
Pre-Processing a Polymer Blend into a Polymer Alloy by KinetiSol Enables Increased Ivacaftor Amorphous Solid Dispersion Drug Loading and Dissolution

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. KinetiSol Compounding
2.3. Dynamic Vapor Sorption (DVS)
2.4. High-Performance Liquid Chromatography (HPLC)
2.5. Modulated Differential Scanning Calorimetry (mDSC)
2.6. Powder X-ray Diffraction (PXRD)
2.7. Fourier Transform Infra-Red (FTIR) Spectroscopy
2.8. Tableting
2.9. Dissolution Study
2.10. Rhodos Particle Size Analysis
2.11. Solid-State Nuclear Magnetic Resonance (ssNMR)
3. Results and Discussion
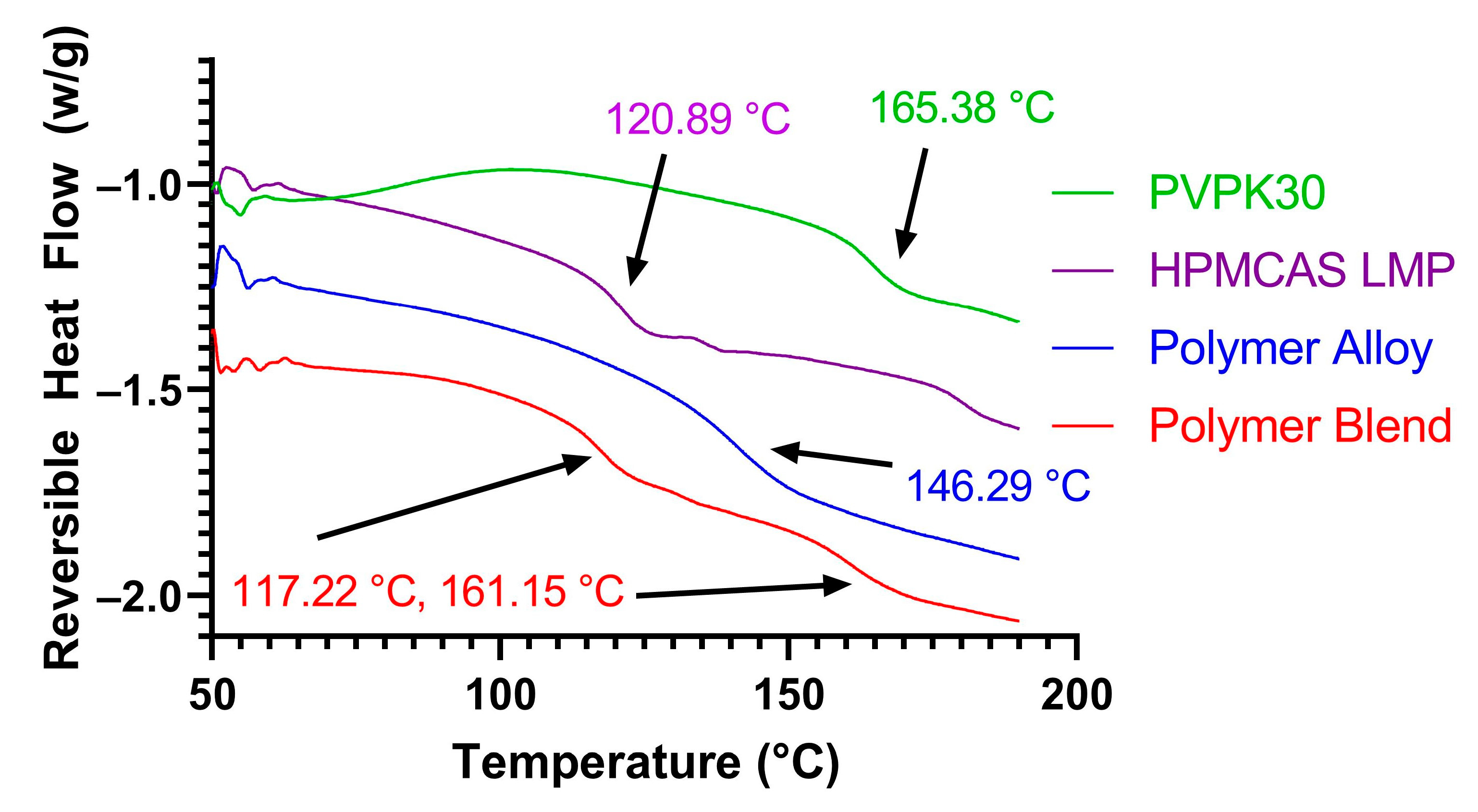
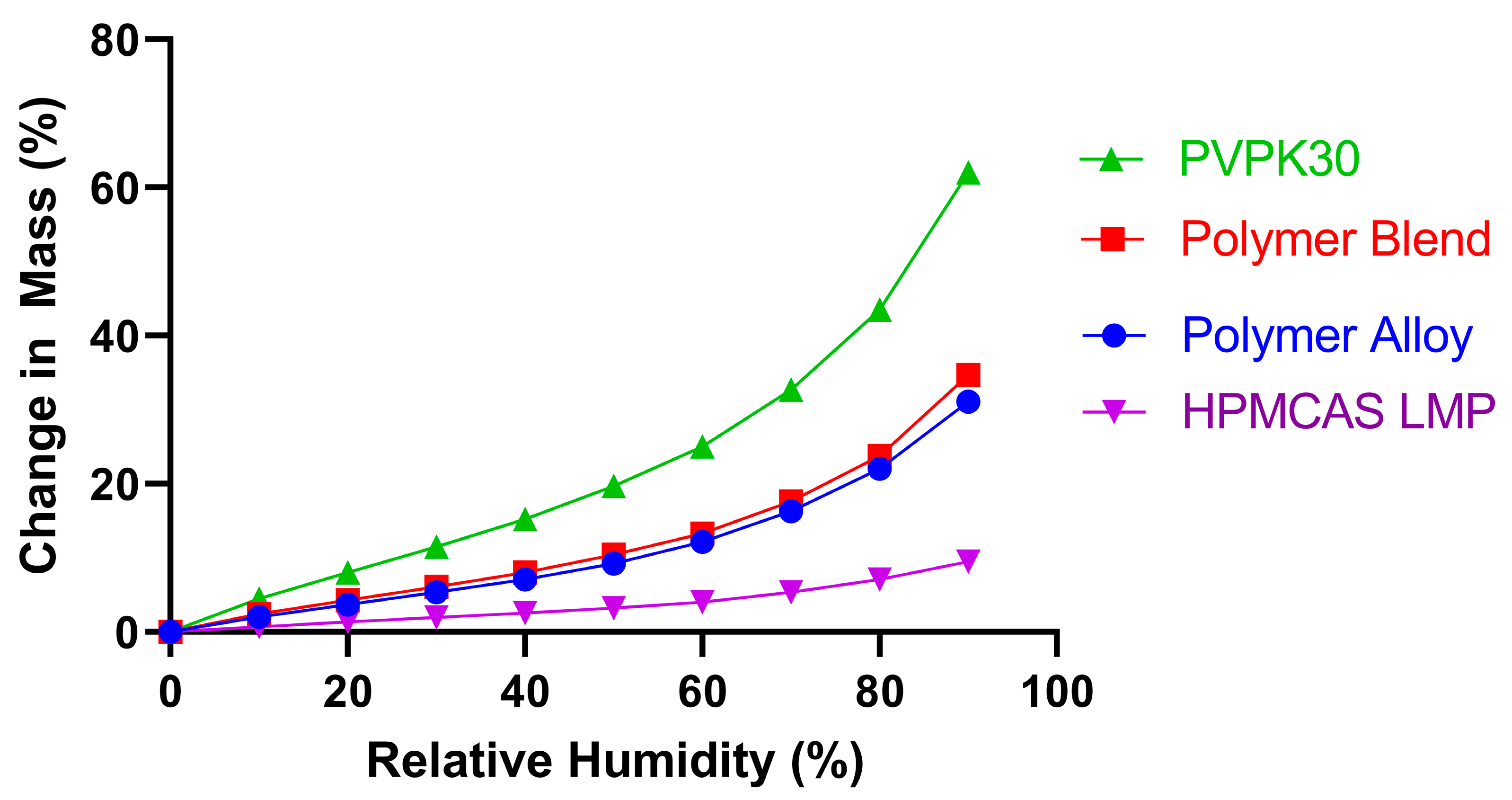
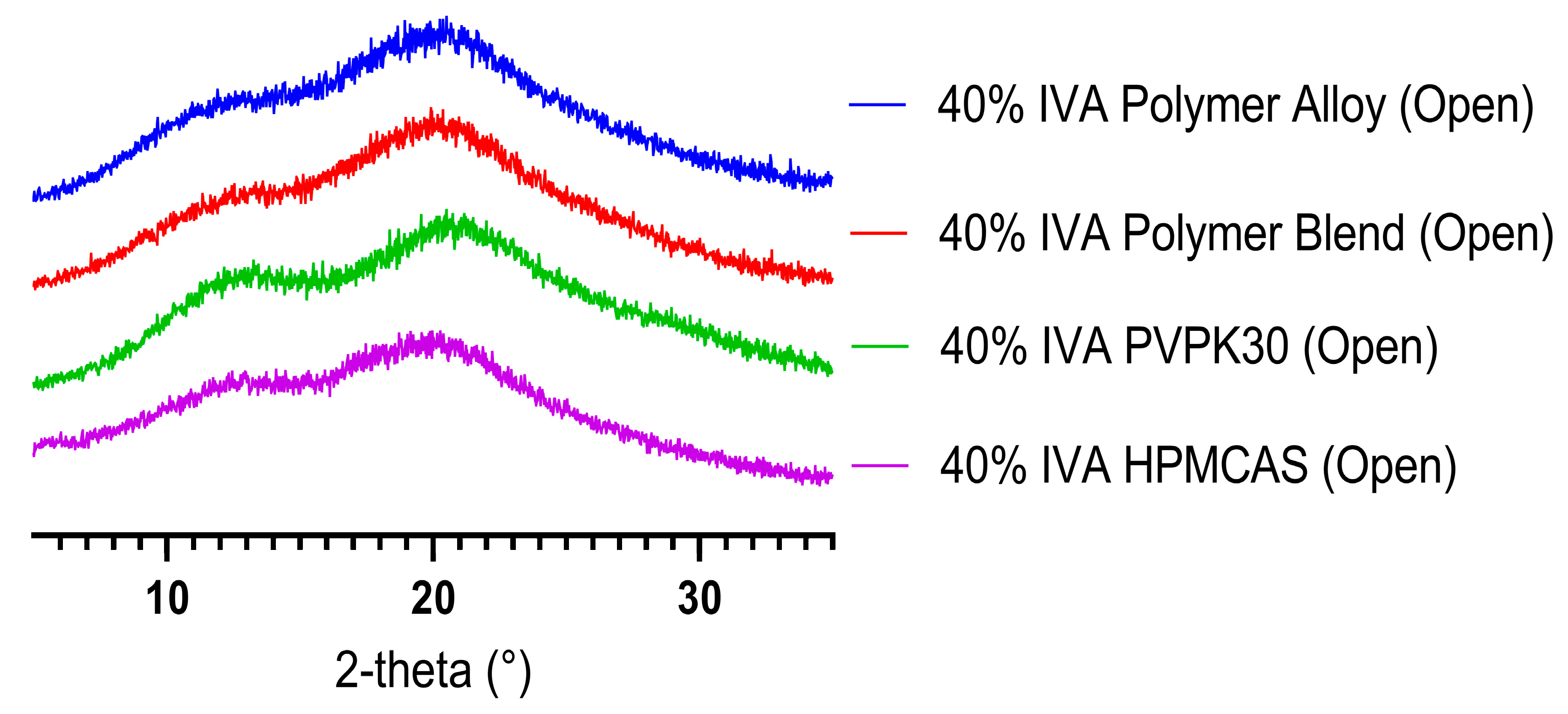
3.1. KinetiSol Compounding Creates a Single-Phase Polymer Alloy
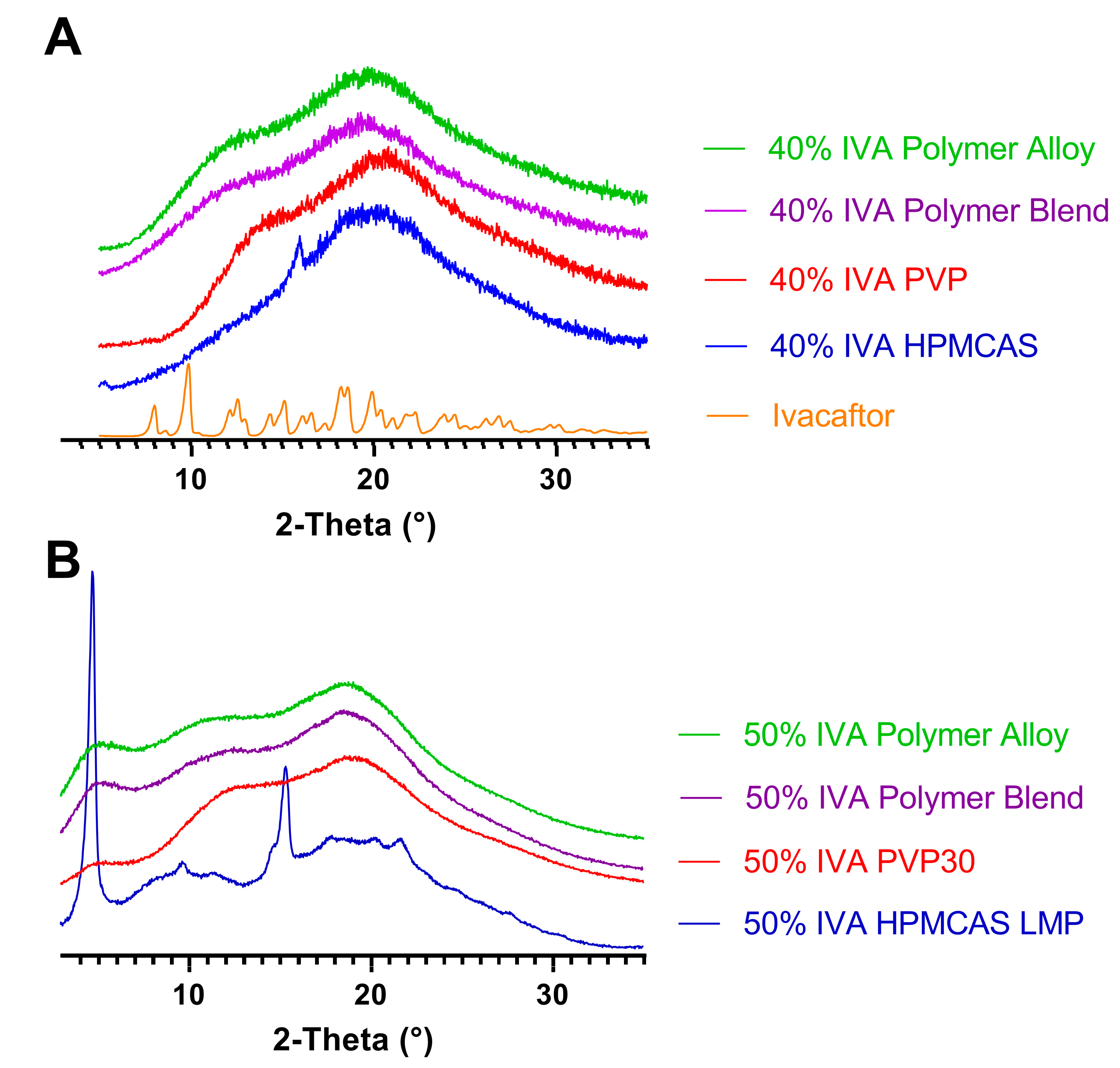
3.2. The Polymer Alloy Method Increases the Maximum Drug Loading in KinetiSol Ivacaftor ASDs
3.3. Increased Particle Size of Polymers during KinetiSol Compounding Increases Maximum Drug Loading
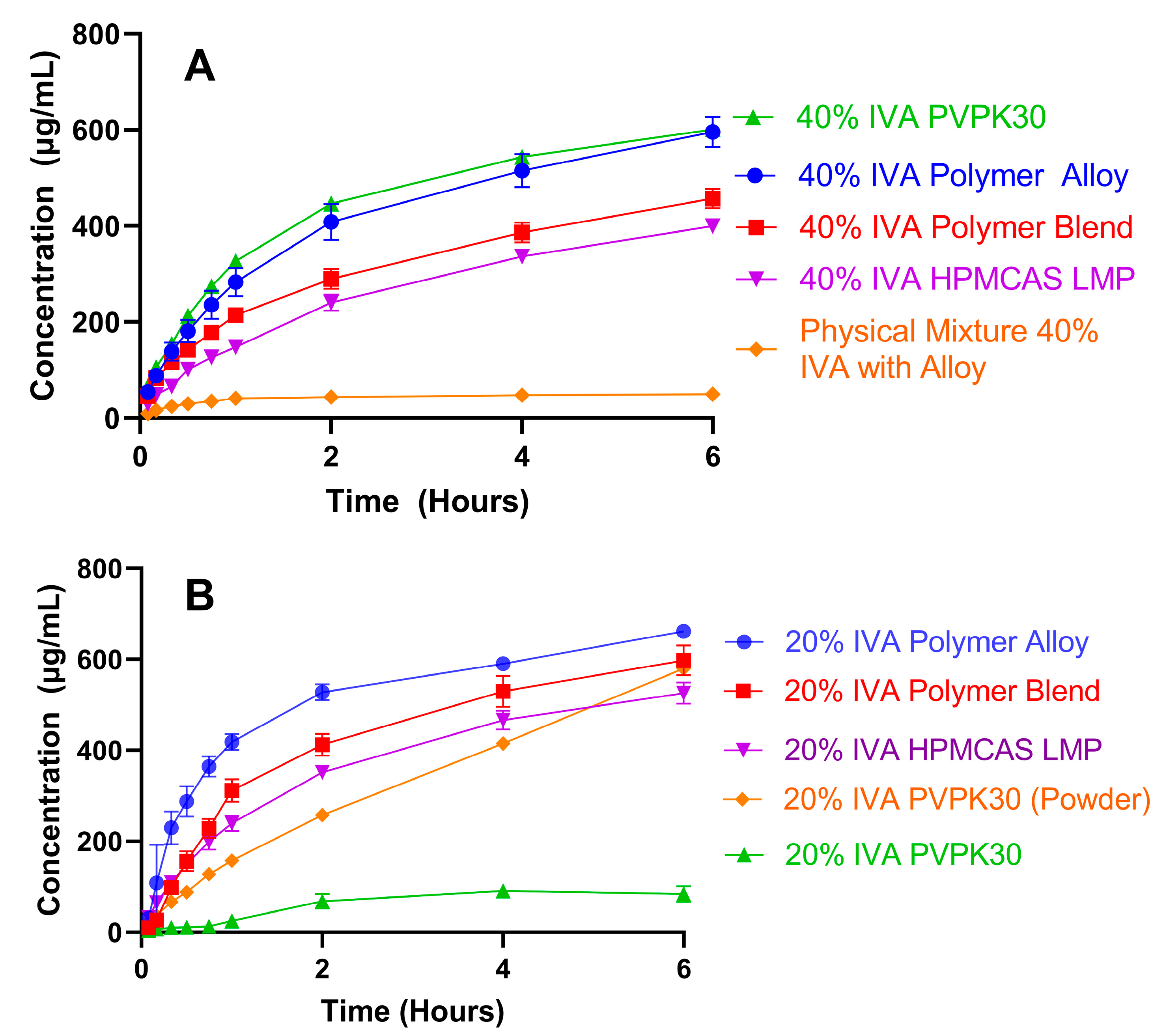
3.4. High Drug-Load KinetiSol ASDs Achieve High and Sustained Supersaturation
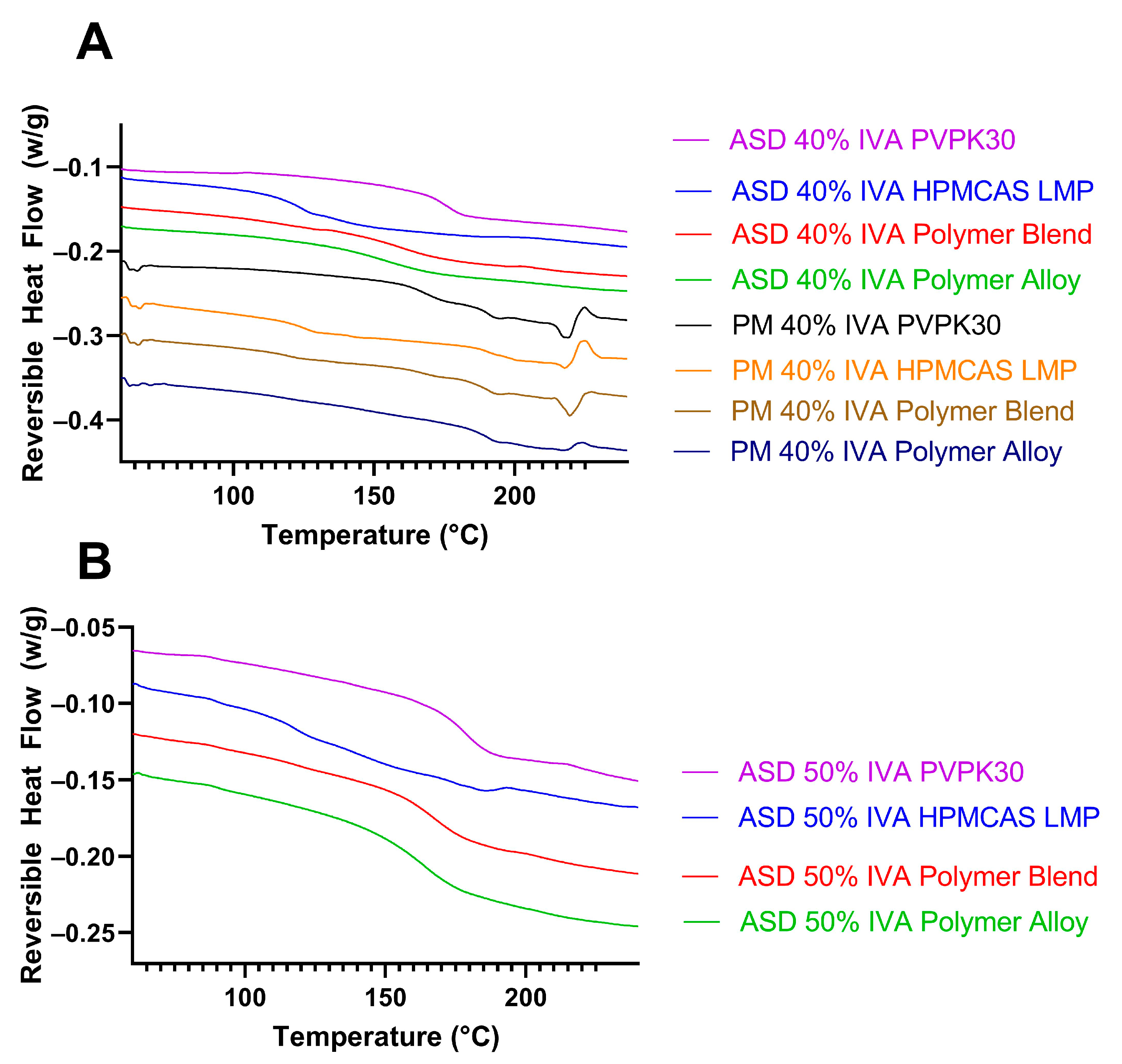
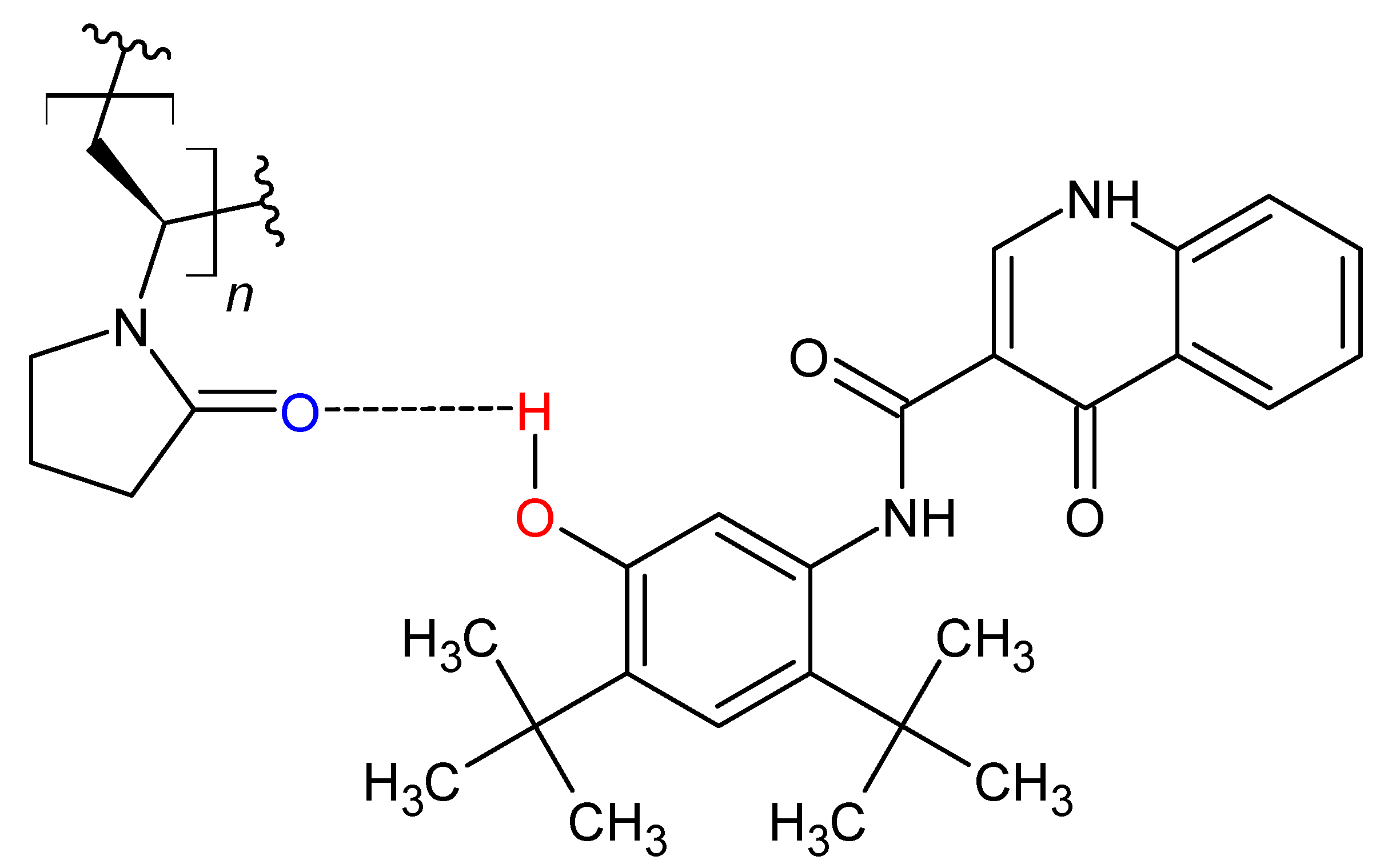
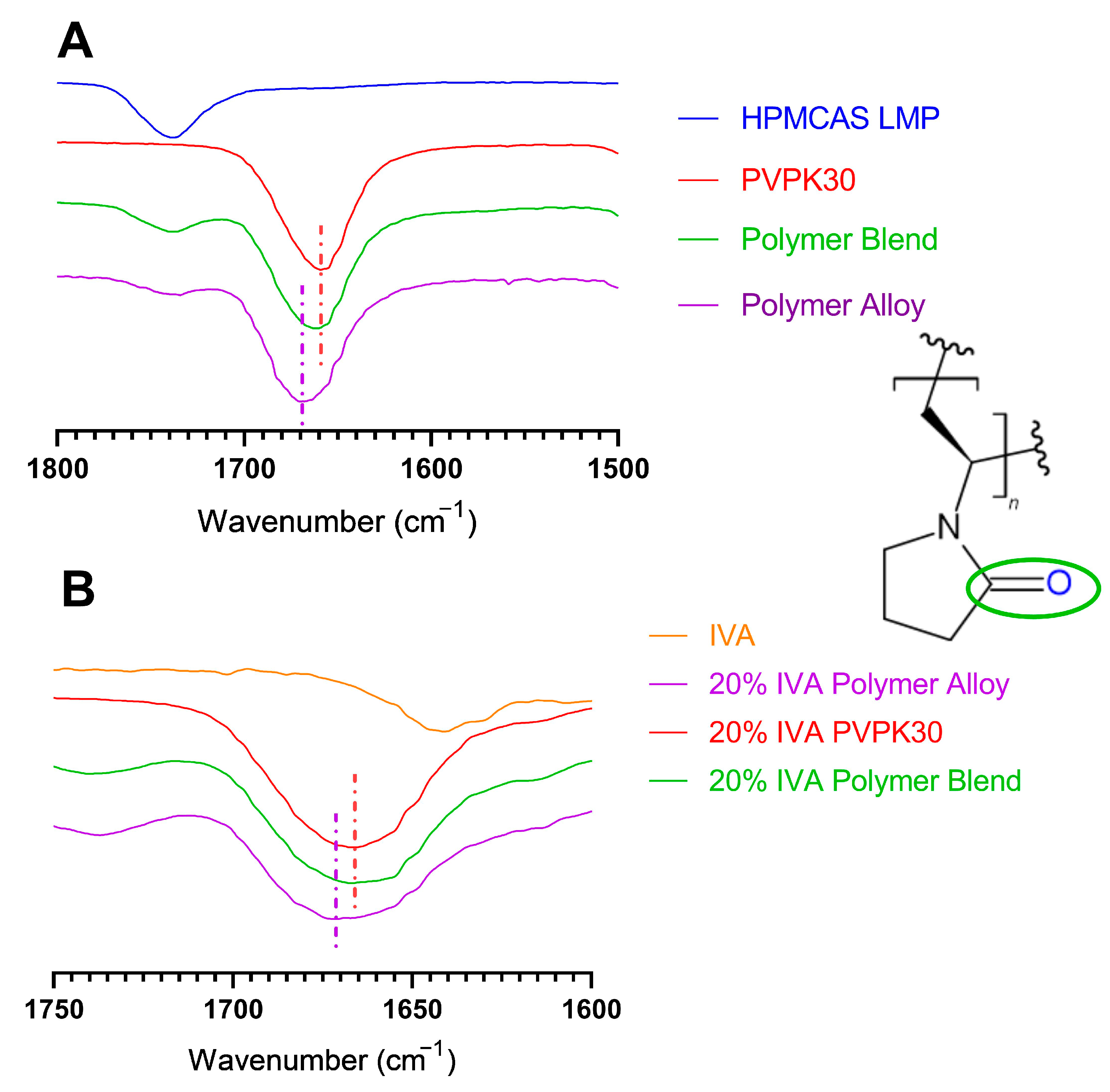
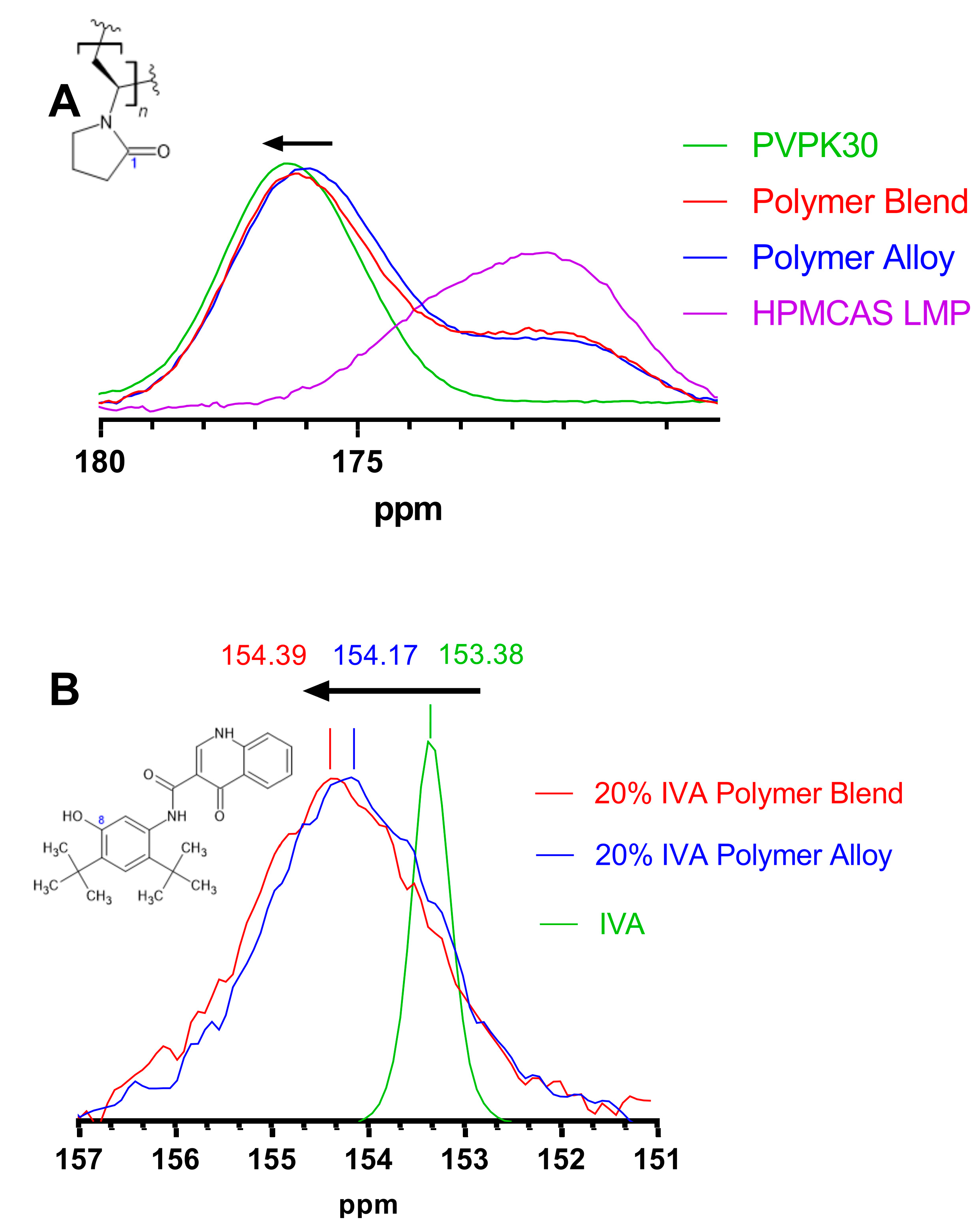
3.5. Modification of Hydrogen Bonding in Polymer Alloy Explains Dissolution Benefit
