; Bismar, T.A.; Evans, A.; Helleday, T.; et al. PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: Implications for radiotherapy and chemotherapy. Clin. Cancer Res. 2012, 18, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, F.; Ren, Y.; Weng, G.; Xu, L.; Xue, X.; Keng, P.C.; Lee, S.O.; Chen, Y. IL-6 signaling contributes to radioresistance of prostate cancer through key DNA repair-associated molecules ATM, ATR, and BRCA 1/2. J. Cancer Res. Clin. Oncol. 2019, 145, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.; Cai, M.Y.; Elmarakeby, H.A.; Park, J.; Ricker, C.; Kochupurakkal, B.S.; Choudhury, A.D.; Hahn, W.C.; et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Haanpää, M.; Hanenberg, H.; Schleutker, J.; Kallioniemi, A.; Kähkönen, M.; Parto, K.; Avela, K.; Aittomäki, K.; von Koskull, H.; et al. Finnish Fanconi anemia mutations and hereditary predisposition to breast and prostate cancer. Clin. Genet. 2015, 88, 68–73. [Google Scholar] [CrossRef]
- Wilkes, D.C.; Sailer, V.; Xue, H.; Cheng, H.; Collins, C.C.; Gleave, M.; Wang, Y.; Demichelis, F.; Beltran, H.; Rubin, M.A.; et al. A germline FANCA alteration that is associated with increased sensitivity to DNA damaging agents. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001487. [Google Scholar] [CrossRef]
- Vietri, M.T.; D’Elia, G.; Caliendo, G.; Resse, M.; Casamassimi, A.; Passariello, L.; Albanese, L.; Cioffi, M.; Molinari, A.M. Hereditary Prostate Cancer: Genes Related, Target Therapy and Prevention. Int. J. Mol. Sci. 2021, 22, 3753. [Google Scholar] [CrossRef]
- Albero-González, R.; Hernández-Llodrà, S.; Juanpere, N.; Lorenzo, M.; Lloret, A.; Segalés, L.; Duran, X.; Fumadó, L.; Cecchini, L.; Lloreta-Trull, J. Immunohistochemical expression of mismatch repair proteins (MSH2, MSH6, MLH1, and PMS2) in prostate cancer: Correlation with grade groups (WHO 2016) and ERG and PTEN status. Virchows Arch. 2019, 475, 223–231. [Google Scholar] [CrossRef]
- Fukuhara, S.; Chang, I.; Mitsui, Y.; Chiyomaru, T.; Yamamura, S.; Majid, S.; Saini, S.; Hirata, H.; Deng, G.; Gill, A.; et al. DNA mismatch repair gene MLH1 induces apoptosis in prostate cancer cells. Oncotarget 2014, 5, 11297–11307. [Google Scholar] [CrossRef]
- Fukuhara, S.; Chang, I.; Mitsui, Y.; Chiyomaru, T.; Yamamura, S.; Majid, S.; Saini, S.; Deng, G.; Gill, A.; Wong, D.K.; et al. Functional role of DNA mismatch repair gene PMS2 in prostate cancer cells. Oncotarget 2015, 6, 16341–16351. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, X.; Wei, Y.; Chen, X.; Li, G.; Lu, Z.; Zhang, X.; Ren, X.; Wang, S.; Qin, C. Prognostic Role of DNA Damage Response Genes Mutations and their Association with the Sensitivity of Olaparib in Prostate Cancer Patients. Cancer Control 2022, 29, 10732748221129451. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef]
- Li, B.; Shi, X.B.; Nori, D.; Chao, C.K.; Chen, A.M.; Valicenti, R.; White Rde, V. Down-regulation of microRNA 106b is involved in p21-mediated cell cycle arrest in response to radiation in prostate cancer cells. Prostate 2011, 71, 567–574. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Li, J. Exendin-4 enhances radiation response of prostate cancer. Prostate 2018, 78, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. Modeling the role of microRNA-449a in the regulation of the G2/M cell cycle checkpoint in prostate LNCaP cells under ionizing radiation. PLoS ONE 2018, 13, e0200768. [Google Scholar] [CrossRef]
- Madhav, A.; Andres, A.; Duong, F.; Mishra, R.; Haldar, S.; Liu, Z.; Angara, B.; Gottlieb, R.; Zumsteg, Z.S.; Bhowmick, N.A. Antagonizing CD105 enhances radiation sensitivity in prostate cancer. Oncogene 2018, 37, 4385–4397. [Google Scholar] [CrossRef]
