

The Effects of Alcohol Intoxication and Withdrawal on Hypothalamic Neurohormones and Extrahypothalamic Neurotransmitters
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Substances
2.3. Surgery
2.4. Treatment
2.5. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
2.6. Sandwich Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Chemofluorescent Assay
2.8. In Vitro Superfusion Assay
3. Statistical Analysis
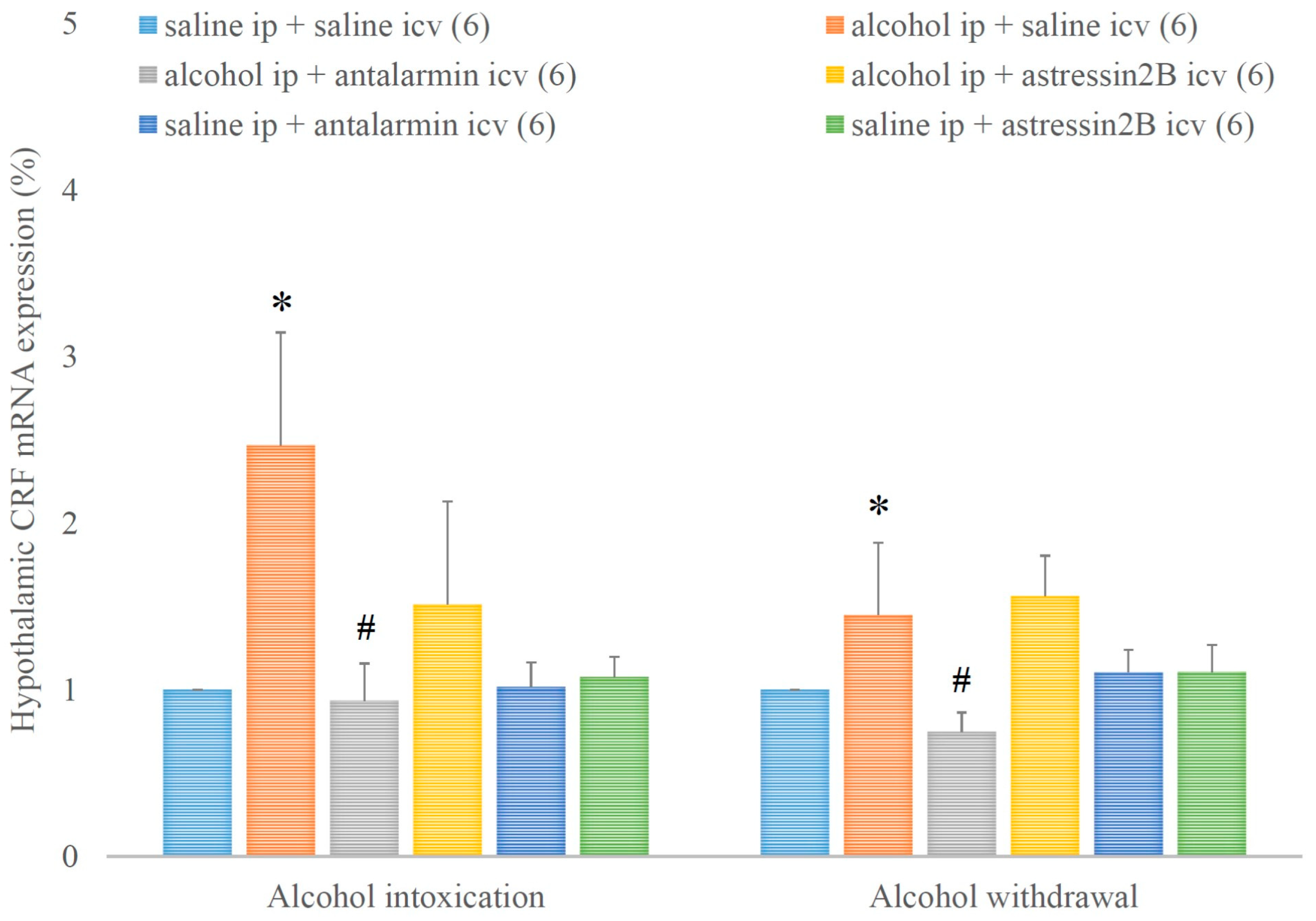
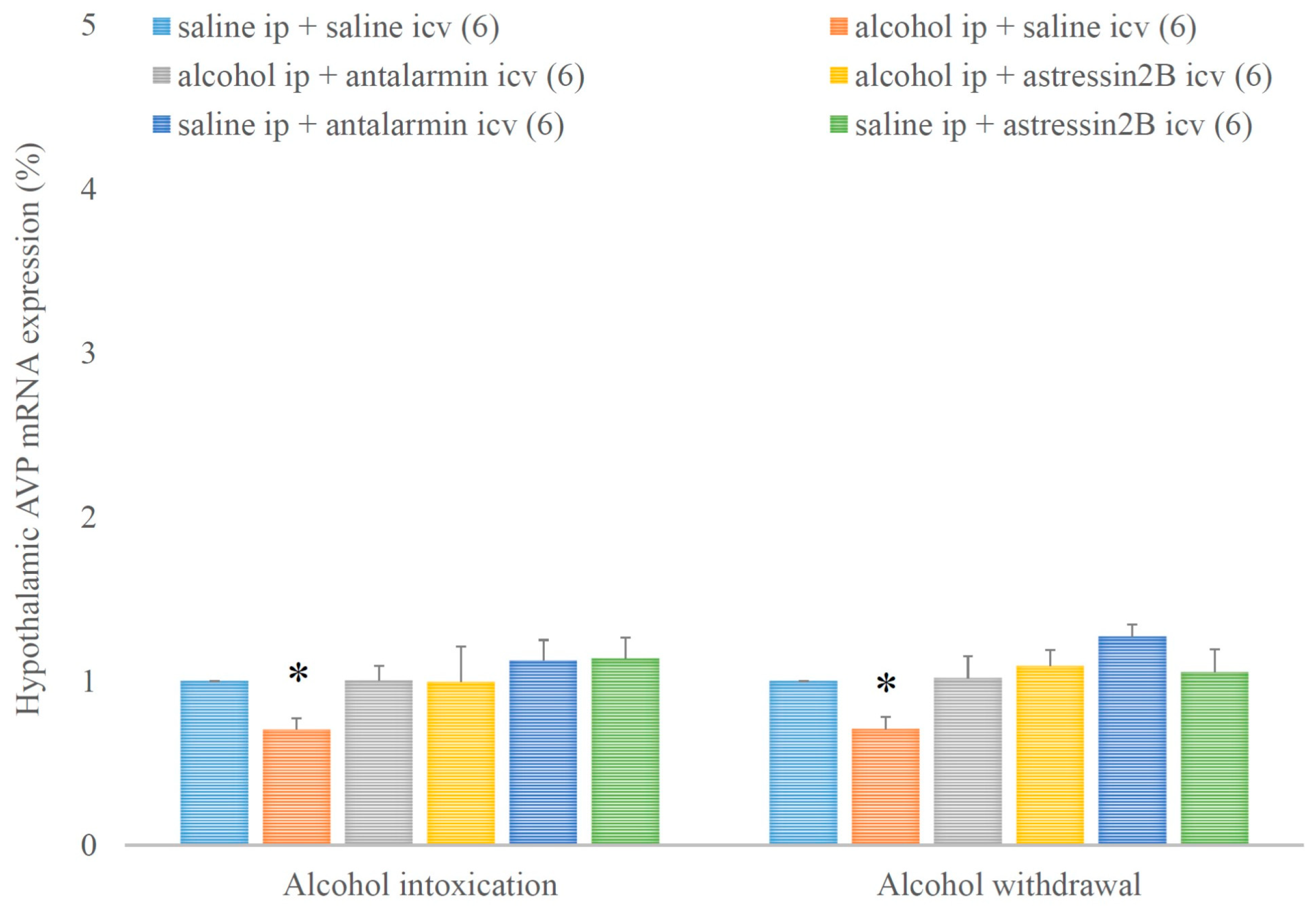
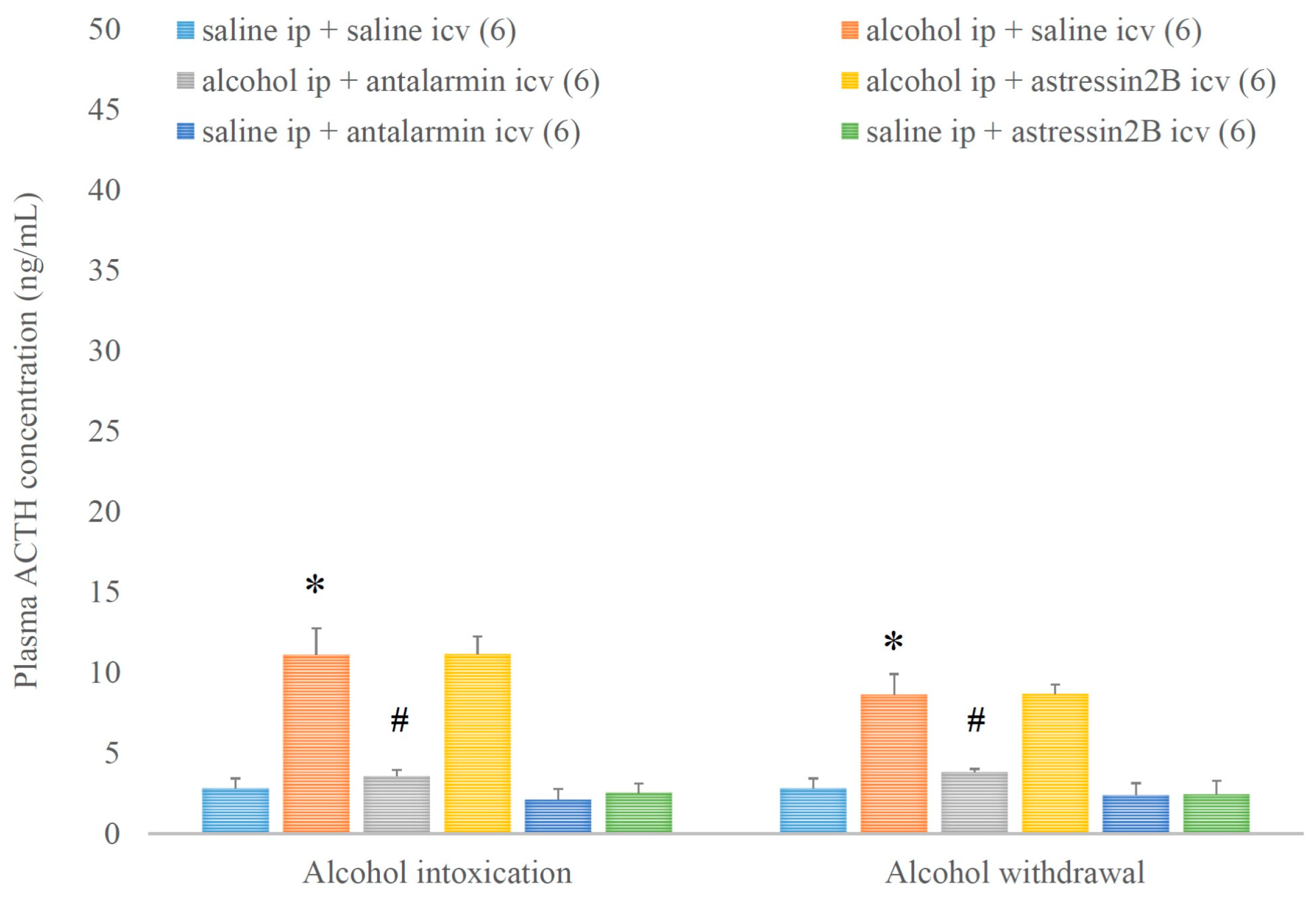
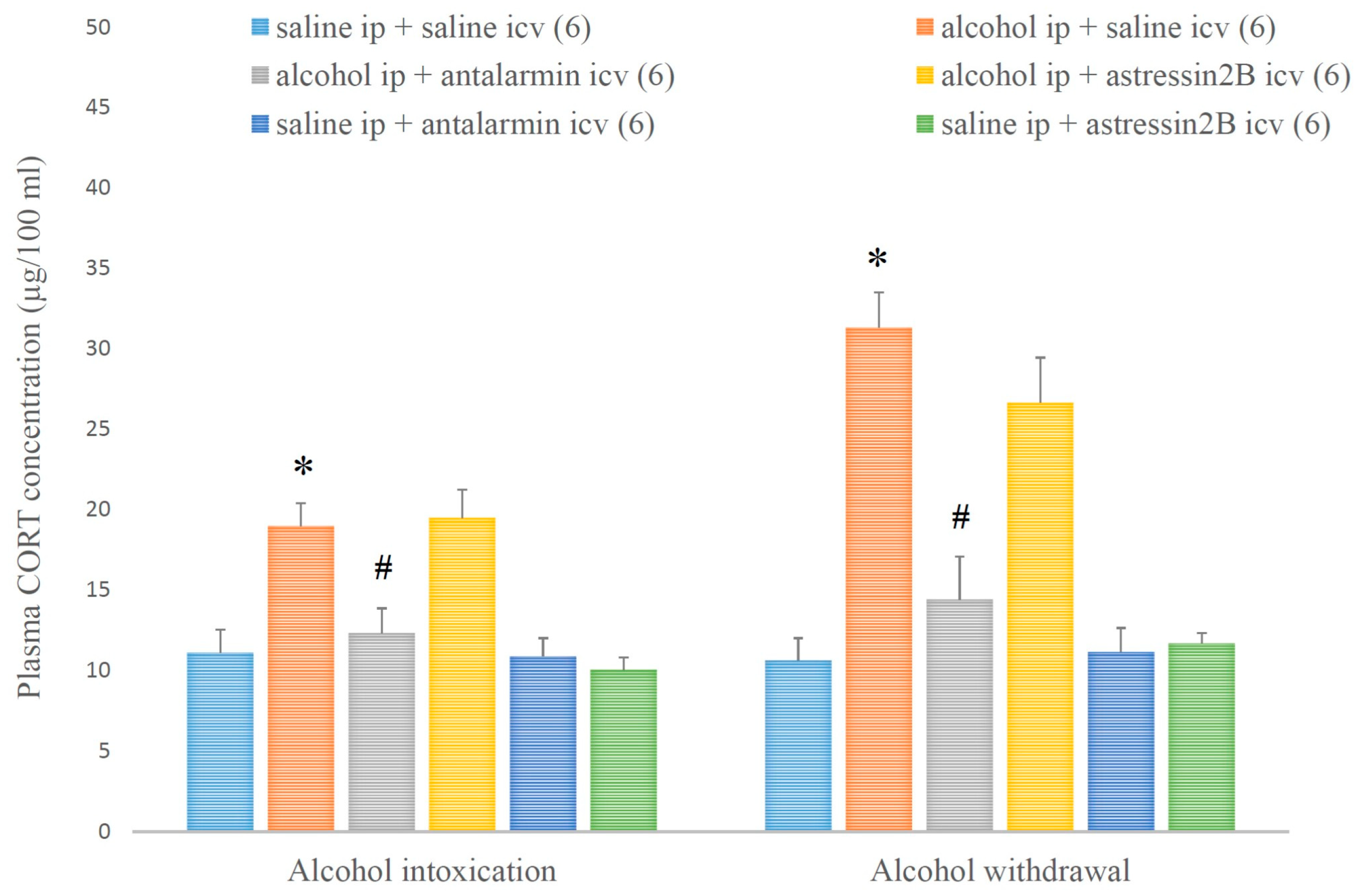
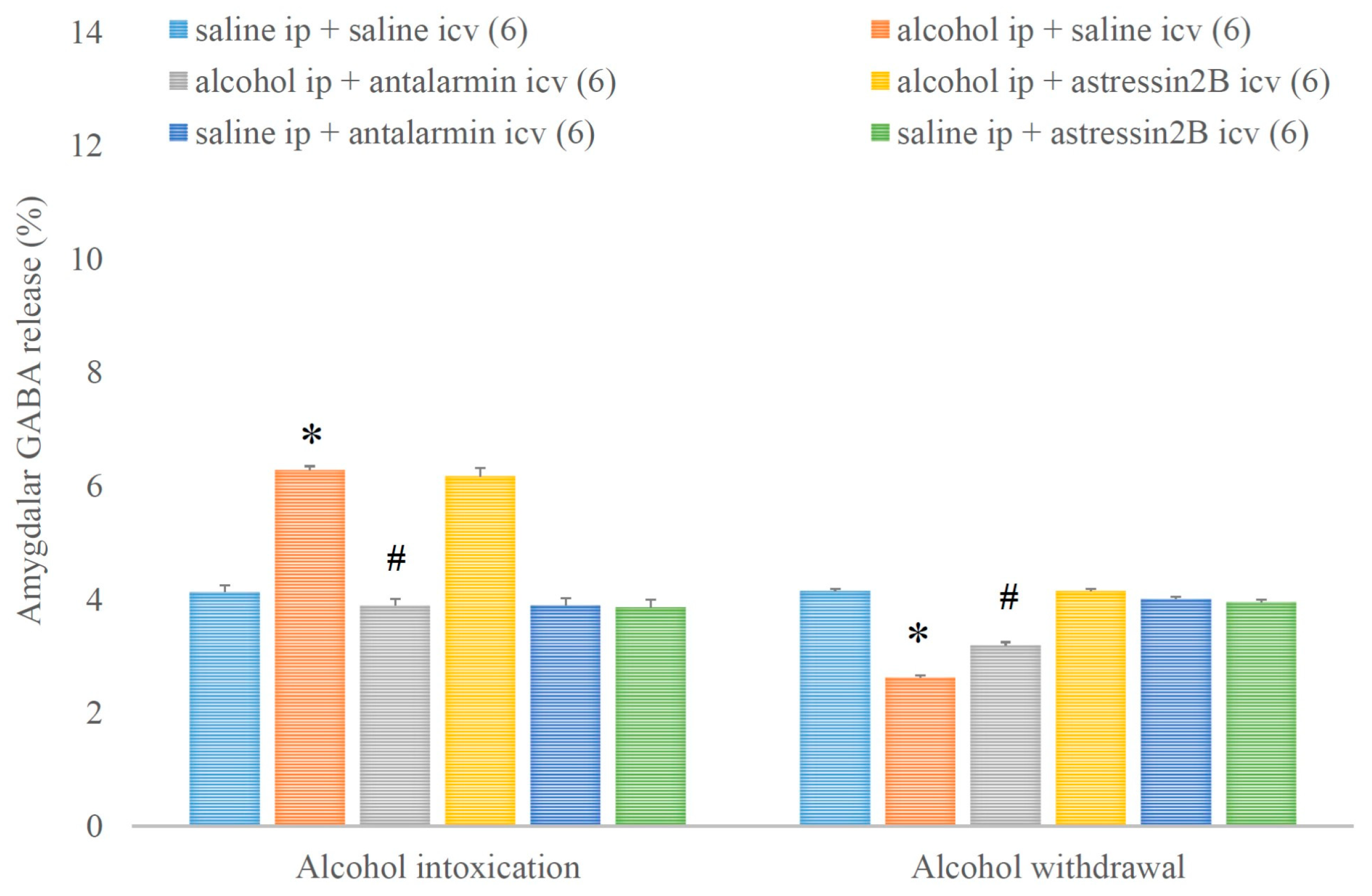
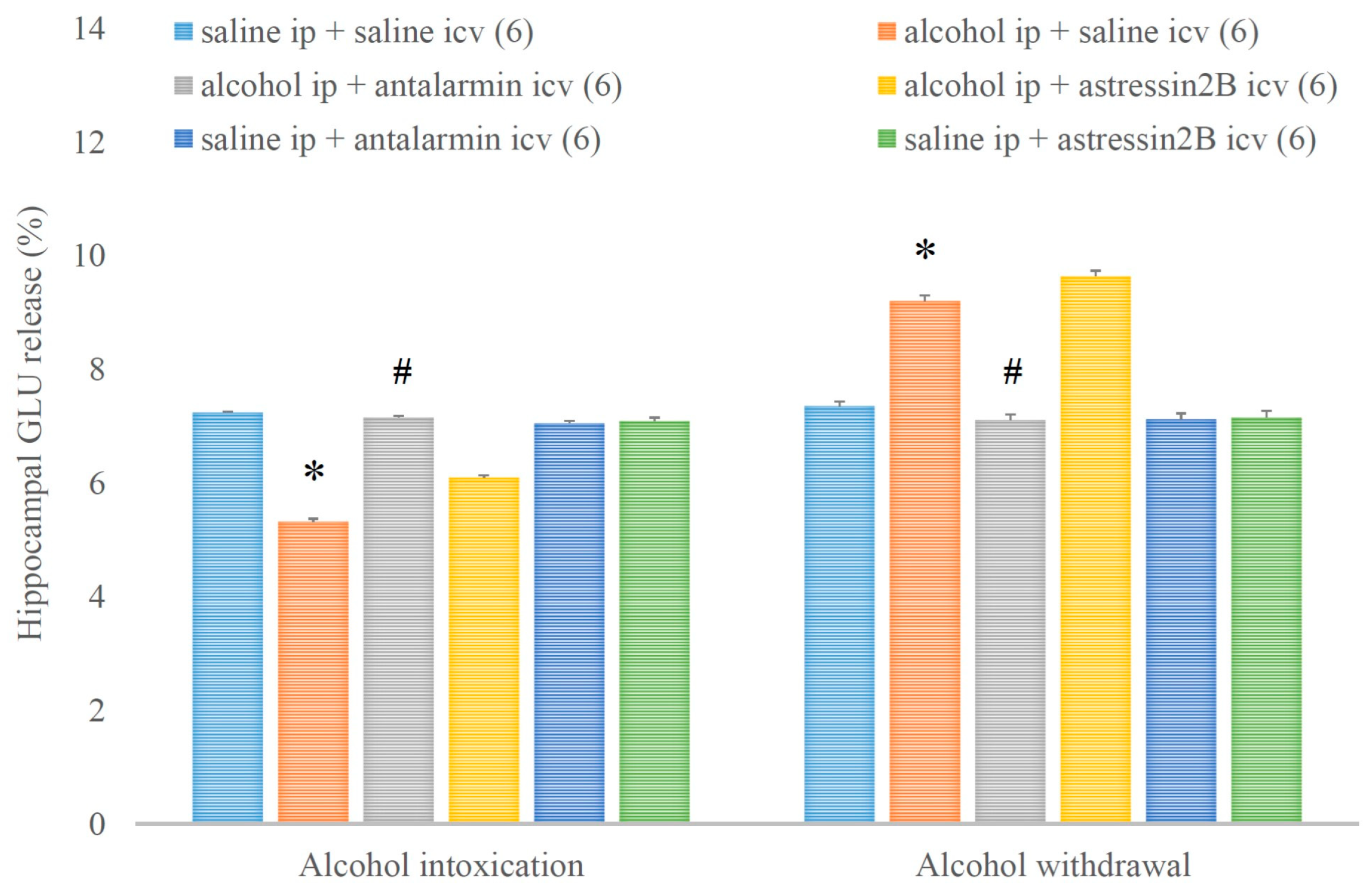
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bruijnzeel, A.W.; Gold, M.S. The role of corticotropin-releasing factor-like peptides in cannabis, nicotine, and alcohol dependence. Brain Res. Rev. 2005, 49, 505–528. [Google Scholar] [CrossRef] [PubMed]
- Sarnyai, Z.; Shaham, Y.; Heinrichs, S.C. The role of corticotropin-releasing factor in drug addiction. Pharmacol. Rev. 2001, 53, 209–243. [Google Scholar]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, G.A.; Van de Kar, L.D. Neuroendocrine pharmacology of stress. Eur. J. Pharmacol. 2003, 463, 235–272. [Google Scholar] [CrossRef]
- Tsigos, C.; Chrousos, G.P. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002, 53, 865–871. [Google Scholar] [CrossRef]
- Koob, G.F. The role of CRF and CRF-related peptides in the dark side of addiction. Brain Res. 2010, 1314, 3–14. [Google Scholar] [CrossRef]
- Van Pett, K.; Viau, V.; Bittencourt, J.C.; Chan, R.K.; Li, H.Y.; Arias, C.; Prins, G.S.; Perrin, M.; Vale, W.; Sawchenko, P.E. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J. Comp. Neurol. 2000, 428, 191–212. [Google Scholar] [CrossRef]