3.6. Ivacaftor ASDs Exhibit High Stability under Accelerated Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jermain, S.V.; Brough, C.; Williams, R.O. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery—An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef]
- Agarwal, P.; Huckle, J.; Newman, J.; Reid, D.L. Trends in small molecule drug properties: A developability molecule assessment perspective. Drug Discov. Today 2022, 27, 103366. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.A.; Davis, D.A.; Moon, C.; Williams, R.O. Increasing Drug Loading of Weakly Acidic Telmisartan in Amorphous Solid Dispersions through pH Modification during Hot-Melt Extrusion. Mol. Pharm. 2022, 19, 318–331. [Google Scholar] [CrossRef]
- Mosquera-Giraldo, L.I.; Taylor, L.S. Glass-liquid phase separation in highly supersaturated aqueous solutions of telaprevir. Mol. Pharm. 2015, 12, 496–503. [Google Scholar] [CrossRef]
- Xiang, T.X.; Anderson, B.D. Effects of Molecular Interactions on Miscibility and Mobility of Ibuprofen in Amorphous Solid Dispersions With Various Polymers. J. Pharm. Sci. 2019, 108, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.; Song, H.; Wang, Y.; Gao, H.; Fu, Q. Lacidipine Amorphous Solid Dispersion Based on Hot Melt Extrusion: Good Miscibility, Enhanced Dissolution, and Favorable Stability. AAPS PharmSciTech 2018, 19, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Lehmkemper, K.; Kyeremateng, S.O.; Bartels, M.; Degenhardt, M.; Sadowski, G. Physical stability of API/polymer-blend amorphous solid dispersions. Eur. J. Pharm. Biopharm. 