- Rashid, A.; Liu, C.; Sanli, T.; Tsiani, E.; Singh, G.; Bristow, R.G.; Dayes, I.; Lukka, H.; Wright, J.; Tsakiridis, T. Resveratrol enhances prostate cancer cell response to ionizing radiation. Modulation of the AMPK, Akt and mTOR pathways. Radiat. Oncol. 2011, 6, 144. [Google Scholar] [CrossRef]
- Sánchez, C.; Mercado, A.; Contreras, H.R.; Carvajal, V.F.; Salgado, A.; Huidobro, C.; Castellón, E.A. Membrane translocation and activation of GnRH receptor sensitize prostate cancer cells to radiation. Int. J. Radiat. Biol. 2021, 97, 1555–1562. [Google Scholar] [CrossRef]
- Hussain, S.S.; Huang, S.B.; Bedolla, R.G.; Rivas, P.; Basler, J.W.; Swanson, G.P.; Hui-Ming Huang, T.; Narayanasamy, G.; Papanikolaou, N.; Miyamoto, H.; et al. Suppression of ribosomal protein RPS6KB1 by Nexrutine increases sensitivity of prostate tumors to radiation. Cancer Lett. 2018, 433, 232–241. [Google Scholar] [CrossRef]
- Wang, F.; Mao, A.; Tang, J.; Zhang, Q.; Yan, J.; Wang, Y.; Di, C.; Gan, L.; Sun, C.; Zhang, H. microRNA-16-5p enhances radiosensitivity through modulating Cyclin D1/E1-pRb-E2F1 pathway in prostate cancer cells. J. Cell. Physiol. 2019, 234, 13182–13190. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Cao, F.; Liu, H. Radiation-induced Cell Death and Its Mechanisms. Health Phys. 2022, 123, 376–386. [Google Scholar] [CrossRef]
- Przybyszewski, W.M.; Wideł, M.; Szurko, A.; Maniakowski, Z. Dose rate-dependent cellular and molecular effects of ionizing radiation. Postep. Hig. Med. Dosw. 2008, 62, 468–477. [Google Scholar]
- Wideł, M.; Przybyszewski, W.; Rzeszowska-Wolny, J. Radiation-induced bystander effect: The important part of ionizing radiation response. Potential clinical implications. Postep. Hig. Med. Dosw. 2009, 63, 377–388. [Google Scholar]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Parga, J.A.; Rodriguez-Perez, A.I.; Garcia-Garrote, M.; Rodriguez-Pallares, J.; Labandeira-Garcia, J.L. NRF2 Activation and Downstream Effects: Focus on Parkinson’s Disease and Brain Angiotensin. Antioxidants 2021, 10, 1649. [Google Scholar] [CrossRef]
- Bayo Jimenez, M.T.; Frenis, K.; Hahad, O.; Steven, S.; Cohen, G.; Cuadrado, A.; Münzel, T.; Daiber, A. Protective actions of nuclear factor erythroid 2-related factor 2 (NRF2) and downstream pathways against environmental stressors. Free Radic. Biol. Med. 2022, 187, 72–91. [Google Scholar] [CrossRef]
- Hellyer, J.A.; Padda, S.K.; Diehn, M.; Wakelee, H.A. Clinical Implications of KEAP1-NFE2L2 Mutations in NSCLC. J. Thorac. Oncol. 2021, 16, 395–403. [Google Scholar] [CrossRef]
- Ge, W.; Zhao, K.; Wang, X.; Li, H.; Yu, M.; He, M.; Xue, X.; Zhu, Y.; Zhang, C.; Cheng, Y.; et al. iASPP Is an Antioxidative Factor and Drives Cancer Growth and Drug Resistance by Competing with Nrf2 for Keap1 Binding. Cancer Cell 2017, 32, 561–573.e566. [Google Scholar] [CrossRef]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A.; Stehr, H.; Newman, A.M.; Gentles, A.J.; Kong, W.; Truong, D.; Martin, S.; Chaudhuri, A.; et al. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Mohammadi, A.T.; Gholami, M.H.; Hashemi, F.; Zarrabi, A.; Zabolian, A.; Hushmandi, K.; Makvandi, P.; Samec, M.; Liskova, A.; et al. Nrf2 signaling pathway in cisplatin chemotherapy: Potential involvement in organ protection and chemoresistance. Pharmacol. Res. 2021, 167, 105575. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Fuentes, F.; Shu, L.; Paredes-Gonzalez, X.; Yang, A.Y.; Liu, Y.; Smiraglia, D.J.; Yegnasubramanian, S.; Nelson, W.G.; Kong, A.N. Epigenetic DNA methylation of antioxidative stress regulator NRF2 in human prostate cancer. Cancer Prev. Res. 2014, 7, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, D.A.; McCabe, M.T.; Arnold, R.S.; Day, M.L. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 2008, 27, 4353–4362. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.C.S.; Morelli, A.P.; Severino, M.B.; Pavan, I.C.B.; Zambalde, É.P.; Góis, M.M.; Silva, L.; Quintero-Ruiz, N.; Romeiro, C.F.; Dos Santos, D.F.G.; et al. Knockout of NRF2 triggers prostate cancer cells death through ROS modulation and sensitizes to cisplatin. J. Cell. Biochem. 2022, 123, 2079–2092. [Google Scholar] [CrossRef]