- Vale, W.; Spiess, J.; Rivier, C.; Rivier, J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 1981, 213, 1394–1397. [Google Scholar] [CrossRef]
- Bale, T.L.; Lee, K.F.; Vale, W.W. The role of corticotropin-releasing factor receptors in stress and anxiety. Integr. Comp. Biol. 2002, 42, 552–555. [Google Scholar] [CrossRef]
- Bale, T.L.; Vale, W.W. CRF and CRF receptors: Role in stress responsivity and other behaviors. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 525–557. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press/Elsevier: Amsterdam, The Netherlands; Boston, MA, USA, 2007. [Google Scholar]
- Silvestre de Ferron, B.; Bennouar, K.E.; Kervern, M.; Alaux-Cantin, S.; Robert, A.; Rabiant, K.; Antol, J.; Naassila, M.; Pierrefiche, O. Two Binges of Ethanol a Day Keep the Memory Away in Adolescent Rats: Key Role for GLUN2B Subunit. Int. J. Neuropsychopharmacol. 2015, 19, pyv087. [Google Scholar] [CrossRef] [PubMed]
- Buzas, A.; Bokor, P.; Balango, B.; Pinter, D.; Palotai, M.; Simon, B.; Csabafi, K.; Telegdy, G.; Szabo, G.; Bagosi, Z. Changes in striatal dopamine release and locomotor activity following acute withdrawal from chronic nicotine are mediated by CRF1, but not CRF2, receptors. Brain Res. 2019, 1706, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Bagosi, Z.; Palotai, M.; Simon, B.; Bokor, P.; Buzas, A.; Balango, B.; Pinter, D.; Jaszberenyi, M.; Csabafi, K.; Szabo, G. Selective CRF2 receptor agonists ameliorate the anxiety- and depression-like state developed during chronic nicotine treatment and consequent acute withdrawal in mice. Brain Res. 2016, 1652, 21–29. [Google Scholar] [CrossRef]
