
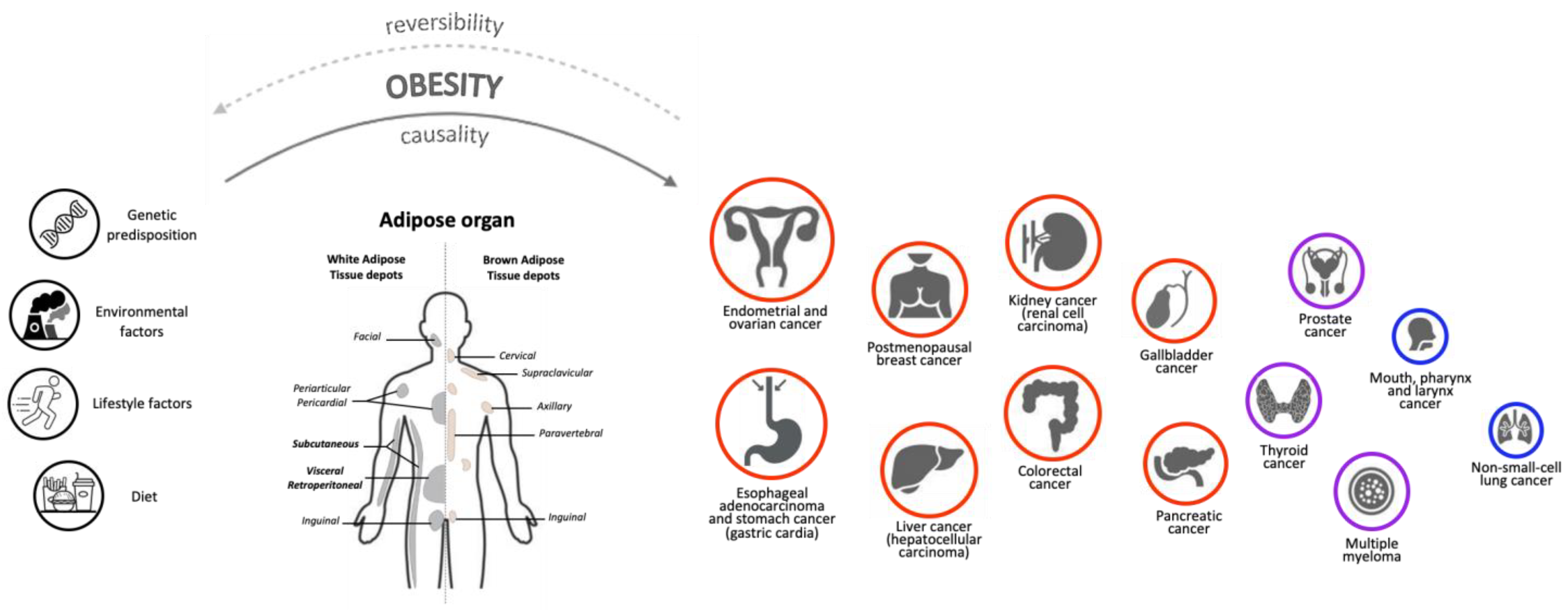
Obesity, the Adipose Organ and Cancer in Humans: Association or Causation?


Abstract
1. Causality Criteria in the Relationship between Obesity and Cancer
2. Obesity and Cancer Risk: Epidemiology and Controversial Evidence
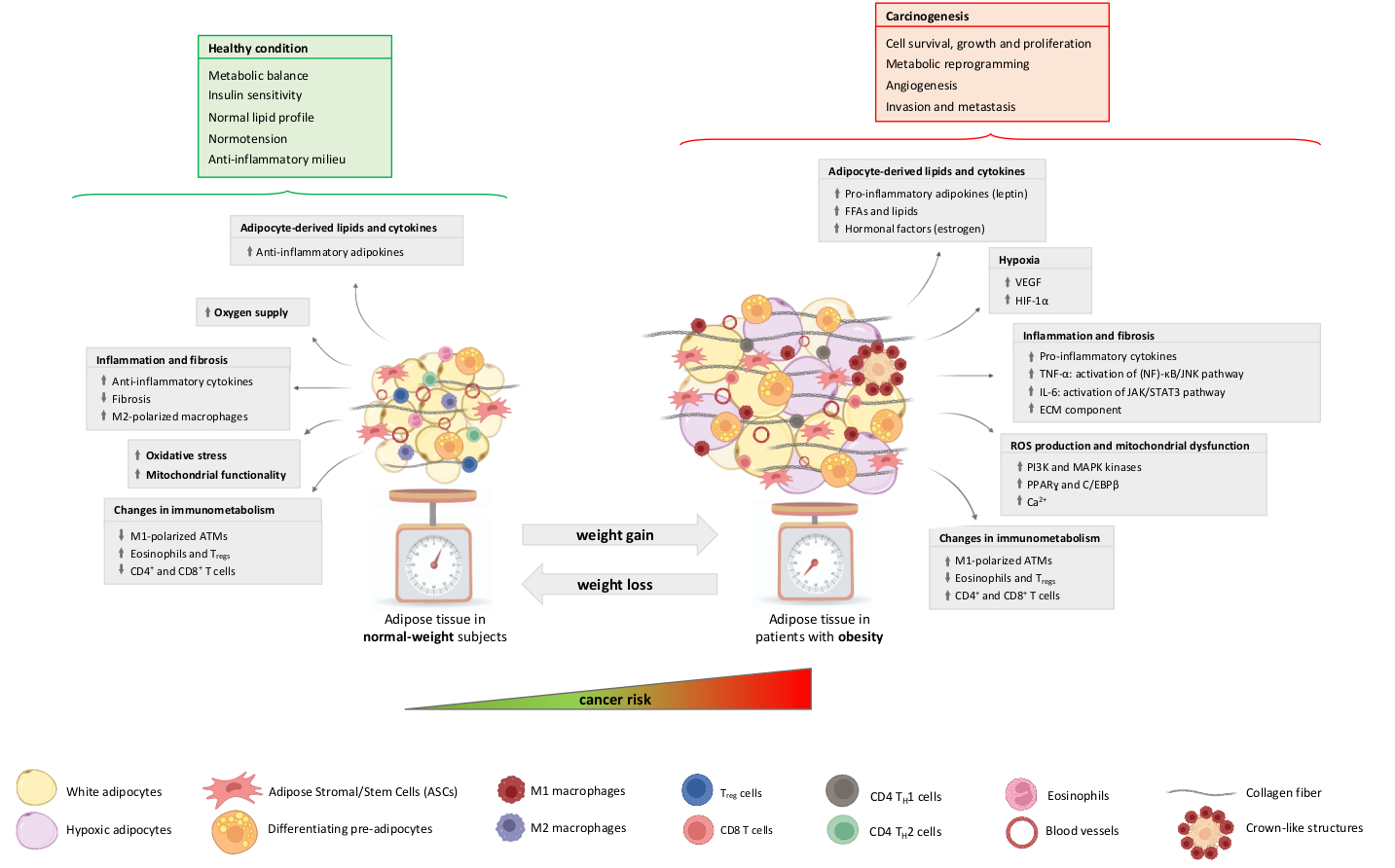
3. The Adipose Organ and Cancer: General Aspects and Specific Mechanisms
3.1. Hypoxia
3.2. Inflammation, Fibrosis and Senescence
3.3. ROS Production and Mitochondrial Alterations
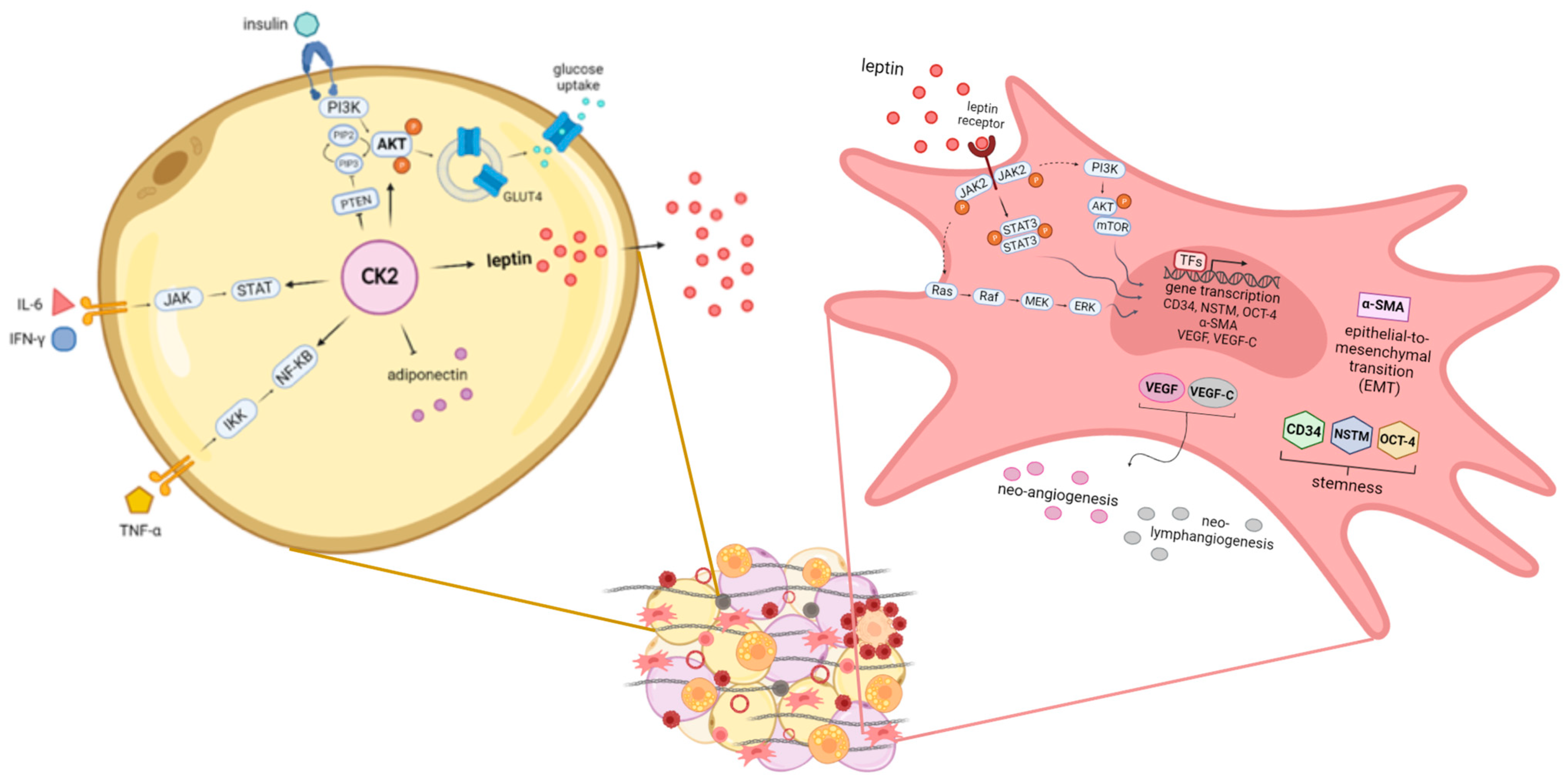
3.4. Altered Secretion of Adipocyte-Derived Factors
3.5. The Protein Kinase CK2
3.6. The Adipose Tissue Stromal Stem Cells
4. The Involvement of Microbiota Dysregulation
5. Circadian Rhythms in Obesity and Cancer Progression
6. Effects of Adipose Tissue on Cancer Cachexia
7. Effects of Obesity Treatment on Cancer Incidence and Prevention
8. Concluding Remarks: Is There Evidence of a Cause–Effect Relationship?
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
References
- Evans, A.S. Causation and disease: The Henle-Koch postulates revisited. Yale J. Biol. Med. 1976, 49, 175–195. [Google Scholar]
- Hill, A.B. The Environment and Disease: Association or Causation? Proc. R. Soc. Med. 1965, 58, 295–300. [Google Scholar] [CrossRef]
- Glass, T.A.; Goodman, S.N.; Hernán, M.A.; Samet, J.M. Causal Inference in Public Health. Annu. Rev. Public Health 2013, 34, 61–75. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Fact Sheets, Obesity and Overweight. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 24 January 2023).
- Bray, G.A.; Kim, K.K.; Wilding, J.P.H.; World Obesity Federation. Obesity: A chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes. Rev. 2017, 18, 715–723. [Google Scholar] [CrossRef]
- Bays, H.; Abate, N.; Chandalia, M. Adiposopathy: Sick fat causes high blood sugar, high blood pressure and dyslipidemia. Futur. Cardiol. 2005, 1, 39–59. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef] [PubMed]
- A Hernán, M.; Taubman, S.L. Does obesity shorten life? The importance of well-defined interventions to answer causal questions. Int. J. Obes. 2008, 32 (Suppl. S3), S8–S14. [Google Scholar] [CrossRef]
- American Institute for Cancer Research (AICR)—The Continuous Update Project (CUP). Available online: https://www.aicr.org/research/the-continuous-update-project (accessed on 24 January 2023).
- Chan, D.S.M.; Abar, L.; Cariolou, M.; Nanu, N.; Greenwood, D.C.; Bandera, E.V.; McTiernan, A.; Norat, T. World Cancer Research Fund International: Continuous Update Project—Systematic literature review and meta-analysis of observational cohort studies on physical activity, sedentary behavior, adiposity, and weight change and breast cancer risk. Cancer Causes Control 2019, 30, 1183–1200. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Cagol, M.; Bettini, S.; Alfieri, R.; Carraro, A.; Cavallin, F.; Trevellin, E.; Saadeh, L.M.; Ruol, A.; Vettor, R.; et al. Overweight Patients Operated on for Cancer of the Esophagus Survive Longer than Normal-Weight Patients. J. Gastrointest. Surg. 2012, 17, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Furberg, H.; Kuo, F.; Vuong, L.; Ged, Y.; Patil, S.; Ostrovnaya, I.; Petruzella, S.; Reising, A.; Patel, P.; et al. Transcriptomic signatures related to the obesity paradox in patients with clear cell renal cell carcinoma: A cohort study. Lancet Oncol. 2020, 21, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, C.; Wu, G.; Yang, W.; Wang, X.; Duan, L.; Niu, L.; Chen, J.; Zhang, Y.; Zhou, W.; et al. The obesity paradox in patients with colorectal cancer: A systematic review and meta-analysis. Nutr. Rev. 2022, 80, 1755–1768. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.H.; Kim, N.; Jang, J.; Choi, Y.; Park, J.; Ahn, S.; Yoon, H.; Shin, C.M.; Lee, D.H.; Oh, H.J.; et al. Impact of Body Mass Index on Survival Depending on Sex in 14,688 Patients with Gastric Cancer in a Tertiary Hospital in South Korea. Gut Liver 2022, 17, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lu, L.; Qu, C.; Gari, A.; Deng, F.; Cai, M.; Chen, W.; Zheng, L.; Chen, J. Body mass index, as a novel predictor of hepatocellular carcinoma patients treated with Anti-PD-1 immunotherapy. Front. Med. 2022, 9, 981001. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, O.; Ghosh, S.; Morrish, D. Impact of baseline body mass index on the outcomes of patients with neuroendocrine neoplasms. J. Endocrinol. Investig. 2022, 45, 1683–1688. [Google Scholar] [CrossRef]
- Takemura, K.; Yonekura, S.; Downey, L.E.; Evangelopoulos, D.; Heng, D.Y. Impact of Body Mass Index on Survival Outcomes of Patients with Metastatic Renal Cell Carcinoma in the Immuno-oncology Era: A Systematic Review and Meta-analysis. Eur. Urol. Open Sci. 2022, 39, 62–71. [Google Scholar] [CrossRef]
- Fuchs, J.; Schellerer, V.S.; Brunner, M.; I Geppert, C.; Grützmann, R.; Weber, K.; Merkel, S. The impact of body mass index on prognosis in patients with colon carcinoma. Int. J. Colorectal. Dis. 2022, 37, 1107–1117. [Google Scholar] [CrossRef]
- Lee, S.; Lee, D.H.; Lee, J.-H.; Shin, S.-J.; Lee, H.S.; Park, E.J.; Baik, S.H.; Lee, K.Y.; Kang, J. Association of Body Mass Index with Survival in Asian Patients with Colorectal Cancer. Cancer Res. Treat. 2022, 54, 860–872. [Google Scholar] [CrossRef]
- Petrelli, F.; Cortellini, A.; Indini, A.; Tomasello, G.; Ghidini, M.; Nigro, O.; Salati, M.; Dottorini, L.; Iaculli, A.; Varricchio, A.; et al. Association of Obesity with Survival Outcomes in Patients with Cancer: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e213520. [Google Scholar] [CrossRef]
- Pati, S.; Irfan, W.; Jameel, A.; Ahmed, S.; Shahid, R.K. Obesity and Cancer: A Current Overview of Epidemiology, Pathogenesis, Outcomes, and Management. Cancers 2023, 15, 485. [Google Scholar] [CrossRef]
- Lennon, H.; Sperrin, M.; Badrick, E.; Renehan, A.G. The Obesity Paradox in Cancer: A Review. Curr. Oncol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef]