- Da, C.; Pu, J.; Liu, Z.; Wei, J.; Qu, Y.; Wu, Y.; Shi, B.; Yang, J.; He, N.; Hou, P. HACE1-mediated NRF2 activation causes enhanced malignant phenotypes and decreased radiosensitivity of glioma cells. Signal Transduct. Target. Ther. 2021, 6, 399. [Google Scholar] [CrossRef]
- Wang, N.; Yu, Y.; Wang, R.; Chen, Y.; Tang, J. mRNA-Modified FUS/NRF2 Signalling Inhibits Ferroptosis and Promotes Prostate Cancer Growth. Comput. Math. Methods Med. 2022, 2022, 8509626. [Google Scholar] [CrossRef]
- Abdel-Wahab, B.A.; Walbi, I.A.; Albarqi, H.A.; Ali, F.E.M.; Hassanein, E.H.M. Roflumilast protects from cisplatin-induced testicular toxicity in male rats and enhances its cytotoxicity in prostate cancer cell line. Role of NF-κB-p65, cAMP/PKA and Nrf2/HO-1, NQO1 signaling. Food Chem. Toxicol. 2021, 151, 112133. [Google Scholar] [CrossRef]
- Ghosh, S.; Dutta, N.; Banerjee, P.; Gajbhiye, R.L.; Sareng, H.R.; Kapse, P.; Pal, S.; Burdelya, L.; Mandal, N.C.; Ravichandiran, V.; et al. Induction of monoamine oxidase A-mediated oxidative stress and impairment of NRF2-antioxidant defence response by polyphenol-rich fraction of Bergenia ligulata sensitizes prostate cancer cells in vitro and in vivo. Free Radic. Biol. Med. 2021, 172, 136–151. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef] [PubMed]
- Chaiswing, L.; Xu, F.; Zhao, Y.; Thorson, J.; Wang, C.; He, D.; Lu, J.; Ellingson, S.R.; Zhong, W.; Meyer, K.; et al. The RelB-BLNK Axis Determines Cellular Response to a Novel Redox-Active Agent Betamethasone during Radiation Therapy in Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 6409. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; St Clair, D.K.; Xu, Y.; Crooks, P.A.; St Clair, W.H. A NADPH oxidase-dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells. Cancer Res. 2010, 70, 2880–2890. [Google Scholar] [CrossRef]
- Shiota, M.; Fujimoto, N.; Itsumi, M.; Takeuchi, A.; Inokuchi, J.; Tatsugami, K.; Yokomizo, A.; Kajioka, S.; Uchiumi, T.; Eto, M. Gene polymorphisms in antioxidant enzymes correlate with the efficacy of androgen-deprivation therapy for prostate cancer with implications of oxidative stress. Ann. Oncol. 2017, 28, 569–575. [Google Scholar] [CrossRef]
- Teoh, J.Y.; Tian, X.Y.; Wong, C.Y.; Lau, C.W.; Cheng, C.K.; Tang, V.W.; Chan, R.C.; Huang, Y.; Ng, C.F. Endothelial dysfunction after androgen deprivation therapy and the possible underlying mechanisms. Prostate 2022, 82, 13–25. [Google Scholar] [CrossRef]
- Schultz, M.A.; Abdel-Mageed, A.B.; Mondal, D. The nrf1 and nrf2 balance in oxidative stress regulation and androgen signaling in prostate cancer cells. Cancers 2010, 2, 1354–1378. [Google Scholar] [CrossRef]
- Feng, T.; Zhao, R.; Sun, F.; Lu, Q.; Wang, X.; Hu, J.; Wang, S.; Gao, L.; Zhou, Q.; Xiong, X.; et al. TXNDC9 regulates oxidative stress-induced androgen receptor signaling to promote prostate cancer progression. Oncogene 2020, 39, 356–367. [Google Scholar] [CrossRef]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef]