- Suda, T.; Yajima, F.; Tomori, N.; Demura, H.; Shizume, K. In vitro study of immunoreactive corticotropin-releasing factor release from the rat hypothalamus. Life Sci. 1985, 37, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Joanny, P.; Steinberg, J.; Zamora, A.J.; Conte-Devolx, B.; Millet, Y.; Oliver, C. Corticotropin-releasing factor release from in vitro superfused and incubated rat hypothalamus. Effect of potassium, norepinephrine, and dopamine. Peptides 1989, 10, 903–911. [Google Scholar] [CrossRef]
- Purves, H.D.; Sirett, N.E. Assay of corticotrophin in dexamethasone-treated rats. Endocrinology 1965, 77, 366–374. [Google Scholar] [CrossRef]
- Zenker, N.; Bernstein, D.E. The estimation of small amounts of corticosterone in rat plasma. J. Biol. Chem. 1958, 231, 695–701. [Google Scholar] [CrossRef]
- Gaddum, J.H. The technique of superfusion. Br. J. Pharmacol. Chemother. 1953, 8, 321–326. [Google Scholar] [CrossRef]
- Harsing, L.G., Jr.; Vizi, E.S. Release of endogenous dopamine from rat isolated striatum: Effect of clorgyline and (-)-deprenyl. Br. J. Pharmacol. 1984, 83, 741–749. [Google Scholar] [CrossRef]
- Rivier, C. Role of hypothalamic corticotropin-releasing factor in mediating alcohol-induced activation of the rat hypothalamic-pituitary-adrenal axis. Front. Neuroendocrinol. 2014, 35, 221–233. [Google Scholar] [CrossRef]
- Rivier, C.; Bruhn, T.; Vale, W. Effect of ethanol on the hypothalamic-pituitary-adrenal axis in the rat: Role of corticotropin-releasing factor (CRF). J. Pharmacol. Exp. Ther. 1984, 229, 127–131. [Google Scholar] [PubMed]
- Rivier, C.; Rivier, J.; Vale, W. Inhibition of adrenocorticotropic hormone secretion in the rat by immunoneutralization of corticotropin-releasing factor. Science 1982, 218, 377–379. [Google Scholar] [CrossRef]
