
Technological Aspects and Evaluation Methods for Polymer Matrices as Dental Drug Carriers


Abstract
1. Introduction
2. Methods for Preparing Polymeric Carriers—Technologies and Parameters
2.1. Solvent-Casting Method (SCM)
2.2. Freeze-Casting Method
2.3. Electrospinning
2.4. 3D Printing
3. Assessment Methods for Polymer-Based Dental Carriers
3.1. Physicochemical Properties of Matrices
3.1.1. Morphology of the Surface of Fabricated Matrices and Component Distribution
3.1.2. Pore Size and Volume Assessment
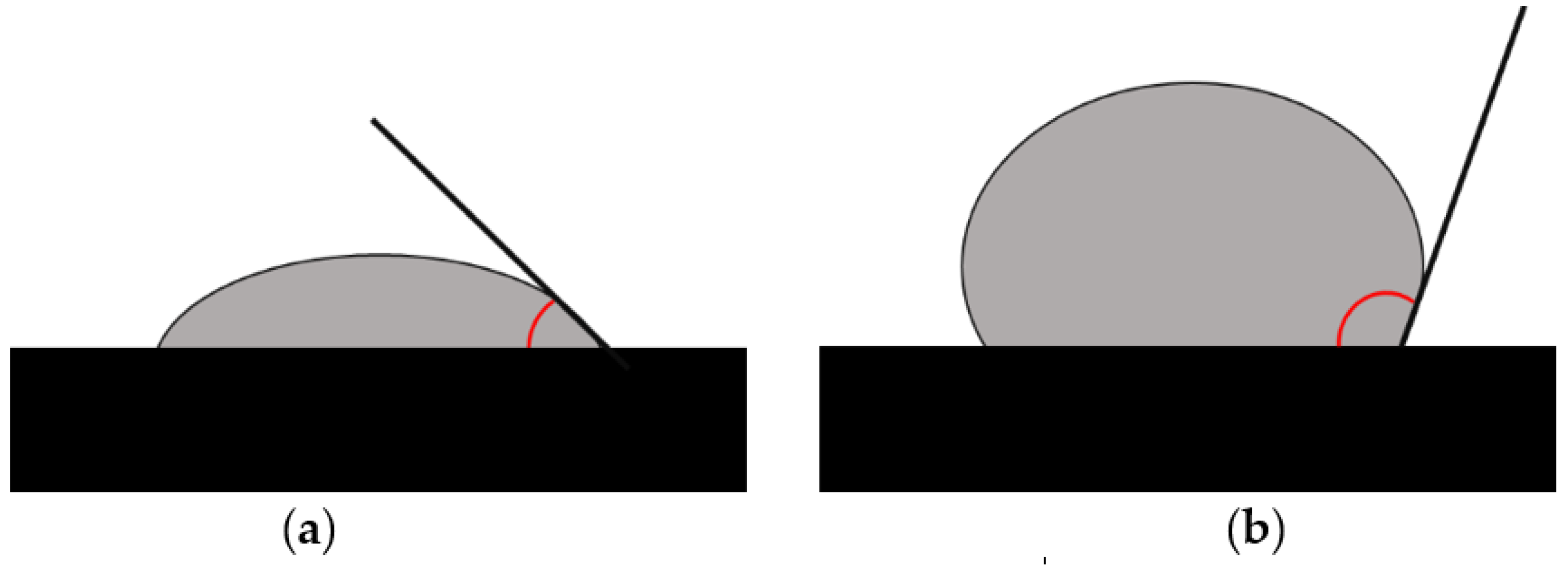
3.1.3. Assessment of Hydrophobic/Hydrophilic Properties
3.1.4. Testing of Mechanical Properties
3.1.5. Matrix Swelling and Leaching/Disintegration Assessment
3.1.6. Mucoadhesion Evaluation
3.1.7. Methods for Assessing Drug–Excipient Interactions and the Stability and Physical State of Drugs in the Formulation
3.2. Pharmaceutical Properties
3.2.1. Drug Load Uniformity
3.2.2. Study of Drug Release/Pharmaceutical Availability of API in Developed Carriers
3.2.3. Stability Study
3.3. Biological Properties
3.3.1. Evaluation of the Antibacterial/Antifungal Activity of the Formulations
3.3.2. Cytotoxicity Testing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | Test Category | Assay/Detection Method |
|---|---|---|
| NR Neutral red | colorimetric |
|
| LDH Dehydrogenase lactate | colorimetric |
|
| NAG N-Acetyl-β-D-Glucosaminidase | colorimetric |
|
| SRB Sulforhodamine B | colorimetric |
|
| ATP Adenosine triphosphate | bioluminescent |
|
| MTT/MTS | colorimetric |
|
4. In Vivo Assessment of Polymer-Based Matrices
4.1. Evaluation of Therapeutic Effects of the Examined Material In Vivo
4.2. Clinical Studies
5. Conclusions
- -
- Homogeneity of their structure;
- -
- API content of the carrier;
- -
- API stability, including temperature-sensitive substances;
- -
- High drug loading;
- -
- Elimination, for example, of residual solvents.