- Park, Y.; Peterson, L.L.; Colditz, G.A. The Plausibility of Obesity Paradox in Cancer—Point. Cancer Res. 2018, 78, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Adinolfi, V.; Barucca, V.; Prinzi, N.; Renzelli, V.; Barrea, L.; Di Giacinto, P.; Ruggeri, R.M.; Sesti, F.; Arvat, E.; et al. Expected and paradoxical effects of obesity on cancer treatment response. Rev. Endocr. Metab. Disord. 2020, 22, 681–702. [Google Scholar] [CrossRef] [PubMed]
- Sperrin, M.; Candlish, J.; Badrick, E.; Renehan, A.; Buchan, I. Collider Bias Is Only a Partial Explanation for the Obesity Paradox. Epidemiology 2016, 27, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Doll, R.; Peto, R. The causes of cancer: Quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 1981, 66, 1191–1308. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- O’sullivan, J.; Lysaght, J.; Donohoe, C.L.; Reynolds, J.V. Obesity and gastrointestinal cancer: The interrelationship of adipose and tumour microenvironments. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 699–714. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef]
- Marzullo, P.; Bettini, S.; Menafra, D.; Aprano, S.; Muscogiuri, G.; Barrea, L.; Savastano, S.; Colao, A.; Magno, S.; Di Nisio, A.; et al. Spot-light on microbiota in obesity and cancer. Int. J. Obes. 2021, 45, 2291–2299. [Google Scholar] [CrossRef]
- Akinyemiju, T.; Moore, J.X.; Pisu, M.; Judd, S.E.; Goodman, M.; Shikany, J.M.; Howard, V.J.; Safford, M.; Gilchrist, S.C. A Prospective Study of Obesity, Metabolic Health, and Cancer Mortality. Obesity 2018, 26, 193–201. [Google Scholar] [CrossRef]
- Dibaba, D.T.; Judd, S.E.; Gilchrist, S.C.; Cushman, M.; Pisu, M.; Safford, M.; Akinyemiju, T. Association between obesity and biomarkers of inflammation and metabolism with cancer mortality in a prospective cohort study. Metabolism 2019, 94, 69–76. [Google Scholar] [CrossRef]
- Pan, K.; Nelson, R.A.; Wactawski-Wende, J.; Lee, D.J.; E Manson, J.; Aragaki, A.K.; E Mortimer, J.; Phillips, L.S.; Rohan, T.; Ho, G.Y.F.; et al. Insulin Resistance and Cancer-Specific and All-Cause Mortality in Postmenopausal Women: The Women’s Health Initiative. J. Natl. Cancer Inst. 2020, 112, 170–178. [Google Scholar] [CrossRef]
- Burgess, S.; Daniel, R.M.; Butterworth, A.S.; Thompson, S.G.; the EPIC-InterAct Consortium. Network Mendelian randomization: Using genetic variants as instrumental variables to investigate mediation in causal pathways. Int. J. Epidemiol. 2014, 44, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. “Mendelian randomization”: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.G.; Sanderson, E.; Elsworth, B.; Tilling, K.; Smith, G.D. Use of genetic variation to separate the effects of early and later life adiposity on disease risk: Mendelian randomisation study. BMJ 2020, 369, m1203. [Google Scholar] [CrossRef]
- Fang, Z.; Song, M.; Lee, D.H.; Giovannucci, E.L. The Role of Mendelian Randomization Studies in Deciphering the Effect of Obesity on Cancer. J. Natl. Cancer Inst. 2021, 114, 361–371. [Google Scholar] [CrossRef] [PubMed]
- World Cancer Research Fund/American Institute for Cancer Research: Continuous Update Project Report. Food: Nutrition, Physical Activity, and the Prevention of Endometrial Cancer. Available online: https://www.wcrf.org/diet-activity-and-cancer/cancer-types/endometrial-cancer/ (accessed on 24 January 2023).
- Reeves, G.K.; Pirie, K.; Beral, V.; Green, J.; Spencer, E.; Bull, D.; Million Women Study Collaboration. Cancer incidence and mortality in relation to body mass index in the Million Women Study: Cohort study. BMJ 2007, 335, 1134. [Google Scholar] [CrossRef]
- Von Gruenigen, V.E.; Tian, C.; Frasure, H.; Waggoner, S.; Keys, H.; Barakat, R.R. Treatment effects, disease recurrence, and survival in obese women with early endometrial carcinoma: A Gynecologic Oncology Group study. Cancer 2006, 107, 2786–2791. [Google Scholar] [CrossRef]
- World Cancer Research Fund/American Institute for Cancer Research: Continuous Update Project Report: Food, Nutrition, Physical Activity, and the Prevention of Ovarian Cancer. Available online: https://www.wcrf.org/diet-activity-and-cancer/cancer-types/ovarian-cancer/ (accessed on 24 January 2023).
- Delort, L.; Kwiatkowski, F.; Chalabi, N.; Satih, S.; Bignon, Y.-J.; Bernard-Gallon, D.J. Central adiposity as a major risk factor of ovarian cancer. Anticancer Res. 2009, 29, 5229–5234. [Google Scholar]
- Beddy, P.; Howard, J.; McMahon, C.; Knox, M.; de Blacam, C.; Ravi, N.; Reynolds, J.V.; Keogan, M.T. Association of visceral adiposity with oesophageal and junctional adenocarcinomas. Br. J. Surg. 2010, 97, 1028–1034. [Google Scholar] [CrossRef]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.T.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.; Tinker, L.F.; et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef]
- Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J. Clin. 2017, 67, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Munsell, M.F.; Sprague, B.L.; Berry, D.A.; Chisholm, G.; Trentham-Dietz, A. Body Mass Index and Breast Cancer Risk According to Postmenopausal Estrogen-Progestin Use and Hormone Receptor Status. Epidemiol. Rev. 2014, 36, 114–136. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ursin, G.; Xu, X.; Lee, E.; Togawa, K.; Malone, K.E.; Marchbanks, P.A.; McDonald, J.A.; Simon, M.S.; Folger, S.G.; et al. Body mass index at age 18 years and recent body mass index in relation to risk of breast cancer overall and ER/PR/HER2-defined subtypes in white women and African-American women: A pooled analysis. Breast Cancer Res. 2018, 20, 5. [Google Scholar] [CrossRef]
- Rose, D.P.; Gracheck, P.J.; Vona-Davis, L. The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer. Cancers 2015, 7, 2147–2168. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Howe, L.R.; Bhardwaj, P.; Du, B.; Gravaghi, C.; Yantiss, R.K.; Zhou, X.K.; Blaho, V.A.; Hla, T.; Yang, P.; et al. Obesity Is Associated with Inflammation and Elevated Aromatase Expression in the Mouse Mammary Gland. Cancer Prev. Res. 2011, 4, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Karagozian, R.; Bhardwaj, G.; Wakefield, D.B.; Baffy, G. Obesity paradox in advanced liver disease: Obesity is associated with lower mortality in hospitalized patients with cirrhosis. Liver Int. 2016, 36, 1450–1456. [Google Scholar] [CrossRef]