- Rivier, C.; Vale, W. Modulation of stress-induced ACTH release by corticotropin-releasing factor, catecholamines and vasopressin. Nature 1983, 305, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, K.M.; Lee, S.; Rivier, C. Role of arginine vasopressin and corticotropin-releasing factor in mediating alcohol-induced adrenocorticotropin and vasopressin secretion in male rats bearing lesions of the paraventricular nuclei. Brain Res. 1997, 744, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Rivest, S.; Rivier, C. Lesions of hypothalamic PVN partially attenuate stimulatory action of alcohol on ACTH secretion in rats. Am. J. Physiol. 1994, 266, R553–R558. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C.; Lee, S. Acute alcohol administration stimulates the activity of hypothalamic neurons that express corticotropin-releasing factor and vasopressin. Brain Res. 1996, 726, 1–10. [Google Scholar] [CrossRef]
- Ogilvie, K.; Lee, S.; Rivier, C. Effect of three different modes of alcohol administration on the activity of the rat hypothalamic-pituitary-adrenal axis. Alcohol. Clin. Exp. Res. 1997, 21, 467–476. [Google Scholar] [CrossRef]
- Rivier, C. Gender, sex steroids, corticotropin-releasing factor, nitric oxide, and the HPA response to stress. Pharmacol. Biochem. Behav. 1999, 64, 739–751. [Google Scholar] [CrossRef]
- Budygin, E.A.; John, C.E.; Mateo, Y.; Daunais, J.B.; Friedman, D.P.; Grant, K.A.; Jones, S.R. Chronic ethanol exposure alters presynaptic dopamine function in the striatum of monkeys: A preliminary study. Synapse 2003, 50, 266–268. [Google Scholar] [CrossRef]
