
The Inability to Disassemble Rad51 Nucleoprotein Filaments Leads to Aberrant Mitosis and Cell Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Yeast Strains, Oligonucleotides and Plasmids
2.2. Spotting Assay
2.3. SSA Plating Assay
2.4. Treatment with α-Factor and PGAL1 Induction during Synchronised Growth Experiments
2.5. Southern Blotting Analysis of the Repair Product Formation during SSA
2.6. qPCR Analysis of Non-Homologous End Cleavage during SSA
2.7. Cell Fractionation
2.8. Western Blotting
2.9. Rad51 Chromatin Immunoprecipitation and DNA Sequencing (ChIP-seq)
2.10. FACS Analysis
2.11. Live Cell Imaging
2.12. Transient RAD55 Expression and HU Treatment
2.13. Statistical Analyses
3. Results
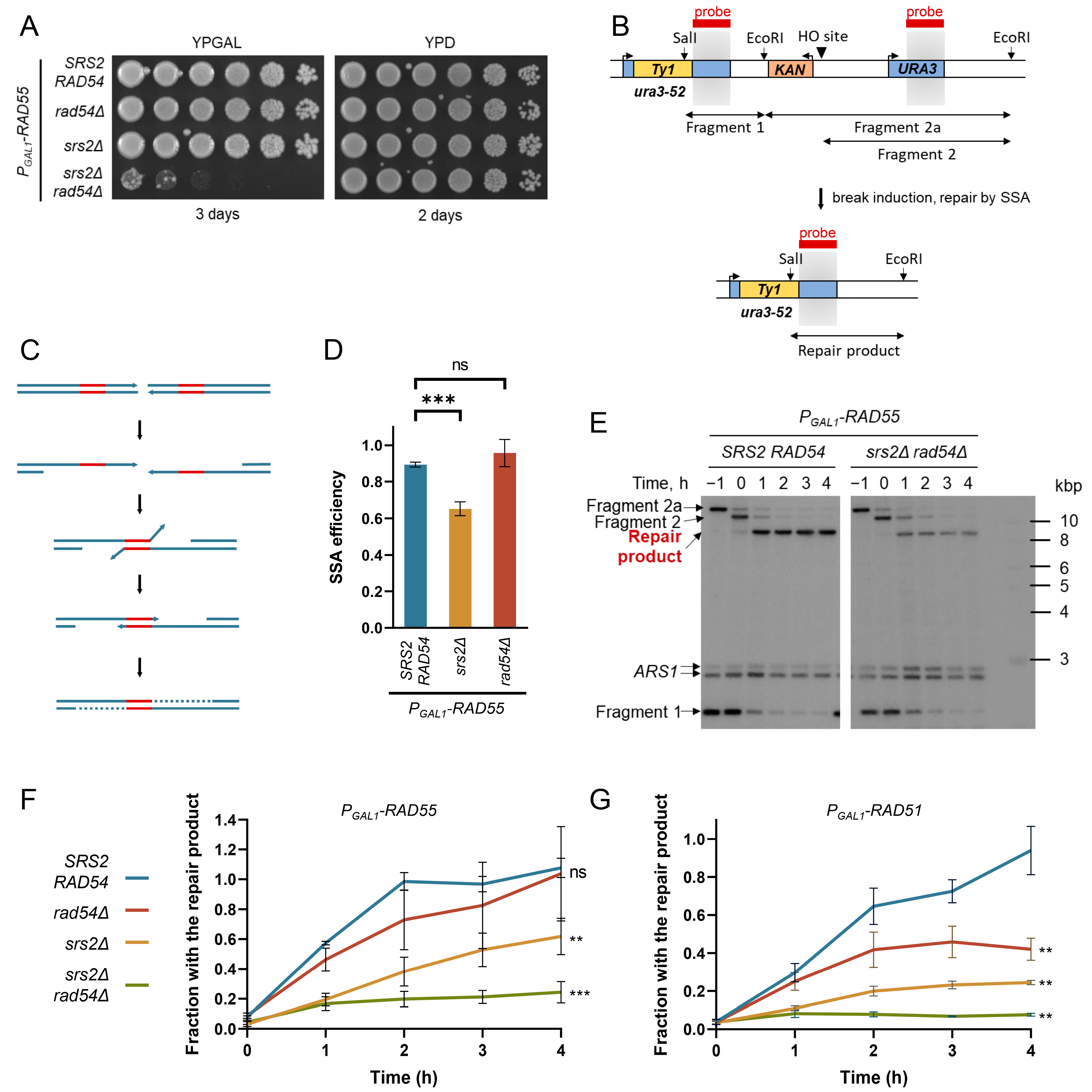
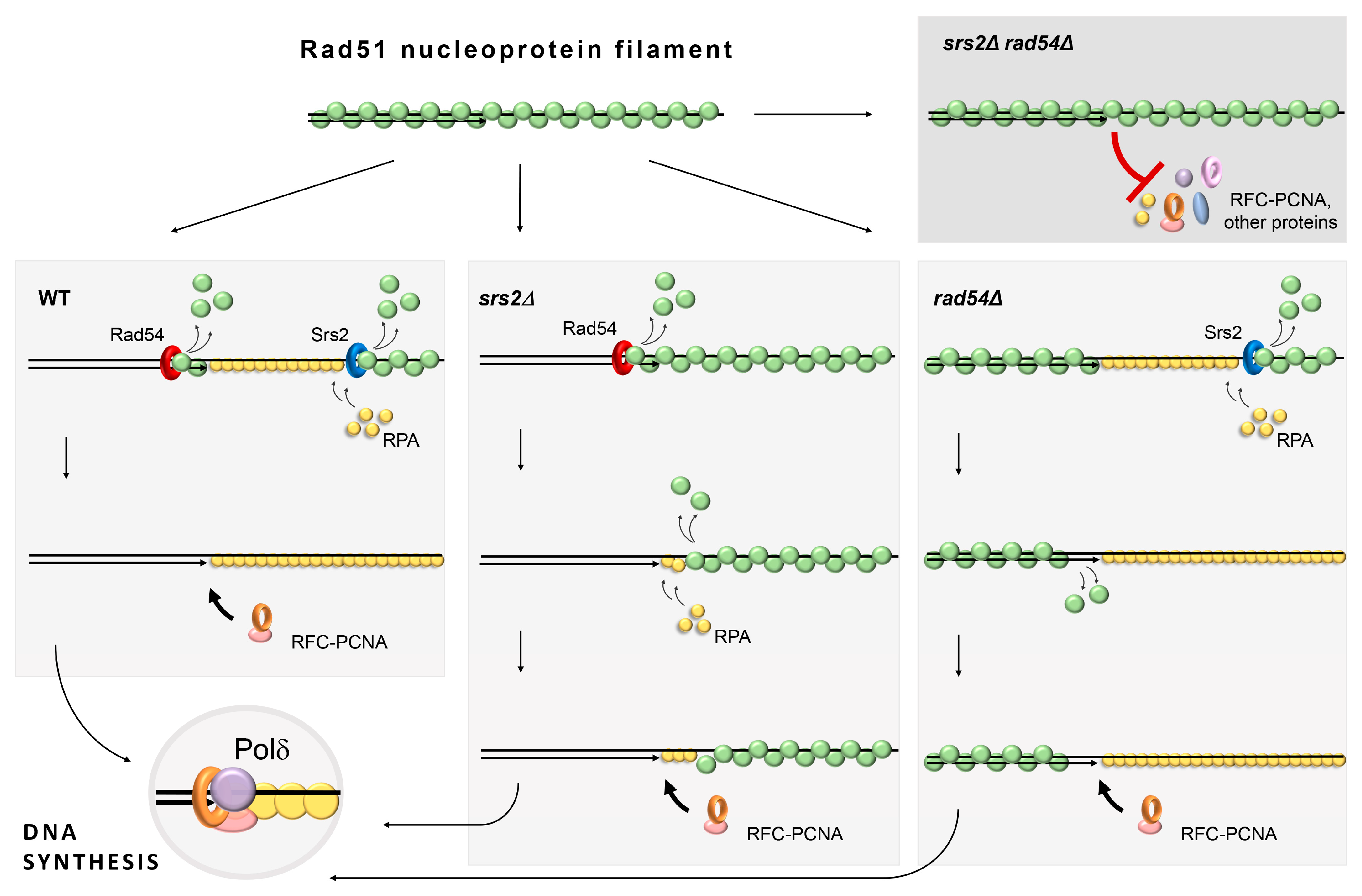
3.1. Rad54 and Srs2 Function to Provide Efficient DNA Synthesis during Repair
3.2. The Role of Rad54 in the Facilitation of DNA Re-Synthesis Can Be Revealed through Rad51 Overproduction
3.3. The C-Terminus of Srs2 Is Not Required for Its Role in Rad51 Nucleoprotein Filament Regulation in rad54Δ Cells
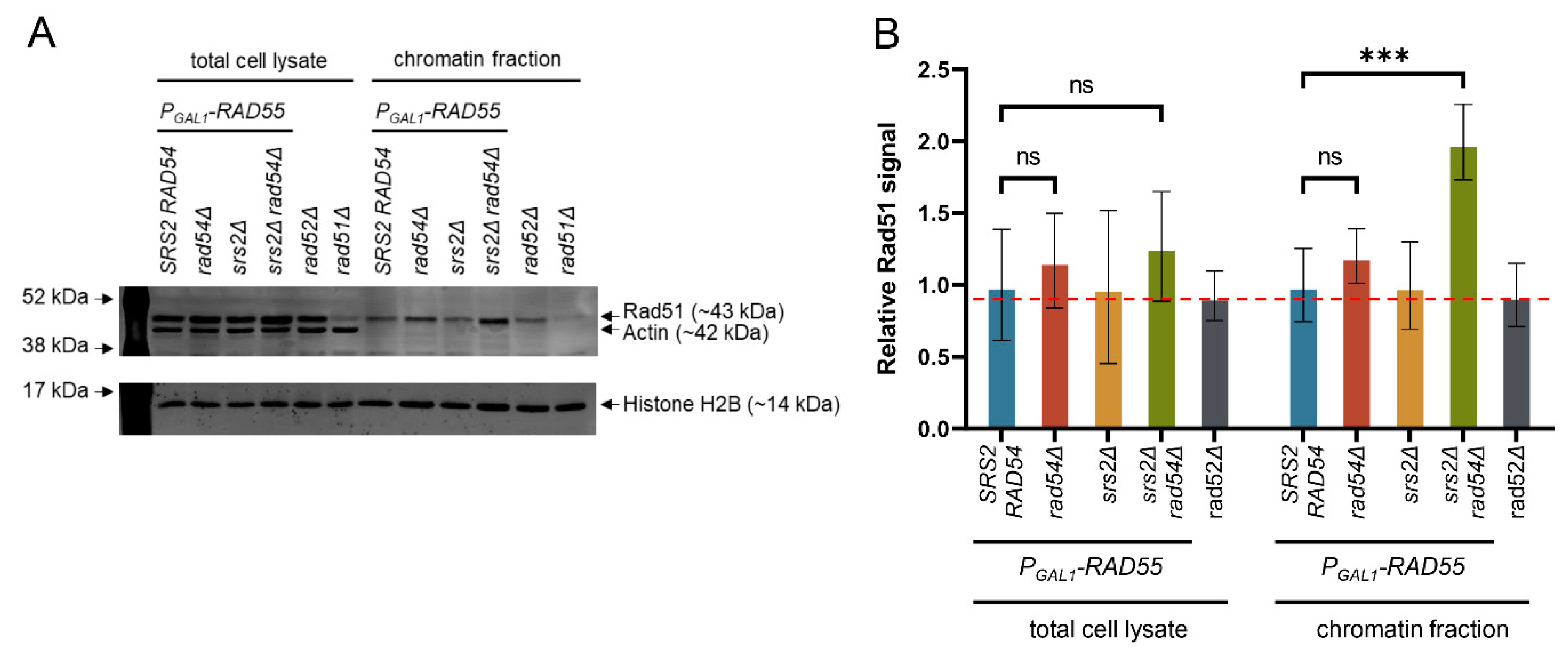
3.4. Chromatin-Bound Rad51 Accumulates in the Absence of Srs2 and Rad54
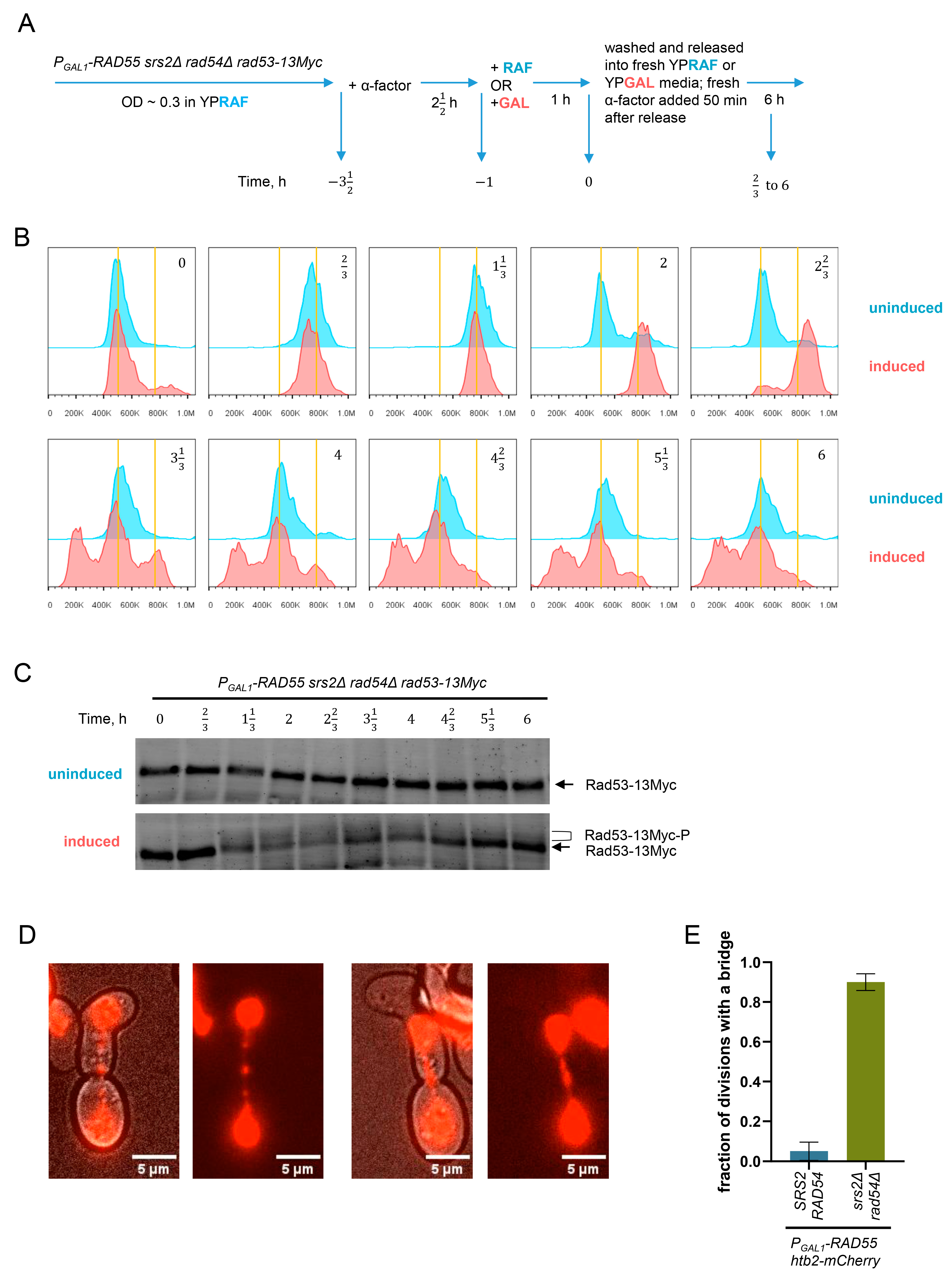
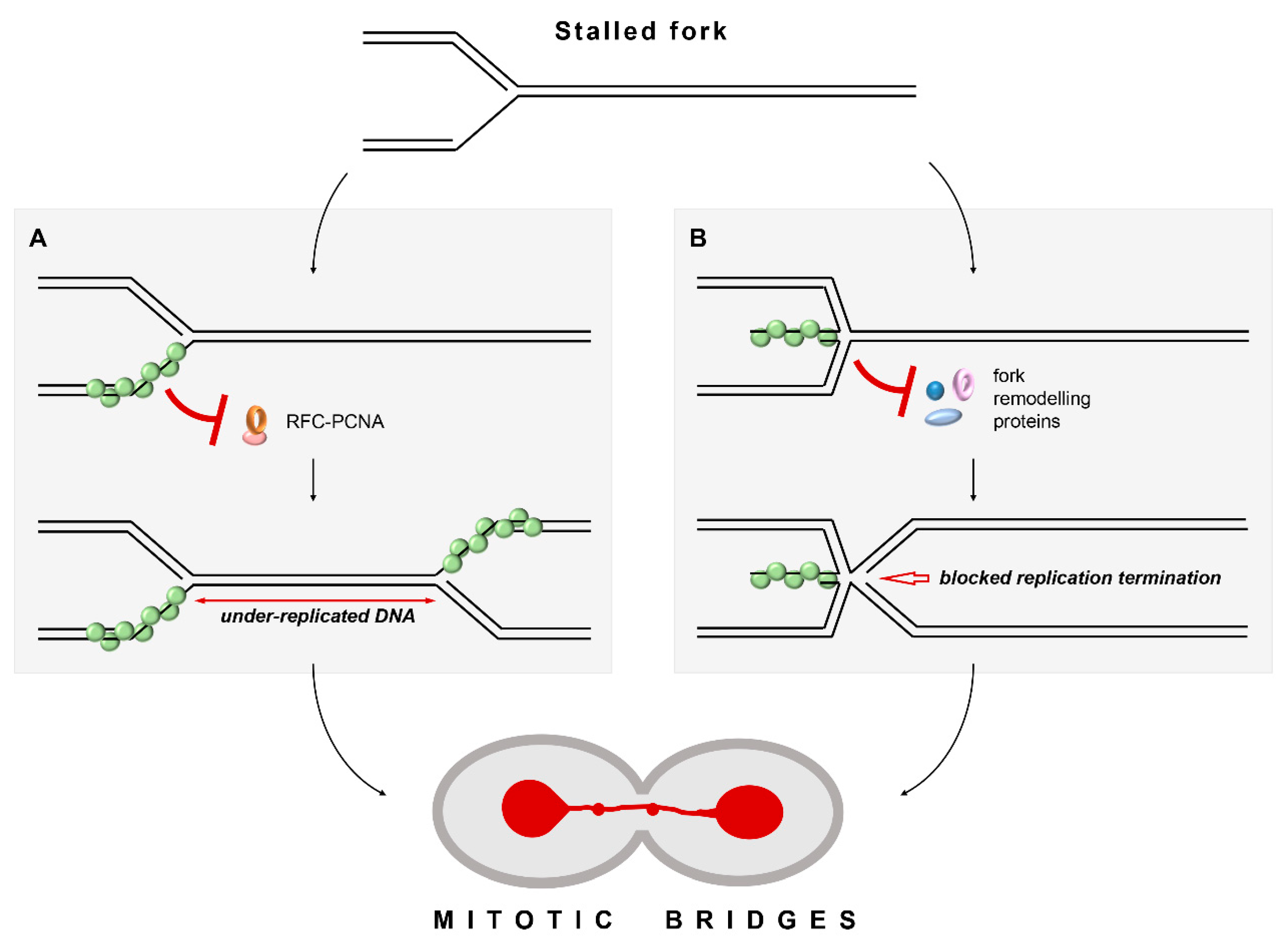
3.5. Unregulated Rad51 Filaments Induce Cell Cycle Arrest, Mitotic Bridges and Aberrant Mitoses
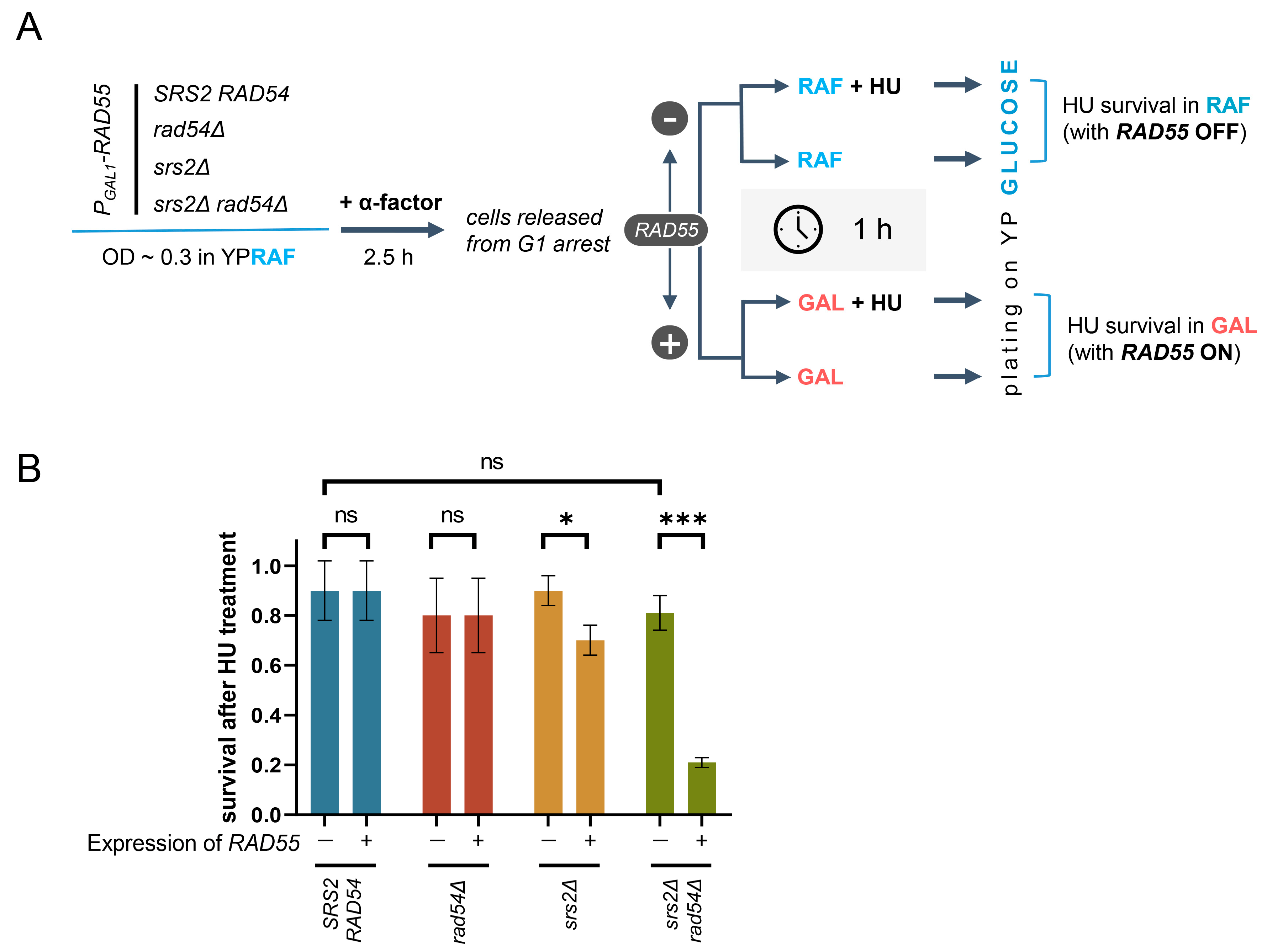
3.6. Unregulated Rad51 Nucleoprotein Filaments Hinder Cell Recovery from Replication Stress
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andriuskevicius, T.; Kotenko, O.; Makovets, S. Putting together and taking apart: Assembly and disassembly of the Rad51 nucleoprotein filament in DNA repair and genome stability. Cell Stress 2018, 2, 96–112. [Google Scholar] [CrossRef]
- Renkawitz, J.; Lademann, C.A.; Kalocsay, M.; Jentsch, S. Monitoring homology search during DNA double-strand break repair in vivo. Mol. Cell 2013, 50, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Crickard, J.B.; Moevus, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Rad54 Drives ATP Hydrolysis-Dependent DNA Sequence Alignment during Homologous Recombination. Cell 2020, 181, 1380–1394.e1318. [Google Scholar] [CrossRef] [PubMed]