- Bettini, S.; Serra, R.; Fabris, R.; Prà, C.D.; Favaretto, F.; Dassie, F.; Duso, C.; Vettor, R.; Busetto, L. Association of obstructive sleep apnea with non-alcoholic fatty liver disease in patients with obesity: An observational study. Eat. Weight Disord. 2021, 27, 335–343. [Google Scholar] [CrossRef]
- Adams, K.F.; Leitzmann, M.F.; Albanes, D.; Kipnis, V.; Moore, S.C.; Schatzkin, A.; Chow, W.-H. Body Size and Renal Cell Cancer Incidence in a Large US Cohort Study. Am. J. Epidemiol. 2008, 168, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Häggström, C.; Rapp, K.; Stocks, T.; Manjer, J.; Bjørge, T.; Ulmer, H.; Engeland, A.; Almqvist, M.; Concin, H.; Selmer, R.; et al. Metabolic Factors Associated with Risk of Renal Cell Carcinoma. PLoS ONE 2013, 8, e57475. [Google Scholar] [CrossRef]
- Larsson, S.C.; Wolk, A. Obesity and colon and rectal cancer risk: A meta-analysis of prospective studies. Am. J. Clin. Nutr. 2007, 86, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, A.A.; Woodward, M.; Huxley, R. Obesity and Risk of Colorectal Cancer: A Meta-analysis of 31 Studies with 70,000 Events. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2533–2547. [Google Scholar] [CrossRef]
- Camilleri, M.; Malhi, H.; Acosta, A. Gastrointestinal Complications of Obesity. Gastroenterology 2017, 152, 1656–1670. [Google Scholar] [CrossRef]
- Larsson, S.C.; Orsini, N.; Wolk, A. Body mass index and pancreatic cancer risk: A meta-analysis of prospective studies. Int. J. Cancer 2007, 120, 1993–1998. [Google Scholar] [CrossRef]
- Bonn, S.E.; Sjölander, A.; Tillander, A.; Wiklund, F.; Grönberg, H.; Bälter, K. Body mass index in relation to serum prostate-specific antigen levels and prostate cancer risk. Int. J. Cancer 2016, 139, 50–57. [Google Scholar] [CrossRef]
- Harrison, S.; Tilling, K.; Turner, E.L.; Martin, R.M.; Lennon, R.; Lane, J.A.; Donovan, J.L.; Hamdy, F.C.; Neal, D.E.; Bosch, J.L.H.R.; et al. Systematic review and meta-analysis of the associations between body mass index, prostate cancer, advanced prostate cancer, and prostate-specific antigen. Cancer Causes Control 2020, 31, 431–449. [Google Scholar] [CrossRef]
- Schmid, D.; Ricci, C.; Behrens, G.; Leitzmann, M.F. Adiposity and risk of thyroid cancer: A systematic review and meta-analysis. Obes. Rev. 2015, 16, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, C.M.; McCullough, M.L.; Franceschi, S.; Rinaldi, S.; Wolk, A.; Neta, G.; Adami, H.O.; Anderson, K.; Andreotti, G.; Freeman, L.E.B.; et al. Anthropometric Factors and Thyroid Cancer Risk by Histological Subtype: Pooled Analysis of 22 Prospective Studies. Thyroid 2016, 26, 306–318. [Google Scholar] [CrossRef]
- Kim, K.-N.; Hwang, Y.; Kim, K.H.; Lee, K.E.; Park, Y.J.; Kim, S.-J.; Kwon, H.; Park, D.J.; Cho, B.; Choi, H.-C.; et al. Adolescent overweight and obesity and the risk of papillary thyroid cancer in adulthood: A large-scale case-control study. Sci. Rep. 2020, 10, 5000. [Google Scholar] [CrossRef]
- Sung, H.; Siegel, R.L.; Rosenberg, P.S.; Jemal, A. Emerging cancer trends among young adults in the USA: Analysis of a population-based cancer registry. Lancet Public Health 2019, 4, e137–e147. [Google Scholar] [CrossRef]
- World Cancer Research Fund/American Institute for Cancer Research: Continuous Update Project Report: Diet, Nutrition, Physical Activity and Cancers of the Mouth, Pharynx and Larynx. Available online: https://www.aicr.org/research/the-continuous-update-project/mouth-pharynx-larynx-cancers/ (accessed on 24 January 2023).
- Gaudet, M.M.; Olshan, A.F.; Chuang, S.-C.; Berthiller, J.; Zhang, Z.-F.; Lissowska, J.; Zaridze, D.; Winn, D.M.; Wei, Q.; Talamini, R.; et al. Body mass index and risk of head and neck cancer in a pooled analysis of case–control studies in the International Head and Neck Cancer Epidemiology (INHANCE) Consortium. Int. J. Epidemiol. 2010, 39, 1091–1102. [Google Scholar] [CrossRef]
- Kim, C.-S.; Park, J.-O.; Nam, I.-C.; Park, S.J.; Lee, D.-H.; Kim, H.-B.; Han, K.-D.; Joo, Y.-H. Associations of Body Mass Index and Waist Circumference with the Risk of Head and Neck Cancer: A National Population-Based Study. Cancers 2022, 14, 3880. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.K.; Bentzen, S.M.; Mohindra, P.; Nichols, E.M.; Bhooshan, N.; Vyfhuis, M.; Scilla, K.A.; Feigenberg, S.J.; Edelman, M.J.; Feliciano, J.L. Obesity is associated with long-term improved survival in definitively treated locally advanced non-small cell lung cancer (NSCLC). Lung Cancer 2016, 104, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Cheung, M.C.; Pedroso, F.E.; Byrne, M.M.; Koniaris, L.G.; Zimmers, T.A. Obesity and Weight Loss at Presentation of Lung Cancer are Associated with Opposite Effects on Survival. J. Surg. Res. 2011, 170, e75–e83. [Google Scholar] [CrossRef] [PubMed]
- Haaf, K.T.; Jeon, J.; Tammemägi, M.C.; Han, S.S.; Kong, C.Y.; Plevritis, S.K.; Feuer, E.J.; de Koning, H.J.; Steyerberg, E.W.; Meza, R. Risk prediction models for selection of lung cancer screening candidates: A retrospective validation study. PLoS Med. 2017, 14, e1002277. [Google Scholar] [CrossRef]
- Kichenadasse, G.; Miners, J.O.; Mangoni, A.A.; Rowland, A.; Hopkins, A.M.; Sorich, M.J. Association between Body Mass Index and Overall Survival with Immune Checkpoint Inhibitor Therapy for Advanced Non–Small Cell Lung Cancer. JAMA Oncol. 2020, 6, 512–518. [Google Scholar] [CrossRef]
- Barbi, J.; Patnaik, S.K.; Pabla, S.; Zollo, R.; Smith, R.J., Jr.; Sass, S.N.; Srinivasan, A.; Petrucci, C.; Seager, R.; Conroy, J.; et al. Visceral Obesity Promotes Lung Cancer Progression—Toward Resolution of the Obesity Paradox in Lung Cancer. J. Thorac. Oncol. 2021, 16, 1333–1348. [Google Scholar] [CrossRef]