- Budygin, E.A.; Phillips, P.E.; Robinson, D.L.; Kennedy, A.P.; Gainetdinov, R.R.; Wightman, R.M. Effect of acute ethanol on striatal dopamine neurotransmission in ambulatory rats. J. Pharmacol. Exp. Ther. 2001, 297, 27–34. [Google Scholar]
- Vengeliene, V.; Bilbao, A.; Molander, A.; Spanagel, R. Neuropharmacology of alcohol addiction. Br. J. Pharmacol. 2008, 154, 299–315. [Google Scholar] [CrossRef]
- Herman, M.A.; Kallupi, M.; Luu, G.; Oleata, C.S.; Heilig, M.; Koob, G.F.; Ciccocioppo, R.; Roberto, M. Enhanced GABAergic transmission in the central nucleus of the amygdala of genetically selected Marchigian Sardinian rats: Alcohol and CRF effects. Neuropharmacology 2013, 67, 337–348. [Google Scholar] [CrossRef]
- McBride, W.J. Central nucleus of the amygdala and the effects of alcohol and alcohol-drinking behavior in rodents. Pharmacol. Biochem. Behav. 2002, 71, 509–515. [Google Scholar] [CrossRef]
- Diaz, M.R.; Morton, R.A. Ethanol untangles the amygdala-anxiety circuit through tonic GABA inhibition. Alcohol. Clin. Exp. Res. 2014, 38, 619–623. [Google Scholar] [CrossRef]
- Roberto, M.; Madamba, S.G.; Stouffer, D.G.; Parsons, L.H.; Siggins, G.R. Increased GABA release in the central amygdala of ethanol-dependent rats. J. Neurosci. 2004, 24, 10159–10166. [Google Scholar] [CrossRef]
- Roberto, M.; Gilpin, N.W.; Siggins, G.R. The central amygdala and alcohol: Role of gamma-aminobutyric acid, glutamate, and neuropeptides. Cold Spring Harb. Perspect. Med. 2012, 2, a012195. [Google Scholar] [CrossRef]
- Chefer, V.; Meis, J.; Wang, G.; Kuzmin, A.; Bakalkin, G.; Shippenberg, T. Repeated exposure to moderate doses of ethanol augments hippocampal glutamate neurotransmission by increasing release. Addict. Biol. 2011, 16, 229–237. [Google Scholar] [CrossRef]