Author Contributions
Funding
Conflicts of Interest
References
- Paderini, C.; Compilato, D.; Giannola, L.I.; Campisi, G. Oral local drug delivery and new perspectives in oral drug formulation. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2012, 114, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Zięba, M.; Chaber, P.; Duale, K.; Martinka Maksymiak, M.; Basczok, M.; Kowalczuk, M.; Adamus, G. Polymeric Carriers for Delivery Systems in the Treatment of Chronic Periodontal Disease. Polymers 2020, 12, 1574. [Google Scholar] [CrossRef] [PubMed]
- Baranov, N.; Popa, M.; Atanase, L.I.; Ichim, D.L. Polysaccharide-Based Drug Delivery Systems for the Treatment of Periodontitis. Molecules 2021, 26, 2735. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Brizuela, M.; Raja, A. Oral Candidiasis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK545282/ (accessed on 13 April 2023).
- Lin, G.S.S.; Luddin, N.; Ghani, H.A.; Lai, J.C.H.; Noorani, T.Y. Dislodgment Resistance, Adhesive Pattern, and Dentinal Tubule Penetration of a Novel Experimental Algin Biopolymer-Incorporated Bioceramic-Based Root Canal Sealer. Polymers 2023, 15, 1317. [Google Scholar] [CrossRef] [PubMed]
- Raj, G.; Raj, M. Oral Lichen Planus. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK578201/ (accessed on 13 April 2023).
- Remiro, P.d.F.R.; Nagahara, M.H.T.; Azoubel, R.A.; Franz-Montan, M.; d’Ávila, M.A.; Moraes, Â.M. Polymeric Biomaterials for Topical Drug Delivery in the Oral Cavity: Advances on Devices and Manufacturing Technologies. Pharmaceutics 2023, 15, 12. [Google Scholar] [CrossRef]
- Szulc, M.; Zakrzewska, A.; Zborowski, J. Local drug delivery in periodontitis treatment: A review of contemporary literature. Dent. Med. Probl. 2018, 55, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Puertas-Bartolomé, M.; Mora-Boza, A.; García-Fernández, L. Emerging Biofabrication Techniques: A Review on Natural Polymers for Biomedical Applications. Polymers 2021, 13, 1209. [Google Scholar] [CrossRef]
- Abouhussein, D.; El Nabarawi, M.A.; Shalaby, S.H.; El-Bary, A.A. Cetylpyridinium chloride chitosan blended mucoadhesive buccal films for treatment of pediatric oral diseases. J. Drug Deliv. Sci. Technol. 2020, 57, 101676. [Google Scholar] [CrossRef]
- Khan, G.; Yadav, S.K.; Patel, R.R.; Kumar, N.; Bansal, M.; Mishra, B. Tinidazole functionalized homogeneous electrospun chitosan/poly(ε-caprolactone) hybrid nanofiber membrane: Development, optimization and its clinical implications. Int. J. Biol. Macromol. 2017, 103, 1311–1326. [Google Scholar] [CrossRef]
- Ribeiro Costa, J.; Oliveira Cruvinel, K.; Oliverira-Nascimento, L. A mini-review on drug delivery through wafer technology: Formulation and manufacturing of buccal and oral lyophilizates. J. Adv. Res. 2019, 20, 33–41. [Google Scholar] [CrossRef]
- El-Feky, G.; Abdulmaguid, R.F.; Zayed, G.M.; Kamel, R. Mucosal co-delivery of ketorolac and lidocaine using polymeric wafers for dental application. Drug Deliv. 2017, 25, 35–42. [Google Scholar] [CrossRef]
- Bromberg, L.; Mraman, V.; Rothstein, D.; Spacciapoli, P.; O’Connor, S.; Nelson, E.; Buxton, D.; Tonetti, M.; Friden, F. Sustained release of silver from periodontal wafers for treatment of periodontitis. J. Control. Release 2000, 68, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yamaguchi, S.; Soga, D.; Ueda, K.; Yoshimoto, T.; Koyama, Y. Water-Absorbing Bioadhesive Poly(Acrylic Acid)/Polyvinylpyrrolidone Complex Sponge for Hemostatic Agents. Bioengineering 2022, 9, 755. [Google Scholar] [CrossRef] [PubMed]
- Maani, S.; Saleh, M.; Melek, L.; Sadaka, M. Evaluation of colloidal silver Gelatin Sponge (Gelatamp) in patients receiving anticoagulant after tooth extraction (clinical study). Alex. Dent. J. 2015, 40, 101–106. [Google Scholar] [CrossRef]
- Mehravaran, M.; Haeri, A.; Rabbani, S.; Mortazavi, S.; Torshabi, M. Preparation and characterization of benzydamine hydrochloride-loaded lyophilized mucoadhesive wafers for the treatment of oral mucositis. J. Drug Deliv. Sci. Technol. 2022, 78, 103944. [Google Scholar] [CrossRef]