2018, 124, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, S.; Wang, S.; Liu, C.; Su, C.; Hageman, M.; Hussain, M.; Haskell, R.; Stefanski, K.; Qian, F. Initial Drug Dissolution from Amorphous Solid Dispersions Controlled by Polymer Dissolution and Drug-Polymer Interaction. Pharm. Res. 2016, 33, 2445–2458. [Google Scholar] [CrossRef]
- Yuan, X.; Sperger, D.; Munson, E.J. Investigating miscibility and molecular mobility of nifedipine-PVP amorphous solid dispersions using solid-state NMR spectroscopy. Mol. Pharm. 2014, 11, 329–337. [Google Scholar] [CrossRef]
- Li, Y.; Pang, H.; Guo, Z.; Lin, L.; Dong, Y.; Li, G.; Lu, M.; Wu, C. Interactions between drugs and polymers influencing hot melt extrusion. J. Pharm. Pharmacol. 2014, 66, 148–166. [Google Scholar] [CrossRef]
- Yang, R.; Mann, A.K.P.; Van Duong, T.; Ormes, J.D.; Okoh, G.A.; Hermans, A.; Taylor, L.S. Drug Release and Nanodroplet Formation from Amorphous Solid Dispersions: Insight into the Roles of Drug Physicochemical Properties and Polymer Selection. Mol. Pharm. 2021, 18, 2066–2081. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, W.; Nightingale, J.A.; Herbig, S.M. Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieu. Pharm. Res. 2009, 26, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.S.; Kumar, S.; Zhou, Q.T. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm. Sin. B 2021, 11, 2505–2536. [Google Scholar] [CrossRef] [PubMed]