- Martin, D.; Swartzwelder, H.S. Ethanol inhibits release of excitatory amino acids from slices of hippocampal area CA1. Eur. J. Pharmacol. 1992, 219, 469–472. [Google Scholar] [CrossRef]
- Sepulveda, C.; Bustos, G.; Gysling, K.; Seguel, M.; Labarca, R. Effects of in vitro ethanol and chronic ethanol consumption on the release of excitatory amino acids in the rat hippocampus. Brain Res. 1995, 674, 104–106. [Google Scholar] [CrossRef]
- Lee, S.; Rivier, C. Alcohol increases the expression of type 1, but not type 2 alpha corticotropin-releasing factor (CRF) receptor messenger ribonucleic acid in the rat hypothalamus. Brain Res. Mol. Brain Res. 1997, 52, 78–89. [Google Scholar] [CrossRef]
- Rivier, C.L.; Grigoriadis, D.E.; Rivier, J.E. Role of corticotropin-releasing factor receptors type 1 and 2 in modulating the rat adrenocorticotropin response to stressors. Endocrinology 2003, 144, 2396–2403. [Google Scholar] [CrossRef]
- Bagosi, Z.; Jaszberenyi, M.; Bujdoso, E.; Telegdy, G. The effects of corticoptropin-releasing factor and the urocortins on striatal dopamine release induced by electrical stimulation-an in vitro superfusion study. Neurochem. Res. 2006, 31, 209–213. [Google Scholar] [CrossRef]
- Bagosi, Z.; Jaszberenyi, M.; Szabo, G.; Telegdy, G. The effects of CRF and the urocortins on [3H]GABA release from the rat amygdala—An in vitro superfusion study. Brain Res. Bull. 2008, 75, 15–17. [Google Scholar] [CrossRef]
- Bagosi, Z.; Balango, B.; Pinter, D.; Csabafi, K.; Jaszberenyi, M.; Szabo, G.; Telegdy, G. The effects of CRF and urocortins on the hippocampal glutamate release. Neurochem. Int. 2015, 90, 67–71. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 |
|---|---|---|---|---|---|---|