- Taki, M.; Abiko, K.; Ukita, M.; Murakami, R.; Yamanoi, K.; Yamaguchi, K.; Hamanishi, J.; Baba, T.; Matsumura, N.; Mandai, M. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clin. Cancer Res. 2021, 27, 4669–4679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, Y.; Wu, Q.; Xie, L.; Barwick, B.; Fu, C.; Li, X.; Wu, D.; Xia, S.; Chen, J.; et al. Acetylation of KLF5 maintains EMT and tumorigenicity to cause chemoresistant bone metastasis in prostate cancer. Nat. Commun. 2021, 12, 1714. [Google Scholar] [CrossRef]
- Nambiar, D.K.; Rajamani, P.; Singh, R.P. Silibinin attenuates ionizing radiation-induced pro-angiogenic response and EMT in prostate cancer cells. Biochem. Biophys. Res. Commun. 2015, 456, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Yu, H.; Wang, F.; Yan, F.; He, X. Inhibition of LOXL2 Enhances the Radiosensitivity of Castration-Resistant Prostate Cancer Cells Associated with the Reversal of the EMT Process. BioMed Res. Int. 2019, 2019, 4012590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef]
- El Bezawy, R.; Cominetti, D.; Fenderico, N.; Zuco, V.; Beretta, G.L.; Dugo, M.; Arrighetti, N.; Stucchi, C.; Rancati, T.; Valdagni, R.; et al. miR-875-5p counteracts epithelial-to-mesenchymal transition and enhances radiation response in prostate cancer through repression of the EGFR-ZEB1 axis. Cancer Lett. 2017, 395, 53–62. [Google Scholar] [CrossRef]
- Fernandez, H.R.; Lindén, S.K. The aspirin metabolite salicylate inhibits lysine acetyltransferases and MUC1 induced epithelial to mesenchymal transition. Sci. Rep. 2017, 7, 5626. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Blanca, A.; Cimadamore, A.; Montironi, R.; Luque, R.J.; Volavšek, M.; Cheng, L. T1 bladder carcinoma with variant histology: Pathological features and clinical significance. Virchows Arch. 2022, 480, 989–998. [Google Scholar] [CrossRef]
- Claps, F.; van de Kamp, M.W.; Mayr, R.; Bostrom, P.J.; Shariat, S.F.; Hippe, K.; Bertz, S.; Neuzillet, Y.; Sanders, J.; Otto, W.; et al. Prognostic impact of variant histologies in urothelial bladder cancer treated with radical cystectomy. BJU Int. 2023. [Google Scholar] [CrossRef]
- Netto, G.J.; Amin, M.B.; Berney, D.M.; Compérat, E.M.; Gill, A.J.; Hartmann, A.; Menon, S.; Raspollini, M.R.; Rubin, M.A.; Srigley, J.R.; et al. The 2022 World Health Organization Classification of Tumors of the Urinary System and Male Genital Organs-Part B: Prostate and Urinary Tract Tumors. Eur. Urol. 2022, 82, 469–482. [Google Scholar] [CrossRef]
- Bergamin, S.; Eade, T.; Kneebone, A.; Kench, J.G.; Sved, P.; Biset, J.F.; Hruby, G. Ductal Carcinoma of the Prostate: An Uncommon Entity With Atypical Behaviour. Clin. Oncol. 2019, 31, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, W.; Shapiro, D.D.; Zhang, M.; Bathala, T.; Navone, N.; Thompson, T.C.; Broom, B.; Aparicio, A.; Tu, S.M.; Tang, C.; et al. Optimizing the diagnosis and management of ductal prostate cancer. Nat. Rev. Urol. 2021, 18, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Van der Kwast, T.; Al Daoud, N.; Collette, L.; Sykes, J.; Thoms, J.; Milosevic, M.; Bristow, R.G.; Van Tienhoven, G.; Warde, P.; Mirimanoff, R.O.; et al. Biopsy diagnosis of intraductal carcinoma is prognostic in intermediate and high risk prostate cancer patients treated by radiotherapy. Eur. J. Cancer 2012, 48, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Kumar, A.; Nalawade, V.; Nonato, T.; Shabaik, A.; Roma, A.; Rose, B.S.; McKay, R.R. Associations Between Intraductal Prostate Cancer and Metastases Following Radical Prostatectomy in Men with Prostate Cancer in the Veterans Affairs Database. Clin. Genitourin. Cancer, 2023; in press. [Google Scholar] [CrossRef]
- Montironi, R.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Scarpelli, M.; Cheng, L. Histopathology of Prostate Cancer and its Precursors. Appl. Immunohistochem. Mol. Morphol. 2022. [Google Scholar] [CrossRef]