- Shepard, K.B.; Pluntze, A.M.; Vodak, D.T. Simultaneous Spray Drying for Combination Dry Powder Inhaler Formulations. Pharmaceutics 2022, 14, 1130. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Meena, A.; Parikh, T.; Serajuddin, A.T.M. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion-I: Polyvinylpyrrolidone and related polymers. J. Excip. Food Chem. 2016, 5, 1001. [Google Scholar]
- Anwer, M.K.; Ahmed, M.M.; Alshetaili, A.; Almutairy, B.K.; Alalaiwe, A.; Fatima, F.; Ansari, M.N.; Iqbal, M. Preparation of spray dried amorphous solid dispersion of diosmin in soluplus with improved hepato-renoprotective activity: In vitro anti-oxidant and in-vivo safety studies. J. Drug Deliv. Sci. Technol. 2020, 60, 102101. [Google Scholar] [CrossRef]
- Brough, C.; Miller, D.A.; Keen, J.M.; Kucera, S.A.; Lubda, D.; Williams, R.O. Use of Polyvinyl Alcohol as a Solubility-Enhancing Polymer for Poorly Water Soluble Drug Delivery (Part 1). AAPS PharmSciTech 2016, 17, 167–179. [Google Scholar] [CrossRef]
- Tan, D.K.; Davis, D.A.; Miller, D.A.; Williams, R.O.; Nokhodchi, A. Innovations in Thermal Processing: Hot-Melt Extrusion and KinetiSol® Dispersing. AAPS PharmSciTech 2020, 21, 1933–1956. [Google Scholar] [CrossRef]
- Ellenberger, D.J.; Miller, D.A.; Williams, R.O. Expanding the Application and Formulation Space of Amorphous Solid Dispersions with KinetiSol®: A Review. AAPS PharmSciTech 2018, 19, 1933–1956. [Google Scholar] [CrossRef]