| Group #1 (N = 6) | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip + saline icv at 8:00 + assays at 8:30 | |
| Group #2 (N = 6) | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip + antalarmin icv at 8:00 + assays at 8:30 | |
| Group #3 (N = 6) | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip + astressin2B icv at 8:00 + assays at 8:30 | |
| Group #4 (N = 6) | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip + saline icv at 8:00 + assays at 8:30 | |
| Group #4 (N = 6) | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip + saline icv at 8:00 + assays at 8:30 | |
| Group #5 (N = 6) | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip + antalarmin icv at 8:00 + assays at 8:30 | |
| Group #6 (N = 6) | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip + astressin2B icv at 8:00 + assays at 8:30 | |
| Group #7 (N = 6) | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 | Saline icv at 8:00 + assays at 8:30 |
| Group #8 (N = 6) | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 | Antalarmin icv at 8:00 + assays at 8:30 |
| Group #9 (N = 6) | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 and 20:00 | Saline ip at 8:00 | Astressin2B icv at 8:00 + assays at 8:30 |
| Group #10 (N = 6) | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 | Saline icv at 8:00 + assays at 8:30 |
| Group #11 (N = 6) | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 | Antalarmin icv at 8:00 + assays at 8:30 |
| Group #12 (N = 6) | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 and 20:00 | Alcohol ip at 8:00 | Astressin2B icv at 8:00 + assays at 8:30 |
| Gene | Forward | Reverse |
|---|---|---|
| CRF | 5′-TGG TGT GGA GAA ACT CAG AGC-3′ | 5′-CAT GTT AGG GGC GCT CTC TTC-3′ |
| AVP | 5′-CTG ACA TGG AGC TGA GAC AGT-3′ | 5′-CGC AGC TCT CGT CGC T-3′ |