- Boateng, J.S.; Auffret, A.D.; Matthews, K.H.; Humphrey, M.J.; Stevens, H.N.; Eccleston, G.M. Characterisation of freeze-dried wafers and solvent evaporated films as potential drug delivery systems to mucosal surfaces. Int. J. Pharm. 2010, 389, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Timur, S.S.; Yüksel, S.; Akca, G.; Şenel, S. Localized drug delivery with mono and bilayered mucoadhesive films and wafers for oral mucosal infections. Int. J. Pharm. 2019, 559, 102–112. [Google Scholar] [CrossRef]
- Paradowska-Stolarz, A.; Wieckiewicz, M.; Owczarek, A.; Wezgowiec, J. Natural Polymers for the Maintenance of Oral Health:Review of Recent Advances and Perspectives. Int. J. Mol. Sci. 2021, 22, 10337. [Google Scholar] [CrossRef]
- Karavasili, C.; Eleftheriadis, G.; Gioumouxouzis, C.; Andriotis, E.; Fatouros, D. Mucosal drug delivery and 3D printing technologies: A focus on special patient populations. Adv. Drug Deliv. Rev. 2021, 176, 113858. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, M.; Yan, C.; Liu, H.; Yu, D.-G. Advances in the Application of Electrospun Drug-Loaded Nanofibers in the Treatment of Oral Ulcers. Biomolecules 2022, 12, 1254. [Google Scholar] [CrossRef]
- Siemann, U. Solvent cast technology—A versatile tool for thin film production. In Scattering Methods and the Properties of Polymer Materials; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–14. [Google Scholar]
- Karki, S.; Kim, H.; Na, S.-J.; Shin, D.; Jo, K.; Lee, J. Thin films as an emerging platform for drug delivery. Asian J. Pharm. Sci. 2016, 11, 559–574. [Google Scholar] [CrossRef]
- Lim, H.; Hoag, S.W. Plasticizer effects on physical-mechanical properties of solvent cast Soluplus® films. AAPS PharmSciTech 2013, 14, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Cindradewi, A.W.; Bandi, R.; Park, C.-W.; Park, J.-S.; Lee, E.-A.; Kim, J.-K.; Kwon, G.-J.; Han, S.-Y.; Lee, S.-H. Preparation and Characterization of Cellulose Acetate Film Reinforced with Cellulose Nanofibril. Polymers 2021, 13, 2990. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.S.; Gowda, D.V.; Kumar, T.P.; Rosenholm, J.M. A Comprehensive Review of Patented Technologies to Fabricate Orodispersible Films: Proof of Patent Analysis (2000–2020). Pharmaceutics 2022, 14, 820. [Google Scholar] [CrossRef]
- Constantin, M.; Lupei, M.; Bucatariu, S.-M.; Pelin, I.M.; Doroftei, F.; Ichim, D.L.; Daraba, O.M.; Fundueanu, G. PVA/Chitosan Thin Films Containing Silver Nanoparticles and Ibuprofen for the Treatment of Periodontal Disease. Polymers 2023, 15, 4. [Google Scholar] [CrossRef]
- Moghadas, B.; Solouk, A.; Sadeghi, D. Development of chitosan membrane using non-toxic crosslinkers for potential wound dressing applications. Polym. Bull. 2021, 78, 4919–4929. [Google Scholar] [CrossRef]
- Shah, R.; Stodulka, P.; Skopalova, K.; Saha, P. Dual Crosslinked Collagen/Chitosan Film for Potential Biomedical Applications. Polymers 2019, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Kida, D.; Gładysz, O.; Szulc, M.; Zborowski, J.; Junka, A.; Janeczek, M.; Lipińska, A.; Skalec, A.; Karolewicz, B. Development and Evaluation of a Polyvinylalcohol-Cellulose Derivative-Based Film with Povidone-Iodine Predicted for Wound Treatment. Polymers 2020, 12, 1271. [Google Scholar] [CrossRef]
- Preis, M.; Breitkreutz, J.; Sandler, N. Perspective: Concepts of printing technologies for oral film formulations. Int. J. Pharm. 2015, 494, 578–584. [Google Scholar] [CrossRef]
- Rohani Shirvan, A.; Hemmatinejad, N.; Bahrami, S.H.; Bashari, A. A comparison between solvent casting and electrospinning methods for the fabrication of neem extract-containing buccal films. J. Ind. Text. 2022, 51, 311S–335S. [Google Scholar] [CrossRef]
- Available online: https://pocketdentistry.com/dental-polymers (accessed on 16 February 2023).