- Nitsche, L.J.; Mukherjee, S.; Cheruvu, K.; Krabak, C.; Rachala, R.; Ratnakaram, K.; Sharma, P.; Singh, M.; Yendamuri, S. Exploring the Impact of the Obesity Paradox on Lung Cancer and Other Malignancies. Cancers 2022, 14, 1440. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Calejman, C.M.; Doxsey, W.; Fazakerley, D.; Guertin, D. Integrating adipocyte insulin signaling and metabolism in the multi-omics era. Trends Biochem. Sci. 2022, 47, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Henriques, F.; Bedard, A.H.; Czech, M.P. Molecular pathways linking adipose innervation to insulin action in obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 207–225. [Google Scholar] [CrossRef]
- Milan, G.; Conci, S.; Sanna, M.; Favaretto, F.; Bettini, S.; Vettor, R. ASCs and their role in obesity and metabolic diseases. Trends Endocrinol. Metab. 2021, 32, 994–1006. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Stambolic, V. Impact of the Obesity Epidemic on Cancer. Annu. Rev. Med. 2015, 66, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, Y.; Yan, X.; Yan, F.; Sun, Y.; Zeng, J.; Waigel, S.; Yin, Y.; Fraig, M.M.; Egilmez, N.K.; et al. Circulating Adipose Fatty Acid Binding Protein Is a New Link Underlying Obesity-Associated Breast/Mammary Tumor Development. Cell Metab. 2018, 28, 689–705.e5. [Google Scholar] [CrossRef]
- Brown, K.A. Metabolic pathways in obesity-related breast cancer. Nat. Rev. Endocrinol. 2021, 17, 350–363. [Google Scholar] [CrossRef]
- Zhang, M.; Di Martino, J.S.; Bowman, R.L.; Campbell, N.R.; Baksh, S.C.; Simon-Vermot, T.; Kim, I.S.; Haldeman, P.; Mondal, C.; Yong-Gonzales, V.; et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov. 2018, 8, 1006–1025. [Google Scholar] [CrossRef] [PubMed]
- Himbert, C.; Delphan, M.; Scherer, D.; Bowers, L.W.; Hursting, S.; Ulrich, C.M. Signals from the Adipose Microenvironment and the Obesity–Cancer Link—A Systematic Review. Cancer Prev. Res. 2017, 10, 494–506. [Google Scholar] [CrossRef]
- Kozlov, A.P. Mammalian tumor-like organs. 2. Mammalian adipose has many tumor features and obesity is a tumor-like process. Infect. Agents Cancer 2022, 17, 15. [Google Scholar] [CrossRef]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2018, 15, 139–154. [Google Scholar] [CrossRef]
- Bays, H. Adiposopathy, “Sick Fat,” Ockham’s Razor, and Resolution of the Obesity Paradox. Curr. Atheroscler. Rep. 2014, 16, 409. [Google Scholar] [CrossRef] [PubMed]
- Paavonsalo, S.; Hariharan, S.; Lackman, M.H.; Karaman, S. Capillary Rarefaction in Obesity and Metabolic Diseases—Organ-Specificity and Possible Mechanisms. Cells 2020, 9, 2683. [Google Scholar] [CrossRef] [PubMed]
- Belligoli, A.; Compagnin, C.; Sanna, M.; Favaretto, F.; Fabris, R.; Busetto, L.; Foletto, M.; Prà, C.D.; Serra, R.; Prevedello, L.; et al. Characterization of subcutaneous and omental adipose tissue in patients with obesity and with different degrees of glucose impairment. Sci. Rep. 2019, 9, 11333. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- van Kruijsdijk, R.C.; van der Wall, E.; Visseren, F.L. Obesity and Cancer: The Role of Dysfunctional Adipose Tissue. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2569–2578. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose Expression of Tumor Necrosis Factor-α: Direct Role in Obesity-Linked Insulin Resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Makowski, L.; Chaib, M.; Rathmell, J.C. Immunometabolism: From basic mechanisms to translation. Immunol. Rev. 2020, 295, 5–14. [Google Scholar] [CrossRef]
- Rathmell, J.C. Obesity, Immunity, and Cancer. N. Engl. J. Med. 2021, 384, 1160–1162. [Google Scholar] [CrossRef]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef]
- Rabhi, N.; Desevin, K.; Belkina, A.C.; Tilston-Lunel, A.; Varelas, X.; Layne, M.D.; Farmer, S.R. Obesity-induced senescent macrophages activate a fibrotic transcriptional program in adipocyte progenitors. Life Sci. Alliance 2022, 5, e202101286. [Google Scholar] [CrossRef]
- Frasca, D. Obesity Accelerates Age Defects in Human B Cells and Induces Autoimmunity. Immunometabolism 2022, 4, e220010. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hagberg, C.E.; Cascales, H.S.; Lang, S.; Hyvönen, M.T.; Salehzadeh, F.; Chen, P.; Alexandersson, I.; Terezaki, E.; Harms, M.J.; et al. Obesity and hyperinsulinemia drive adipocytes to activate a cell cycle program and senesce. Nat. Med. 2021, 27, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, I.R.; Rodrigues, A.D.S.; Flores, M.S.; Cunha, E.L.V.; Goldberg, M.; Harmon, B.; Batabyal, R.; Freishtat, R.J.; Cechinel, L.R. Circulating Extracellular Vesicles and Particles Derived from Adipocytes: The Potential Role in Spreading MicroRNAs Associated with Cellular Senescence. Front. Aging 2022, 3, 867100. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Tomás, T.; Rueda-Robles, A.; Plaza-Díaz, J.; Álvarez-Mercado, A.I. Nutrition and cellular senescence in obesity-related disorders. J. Nutr. Biochem. 2021, 99, 108861. [Google Scholar] [CrossRef] [PubMed]
- Maliniak, M.L.; Miller-Kleinhenz, J.; Cronin-Fenton, D.P.; Lash, T.L.; Gogineni, K.; Janssen, E.A.M.; McCullough, L.E. Crown-Like Structures in Breast Adipose Tissue: Early Evidence and Current Issues in Breast Cancer. Cancers 2021, 13, 2222. [Google Scholar] [CrossRef] [PubMed]
- Pellegata, N.S.; Diaz, M.B.; Rohm, M.; Herzig, S. Obesity and cancer—Extracellular matrix, angiogenesis, and adrenergic signaling as unusual suspects linking the two diseases. Cancer Metastasis Rev. 2022, 41, 517–547. [Google Scholar] [CrossRef]
- Ahechu, P.; Zozaya, G.; Martí, P.; Hernández-Lizoáin, J.L.; Baixauli, J.; Unamuno, X.; Frühbeck, G.; Catalán, V. NLRP3 Inflammasome: A Possible Link between Obesity-Associated Low-Grade Chronic Inflammation and Colorectal Cancer Development. Front. Immunol. 2018, 9, 02918. [Google Scholar] [CrossRef]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.; Rensen, P.C.N.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The Inflammasome-Mediated Caspase-1 Activation Controls Adipocyte Differentiation and Insulin Sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Shahbaz, S.K.; Koushki, K.; Ayati, S.H.; Bland, A.R.; Bezsonov, E.E.; Sahebkar, A. Inflammasomes and Colorectal Cancer. Cells 2021, 10, 2172. [Google Scholar] [CrossRef] [PubMed]