| Gapdh | 5′-CGG CCA AAT CTG AGG CAA GA-3′ | 5′-TTT TGT GAT GCG TGT GTA GCG-3′ |
| Phase | Temperature °C | Time | Number of Cycles |
|---|---|---|---|
| UDG pre-treatment | 50 | 2 min | 1 |
| Initial denaturation | 95 | 10 min | 1 |
| Denaturation | 95 | 15 s | 40 |
| Annealing | 60 | 30 s | 40 |
| Extension | 72 | 30 s | 40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, B.; Buzás, A.; Bokor, P.; Csabafi, K.; Ibos, K.E.; Bodnár, É.; Török, L.; Földesi, I.; Siska, A.; Bagosi, Z. The Effects of Alcohol Intoxication and Withdrawal on Hypothalamic Neurohormones and Extrahypothalamic Neurotransmitters. Biomedicines 2023, 11, 1288. https://doi.org/10.3390/biomedicines11051288
Simon B, Buzás A, Bokor P, Csabafi K, Ibos KE, Bodnár É, Török L, Földesi I, Siska A, Bagosi Z. The Effects of Alcohol Intoxication and Withdrawal on Hypothalamic Neurohormones and Extrahypothalamic Neurotransmitters. Biomedicines. 2023; 11(5):1288. https://doi.org/10.3390/biomedicines11051288
Chicago/Turabian StyleSimon, Balázs, András Buzás, Péter Bokor, Krisztina Csabafi, Katalin Eszter Ibos, Éva Bodnár, László Török, Imre Földesi, Andrea Siska, and Zsolt Bagosi. 2023. "The Effects of Alcohol Intoxication and Withdrawal on Hypothalamic Neurohormones and Extrahypothalamic Neurotransmitters" Biomedicines 11, no. 5: 1288. https://doi.org/10.3390/biomedicines11051288
APA StyleSimon, B., Buzás, A., Bokor, P., Csabafi, K., Ibos, K. E., Bodnár, É., Török, L., Földesi, I., Siska, A., & Bagosi, Z. (2023). The Effects of Alcohol Intoxication and Withdrawal on Hypothalamic Neurohormones and Extrahypothalamic Neurotransmitters. Biomedicines, 11(5), 1288. https://doi.org/10.3390/biomedicines11051288