- Game, J.C.; Mortimer, R.K. A genetic study of x-ray sensitive mutants in yeast. Mutat. Res. 1974, 24, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.W. Responses of radiation-sensitive mutants of Saccharomyces cerevisiae to lethal effects of bleomycin. Mutat. Res. 1978, 51, 165–180. [Google Scholar] [CrossRef]
- Abe, H.; Wada, M.; Kohno, K.; Kuwano, M. Altered drug sensitivities to anticancer agents in radiation-sensitive DNA repair deficient yeast mutants. Anticancer Res. 1994, 14, 1807–1810. [Google Scholar]
- Donovan, J.W.; Milne, G.T.; Weaver, D.T. Homotypic and heterotypic protein associations control Rad51 function in double-strand break repair. Genes Dev. 1994, 8, 2552–2562. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Techer, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e867. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Wassing, I.E.; Esashi, F. RAD51: Beyond the break. Semin. Cell Dev. Biol. 2021, 113, 38–46. [Google Scholar] [CrossRef]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-strand break repair in the absence of RAD51 in yeast: A possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.S.; Hasty, P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol. Cell Biol. 1996, 16, 7133–7143. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, E.; Sasaki, M.S.; Buerstedde, J.M.; Bezzubova, O.; Shinohara, A.; Ogawa, H.; Takata, M.; Yamaguchi-Iwai, Y.; Takeda, S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998, 17, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell. Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Sannino, V.; de Antoni, A.; Techer, H.; Baldi, G.; Costanzo, V. Moonlighting at replication forks—A new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017, 591, 1083–1100. [Google Scholar] [CrossRef]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef]
- Sung, P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem. 1997, 272, 28194–28197. [Google Scholar] [CrossRef]
- Hays, S.L.; Firmenich, A.A.; Massey, P.; Banerjee, R.; Berg, P. Studies of the interaction between Rad52 protein and the yeast single-stranded DNA binding protein RPA. Mol. Cell. Biol. 1998, 18, 4400–4406. [Google Scholar] [CrossRef]
- Song, B.; Sung, P. Functional interactions among yeast Rad51 recombinase, Rad52 mediator, and replication protein A in DNA strand exchange. J. Biol. Chem. 2000, 275, 15895–15904. [Google Scholar] [CrossRef]