- Huan, Y.; Idrees, M.; Gribetz, M.E.; Unger, P.D. Sarcomatoid carcinoma after radiation treatment of prostatic adenocarcinoma. Ann. Diagn. Pathol. 2008, 12, 142–145. [Google Scholar] [CrossRef]
- Hansel, D.E.; Epstein, J.I. Sarcomatoid carcinoma of the prostate: A study of 42 cases. Am. J. Surg. Pathol. 2006, 30, 1316–1321. [Google Scholar] [CrossRef]
- Alhamar, M.; Tudor Vladislav, I.; Smith, S.C.; Gao, Y.; Cheng, L.; Favazza, L.A.; Alani, A.M.; Ittmann, M.M.; Riddle, N.D.; Whiteley, L.J.; et al. Gene fusion characterisation of rare aggressive prostate cancer variants-adenosquamous carcinoma, pleomorphic giant-cell carcinoma, and sarcomatoid carcinoma: An analysis of 19 cases. Histopathology 2020, 77, 890–899. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Li, J.J.; Shen, M.M. Prostate Stem Cells and Cancer Stem Cells. Cold Spring Harb. Perspect. Med. 2019, 9, a030395. [Google Scholar] [CrossRef]
- Cucchiara, V.; Cooperberg, M.R.; Dall’Era, M.; Lin, D.W.; Montorsi, F.; Schalken, J.A.; Evans, C.P. Genomic Markers in Prostate Cancer Decision Making. Eur. Urol. 2018, 73, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Elliott, J.; Salamone, R.J.; Kim, S.; Aimone, L.J.; Walker, J.R.; Watson, J.; Sauveur-Michel, M.; Garcia-Echeverria, C.; Cho, C.Y.; et al. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clin. Cancer Res. 2010, 16, 5692–5702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, D.G.; Rycaj, K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin. Cancer Biol. 2018, 52, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013, 4, e875. [Google Scholar] [CrossRef] [PubMed]
- Cojoc, M.; Peitzsch, C.; Kurth, I.; Trautmann, F.; Kunz-Schughart, L.A.; Telegeev, G.D.; Stakhovsky, E.A.; Walker, J.R.; Simin, K.; Lyle, S.; et al. Aldehyde Dehydrogenase Is Regulated by β-Catenin/TCF and Promotes Radioresistance in Prostate Cancer Progenitor Cells. Cancer Res. 2015, 75, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Novak, D.; Hüser, L.; Elton, J.J.; Umansky, V.; Altevogt, P.; Utikal, J. SOX2 in development and cancer biology. Semin. Cancer Biol. 2020, 67, 74–82. [Google Scholar] [CrossRef]
- Jia, X.; Li, X.; Xu, Y.; Zhang, S.; Mou, W.; Liu, Y.; Liu, Y.; Lv, D.; Liu, C.H.; Tan, X.; et al. SOX2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cell. J. Mol. Cell. Biol. 2011, 3, 230–238. [Google Scholar] [CrossRef]
- Ni, J.; Cozzi, P.J.; Hao, J.L.; Beretov, J.; Chang, L.; Duan, W.; Shigdar, S.; Delprado, W.J.; Graham, P.H.; Bucci, J.; et al. CD44 variant 6 is associated with prostate cancer metastasis and chemo-/radioresistance. Prostate 2014, 74, 602–617. [Google Scholar] [CrossRef]
- Yadav, S.; Kowolik, C.M.; Lin, M.; Zuro, D.; Hui, S.K.; Riggs, A.D.; Horne, D.A. SMC1A is associated with radioresistance in prostate cancer and acts by regulating epithelial-mesenchymal transition and cancer stem-like properties. Mol. Carcinog. 2019, 58, 113–125. [Google Scholar] [CrossRef]
- Zhang, Y.; He, L.; Sadagopan, A.; Ma, T.; Dotti, G.; Wang, Y.; Zheng, H.; Gao, X.; Wang, D.; DeLeo, A.B.; et al. Targeting Radiation-Resistant Prostate Cancer Stem Cells by B7-H3 CAR T Cells. Mol. Cancer Ther. 2021, 20, 577–588. [Google Scholar] [CrossRef]
- Duncan, W. Exploitation of the oxygen enhancement ratio in clinical practice. Br. Med. Bull. 1973, 29, 33–38. [Google Scholar] [CrossRef]
- Wenzl, T.; Wilkens, J.J. Modelling of the oxygen enhancement ratio for ion beam radiation therapy. Phys. Med. Biol. 2011, 56, 3251–3268. [Google Scholar] [CrossRef] [PubMed]