- Raja, R.; Wu, C.; Limbeck, F.; Butler, K.; Acharya, A.P.; Curtis, M. Instruction of Immunometabolism by Adipose Tissue: Implications for Cancer Progression. Cancers 2021, 13, 3327. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Castrejón-Téllez, V.; Soto, M.E.; Rubio-Ruiz, M.E.; Manzano-Pech, L.; Guarner-Lans, V. Oxidative Stress, Plant Natural Antioxidants, and Obesity. Int. J. Mol. Sci. 2021, 22, 1786. [Google Scholar] [CrossRef] [PubMed]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef]
- Zhang, Z.; Scherer, P.E. Adipose tissue: The dysfunctional adipocyte—A cancer cell’s best friend. Nat. Rev. Endocrinol. 2018, 14, 132–134. [Google Scholar] [CrossRef]
- Zahid, H.; Simpson, E.R.; A Brown, K. Inflammation, dysregulated metabolism and aromatase in obesity and breast cancer. Curr. Opin. Pharmacol. 2016, 31, 90–96. [Google Scholar] [CrossRef]
- Andò, S.; Catalano, S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 2011, 8, 263–275. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Au, C.C.; Benito-Martin, A.; Ladumor, H.; Oshchepkova, S.; Moges, R.; Brown, K.A. Estrogens and breast cancer: Mechanisms involved in obesity-related development, growth and progression. J. Steroid Biochem. Mol. Biol. 2019, 189, 161–170. [Google Scholar] [CrossRef]
- Mutoh, M.; Teraoka, N.; Takasu, S.; Takahashi, M.; Onuma, K.; Yamamoto, M.; Kubota, N.; Iseki, T.; Kadowaki, T.; Sugimura, T.; et al. Loss of Adiponectin Promotes Intestinal Carcinogenesis in Min and Wild-type Mice. Gastroenterology 2011, 140, 2000–2008.e2. [Google Scholar] [CrossRef]
- Liu, Z.; Brooks, R.S.; Ciappio, E.D.; Kim, S.J.; Crott, J.W.; Bennett, G.; Greenberg, A.S.; Mason, J.B. Diet-induced obesity elevates colonic TNF-α in mice and is accompanied by an activation of Wnt signaling: A mechanism for obesity-associated colorectal cancer. J. Nutr. Biochem. 2011, 23, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Fazolini, N.P.; Cruz, A.L.; Werneck, M.B.; Viola, J.P.; Maya-Monteiro, C.M.; Bozza, P.T. Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation, cytokine production and increased cell proliferation. Cell Cycle 2015, 14, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.H.; Thrift, A.P. Risk factors and populations at risk: Selection of patients for screening for Barrett’s oesophagus. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Narita, S.; Huang, M.; Nara, T.; Numakura, K.; Takayama, K.; Tsuruta, H.; Maeno, A.; Saito, M.; Inoue, T.; et al. The impact of obesity and adiponectin signaling in patients with renal cell carcinoma: A potential mechanism for the “obesity paradox”. PLoS ONE 2017, 12, e0171615. [Google Scholar] [CrossRef]
- Li, Z.; Liu, H.; He, J.; Wang, Z.; Yin, Z.; You, G.; Wang, Z.; Davis, R.E.; Lin, P.; Bergsagel, P.L.; et al. Acetyl-CoA Synthetase 2: A Critical Linkage in Obesity-Induced Tumorigenesis in Myeloma. Cell Metab. 2021, 33, 78–93.e7. [Google Scholar] [CrossRef] [PubMed]
- Trevellin, E.; Scarpa, M.; Carraro, A.; Lunardi, F.; Kotsafti, A.; Porzionato, A.; Saadeh, L.; Cagol, M.; Alfieri, R.; Tedeschi, U.; et al. Esophageal adenocarcinoma and obesity: Peritumoral adipose tissue plays a role in lymph node invasion. Oncotarget 2015, 6, 11203–11215. [Google Scholar] [CrossRef]
- Carraro, A.; Trevellin, E.; Fassan, M.; Kotsafti, A.; Lunardi, F.; Porzionato, A.; Dall’Olmo, L.; Cagol, M.; Alfieri, R.; Macchi, V.; et al. Esophageal adenocarcinoma microenvironment: Peritumoral adipose tissue effects associated with chemoresistance. Cancer Sci. 2017, 108, 2393–2404. [Google Scholar] [CrossRef]
- Guerra, B.; Issinger, O.-G. Role of Protein Kinase CK2 in Aberrant Lipid Metabolism in Cancer. Pharmaceuticals 2020, 13, 292. [Google Scholar] [CrossRef]
- Borgo, C.; D’amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef]
- Husain, K.; Williamson, T.T.; Nelson, N.; Ghansah, T. Protein kinase 2 (CK2): A potential regulator of immune cell development and function in cancer. Immunol. Med. 2020, 44, 159–174. [Google Scholar] [CrossRef]
- Borgo, C.; Milan, G.; Favaretto, F.; Stasi, F.; Fabris, R.; Salizzato, V.; Cesaro, L.; Belligoli, A.; Sanna, M.; Foletto, M.; et al. CK2 modulates adipocyte insulin-signaling and is up-regulated in human obesity. Sci. Rep. 2017, 7, 17569. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, K.; Ohyama, K.; Hasegawa, Y.; Chang, H.-Y.; Ogura, M.; Sato, A.; Hong, H.; Hosono, T.; Sharp, L.Z.; Scheel, D.W.; et al. Phosphoproteomics Identifies CK2 as a Negative Regulator of Beige Adipocyte Thermogenesis and Energy Expenditure. Cell Metab. 2015, 22, 997–1008. [Google Scholar] [CrossRef]
- Schwind, L.; Wilhelm, N.; Kartarius, S.; Montenarh, M.; Gorjup, E.; Götz, C. Protein kinase CK2 is necessary for the adipogenic differentiation of human mesenchymal stem cells. Biochim. Et Biophys. Acta BBA-Mol. Cell Res. 2015, 1853, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Sanna, M.; Borgo, C.; Compagnin, C.; Favaretto, F.; Vindigni, V.; Trento, M.; Bettini, S.; Comin, A.; Belligoli, A.; Rugge, M.; et al. White Adipose Tissue Expansion in Multiple Symmetric Lipomatosis Is Associated with Upregulation of CK2, AKT and ERK1/2. Int. J. Mol. Sci. 2020, 21, 7933. [Google Scholar] [CrossRef]
- Roy, B.; Palaniyandi, S.S. Tissue-specific role and associated downstream signaling pathways of adiponectin. Cell Biosci. 2021, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Daquinag, A.C.; Amaya-Manzanares, F.; Sirin, O.; Tseng, C.; Kolonin, M.G. Stromal Progenitor Cells from Endogenous Adipose Tissue Contribute to Pericytes and Adipocytes That Populate the Tumor Microenvironment. Cancer Res. 2012, 72, 5198–5208. [Google Scholar] [CrossRef]
- Bellows, C.F.; Zhang, Y.; Chen, J.; Frazier, M.L.; Kolonin, M.G. Circulation of Progenitor Cells in Obese and Lean Colorectal Cancer Patients. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2461–2468. [Google Scholar] [CrossRef]