- Shinohara, A.; Ogawa, T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature 1998, 391, 404–407. [Google Scholar] [CrossRef]
- New, J.H.; Sugiyama, T.; Zaitseva, E.; Kowalczykowski, S.C. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature 1998, 391, 407–410. [Google Scholar] [CrossRef]
- Milne, G.T.; Weaver, D.T. Dominant negative alleles of RAD52 reveal a DNA repair/recombination complex including Rad51 and Rad52. Genes Dev. 1993, 7, 1755–1765. [Google Scholar] [CrossRef]
- Sung, P. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev. 1997, 11, 1111–1121. [Google Scholar] [CrossRef]
- Roy, U.; Kwon, Y.; Marie, L.; Symington, L.; Sung, P.; Lisby, M.; Greene, E.C. The Rad51 paralog complex Rad55-Rad57 acts as a molecular chaperone during homologous recombination. Mol. Cell 2021, 81, 1043–1057.e1048. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, N.; Wang, X.; Haber, J.E. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell 2003, 12, 209–219. [Google Scholar] [CrossRef]
- Hays, S.L.; Firmenich, A.A.; Berg, P. Complex formation in yeast double-strand break repair: Participation of Rad51, Rad52, Rad55, and Rad57 proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 6925–6929. [Google Scholar] [CrossRef]
- Gasior, S.L.; Wong, A.K.; Kora, Y.; Shinohara, A.; Bishop, D.K. Rad52 associates with RPA and functions with rad55 and rad57 to assemble meiotic recombination complexes. Genes Dev. 1998, 12, 2208–2221. [Google Scholar] [CrossRef]
- Mazin, A.V.; Alexeev, A.A.; Kowalczykowski, S.C. A novel function of Rad54 protein. Stabilization of the Rad51 nucleoprotein filament. J. Biol. Chem. 2003, 278, 14029–14036. [Google Scholar] [CrossRef] [PubMed]
- Wolner, B.; van Komen, S.; Sung, P.; Peterson, C.L. Recruitment of the recombinational repair machinery to a DNA double-strand break in yeast. Mol. Cell 2003, 12, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Gasior, S.L.; Bishop, D.K.; Shinohara, A. Tid1/Rdh54 promotes colocalization of rad51 and dmc1 during meiotic recombination. Proc. Natl. Acad. Sci. USA 2000, 97, 10814–10819. [Google Scholar] [CrossRef]
- Agarwal, S.; van Cappellen, W.A.; Guenole, A.; Eppink, B.; Linsen, S.E.; Meijering, E.; Houtsmuller, A.; Kanaar, R.; Essers, J. ATP-dependent and independent functions of Rad54 in genome maintenance. J. Cell Biol. 2011, 192, 735–750. [Google Scholar] [CrossRef]