- Korelc, K.; Larsen, B.S.; Gašperlin, M.; Tho, I. Water-soluble chitosan eases development of mucoadhesive buccal films and wafers for children. Int. J. Pharm. 2023, 631, 122534. [Google Scholar] [CrossRef]
- Morales, J.O.; McCornville, J.T. Manufacture and characterization ofmucoadhesive buccal films. Eur. J. Pharm. Biopharm. 2011, 77, 187–199. [Google Scholar] [CrossRef]
- Borges, A.F.; Silva, C.; Coelho, J.F.; Simões, S. Oral films: Current status and future perspectives: I—Galenical development and quality attributes. J. Control. Release 2015, 206, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.P.; Fortes, A.C.; Gonsalves da Cruz Fonseca, S.; Breitkreutz, J.; Ferraz, H.G. Manufacture and Characterization of Mucoadhesive Buccal Films Based on Pectin and Gellan Gum Containing Triamcinolone Acetonide. Int. J. Polym. Sci. 2018, 2018, 2403802. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Y.; Li, W.; Gao, P.; Xiang, D.; Ren, X.; Liu, D. Mucoadhesive buccal film containing ornidazole and dexamethasone for oral ulcers: In vitro and in vivo studies. Pharm. Dev. Technol. 2019, 24, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Serrano, D.R.; Fernandez-Garcia, R.; Mele, M.; Healy, A.M.; Lalatsa, A. Designing Fast-Dissolving Orodispersible Films of Amphotericin B for Oropharyngeal Candidiasis. Pharmaceutics 2019, 11, 369. [Google Scholar] [CrossRef]
- Potaś, J.; Szymańska, E.; Wróblewska, M.; Kurowska, I.; Maciejczyk, M.; Basa, A.; Wolska, E.; Wilczewska, A.Z.; Winnicka, K. Multilayer films based on chitosan/pectin polyelectrolyte complexes as novel platforms for buccal administration of clotrimazole. Pharmaceutics 2021, 13, 1588. [Google Scholar] [CrossRef]
- Khan, G.; Yadav, S.K.; Patel, R.R.; Nath, G.; Bansal, M.; Mishra, B. Development and Evaluation of Biodegradable Chitosan Films of Metronidazole and Levofloxacin for the Management of Periodontitis. AAPS Pharm. Sci. Tech. 2015, 17, 1312–1325. [Google Scholar] [CrossRef]
- Sionkowska, A.; Lewandowska, K.; Michalska-Sionkowska, M.; Walczak, M. Characterization of silk fibroin 3D composites modified by collagen. J. Mol. Liq. 2016, 215, 323–327. [Google Scholar] [CrossRef]
- Collins, M.N.; Birkinshaw, C. Morphology of crosslinked hyaluronic acid. J. Appl. Polym. Sci. 2011, 120, 1040–1049. [Google Scholar] [CrossRef]
- Szepes, A.; Ulrich, J.; Farkas, Z.; Kovacs, J.; Szabo-Revesz, P. Freeze-casting technique in the development of solid drug delivery systems. Chem. Eng. Process. 2007, 46, 230–238. [Google Scholar] [CrossRef]
- Scotti, K.L.; Dunand, D.C. Freeze casting—A review of processing, microstructure and properties via the open data repository, FreezeCasting.net. Prog. Mater. Sci. 2018, 94, 243–305. [Google Scholar] [CrossRef]
- Merivaara, A.; Zini, J.; Koivunotko, E.; Valkonen, S.; Korhonen, O.; Fernandes, F.M.; Yliperttula, M. Preservation of biomaterials and cells by freeze-drying: Change of paradigm. J. Control. Release 2021, 336, 480–498. [Google Scholar] [CrossRef]
- Peiren, J.; Hellemans, A.; De Vos, P. Impact of the freeze-drying process on product appearance, residual moisture content, viability, and batch uniformity of freeze-dried bacterial cultures safeguarded at culture collections. Appl. Microbiol. Biotechnol. 2016, 100, 6239–6249. [Google Scholar] [CrossRef] [PubMed]
- Catanzano, O.; D’Esposito, V.; Formisano, P.; Boateng, J.S.; Quaglia, F. Composite Alginate-Hyaluronan Sponges for the Delivery of Tranexamic Acid in Postextractive Alveolar Wounds. J. Pharm. Sci. 2017, 2, 654–661. [Google Scholar] [CrossRef]