- Bunnell, B.A.; Martin, E.C.; Matossian, M.D.; Brock, C.K.; Nguyen, K.; Collins-Burow, B.; Burow, M.E. The effect of obesity on adipose-derived stromal cells and adipose tissue and their impact on cancer. Cancer Metastasis Rev. 2022, 41, 549–573. [Google Scholar] [CrossRef]
- Sabol, R.A.; Giacomelli, P.; Beighley, A.; Bunnell, B.A. Adipose stem cells and cancer: Concise review. Stem Cells 2019, 37, 1261–1266. [Google Scholar] [CrossRef]
- Druso, J.E.; Fischbach, C. Biophysical Properties of Extracellular Matrix: Linking Obesity and Cancer. Trends Cancer 2018, 4, 271–273. [Google Scholar] [CrossRef]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, R. Obesity-Associated ECM Remodeling in Cancer Progression. Cancers 2022, 14, 5684. [Google Scholar] [CrossRef] [PubMed]
- Hamel, K.M.; Liimatta, K.Q.; Belgodere, J.A.; Bunnell, B.A.; Gimble, J.M.; Martin, E.C. Adipose-Derived Stromal/Stem Cell Response to Tumors and Wounds: Evaluation of Patient Age. Stem Cells Dev. 2022, 31, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Toledo, B.; Picon-Ruiz, M.; Marchal, J.A.; Perán, M. Dual Role of Fibroblasts Educated by Tumour in Cancer Behavior and Therapeutic Perspectives. Int. J. Mol. Sci. 2022, 23, 15576. [Google Scholar] [CrossRef] [PubMed]
- Airuddin, S.S.; Halim, A.S.; Sulaiman, W.A.W.; Kadir, R.; Nasir, N.A.M. Adipose-Derived Stem Cell: “Treat or Trick”. Biomedicines 2021, 9, 1624. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef]
- Cox, A.J.; West, N.P.; Cripps, A.W. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. 2015, 3, 207–215. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef]
- Schulz, M.D.; Atay, C.; Heringer, J.; Romrig, F.K.; Schwitalla, S.; Aydin, B.; Ziegler, P.K.; Varga, J.; Reindl, W.; Pommerenke, C.; et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 2014, 514, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Jordan, B.F. Gut microbiota-mediated inflammation in obesity: A link with gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 671–682. [Google Scholar] [CrossRef]
- Pradere, J.-P.; Dapito, D.H.; Schwabe, R.F. The Yin and Yang of Toll-like receptors in cancer. Oncogene 2013, 33, 3485–3495. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y. Roles of circadian clocks in cancer pathogenesis and treatment. Exp. Mol. Med. 2021, 53, 1529–1538. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Poggiogalle, E.; Barrea, L.; Tarsitano, M.G.; Garifalos, F.; Liccardi, A.; Pugliese, G.; Savastano, S.; Colao, A.; Alviggi, C.; et al. Exposure to artificial light at night: A common link for obesity and cancer? Eur. J. Cancer 2022, 173, 263–275. [Google Scholar] [CrossRef]
- Orihara, K.; Haraguchi, A.; Shibata, S. Crosstalk among Circadian Rhythm, Obesity and Allergy. Int. J. Mol. Sci. 2020, 21, 1884. [Google Scholar] [CrossRef]
- Diamantopoulou, Z.; Castro-Giner, F.; Schwab, F.D.; Foerster, C.; Saini, M.; Budinjas, S.; Strittmatter, K.; Krol, I.; Seifert, B.; Heinzelmann-Schwarz, V.; et al. The metastatic spread of breast cancer accelerates during sleep. Nature 2022, 607, 156–162. [Google Scholar] [CrossRef]
- Numata, M.; Hirano, A.; Yamamoto, Y.; Yasuda, M.; Miura, N.; Sayama, K.; Shibata, M.-A.; Asai, T.; Oku, N.; Miyoshi, N.; et al. Metastasis of Breast Cancer Promoted by Circadian Rhythm Disruption due to Light/Dark Shift and its Prevention by Dietary Quercetin in Mice. J. Circadian Rhythm. 2021, 19, 2. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef]
- Weber, B.Z.C.; Arabaci, D.H.; Kir, S. Metabolic Reprogramming in Adipose Tissue during Cancer Cachexia. Front. Oncol. 2022, 12, 848394. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; López-Soriano, F.J.; Stemmler, B.; Busquets, S. Cancer-associated cachexia—Understanding the tumour macroenvironment and microenvironment to improve management. Nat. Rev. Clin. Oncol. 2023, 20, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Patel, B.M. The burning furnace: Alteration in lipid metabolism in cancer-associated cachexia. Mol. Cell Biochem. 2022, 477, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Feng, X.; Wu, X.; Lu, Y.; Chen, K.; Ye, Y. Fat Wasting Is Damaging: Role of Adipose Tissue in Cancer-Associated Cachexia. Front. Cell Dev. Biol. 2020, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, M.; Gamberi, T.; Magherini, F.; Fiaschi, T. The Adipokines in Cancer Cachexia. Int. J. Mol. Sci. 2020, 21, 4860. [Google Scholar] [CrossRef]
- Stephens, N.A.; Skipworth, R.J.E.; MacDonald, A.J.; Greig, C.A.; Ross, J.A.; Fearon, K.C.H. Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. J. Cachexia Sarcopenia Muscle 2011, 2, 111–117. [Google Scholar] [CrossRef]
- Prado, C.M.; Cushen, S.J.; Orsso, C.E.; Ryan, A. Sarcopenia and cachexia in the era of obesity: Clinical and nutritional impact. Proc. Nutr. Soc. 2016, 75, 188–198. [Google Scholar] [CrossRef]
- A Vaitkus, J.; Celi, F.S. The role of adipose tissue in cancer-associated cachexia. Exp. Biol. Med. 2016, 242, 473–481. [Google Scholar] [CrossRef]
- Tan, B.H.; Birdsell, L.A.; Martin, L.; Baracos, V.E.; Fearon, K.C. Sarcopenia in an Overweight or Obese Patient Is an Adverse Prognostic Factor in Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 6973–6979. [Google Scholar] [CrossRef]
- Martin, L.; Birdsell, L.; MacDonald, N.; Reiman, T.; Clandinin, M.T.; McCargar, L.J.; Murphy, R.; Ghosh, S.; Sawyer, M.B.; Baracos, V.E. Cancer Cachexia in the Age of Obesity: Skeletal Muscle Depletion Is a Powerful Prognostic Factor, Independent of Body Mass Index. J. Clin. Oncol. 2013, 31, 1539–1547. [Google Scholar] [CrossRef]
- Di Sebastiano, K.M.; Yang, L.; Zbuk, K.; Wong, R.K.; Chow, T.; Koff, D.; Moran, G.R.; Mourtzakis, M. Accelerated muscle and adipose tissue loss may predict survival in pancreatic cancer patients: The relationship with diabetes and anaemia. Br. J. Nutr. 2013, 109, 302–312. [Google Scholar] [CrossRef]
- Erdem, M.; Möckel, D.; Jumpertz, S.; John, C.; Fragoulis, A.; Rudolph, I.; Wulfmeier, J.; Springer, J.; Horn, H.; Koch, M.; et al. Macrophages protect against loss of adipose tissue during cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 1128–1142. [Google Scholar] [CrossRef]