- van Veelen, L.R.; Essers, J.; van de Rakt, M.W.; Odijk, H.; Pastink, A.; Zdzienicka, M.Z.; Paulusma, C.C.; Kanaar, R. Ionizing radiation-induced foci formation of mammalian Rad51 and Rad54 depends on the Rad51 paralogs, but not on Rad52. Mutat. Res. 2005, 574, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.L.; Essers, J.; Citterio, E.; Swagemakers, S.M.; de Wit, J.; Benson, F.E.; Hoeijmakers, J.H.; Kanaar, R. Mouse Rad54 affects DNA conformation and DNA-damage-induced Rad51 foci formation. Curr. Biol. 1999, 9, 325–328. [Google Scholar] [CrossRef]
- Tavares, E.M.; Wright, W.D.; Heyer, W.D.; Le Cam, E.; Dupaigne, P. In vitro role of Rad54 in Rad51-ssDNA filament-dependent homology search and synaptic complexes formation. Nat. Commun. 2019, 10, 4058. [Google Scholar] [CrossRef] [PubMed]
- Petukhova, G.; Van Komen, S.; Vergano, S.; Klein, H.; Sung, P. Yeast Rad54 promotes Rad51-dependent homologous DNA pairing via ATP hydrolysis-driven change in DNA double helix conformation. J. Biol. Chem. 1999, 274, 29453–29462. [Google Scholar] [CrossRef] [PubMed]
- Ristic, D.; Wyman, C.; Paulusma, C.; Kanaar, R. The architecture of the human Rad54-DNA complex provides evidence for protein translocation along DNA. Proc. Natl. Acad. Sci. USA 2001, 98, 8454–8460. [Google Scholar] [CrossRef] [PubMed]
- Van Komen, S.; Petukhova, G.; Sigurdsson, S.; Stratton, S.; Sung, P. Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol. Cell 2000, 6, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Li, X.; Rolfsmeier, M.; Zhang, X.P. Rad54: The Swiss Army knife of homologous recombination? Nucleic Acids Res. 2006, 34, 4115–4125. [Google Scholar] [CrossRef] [PubMed]
- Marini, V.; Krejci, L. Srs2: The “Odd-Job Man” in DNA repair. DNA Repair. 2010, 9, 268–275. [Google Scholar] [CrossRef]
- Fabre, F.; Chan, A.; Heyer, W.D.; Gangloff, S. Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. USA 2002, 99, 16887–16892. [Google Scholar] [CrossRef] [PubMed]
- Antony, E.; Tomko, E.J.; Xiao, Q.; Krejci, L.; Lohman, T.M.; Ellenberger, T. Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol. Cell 2009, 35, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Veaute, X.; Jeusset, J.; Soustelle, C.; Kowalczykowski, S.C.; Le Cam, E.; Fabre, F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 2003, 423, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Van Komen, S.; Li, Y.; Villemain, J.; Reddy, M.S.; Klein, H.; Ellenberger, T.; Sung, P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 2003, 423, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Kaniecki, K.; De Tullio, L.