- Grimes, D.R.; Partridge, M. A mechanistic investigation of the oxygen fixation hypothesis and oxygen enhancement ratio. Biomed. Phys. Eng. Express 2015, 1, 045209. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Mennerich, D.; Kubaichuk, K.; Kietzmann, T. DUBs, Hypoxia, and Cancer. Trends Cancer 2019, 5, 632–653. [Google Scholar] [CrossRef]
- Kwon, Y.D.; Lee, J.Y.; La, M.T.; Lee, S.J.; Lee, S.H.; Park, J.H.; Kim, H.K. Novel multifunctional (18)F-labelled PET tracer with prostate-specific membrane antigen-targeting and hypoxia-sensitive moieties. Eur. J. Med. Chem. 2020, 189, 112099. [Google Scholar] [CrossRef]
- Rios-Colon, L.; Kim, S.; Su, Y.; Kumar, D.; Deep, G. Immunofluorescence-Based Method to Assess Cancer Biomarker in the Hypoxic Region of the Tumor. Methods Mol. Biol. 2022, 2413, 37–43. [Google Scholar] [CrossRef]
- Wadsworth, B.J.; Decotret, L.R.; Villamil, C.; Yapp, D.; Wilson, D.; Benard, F.; McKenzie, M.; Bennewith, K.L. Evaluation of (18)F-EF5 for detection of hypoxia in localized adenocarcinoma of the prostate. Acta Oncol. 2021, 60, 1489–1498. [Google Scholar] [CrossRef]
- Mainta, I.C.; Zilli, T.; Tille, J.C.; De Perrot, T.; Vallée, J.P.; Buchegger, F.; Garibotto, V.; Miralbell, R. The Effect of Neoadjuvant Androgen Deprivation Therapy on Tumor Hypoxia in High-Grade Prostate Cancer: An (18)F-MISO PET-MRI Study. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 1210–1218. [Google Scholar] [CrossRef]
- Gulaka, P.K.; Rojas-Quijano, F.; Kovacs, Z.; Mason, R.P.; Sherry, A.D.; Kodibagkar, V.D. GdDO3NI, a nitroimidazole-based T1 MRI contrast agent for imaging tumor hypoxia in vivo. J. Biol. Inorg. Chem. 2014, 19, 271–279. [Google Scholar] [CrossRef]
- Li, Y.; Gong, T.; Gao, H.; Chen, Y.; Li, H.; Zhao, P.; Jiang, Y.; Wang, K.; Wu, Y.; Zheng, X.; et al. ZIF-Based Nanoparticles Combine X-Ray-Induced Nitrosative Stress with Autophagy Management for Hypoxic Prostate Cancer Therapy. Angew. Chem. Int. Ed. 2021, 60, 15472–15481. [Google Scholar] [CrossRef] [PubMed]
- Bui, B.P.; Nguyen, P.L.; Lee, K.; Cho, J. Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers 2022, 14, 6054. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Samanta, P.; Sarkar, R.; Biswas, S.; Saha, P.; Hajra, S.; Bhowmik, A. Targeting HIF-1α by Natural and Synthetic Compounds: A Promising Approach for Anti-Cancer Therapeutics Development. Molecules 2022, 27, 5192. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, J.; Yan, C.; Hou, J.; Pu, J.; Zhang, G.; Fu, Z.; Wang, X. Effect of small interfering RNA targeting hypoxia-inducible factor-1α on radiosensitivity of PC3 cell line. Urology 2012, 79, 744.e17–744.e24. [Google Scholar] [CrossRef]
- Azorín-Vega, E.P.; Letechípia de León, C.; García-Reyna, M.G.; Vega-Carrillo, H.R. Mathematical description of the effect of HIF inhibition on the radiobiological response of LNCaP cells. Appl. Radiat. Isot. 2022, 184, 110157. [Google Scholar] [CrossRef]
- Luo, Y.; Li, M.C.; Zhao, J.H.; Han, Y.L.; Lin, Y.H.; Wang, Y.X.; Jiang, Y.G.; Lu, Q.; Lan, L. Activation of HIF-1α/β-catenin signal pathway leads to radioresistance of prostate cancer cells. Zhonghua Yi Xue Za Zhi 2018, 98, 2552–2558. [Google Scholar]
- Han, H.; Lee, S.O.; Xu, Y.; Kim, J.E.; Lee, H.J. SPHK/HIF-1α Signaling Pathway Has a Critical Role in Chrysin-Induced Anticancer Activity in Hypoxia-Induced PC-3 Cells. Cells 2022, 11, 2787. [Google Scholar] [CrossRef]
- Lip, H.; Amini, M.A.; Zetrini, A.; Cai, P.; Abbasi, A.Z.; Bristow, R.G.; Rauth, A.M.; Wu, X.Y. Redox-responsive nanoparticles enhance radiation therapy by altering multifaceted radio-resistance mechanisms in human castration-resistant prostate cancer cells and xenografts. Radiother. Oncol. 2022, 170, 213–223. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, M.; Xing, D.; Feng, Y. Atorvastatin enhances radiosensitivity in hypoxia-induced prostate cancer cells related with HIF-1α inhibition. Biosci. Rep. 2017, 37, BSR20170340. [Google Scholar] [CrossRef]