- Albadarin, A.B.; Potter, C.B.; Davis, M.T.; Iqbal, J.; Korde, S.; Pagire, S.; Paradkar, A.; Walker, G. Development of stability-enhanced ternary solid dispersions via combinations of HPMCP and Soluplus® processed by hot melt extrusion. Int. J. Pharm. 2017, 532, 603–611. [Google Scholar] [CrossRef]
- Hörmann, T.R.; Jäger, N.; Funke, A.; Mürb, R.K.; Khinast, J.G.; Paudel, A. Formulation performance and processability window for manufacturing a dual-polymer amorphous solid dispersion via hot-melt extrusion and strand pelletization. Int. J. Pharm. 2018, 553, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.A.; Wegiel, L.A.; Taylor, L.S.; Edgar, K.J. Pairwise polymer blends for oral drug delivery. J. Pharm. Sci. 2014, 103, 2871–2883. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nollenberger, K.; Albers, J.; Craig, D.; Qi, S. Microstructure of an immiscible polymer blend and its stabilization effect on amorphous solid dispersions. Mol. Pharm. 2013, 10, 2767–2780. [Google Scholar] [CrossRef] [PubMed]
- Danda, L.J.D.A.; Batista, L.D.M.; Melo, V.C.S.; Sobrinho, J.L.S.; Soares, M.F.D.L.R. Combining amorphous solid dispersions for improved kinetic solubility of posaconazole simultaneously released from soluble PVP/VA64 and an insoluble ammonio methacrylate copolymer. Eur. J. Pharm. Sci. 2019, 133, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Meckel, J.; Zhang, F. Investigation of itraconazole ternary amorphous solid dispersions based on povidone and Carbopol. Eur. J. Pharm. Sci. 2017, 106, 413–421. [Google Scholar] [CrossRef]