- Chang, S.J.; Huan, Y.T.; Yang, S.C.; Kuo, S.M.; Lee, M.W. In vitro properties of gellan gum sponge as the dental filling to maintain alveolar space. Carbohydr. Polym. 2012, 88, 684–689. [Google Scholar] [CrossRef]
- Kida, D.; Karolewicz, B.; Junka, A.; Sender-Janeczek, A.; Duś, I.; Marciniak, D.; Szulc, M. Metronidazole-Loaded Porous Matrices for Local Periodontitis Treatment: In vitro Evaluation and in vivo pilot study. Appl. Sci. 2019, 9, 4545. [Google Scholar] [CrossRef]
- Labovitiadi, O.; Lamb, A.J.; Matthews, K.H. Lyophilised wafers as vehicles for the topical release of chlorhexidine digluconate--release kinetics and efficacy against Pseudomonas aeruginosa. Int. J. Pharm. 2012, 439, 157–164. [Google Scholar] [CrossRef]
- Ayensu, I.; Mitchell, J.C.; Boateng, J.S. Development and physico-mechanical characterisation of lyophilised chitosan wafers as potential protein drug delivery systems via the buccal mucosa. Colloids Surf. B Biointerfaces 2012, 91, 258–265. [Google Scholar] [CrossRef]
- Berton, F.; Porrelli, D.; Di Lenarda, R.; Turco, G. A Critical Review on the Production of Electrospun Nanofibres for Guided Bone Regeneration in Oral Surgery. Nanomaterials 2019, 10, 16. [Google Scholar] [CrossRef]
- Ghosal, K.; Chandra, A.G.; Snigdha, S.; Roy, S.; Agatemor, C.; Thomas, S.; Provaznik, I. Electrospinning over Solvent Casting: Tuning of Mechanical Properties of Membranes. Sci. Rep. 2018, 8, 5058. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Kundu, S.C. Electrospinning: A fascinating fiber fabrication technique. Biotechnol. Adv. 2010, 28, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.R.; Khan, N.; Vivekanandhan, S.; Satyanarayana, N.; Mohanty, A.K.; Misra, M. Nanofibers: Effective generation by electrospinning and their applications. J. Nanosci. Nanotechnol. 2012, 12, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Procopio, A.; Lagreca, E.; Jamaledin, R.; La Manna, S.; Corrado, B.; Di Natale, C.; Onesto, V. Recent Fabrication Methods to Produce Polymer-Based Drug Delivery Matrices (Experimental and In Silico Approaches). Pharmaceutics 2022, 14, 872. [Google Scholar] [CrossRef]
- Sill, T.J.; von Recum, H.A. Electrospinning: Applications in drug delivery and tissue engineering. Biomaterials 2008, 29, 1989–2006. [Google Scholar] [CrossRef]
- Shamim, Z.; Saeed, B.; Amir, T.; Abo Saied, R.; Rogheih, D. The effect of flow rate on morphology and deposition area of electrospun nylon 6 nanofiber. J. Eng. Fabr. Fibers 2012, 7, 42. [Google Scholar]
- Reda, R.I.; Wen, M.M.; El-Kamel, A.H. Ketoprofen-loaded Eudragit electrospun nanofibers for the treatment of oral mucositis. Int. J. Nanomed. 2017, 12, 2335–2351. [Google Scholar] [CrossRef]
- Dos Santos, D.M.; Chagas, P.A.M.; Leite, I.S.; Inada, N.M.; de Annunzio, S.R.; Fontana, C.R.; Campana-Filho, S.P.; Correa, D.S. Core-sheath nanostructured chitosan-based nonwovens as a potential drug delivery system for periodontitis treatment. Int. J. Biol. Macromol. 2020, 142, 521–534. [Google Scholar] [CrossRef]
- Nasajpour, A.; Ansari, S.; Rinoldi, C.; Rad, A.S.; Aghaloo, T.; Shin, S.R.; Mishra, Y.K.; Adelung, R.; Swieszkowski, W.; Annabi, N.; et al. Multifunctional Polymeric Periodontal Membrane with Osteogenic and Antibacterial Characteristics. Adv. Funct. Mater. 2018, 28, 1703437. [Google Scholar] [CrossRef]
- Lee, F.Y.; Lee, D.; Lee, T.C.; Chen, J.K.; Wu, R.C.; Liu, K.C.; Liu, S.J. Fabrication of Multi-Layered Lidocaine and Epinephrine-Eluting PLGA/Collagen Nanofibers: In Vitro and In Vivo Study. Polymers 2017, 5, 416. [Google Scholar] [CrossRef]
- Budai-Szűcs, M.; Ruggeri, M.; Faccendini, A.; Léber, A.; Rossi, S.; Varga, G.; Bonferoni, M.C.; Vályi, P.; Burián, K.; Csányi, E.; et al. Electrospun Scaffolds in Periodontal Wound Healing. Polymers 2021, 13, 307. [Google Scholar] [CrossRef]