- Tsakiridis, E.E.; Broadfield, L.; Marcinko, K.; Biziotis, O.D.; Ali, A.; Mekhaeil, B.; Ahmadi, E.; Singh, K.; Mesci, A.; Zacharidis, P.G.; et al. Combined metformin-salicylate treatment provides improved anti-tumor activity and enhanced radiotherapy response in prostate cancer; drug synergy at clinically relevant doses. Transl. Oncol. 2021, 14, 101209. [Google Scholar] [CrossRef]
- Yang, J.; Ruan, Y.; Wang, D.; Fan, J.; Luo, N.; Chen, H.; Li, X.; Chen, W.; Wang, X. VHL-recruiting PROTAC attenuates renal fibrosis and preserves renal function via simultaneous degradation of Smad3 and stabilization of HIF-2α. Cell Biosci. 2022, 12, 203. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L. The Rise of Molecular Glues. Cell 2021, 184, 3–9. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Applicable Conditions | Relevant Research Progress |
|---|---|---|
| Base Excision Repair (BER) | A small amount of base damage | Soy isoflavones make PCa PC-3 cells more sensitive to radiation by inhibiting the expression of DNA repair enzyme APE1/Ref-1 in the nucleus [27]. XRCC1 R194W SNP information is a predisposing factor for PCa patients [28,29]. The presence of XRCC3 rs1799794 SNP information predicts the likelihood of gastrointestinal (GI) toxicity in PCa patients following radiotherapy [30]. |
| Nucleotide Excision Repair (NER) | Larger pyrimidine dimers formed after excision of DNA damage | Numerous studies have investigated the SNPs in NER-related genes [31]. The PAT+/- polymorphism in the XPC gene is a predisposing factor for PCa susceptibility [32]. Cadmium exposure affects the expression of NER-related genes, including XPA [33]. The ERCC2 mutations (G > A, Asp (711) Asp) are predictive markers of toxic reactions to radiotherapy in PCa patients [34]. |
| NHEJ | DNA double-strand break (DSB), independent of the cell cycle | The LIG4 (T > C, Asp (568) Asp) variant serves as a predictor of toxic reactions to radiotherapy in PCa patients [34]. The inhibitor of DNA PKcs and ATM, silymarin, accelerates the radiosensitivity of DU145 cells in PCa [35]. The expression level of Ku70 in PCa tissue can predict the treatment response of radiotherapy [36]. By targeting LITAF, miR-106 increases the expression of ATM, promoting radiotherapy resistance in PC-3 and DU145 cells of PCa [37]. The catalytic activity of Tip60 on ATM acetylation and phosphorylation in PCa cells makes it a candidate marker for radiotherapy resistance [38]. |
| Homologous Recombination Repair (HRR) | DNA double-strand break (DSB), dependent on the existence of sister chromatids in the cell cycle | Approximately 30.7% of PCa patients harbor BRCA1/2 mutations [39]. In patients with mCRPC and BRCA1/2 or HRR mutations, the FDA has authorized the use of the PARP inhibitor Rucaparib [40]. Patients with mCRPC and the HRR mutation may benefit from Olaparib [41]. Silencing of RAD51 in PCa DU145 cells improves their radiosensitivity [42]. IL-6 promotes resistance to radiation in PCa C4-2 and 22Rv1 cells via DNA-damage-repair-related molecules (ATM, ATR, and BRCA1/2), and this effect can be counteracted with JAK and STAT3 inhibitors [43]. After ATM deletion, PCa cells (DU145, LNCaP, and 22Rv1) display increased sensitivity to radiation and ATR inhibition [44]. |
| Cross-Link Repair | Cross-linking between DNA-DNA and DNA-protein due to IR | PCa harbors mutations in genes that encode the core complex of Fanconi anemia (FA), including FANCA ex1-12del and FANCA c.3384-1 G > A, but the relationship between these mutations and radiotherapy remains unexplored [45]. The S1088F mutant protein of FANCA enhances the susceptibility of cells to DNA damage induced by cis platinum [46]. |