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Dissociation of Rad51 Presynaptic Complexes and Heteroduplex DNA Joints by Tandem Assemblies of Srs2. Cell Rep. 2017, 21, 3166–3177. [Google Scholar] [CrossRef]
- Vasianovich, Y.; Altmannova, V.; Kotenko, O.; Newton, M.D.; Krejci, L.; Makovets, S. Unloading of homologous recombination factors is required for restoring double-stranded DNA at damage repair loci. EMBO J. 2017, 36, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Holzschu, D.L.; Sugiyama, T. PCNA is efficiently loaded on the DNA recombination intermediate to modulate polymerase delta, eta, and zeta activities. Proc. Natl. Acad. Sci. USA 2013, 110, 7672–7677. [Google Scholar] [CrossRef]
- Cai, J.; Uhlmann, F.; Gibbs, E.; Flores-Rozas, H.; Lee, C.G.; Phillips, B.; Finkelstein, J.; Yao, N.; O’Donnell, M.; Hurwitz, J. Reconstitution of human replication factor C from its five subunits in baculovirus-infected insect cells. Proc. Natl. Acad. Sci. USA 1996, 93, 12896–12901. [Google Scholar] [CrossRef]
- Waga, S.; Stillman, B. Cyclin-dependent kinase inhibitor p21 modulates the DNA primer-template recognition complex. Mol. Cell Biol. 1998, 18, 4177–4187. [Google Scholar] [CrossRef]
- Yuzhakov, A.; Kelman, Z.; Hurwitz, J.; O’Donnell, M. Multiple competition reactions for RPA order the assembly of the DNA polymerase delta holoenzyme. EMBO J. 1999, 18, 6189–6199. [Google Scholar] [CrossRef]
- Tsurimoto, T.; Stillman, B. Functions of replication factor C and proliferating-cell nuclear antigen: Functional similarity of DNA polymerase accessory proteins from human cells and bacteriophage T4. Proc. Natl. Acad. Sci. USA 1990, 87, 1023–1027. [Google Scholar] [CrossRef]
- Hingorani, M.M.; Coman, M.M. On the specificity of interaction between the Saccharomyces cerevisiae clamp loader replication factor C and primed DNA templates during DNA replication. J. Biol. Chem. 2002, 277, 47213–47224. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.M.; Gildenberg, M.S.; Washington, M.T. The Many Roles of PCNA in Eukaryotic DNA Replication. Enzymes 2016, 39, 231–254. [Google Scholar] [CrossRef] [PubMed]