- Colley, H.E.; Said, Z.; Santocildes-Romero, M.E.; Baker, S.R.; D’Apice, K.; Hansen, J.; Madsen, L.S.; Thornhill, M.H.; Hatton, P.V.; Murdoch, C. Pre-clinical evaluation of novel mucoadhesive bilayer patches for local delivery of clobetasol-17-propionate to the oral mucosa. Biomaterials 2018, 178, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Clitherow, K.; Murdoch, C.; Spain, S.G.; Handler, A.M.; Colley, H.E.; Stie, M.B.; Mørck Nielsen, H.; Janfelt, C.; Hatton, P.V.; Jacobsen, J. Mucoadhesive Electrospun Patch Delivery of Lidocaine to the Oral Mucosa and Investigation of Spatial Distribution in a Tissue Using MALDI-Mass Spectrometry Imaging. Mol. Pharm. 2019, 16, 3948–3956. [Google Scholar] [CrossRef]
- Trenfield, S.J.; Awad, A.; Goyanes, A.; Gaisford, S.; Basit, A.W. 3D Printing Pharmaceuticals: Drug Development to Frontline Care. Trends Pharmacol. Sci. 2018, 39, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Goole, J.; Amighi, K. 3D printing in pharmaceutics: A new tool for designing customized drug delivery systems. Int. J. Pharm. 2016, 499, 376–394. [Google Scholar] [CrossRef]
- Sadia, M.; So, A.; Arafat, B.; Isreb, A.; Ahmed, W. Adaptation of pharmaceutical excipients to FDM 3D printing for the fabrication of patient-tailored immediate release tablets. Int. J. Pharm. 2016, 513, 659–668. [Google Scholar] [CrossRef]
- Aguilar-de-Leyva, Á.; Linares, V.; Casas, M.; Caraballo, I. 3D Printed Drug Delivery Systems Based on Natural Products. Pharmaceutics 2020, 12, 620. [Google Scholar] [CrossRef]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Yang, J.; Roberts, C.J. 3D printing of five-in-one dose combination polypill with defined immediate and sustained release profiles. J. Control. Release 2015, 217, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Boateng, J.S.; Snowden, M.J.; Douroumis, D. A Review of Hot-Melt Extrusion: Process Technology to Pharmaceutical Products. ISRN Pharm. 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Seoane-Viaño, I.; Januskaite, P.; Alvarez-Lorenzo, C.; Basit, A.W.; Goyanes, A. Semi-solid extrusion 3D printing in drug delivery and biomedicine: Personalised solutions for healthcare challenges. J. Control. Release 2021, 332, 367–389. [Google Scholar] [CrossRef]
- El Aita, I.; Breitkreutz, J.; Quodbach, J. On-demand manufacturing of immediate release levetiracetam tabletsusing pressure-assisted microsyringe printing. Eur. J. Pharm. Biopharm. 2019, 134, 29–36. [Google Scholar] [CrossRef]
- Zhang, L.; Forgham, H.; Shen, A.; Wang, J.; Zhu, J.; Huang, X.; Tang, S.Y.; Xu, C.; Davis, T.P.; Qiao, R. Nanomaterial integrated 3D printing for biomedical applications. J. Mat. Chem. 2022, 10, 7473–7490. [Google Scholar] [CrossRef]
- Berger, V.; Luo, Z.; Leroux, J.C. 3D printing of a controlled fluoride delivery device for the prevention and treatment of tooth decay. J. Control. Release 2022, 348, 870–880. [Google Scholar] [CrossRef]
- Tagami, T.; Yoshimura, N.; Goto, E.; Noda, T.; Ozeki, T. Fabrication of Muco-Adhesive Oral Films by the 3D Printing of Hydroxypropyl Methylcellulose-Based Catechin-Loaded Formulations. Biol. Pharm. Bull. 2019, 42, 1898–1905. [Google Scholar] [CrossRef]
- Tagami, T.; Okamura, M.; Ogawa, K.; Ozeki, T. Fabrication of Mucoadhesive Films Containing Pharmaceutical Ionic Liquid and Eudragit Polymer Using Pressure-Assisted Microsyringe-Type 3D Printer for Treating Oral Mucositis. Pharmaceutics 2022, 14, 1930. [Google Scholar] [CrossRef]