- Henriques, F.; Lopes, M.A.; Franco, F.O.; Knobl, P.; Santos, K.B.; Bueno, L.L.; Correa, V.A.; Bedard, A.H.; Guilherme, A.; Birbrair, A.; et al. Toll-Like Receptor-4 Disruption Suppresses Adipose Tissue Remodeling and Increases Survival in Cancer Cachexia Syndrome. Sci. Rep. 2018, 8, 18024. [Google Scholar] [CrossRef]
- The Look AHEAD Research Group Eight-year weight losses with an intensive lifestyle intervention: The look AHEAD study. Obesity 2014, 22, 5–13. [CrossRef]
- Singh, P.; Subramanian, A.; Adderley, N.; Gokhale, K.; Singhal, R.; Bellary, S.; Nirantharakumar, K.; Tahrani, A.A. Impact of bariatric surgery on cardiovascular outcomes and mortality: A population-based cohort study. Br. J. Surg. 2020, 107, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Mingrone, G.; Bornstein, S.R. Gain in survival after metabolic–bariatric surgery. Lancet 2021, 397, 1785–1787. [Google Scholar] [CrossRef] [PubMed]
- Teras, L.R.; Patel, A.V.; Wang, M.; Yaun, S.-S.; Anderson, K.; Brathwaite, R.; Caan, B.; Chen, Y.; E Connor, A.; Eliassen, A.H.; et al. Sustained Weight Loss and Risk of Breast Cancer in Women 50 Years and Older: A Pooled Analysis of Prospective Data. J. Natl. Cancer Inst. 2019, 112, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Chlebowski, R.T.; Hendryx, M.; Rohan, T.; Wactawski-Wende, J.; Thomson, C.A.; Felix, A.S.; Chen, C.; Barrington, W.; Coday, M.; et al. Intentional Weight Loss and Endometrial Cancer Risk. J. Clin. Oncol. 2017, 35, 1189–1193. [Google Scholar] [CrossRef]
- Rodriguez, C.; Freedland, S.J.; Deka, A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Thun, M.J.; Calle, E.E. Body Mass Index, Weight Change, and Risk of Prostate Cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol. Biomark. Prev. 2007, 16, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Look AHEAD Research Group; Yeh, H.; Bantle, J.P.; Cassidy-Begay, M.; Blackburn, G.; Bray, G.A.; Byers, T.; Clark, J.M.; Coday, M.; Egan, C.; et al. Intensive Weight Loss Intervention and Cancer Risk in Adults with Type 2 Diabetes: Analysis of the Look AHEAD Randomized Clinical Trial. Obesity 2020, 28, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, N.; Markozannes, G.; Kanellopoulou, A.; Critselis, E.; Alhardan, S.; Karafousia, V.; Kasimis, J.C.; Katsaraki, C.; Papadopoulou, A.; Zografou, M.; et al. An umbrella review of the evidence associating diet and cancer risk at 11 anatomical sites. Nat. Commun. 2021, 12, 4579. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2019, 33, 102–121. [Google Scholar] [CrossRef]
- Sutton, E.F.; Beyl, R.; Early, K.S.; Cefalu, W.T.; Ravussin, E.; Peterson, C.M. Early time-restricted feeding improves insulin sensitivity, blood pressure, and oxidative stress even without weight loss in men with prediabetes. Cell Metab. 2018, 27, 1212–1221.e3. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, L. Review of the key results from the Swedish Obese Subjects (SOS) trial—A prospective controlled intervention study of bariatric surgery. J. Intern. Med. 2013, 273, 219–234. [Google Scholar] [CrossRef]
- Belligoli, A.; Bettini, S.; Busetto, L. Bariatric surgery: Is a matter of cutting calories or cutting metabolic regulators? Curr. Opin. Endocr. Metab. Res. 2018, 4, 83–88. [Google Scholar] [CrossRef]
- Schauer, D.P.; Feigelson, H.S.; Koebnick, C.; Caan, B.; Weinmann, S.; Leonard, A.C.; Powers, J.D.; Yenumula, P.R.; Arterburn, D.E. Association between Weight Loss and the Risk of Cancer after Bariatric Surgery. Obesity 2017, 25, S52–S57. [Google Scholar] [CrossRef]
- Lazzati, A.; Epaud, S.; Ortala, M.; Katsahian, S.; Lanoy, E. Effect of bariatric surgery on cancer risk: Results from an emulated target trial using population-based data. Br. J. Surg. 2022, 109, 433–438. [Google Scholar] [CrossRef]
- Lovrics, O.; Butt, J.; Lee, Y.; Boudreau, V.; Anvari, M.; Hong, D.; Doumouras, A. The effect of bariatric surgery on breast cancer incidence and characteristics: A meta-analysis and systematic review. Am. J. Surg. 2021, 222, 715–722. [Google Scholar] [CrossRef]
- Adams, T.D.; Stroup, A.M.; Gress, R.E.; Adams, K.F.; Calle, E.E.; Smith, S.C.; Halverson, R.C.; Simper, S.C.; Hopkins, P.N.; Hunt, S.C. Cancer Incidence and Mortality after Gastric Bypass Surgery. Obesity 2009, 17, 796–802. [Google Scholar] [CrossRef]
- Adams, T.D.; Mehta, T.S.; Davidson, L.E.; Hunt, S.C. All-Cause and Cause-Specific Mortality Associated with Bariatric Surgery: A Review. Curr. Atheroscler. Rep. 2015, 17, 74. [Google Scholar] [CrossRef]
- Tao, W.; Santoni, G.; Von Euler-Chelpin, M.; Ljung, R.; Lynge, E.; Pukkala, E.; Ness-Jensen, E.; Romundstad, P.; Tryggvadottir, L.; Lagergren, J. Cancer Risk after Bariatric Surgery in a Cohort Study from the Five Nordic Countries. Obes. Surg. 2020, 30, 3761–3767. [Google Scholar] [CrossRef]
- Hussan, H.; Akinyeye, S.; Mihaylova, M.; McLaughlin, E.; Chiang, C.; Clinton, S.K.; Lieberman, D. Colorectal Cancer Risk Is Impacted by Sex and Type of Surgery after Bariatric Surgery. Obes. Surg. 2022, 32, 2880–2890. [Google Scholar] [CrossRef]
- Taube, M.; Peltonen, M.; Sjöholm, K.; Palmqvist, R.; Andersson-Assarsson, J.C.; Jacobson, P.; Svensson, P.-A.; Carlsson, L.M.S. Long-term incidence of colorectal cancer after bariatric surgery or usual care in the Swedish Obese Subjects study. PLoS ONE 2021, 16, e0248550. [Google Scholar] [CrossRef] [PubMed]
- Derogar, M.; Hull, M.A.; Kant, P.; Östlund, M.; Lu, Y.; Lagergren, J. Increased Risk of Colorectal Cancer after Obesity Surgery. Ann. Surg. 2013, 258, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Ahmed, K.; Rowland, S.P.; Patel, V.M.; Gooderham, N.J.; Holmes, E.; Darzi, A.; Athanasiou, T. Metabolic surgery and cancer: Protective effects of bariatric procedures. Cancer 2010, 117, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Upala, S.; Sanguankeo, A. Bariatric surgery reduces risk of endometrial cancer. Surg. Obes. Relat. Dis. 2015, 11, 1410. [Google Scholar] [CrossRef] [PubMed]