- Mazin, A.V.; Zaitseva, E.; Sung, P.; Kowalczykowski, S.C. Tailed duplex DNA is the preferred substrate for Rad51 protein-mediated homologous pairing. EMBO J. 2000, 19, 1148–1156. [Google Scholar] [CrossRef]
- Ramos, F.; Duran, L.; Sanchez, M.; Campos, A.; Hernandez-Villamor, D.; Antequera, F.; Clemente-Blanco, A. Genome-wide sequencing analysis of Sgs1, Exo1, Rad51, and Srs2 in DNA repair by homologous recombination. Cell Rep. 2022, 38, 110201. [Google Scholar] [CrossRef] [PubMed]
- Solinger, J.A.; Kiianitsa, K.; Heyer, W.D. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol. Cell 2002, 10, 1175–1188. [Google Scholar] [CrossRef]
- Spies, J.; Waizenegger, A.; Barton, O.; Surder, M.; Wright, W.D.; Heyer, W.D.; Lobrich, M. Nek1 Regulates Rad54 to Orchestrate Homologous Recombination and Replication Fork Stability. Mol. Cell 2016, 62, 903–917. [Google Scholar] [CrossRef]
- Lengert, N.; Spies, J.; Drossel, B. Rad54 Phosphorylation Promotes Homologous Recombination by Balancing Rad54 Mobility and DNA Binding. Biophys. J. 2019, 116, 1406–1419. [Google Scholar] [CrossRef]
- Shah, P.P.; Zheng, X.; Epshtein, A.; Carey, J.N.; Bishop, D.K.; Klein, H.L. Swi2/Snf2-related translocases prevent accumulation of toxic Rad51 complexes during mitotic growth. Mol. Cell 2010, 39, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Dusad, K.; Wright, W.D.; Grubb, J.; Budke, B.; Heyer, W.D.; Connell, P.P.; Weichselbaum, R.R.; Bishop, D.K. RAD54 family translocases counter genotoxic effects of RAD51 in human tumor cells. Nucleic Acids Res. 2015, 43, 3180–3196. [Google Scholar] [CrossRef]
- Klein, H.L. Mutations in recombinational repair and in checkpoint control genes suppress the lethal combination of srs2Delta with other DNA repair genes in Saccharomyces cerevisiae. Genetics 2001, 157, 557–565. [Google Scholar] [CrossRef]
- Schild, D. Suppression of a new allele of the yeast RAD52 gene by overexpression of RAD51, mutations in srs2 and ccr4, or mating-type heterozygosity. Genetics 1995, 140, 115–127. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A. ASTQ/A Short-Reads Pre-Processing Tools. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 15 February 2023).
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Schuster-Boeckler, B. TrimGalore. Available online: https://zenodo.org/record/5127899#.Y-PVxHbP238 (accessed on 15 February 2023).