- Takashima, H.; Tagami, T.; Kato, S.; Pae, H.; Ozeki, T.; Shibuya, Y. Three-Dimensional Printing of an Apigenin-Loaded Mucoadhesive Film for Tailored Therapy to Oral Leukoplakia and the Chemopreventive Effect on a Rat Model of Oral Carcinogenesis. Pharmaceutics 2022, 14, 1575. [Google Scholar] [CrossRef]
- Sarna-Boś, K.; Skic, K.; Sobieszczański, J.; Boguta, P.; Chałas, R. Contemporary Approach to the Porosity of Dental Materials and Methods of Its Measurement. Int. J. Mol. Sci. 2021, 22, 8903. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.; Saudino, G.; Musteata, V.E.; Madhavan, P.; Genovese, A.; Behzad, A.R.; Sougrat, R.; Boi, C.; Peinemann, K.; Nunes, S.P. 3D Analysis of Ordered Porous Polymeric Particles using Complementary Electron Microscopy Methods. Sci. Rep. 2019, 9, 13987. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthi, G.; Hadwiger, M.; Ben-Romdhane, M.; Madhavan, P.; Nunes, S.P. 3D membrane imaging and porosity visualization. Ind. Eng. Chem. Res. 2016, 55, 3689–3695. [Google Scholar] [CrossRef]
- Son, D.; Cho, S.; Nam, J.; Lee, H.; Kim, M. X-ray-Based Spectroscopic Techniques for Characterization of Polymer Nanocomposite Materials at a Molecular Level. Polymers 2020, 12, 1053. [Google Scholar] [CrossRef]
- Ye, S.; Jiang, L.; Su, C.; Zhu, Z.; Wen, Y.; Shao, W. Development of gelatin/bacterial cellulose composite sponges as potential natural wound dressings. Int. J. Biol. Macromol. 2019, 133, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Zdravkov, B.D.; Čermák, J.J.; Šefara, M.; Janku, J. Pore classification in the characterization of porous materials: A perspective. Cent. Eur. J. Chem. 2007, 5, 385–395. [Google Scholar]
- Dollimore, D.; Heal, G.R. The analysis of gas adsorption data to determine pore structure. Surf. Technol. 1978, 6, 231–258. [Google Scholar] [CrossRef]
- Huhtamäki, T.; Tian, X.; Korhonen, J.T.; Ras, R.H.A. Surface-wetting characterization using contact-angle measurements. Nat. Protoc. 2018, 13, 1521–1538. [Google Scholar] [CrossRef]
- Amorós-Galicia, L.; Nardi-Ricart, A.; Verdugo-González, C.; Arroyo-García, C.M.; García-Montoya, E.; Pérez-Lozano, P.; Suñé-Negre, J.M.; Suñé-Pou, M. Development of a Standardized Method for Measuring Bioadhesion and Mucoadhesion That Is Applicable to Various Pharmaceutical Dosage Forms. Pharmaceutics 2022, 14, 1995. [Google Scholar] [CrossRef] [PubMed]
- Bassi da Silva, J.; Ferreira, S.B.d.S.; Reis, A.V.; Cook, M.T.; Bruschi, M.L. Assessing Mucoadhesion in Polymer Gels: The Effect of Method Type and Instrument Variables. Polymers 2018, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.L.; Bull, S.B.; Methven, L.; Parker, J.K.; Khutoryanskiy, V.V. Mucoadhesion: A food perspective. Food Hydrocoll. 2017, 72, 281–296. [Google Scholar] [CrossRef]
- Kianfar, F.; Ayensu, I.; Boateng, J.S. Development and physico-mechanical characterization of carrageenan and poloxamer-based lyophilized matrix as a potential buccal drug delivery system. Drug Dev. Ind. Pharm. 2014, 40, 361–369. [Google Scholar] [CrossRef]
- Kalachandra, S.; Lin, D.M.; Stejskal, E.O.; Prakki, A.; Offenbacher, S. Drug release from cast films of ethylene vinyl acetate (EVA) copolymer: Stability of drugs by 1H NMR and solid state 13C CP/MAS NMR. J. Mater. Sci. Mater. Med. 2005, 16, 597–605. [Google Scholar] [CrossRef]
- Registration of Medicinal Products, Medical Devices and Biocidal Products. Polish Pharmacopoeia, 12th ed.; Registration of Medicinal Products, Medical Devices and Biocidal Products: Warsaw, Poland, 2020.
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar]
- ICH. Q1A(R2): Stability testing of new drug substances and products. In ICH Harmonised Tripartite Guideline Stability; ICH: Geneva, Switzerland, 2003; pp. 1–18.