- Deeks, E.D. Ivacaftor: A review of its use in patients with cystic fibrosis. Drugs 2013, 73, 1595–1604. [Google Scholar] [CrossRef]
- Rowe, W.; Hurter, P.; Young, C.; Dinehart, K.; Verwijs, M.J.; Overhoff, K.; Grootenhuis, P.D.J.; Botfield, M.; Grossi, A. Pharmaceutical Composition and Administrations Thereof. U.S. Patent 10646481 B2, 12 May 2020. [Google Scholar]
- LaFountaine, J.S.; McGinity, J.W.; Williams, R.O. Challenges and Strategies in Thermal Processing of Amorphous Solid Dispersions: A Review. AAPS PharmSciTech 2016, 17, 43–55. [Google Scholar] [CrossRef]
- Matsumoto, A.; Matsukawa, Y.; Suzuki, T.; Yoshino, H.; Kobayashi, M. The polymer-alloys method as a new preparation method of biodegradable microspheres: Principle and application to cisplatin-loaded microspheres. J. Control. Release 1997, 48, 19–27. [Google Scholar] [CrossRef]
- Janssens, S.; De Zeure, A.; Paudel, A.; Van Humbeeck, J.; Rombaut, P.; Van Den Mooter, G. Influence of preparation methods on solid state supersaturation of amorphous solid dispersions: A case study with itraconazole and eudragit E100. Pharm. Res. 2010, 27, 775–785. [Google Scholar] [CrossRef]
- Saboo, S.; Moseson, D.E.; Kestur, U.S.; Taylor, L.S. Patterns of drug release as a function of drug loading from amorphous solid dispersions: A comparison of five different polymers. Eur. J. Pharm. Sci. 2020, 155, 105514. [Google Scholar] [CrossRef]