- Christou, N.V.; Lieberman, M.; Sampalis, F.; Sampalis, J.S. Bariatric surgery reduces cancer risk in morbidly obese patients. Surg. Obes. Relat. Dis. 2008, 4, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Aminian, A.; Wilson, R.; Al-Kurd, A.; Tu, C.; Milinovich, A.; Kroh, M.; Rosenthal, R.J.; Brethauer, S.A.; Schauer, P.R.; Kattan, M.W.; et al. Association of Bariatric Surgery with Cancer Risk and Mortality in Adults with Obesity. JAMA 2022, 327, 2423–2433. [Google Scholar] [CrossRef]
- Sharma, A.M.; Kushner, R.F. A proposed clinical staging system for obesity. Int. J. Obes. 2009, 33, 289–295. [Google Scholar] [CrossRef]
- Bettini, S.; Quinto, G.; Neunhaeuserer, D.; Battista, F.; Belligoli, A.; Milan, G.; Gasperetti, A.; Vettor, R.; Ermolao, A.; Busetto, L. Edmonton Obesity Staging System: An improvement by cardiopulmonary exercise testing. Int. J. Obes. 2021, 45, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Norton, K.-A.; Gong, C.; Jamalian, S.; Popel, A.S. Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment. Processes 2019, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Belenchia, M.; Rocchetti, G.; Maestri, S.; Cimadamore, A.; Montironi, R.; Santoni, M.; Merelli, E. Agent-Based Learning Model for the Obesity Paradox in RCC. Front. Bioeng. Biotechnol. 2021, 9, 642760. [Google Scholar] [CrossRef] [PubMed]
- Schutz, D.D.; Busetto, L.; Dicker, D.; Farpour-Lambert, N.; Pryke, R.; Toplak, H.; Widmer, D.; Yumuk, V.; Schutz, Y. European Practical and Patient-Centred Guidelines for Adult Obesity Management in Primary Care. Obes. Facts 2019, 12, 40–66. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cancer Site or Type | RR for Cancer Incidence (CI) | RR of Mortality or HR (CI) | Further Features | Controversial Matters with Obesity | Association with Obesity | References |
|---|---|---|---|---|---|---|
| Endometrial cancer | 2.89 (2.62–3.18) * 1.59 (1.50–1.68) § | BMI 35–39.99 Kg/m2: 2.77 (1.83–4.18) BMI ≥ 40 Kg/m2: 6.25 (3.75–10.42) ° | Worse outcome | Higher degree | [27,28,39,40,41] | |
| Ovarian cancer | 1.14 (1.03–1.27) * 1.03 (0.99–1.08) § | BMI 35–39.99 Kg/m2: 1.51 (1.12–2.02) ° | Association with visceral adiposity | Higher degree | [28,42,43] | |
| Esophageal adenocarcinoma and stomach cancer (gastric cardia) | M: 1.52 (1.33–1.74) § F: 1.51 (1.31–1.74) § | Hazard Ratio 1.08 (0.77–1.52) ^ | Most important risk factor: GERD; association with visceral adiposity | Better survival | Higher degree | [11,20,28,44] |
| Postmenopausal Breast cancer | 1.40 (1.31–1.49) * 1.12 (1.08–1.16) § | BMI 35–39.99 Kg/m2: 1.70 (1.33–2.17) BMI ≥ 40 Kg/m2: 2.12 (1.41–3.19) ° | The inflammation combined with a higher aromatase activity increases estrogen production and insulin-resistance | Protective effect of BMI during premenopausal period | Higher degree | [28,40,45,46,47,48,49,50] |
| Liver cancer (Hepatocelllular carcinoma) | M: 1.24 (0.95–1.62) § F: 1.07 (0.55–2.08) § | BMI 35–39.99 Kg/m2: M: 4.52 (2.94–6.94) F: 1.68 (0.93–3.05) ° | Develop from MAFLD | Higher degree | [27,29,51,52,53,54,55] | |
| Kidney cancer (renal cell carcinoma) | M: 1.24 (1.15–1.34) § F: 1.34 (1.25–1.43) § | BMI 35–39.99 Kg/m2: M: 1.70 (0.99–2.92) F: 1.70 (0.94–3.05) ° | M: association with increased levels of BMI, blood pressure, glucose and triglycerides F: association with BMI | Better survival | Higher degree | [28,29,56,57] |
| Colorectal cancer | M: 1.24 (1.20–1.28) § F: 1.09 (1.05–1.13) § | BMI 35–39.99 Kg/m2: M: 1.84 (1.39–2.41) F: 1.36 (1.06–1.74) ° | Best outcome | Higher degree | [13,27,28,29,58,59] | |
| Gallbladder cancer | M: 1.09 (0.99–1.21) § F:1.59 (1.02–2.47) § | BMI 30–34.99 kg/m2: M: 1.76 (1.06–2.94) F: 2.13 (1.56–2.90) ° | Obesity increases the risk of gallstones | Higher degree | [27,29,60] | |
| Pancreatic cancer | M: 1.16 (1.05–1.28)§ F: 1.10 (1.02–1.09)§ | BMI 30–34.99 kg/m2: M: 1.49 (0.99–2.22) F: 1.41 (1.01–1.99) BMI ≥ 40 Kg/m2: F: 2.76 (1.74–4.36) ° | Higher degree | [28,29,61] | ||
| Prostate cancer | 1.03 (1.00–1.07) § | BMI 35–39.99 Kg/m2: 1.34 (0.98–1.83) ° | High BMI was positively related with non-metastatic high-grade cancer | Inverse association between BMI and PSA | Lower degree | [27,28,62,63] |
| Thyroid cancer | M: 1.33 (1.04–1.70)§ F: 1.14 (1.06–1.23) § | Lack of studies | Associated with both general and visceral adiposity | Inverse association with medullary thyroid cancer | Lower degree | [28,64,65,66] |
| Multiple myeloma | M: 1.11 (1.05–1.18) § F: 1.11 (1.07–1.15) § | BMI 30–34.99 kg/m2: M: 1.71 (0.93–3.14) F: 1.44 (0.91–2.28) ° | Worst outcome | Lower degree | [28,30,67] | |
| Mouth, pharynx and larynx cancer | 0.81 (0.74–0.89) § 0.94 (0.57–1.56)° Adjusted for sex | HR 0.59 (0.33–1.05) ^ | Data were not adjusted for smoking/drinking alcohol | Negative association | [20,68,69,70] | |
| Non-small-cell lung cancer | Lack of studies with this specific histotype | Lung in general: HR 0.86 (0.76–0.98) ^ | Positive prognostic factor, higher survival, metformin use | Negative association | [20,71,72,73,74,75,76] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trevellin, E.; Bettini, S.; Pilatone, A.; Vettor, R.; Milan, G. Obesity, the Adipose Organ and Cancer in Humans: Association or Causation? Biomedicines 2023, 11, 1319. https://doi.org/10.3390/biomedicines11051319
Trevellin E, Bettini S, Pilatone A, Vettor R, Milan G. Obesity, the Adipose Organ and Cancer in Humans: Association or Causation? Biomedicines. 2023; 11(5):1319. https://doi.org/10.3390/biomedicines11051319
Chicago/Turabian StyleTrevellin, Elisabetta, Silvia Bettini, Anna Pilatone, Roberto Vettor, and Gabriella Milan. 2023. "Obesity, the Adipose Organ and Cancer in Humans: Association or Causation?" Biomedicines 11, no. 5: 1319. https://doi.org/10.3390/biomedicines11051319
APA StyleTrevellin, E., Bettini, S., Pilatone, A., Vettor, R., & Milan, G. (2023). Obesity, the Adipose Organ and Cancer in Humans: Association or Causation? Biomedicines, 11(5), 1319. https://doi.org/10.3390/biomedicines11051319