- seqtk. Available online: https://bio.tools/seqtk (accessed on 15 February 2023).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Sasanuma, H.; Furihata, Y.; Shinohara, M.; Shinohara, A. Remodeling of the Rad51 DNA strand-exchange protein by the Srs2 helicase. Genetics 2013, 194, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef]
- Sugawara, N.; Ira, G.; Haber, J.E. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol. Cell Biol. 2000, 20, 5300–5309. [Google Scholar] [CrossRef]
- Pohl, T.J.; Nickoloff, J.A. Rad51-independent interchromosomal double-strand break repair by gene conversion requires Rad52 but not Rad55, Rad57, or Dmc1. Mol. Cell Biol. 2008, 28, 897–906. [Google Scholar] [CrossRef]
- Fortin, G.S.; Symington, L.S. Mutations in yeast Rad51 that partially bypass the requirement for Rad55 and Rad57 in DNA repair by increasing the stability of Rad51-DNA complexes. EMBO J. 2002, 21, 3160–3170. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kantake, N.; Sugiyama, T.; Kowalczykowski, S.C. Rad51 protein controls Rad52-mediated DNA annealing. J. Biol. Chem. 2008, 283, 14883–14892. [Google Scholar] [CrossRef] [PubMed]
- Pfander, B.; Moldovan, G.L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef]
- Kolesar, P.; Altmannova, V.; Silva, S.; Lisby, M.; Krejci, L. Pro-recombination Role of Srs2 Protein Requires SUMO (Small Ubiquitin-like Modifier) but Is Independent of PCNA (Proliferating Cell Nuclear Antigen) Interaction. J. Biol. Chem. 2016, 291, 7594–7607. [Google Scholar] [CrossRef] [PubMed]
- Le Breton, C.; Dupaigne, P.; Robert, T.; Le Cam, E.; Gangloff, S.; Fabre, F.; Veaute, X. Srs2 removes deadly recombination intermediates independently of its interaction with SUMO-modified PCNA. Nucleic Acids Res. 2008, 36, 4964–4974. [Google Scholar] [CrossRef] [PubMed]
- Coic, E.; Feldman, T.; Landman, A.S.; Haber, J.E. Mechanisms of Rad52-independent spontaneous and UV-induced mitotic recombination in Saccharomyces cerevisiae. Genetics 2008, 179, 199–211. [Google Scholar] [CrossRef]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef]
- Claussin, C.; Porubsky, D.; Spierings, D.C.; Halsema, N.; Rentas, S.; Guryev, V.; Lansdorp, P.M.; Chang, M. Genome-wide mapping of sister chromatid exchange events in single yeast cells using Strand-seq. Elife 2017, 6, e30560. [Google Scholar] [CrossRef]
- Donaldson, A.D.; Fangman, W.L.; Brewer, B.J. Cdc7 is required throughout the yeast S phase to activate replication origins. Genes Dev. 1998, 12, 491–501. [Google Scholar] [CrossRef]
- Vogelauer, M.; Rubbi, L.; Lucas, I.; Brewer, B.J.; Grunstein, M. Histone acetylation regulates the time of replication origin firing. Mol. Cell 2002, 10, 1223–1233. [Google Scholar] [CrossRef]
- Osborn, A.J.; Elledge, S.J. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003, 17, 1755–1767. [Google Scholar] [CrossRef]
- Amin, A.; Wu, R.; Cheung, M.H.; Scott, J.F.; Wang, Z.; Zhou, Z.; Liu, C.; Zhu, G.; Wong, C.K.; Yu, Z.; et al. An Essential and Cell-Cycle-Dependent ORC Dimerization Cycle Regulates Eukaryotic Chromosomal DNA Replication. Cell Rep. 2020, 30, 3323–3338.e3326. [Google Scholar] [CrossRef]
- Finardi, A.; Massari, L.F.; Visintin, R. Anaphase Bridges: Not All Natural Fibers Are Healthy. Genes 2020, 11, 902. [Google Scholar] [CrossRef]
- Mankouri, H.W.; Ngo, H.P.; Hickson, I.D. Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1-Rmi1-Top3. Mol. Biol. Cell 2007, 18, 4062–4073. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, T.; Maier, M.; Missarova, A.; Ziegler-Birling, C.; Dam, M.; Gomar-Alba, M.; Carey, L.B.; Mendoza, M. Budding yeast complete DNA synthesis after chromosome segregation begins. Nat. Commun. 2020, 11, 2267. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Carrington, J.T.; Albergante, L.; Al Mamun, M.; Haagensen, E.J.; Komseli, E.S.; Gorgoulis, V.G.; Newman, T.J.; Blow, J.J. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5757–E5764. [Google Scholar] [CrossRef]
- Ait Saada, A.; Teixeira-Silva, A.; Iraqui, I.; Costes, A.; Hardy, J.; Paoletti, G.; Freon, K.; Lambert, S.A.E. Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol. Cell 2017, 66, 398–410.e394. [Google Scholar] [CrossRef]
- Solinger, J.A.; Heyer, W.D. Rad54 protein stimulates the postsynaptic phase of Rad51 protein-mediated DNA strand exchange. Proc. Natl. Acad. Sci. USA 2001, 98, 8447–8453. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, T.; Seidel, R.; Modesti, M.; Kanaar, R.; Wyman, C.; Dekker, C. Real-time assembly and disassembly of human RAD51 filaments on individual DNA molecules. Nucleic Acids Res. 2007, 35, 5646–5657. [Google Scholar] [CrossRef]
- Meir, A.; Crickard, J.B.; Kwon, Y.; Sung, P.; Greene, E.C. Rad54 and Rdh54 prevent Srs2-mediated disruption of Rad51 presynaptic filaments. Proc. Natl. Acad. Sci. USA 2022, 119, e2113871119. [Google Scholar] [CrossRef]
- Kanoh, Y.; Tamai, K.; Shirahige, K. Different requirements for the association of ATR-ATRIP and 9-1-1 to the stalled replication forks. Gene 2006, 377, 88–95. [Google Scholar] [CrossRef]
- Deshpande, I.; Seeber, A.; Shimada, K.; Keusch, J.J.; Gut, H.; Gasser, S.M. Structural Basis of Mec1-Ddc2-RPA Assembly and Activation on Single-Stranded DNA at Sites of Damage. Mol. Cell 2017, 68, 431–445.e435. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Liu, D.; Elledge, S.J. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 13827–13832. [Google Scholar] [CrossRef] [PubMed]