- Hadida, S.; Van Goor, F.; Dinehart, K.; Looker, A.R.; Mueller, P.; Grootenhuis, P.D.J. Case History: Kalydeco® (VX-770, Ivacaftor), a CFTR potentiator for the treatment of patients with cystic fibrosis and the G551D-CFTR mutation. Annu. Rep. Med. Chem. 2014, 49, 383–398. [Google Scholar] [CrossRef]
- Dedroog, S.; Pas, T.; Vergauwen, B.; Huygens, C.; Van den Mooter, G. Solid-state analysis of amorphous solid dispersions: Why DSC and XRPD may not be regarded as stand-alone techniques. J. Pharm. Biomed. Anal. 2020, 178, 112937. [Google Scholar] [CrossRef] [PubMed]
- Chasse, T.; Conway, S.L.; Danzer, G.D.; Feng, L.; Leone, A.M.; McNevin, M.; Smoliga, J.; Stroud, P.A.; van Lishaut, H. Industry white paper: Contemporary opportunities and challenges in characterizing crystallinity in amorphous solid dispersions. J. Pharm. Sci. 2022, 111, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- Bookwala, M.; Buckner, I.S.; Wildfong, P.L.D. Implications of Coexistent Halogen and Hydrogen Bonds in Amorphous Solid Dispersions on Drug Solubility, Miscibility, and Mobility. Mol. Pharm. 2022, 19, 3959–3972. [Google Scholar] [CrossRef]
- BASF. Technical Information (WF-No. 137192). Kollidon ® 25 Kollidon ® 30 Kollidon ® 30 LP Kollidon ® 90 F. 2019, pp. 1–9. Available online: https://pharma.basf.com/products/kollidon-30-1 (accessed on 12 April 2023).