| Mismatch Repair (MMR) | Mismatch while removing replication and mismatch of small insertions | Although infrequent, the occurrence of MMR gene mutations in PCa serves as an unfavorable prognostic marker [47]. The level of MS1H6 expression is linked to Gleason Grade 5 [48]. PMS2 and MLH1 induce downregulation of BCL2A1- and c-Abl-mediated apoptosis in PCa DU145 cells, suggesting their potential as targets for radiosensitization [49,50]. Mutations in MLH1 and PMS1 impact the sensitivity of Olaparib [51]. |
| Target | Phase | Molecular Mechanism |
|---|---|---|
| miR-106b [53] | G2/M arrest | miR-106b can inhibit the proliferation of the PCa LNCaP cells by activating P21-mediated cell cycle arrest. |
| The receptor of Exendin-4, namely, GLP-1R [54] | G2/M arrest | Exendin-4 promotes AMPK phosphorylation and activates downstream signaling pathways, while also inhibiting the expression of mTOR, cyclinB, and p34. |
| miR-449a [55] | Cdc25A, Cdc2/CyclinB | Induction of G2/M arrest. |
| CD105 [56] | G2/M arrest | CD105 has been shown to promote radiotherapy resistance in PCa by depleting intracellular ATP and upregulating SIRT1, which activates the BMP and TGF-β/Smad pathways. However, targeting CD105 with the TRC105 antibody has been demonstrated to enhance radiosensitivity. |
| Resveratrol [57] | G1/S arrest | Resveratrol has been shown to inhibit the phosphorylation of PI3K/Akt, which is an important cell survival signaling pathway in PCa 22Rv1 and PC-3 cells following radiation treatment. Additionally, resveratrol induces the phosphorylation of AMPK and promotes cell cycle arrest in a P21-dependent manner. |
| GnRHR [58] | G2/M arrest | Redistribution of GnRHR expression using IN3 repositioned it on the membrane surface of PC-3 cells, promoting their radiosensitivity in the recoverable phase, while IN3 had a pro-apoptotic effect. |
| RPS6KB1 [59] | cyclinD1, Cdc25C, G2/M arrest | The expression of ChK1, p-Cdc25C, and cyclinD1 in PCa PC-3 cells can be downregulated after RPS6KB1 is inhibited by Nexrutine. When RPS6KB1 is inhibited, and the process of NHEJ is also inhibited. |
| miR-16-5p [60] | Cyclin D1/Cyclin E1/pRb/E2F1 | MiR-16-5p has been shown to downregulate cyclinD1 and E1 expression in PCa LNCaP cells by directly binding with their 3′UTR region, leading to cell cycle arrest at the G0/G1 phase. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, F.; Shang, S.-Y.; Gao, X.-S.; Ma, M.-W.; Xie, M.; Ren, X.-Y.; Liu, M.-Z.; Chen, J.-Y.; Li, S.-S.; Huang, L. Uncovering the Secrets of Prostate Cancer’s Radiotherapy Resistance: Advances in Mechanism Research. Biomedicines 2023, 11, 1628. https://doi.org/10.3390/biomedicines11061628
Lyu F, Shang S-Y, Gao X-S, Ma M-W, Xie M, Ren X-Y, Liu M-Z, Chen J-Y, Li S-S, Huang L. Uncovering the Secrets of Prostate Cancer’s Radiotherapy Resistance: Advances in Mechanism Research. Biomedicines. 2023; 11(6):1628. https://doi.org/10.3390/biomedicines11061628
Chicago/Turabian StyleLyu, Feng, Shi-Yu Shang, Xian-Shu Gao, Ming-Wei Ma, Mu Xie, Xue-Ying Ren, Ming-Zhu Liu, Jia-Yan Chen, Shan-Shi Li, and Lei Huang. 2023. "Uncovering the Secrets of Prostate Cancer’s Radiotherapy Resistance: Advances in Mechanism Research" Biomedicines 11, no. 6: 1628. https://doi.org/10.3390/biomedicines11061628
APA StyleLyu, F., Shang, S.-Y., Gao, X.-S., Ma, M.-W., Xie, M., Ren, X.-Y., Liu, M.-Z., Chen, J.-Y., Li, S.-S., & Huang, L. (2023). Uncovering the Secrets of Prostate Cancer’s Radiotherapy Resistance: Advances in Mechanism Research. Biomedicines, 11(6), 1628. https://doi.org/10.3390/biomedicines11061628