- Parplys, A.C.; Seelbach, J.I.; Becker, S.; Behr, M.; Wrona, A.; Jend, C.; Mansour, W.Y.; Joosse, S.A.; Stuerzbecher, H.W.; Pospiech, H.; et al. High levels of RAD51 perturb DNA replication elongation and cause unscheduled origin firing due to impaired CHK1 activation. Cell Cycle 2015, 14, 3190–3202. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Luis, J.; Machin, F. Fanconi Anaemia-Like Mph1 Helicase Backs up Rad54 and Rad5 to Circumvent Replication Stress-Driven Chromosome Bridges. Genes 2018, 9, 558. [Google Scholar] [CrossRef]
- Urulangodi, M.; Sebesta, M.; Menolfi, D.; Szakal, B.; Sollier, J.; Sisakova, A.; Krejci, L.; Branzei, D. Local regulation of the Srs2 helicase by the SUMO-like domain protein Esc2 promotes recombination at sites of stalled replication. Genes Dev. 2015, 29, 2067–2080. [Google Scholar] [CrossRef]
- Carter, S.D.; Vigasova, D.; Chen, J.; Chovanec, M.; Astrom, S.U. Nej1 recruits the Srs2 helicase to DNA double-strand breaks and supports repair by a single-strand annealing-like mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 12037–12042. [Google Scholar] [CrossRef]
- Chen, X.; Tomkinson, A.E. Yeast Nej1 is a key participant in the initial end binding and final ligation steps of nonhomologous end joining. J. Biol. Chem. 2011, 286, 4931–4940. [Google Scholar] [CrossRef]
- Hu, J.; Sun, L.; Shen, F.; Chen, Y.; Hua, Y.; Liu, Y.; Zhang, M.; Hu, Y.; Wang, Q.; Xu, W.; et al. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 2012, 149, 1221–1232. [Google Scholar] [CrossRef]
- Teixeira-Silva, A.; Ait Saada, A.; Hardy, J.; Iraqui, I.; Nocente, M.C.; Freon, K.; Lambert, S.A.E. The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun. 2017, 8, 1982. [Google Scholar] [CrossRef]
- Tye, S.; Ronson, G.E.; Morris, J.R. A fork in the road: Where homologous recombination and stalled replication fork protection part ways. Semin. Cell Dev. Biol. 2021, 113, 14–26. [Google Scholar] [CrossRef]
- Bugreev, D.V.; Rossi, M.J.; Mazin, A.V. Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 2011, 39, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Daee, D.L.; Ferrari, E.; Longerich, S.; Zheng, X.F.; Xue, X.; Branzei, D.; Sung, P.; Myung, K. Rad5-dependent DNA repair functions of the Saccharomyces cerevisiae FANCM protein homolog Mph1. J. Biol. Chem. 2012, 287, 26563–26575. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Hyun, K.; Kim, J.; Hohng, S. ATP Binding to Rad5 Initiates Replication Fork Reversal by Inducing the Unwinding of the Leading Arm and the Formation of the Holliday Junction. Cell Rep. 2018, 23, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Marie, L.; Symington, L.S. Mechanism for inverted-repeat recombination induced by a replication fork barrier. Nat. Commun. 2022, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef]
- Mazin, A.V.; Mazina, O.M.; Bugreev, D.V.; Rossi, M.J. Rad54, the motor of homologous recombination. DNA Repair. 2010, 9, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Morati, F.; Modesti, M. Insights into the control of RAD51 nucleoprotein filament dynamics from single-molecule studies. Curr. Opin. Genet. Dev. 2021, 71, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Bugreev, D.V.; Yu, X.; Egelman, E.H.; Mazin, A.V. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 2007, 21, 3085–3094. [Google Scholar] [CrossRef]
- Simandlova, J.; Zagelbaum, J.; Payne, M.J.; Chu, W.K.; Shevelev, I.; Hanada, K.; Chatterjee, S.; Reid, D.A.; Liu, Y.; Janscak, P.; et al. FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J. Biol. Chem. 2013, 288, 34168–34180. [Google Scholar] [CrossRef]
- Hu, Y.; Raynard, S.; Sehorn, M.G.; Lu, X.; Bussen, W.; Zheng, L.; Stark, J.M.; Barnes, E.L.; Chi, P.; Janscak, P.; et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007, 21, 3073–3084. [Google Scholar] [CrossRef] [PubMed]
- Sommers, J.A.; Rawtani, N.; Gupta, R.; Bugreev, D.V.; Mazin, A.V.; Cantor, S.B.; Brosh, R.M., Jr. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J. Biol. Chem. 2009, 284, 7505–7517. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; Dejsuphong, D.; Petalcorin, M.I.; Hofmann, K.; Takeda, S.; Boulton, S.J.; D’Andrea, A.D. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol. Cell 2012, 45, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Logan, H.L.; Budke, B.; Wu, M.; Pawlowski, M.; Weichselbaum, R.R.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. The RAD51-stimulatory compound RS-1 can exploit the RAD51 overexpression that exists in cancer cells and tumors. Cancer Res. 2014, 74, 3546–3555. [Google Scholar] [CrossRef] [PubMed]
- Makovets, S.; Herskowitz, I.; Blackburn, E.H. Anatomy and Dynamics of DNA Replication Fork Movement in Yeast Telomeric Regions. Mol. Cell. Biol. 2004, 24, 4019–4031. [Google Scholar] [CrossRef] [PubMed]
- Makovets, S.; Blackburn, E.H. DNA damage signalling prevents deleterious telomere addition at DNA breaks. Nat. Cell Biol. 2009, 11, 1383–1386. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andriuskevicius, T.; Dubenko, A.; Makovets, S. The Inability to Disassemble Rad51 Nucleoprotein Filaments Leads to Aberrant Mitosis and Cell Death. Biomedicines 2023, 11, 1450. https://doi.org/10.3390/biomedicines11051450
Andriuskevicius T, Dubenko A, Makovets S. The Inability to Disassemble Rad51 Nucleoprotein Filaments Leads to Aberrant Mitosis and Cell Death. Biomedicines. 2023; 11(5):1450. https://doi.org/10.3390/biomedicines11051450
Chicago/Turabian StyleAndriuskevicius, Tadas, Anton Dubenko, and Svetlana Makovets. 2023. "The Inability to Disassemble Rad51 Nucleoprotein Filaments Leads to Aberrant Mitosis and Cell Death" Biomedicines 11, no. 5: 1450. https://doi.org/10.3390/biomedicines11051450
APA StyleAndriuskevicius, T., Dubenko, A., & Makovets, S. (2023). The Inability to Disassemble Rad51 Nucleoprotein Filaments Leads to Aberrant Mitosis and Cell Death. Biomedicines, 11(5), 1450. https://doi.org/10.3390/biomedicines11051450