- Honick, M.; Das, S.; Hoag, S.W.; Muller, F.X.; Alayoubi, A.; Feng, X.; Zidan, A.; Ashraf, M.; Polli, J.E. The effects of spray drying, HPMCAS grade, and compression speed on the compaction properties of itraconazole-HPMCAS spray dried dispersions. Eur. J. Pharm. Sci. 2020, 155, 105556. [Google Scholar] [CrossRef]
- Thompson, S.A.; Williams, R.O. Specific mechanical energy—An essential parameter in the processing of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2021, 173, 374–393. [Google Scholar] [CrossRef]
- Goh, H.P.; Heng, P.W.S.; Liew, C.V. Comparative evaluation of powder flow parameters with reference to particle size and shape. Int. J. Pharm. 2018, 547, 133–141. [Google Scholar] [CrossRef]
- Butreddy, A.; Sarabu, S.; Bandari, S.; Batra, A.; Lawal, K.; Chen, N.N.; Bi, V.; Durig, T.; Repka, M.A. Influence of PlasdoneTM S630 Ultra—An Improved Copovidone on the Processability and Oxidative Degradation of Quetiapine Fumarate Amorphous Solid Dispersions Prepared via Hot-Melt Extrusion Technique. AAPS PharmSciTech 2021, 22, 818–827. [Google Scholar] [CrossRef]
- Shiyani, B.; Gattani, S.; Surana, S. Formulation and Evaluation of Bi-layer Tablet of Metoclopramide Hydrochloride and Ibuprofen. AAPS PharmSciTech 2008, 9, 818–827. [Google Scholar] [CrossRef]
- Keen, J.M.; Hughey, J.R.; Bennett, R.C.; Jannin, V.; Rosiaux, Y.; Marchaud, D.; McGinity, J.W. Effect of tablet structure on controlled release from supersaturating solid dispersions containing glyceryl behenate. Mol. Pharm. 2015, 12, 120–126. [Google Scholar] [CrossRef]
- Que, C.; Deac, A.; Zemlyanov, D.Y.; Qi, Q.; Indulkar, A.S.; Gao, Y.; Zhang, G.G.Z.; Taylor, L.S. Impact of Drug–Polymer Intermolecular Interactions on Dissolution Performance of Copovidone-Based Amorphous Solid Dispersions. Mol. Pharm. 2021, 18, 3496–3508. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, L.A.; Zhao, Y.; Mauer, L.J.; Edgar, K.J.; Taylor, L.S. Curcumin amorphous solid dispersions: The influence of intra and intermolecular bonding on physical stability. Pharm. Dev. Technol. 2014, 19, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.S.; Zografi, G. Spectroscopic Characterization of Interactions Between PVP and Indomethacin in Amorphous Molecular Dispersions. Pharm. Res. 1997, 14, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Yuan, H.; Yu, S.; Mao, S.; Tony Zhou, Q. Spray dried inhalable ivacaftor co-amorphous microparticle formulations with leucine achieved enhanced in vitro dissolution and superior aerosol performance. Int. J. Pharm. 2022, 622, 121859. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Meng, F.; Tsutsumi, Y.; Amoureux, J.-P.; Xu, W.; Lu, X.; Zhang, F.; Su, Y. Understanding Molecular Interactions in Rafoxanide–Povidone Amorphous Solid Dispersions from Ultrafast Magic Angle Spinning NMR. Mol. Pharm. 2020, 17, 2196–2207. [Google Scholar] [CrossRef]
- Wang, J.; Cheung, M.K.; Mi, Y. Miscibility and morphology in crystalline/amorphous blends of poly (caprolactone)/poly(4-vinylphenol) as studied by DSC, FTIR, and 13C solid state NMR. Polymer 2002, 43, 1357–1364. [Google Scholar] [CrossRef]
- Yuan, X.; Xiang, T.X.; Anderson, B.D.; Munson, E.J. Hydrogen Bonding Interactions in Amorphous Indomethacin and Its Amorphous Solid Dispersions with Poly(vinylpyrrolidone) and Poly(vinylpyrrolidone-co-vinyl acetate) Studied Using 13C Solid-State NMR. Mol. Pharm. 2015, 12, 4518–4528. [Google Scholar] [CrossRef]
- Saboo, S.; Kestur, U.S.; Flaherty, D.P.; Taylor, L.S. Congruent Release of Drug and Polymer from Amorphous Solid Dispersions: Insights into the Role of Drug-Polymer Hydrogen Bonding, Surface Crystallization, and Glass Transition. Mol. Pharm. 2020, 17, 1261–1275. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Long-Term Physical Stability of PVP- and PVPVA-Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 157–171. [Google Scholar] [CrossRef]
- Patel, N.G.; Serajuddin, A.T.M. Moisture sorption by polymeric excipients commonly used in amorphous solid dispersion and its effect on glass transition temperature: I. Polyvinylpyrrolidone and related copolymers. Int. J. Pharm. 2022, 616, 121532. [Google Scholar] [CrossRef]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular Mobility of Amorphous Pharmaceutical Solids Below Their Glass Transition Temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Composition (% w/w) | ||||
|---|---|---|---|---|---|
| IVA | HPMCAS LMP | PVPK30 | SLS | Polymer Alloy | |
| Polymer Alloy | - | 49.5 | 49.5 | 1 | - |
| 20% IVA HPMCAS | 20 | 79.5 | - | 0.5 | - |
| 20% IVA PVPK30 | 20 | - | 79.5 | 0.5 | - |
| 20% IVA Polymer Blend | 20 | 39.75 | 39.75 | 0.5 | - |
| 20% IVA Polymer Alloy | 20 | - | - | - | 80 |
| 40% IVA HPMCAS | 40 | 59.5 | - | 0.5 | - |
| 40% IVA PVPK30 | 40 | - | 59.5 | 0.5 | - |
| 40% IVA Polymer Blend | 40 | 29.75 | 29.75 | 0.5 | - |
| 40% IVA Polymer Alloy | 40 | - | - | - | 60 |
| 50% IVA HPMCAS | 50 | 49.5 | - | 0.5 | - |
| 50% IVA PVPK30 | 50 | - | 49.5 | 0.5 | - |
| 50% IVA Polymer Blend | 50 | 24.75 | 24.75 | 0.5 | - |
| 50% IVA Polymer Alloy | 50 | - | - | - | 50 |
| Time (min) | Mobile Phase A (%) | Mobile Phase B (%) |
|---|---|---|
| 0.0 | 75 | 25 |
| 6.0 | 75 | 25 |
| 24.0 | 30 | 70 |
| 36.0 | 10 | 90 |
| 42.0 | 10 | 90 |
| 43.0 | 75 | 25 |
| 48.0 | 75 | 25 |
| Identity | Polymer Alloy (Tube-Milled) | Polymer Alloy (Cryo-Milled) |
|---|---|---|
| D10 (µm) | 31.04 | 1.12 |
| D50 (µm) | 112.57 | 10.09 |
| D90 (µm) | 166.83 | 54.71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, S.A.; Davis, D.A., Jr.; Miller, D.A.; Kucera, S.U.; Williams, R.O., III. Pre-Processing a Polymer Blend into a Polymer Alloy by KinetiSol Enables Increased Ivacaftor Amorphous Solid Dispersion Drug Loading and Dissolution. Biomedicines 2023, 11, 1281. https://doi.org/10.3390/biomedicines11051281
Thompson SA, Davis DA Jr., Miller DA, Kucera SU, Williams RO III. Pre-Processing a Polymer Blend into a Polymer Alloy by KinetiSol Enables Increased Ivacaftor Amorphous Solid Dispersion Drug Loading and Dissolution. Biomedicines. 2023; 11(5):1281. https://doi.org/10.3390/biomedicines11051281
Chicago/Turabian StyleThompson, Stephen A., Daniel A. Davis, Jr., Dave A. Miller, Sandra U. Kucera, and Robert O. Williams, III. 2023. "Pre-Processing a Polymer Blend into a Polymer Alloy by KinetiSol Enables Increased Ivacaftor Amorphous Solid Dispersion Drug Loading and Dissolution" Biomedicines 11, no. 5: 1281. https://doi.org/10.3390/biomedicines11051281
APA StyleThompson, S. A., Davis, D. A., Jr., Miller, D. A., Kucera, S. U., & Williams, R. O., III. (2023). Pre-Processing a Polymer Blend into a Polymer Alloy by KinetiSol Enables Increased Ivacaftor Amorphous Solid Dispersion Drug Loading and Dissolution. Biomedicines, 11(5), 1281. https://doi.org/10.3390/biomedicines11051281