- Ali, M.S.; Ali, J.; Ahuja, A.; Alam, M.S. Formulation and Characterization of Dental Film Containing Ofloxacin. J. Appl. Pharm. Sci. 2012, 2, 116–119. [Google Scholar]
- Park, J.B.; Prodduturi, S.; Morott, J.; Kulkarni, V.I.; Jacob, M.R.; Khan, S.I.; Stodghill, S.P.; Repka, M.A. Development of an antifungal denture adhesive film for oral candidiasis utilizing hot melt extrusion technology. Expert Opin. Drug Deliv. 2015, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bopp, S.K.; Lettieri, T. Comparison of four different colorimetric and fluorometric cytotoxicity assays in a zebrafish liver cell line. BMC Pharmacol. 2008, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Pourhajibagher, M.; Chiniforush, N.; Parkerc, N.; Shahabid, S.; Ghorbanzadehe, R.; Kharazifardf, M.J.; Bahador, A. Evaluation of antimicrobial photodynamic therapy with indocyaninegreen and curcumin on human gingival fibroblast cells: An in vitro photocytotoxicity investigation. Photodiagnosis Photodyn. Ther. 2016, 15, 13–18. [Google Scholar] [CrossRef]
- ISO 10993; Biological Evaluation of Medical Devices—Part 5: Tests for Cytotoxicity: In Vitro Methods. ISO: Geneva, Switzerland, 2009.
- Li, W.; Zhou, J.; Xu, Y. Study of the in vitro cytotoxicity testing of medical devices. Biomed. Rep. 2015, 3, 617–620. [Google Scholar] [CrossRef]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio Protoc. 2016, 6, e1984. [Google Scholar] [CrossRef]
- Available online: https://www.bmglabtech.com/en/application-notes/the-use-of-an-atp-bioluminescence-assay-to-quantify-cell-cytotoxicity/ (accessed on 12 April 2023).
- Hartrick, C.T.; Kovan, J.P.; Shapiro, S. The numeric rating scale for clinical pain measurement: A ratio measure? Pain Pract. 2003, 3, 310–316. [Google Scholar] [CrossRef]
- Haag, D.G.; Peres, K.G.; Balasubramanian, M.; Brennan, D.S. Oral conditions and health-related quality of life: A systematic review. J. Dent. Res. 2017, 96, 864–874. [Google Scholar] [CrossRef]
- Slade, G.D.; Spencer, A.J. Development and evaluation of the Oral Health Impact Profile. Community Dent. Health. 1994, 11, 3–11. [Google Scholar]
- Slade, G.D. Derivation and validation of a short-form oral health impact profile. Community Dent. Oral Epidemiol. 1997, 25, 284–290. [Google Scholar] [CrossRef]
- Pfütze, M.; Niedermeier, A.; Hertl, M.; Eming, R. Introducing a novel Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) in pemphigus. Eur. J. Dermatol. 2007, 17, 4–11. [Google Scholar] [PubMed]
- Liu, J.; Zeng, X.; Chen, Q.; Cai, Y.; Chen, F.; Wang, Y. An evaluation on the efficacy and safety of amlexanox oral adhesive tablets in the treatment of recurrent minor aphthous ulceration in a Chinese cohort: A randomized, double-blind, vehicle-controlled, unparallel multicenter clinical trial. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2006, 102, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Wessels, R.; De Roose, S.; De Bruyckere, T.; Eghbali, A.; Jacquet, W.; De Rouck, T.; Cosyn, J. The Mucosal Scarring Index: Reliability of a new composite index for assessing scarring following oral surgery. Clin. Oral Investig. 2019, 23, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Palaia, G.; Tenore, G.; Tribolati, L.; Russo, C.; Gaimari, G.; Del Vecchio, A.; Romeo, U. Evaluation of wound healing and postoperative pain after oral mucosa laser biopsy with the aid of compound with chlorhexidine and sodium hyaluronate: A randomized double blind clinical trial. Clin. Oral Investig. 2019, 23, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Jurczyszyn, K.; Trzeciakowski, W.; Kozakiewicz, M.; Kida, D.; Malec, K.; Karolewicz, B.; Konopka, T.; Zborowski, J. Fractal dimension and texture analysis of lesion autofluorescence in the evaluation of oral lichen planus treatment effectiveness. Materials 2021, 14, 5448. [Google Scholar] [CrossRef]






| Type of Carrier | Polymers/ Excipients | Processing Parameters | API | Ref. |
|---|---|---|---|---|
| Film | PC, GG | Solvent/temperature: water/50 °C Stirring speed/time: 150 rpm/12 h Drying time/temperature: 24 h/40 °C | Triamcinolone acetonide | [38] |
| Film | PVA, HPMC | Drying time: 24 h Temperature: 40 °C | Povidone-iodine | [31] |
| Bilayer film | EC, HPMC, PVA, CS | Anhydrous solvent: ethanol | Ornidazole and dexamethasone sodium phosphate | [39] |
| Fast-dissolving orodispersible films | DX, MT, HMPC-AS, HPC, Microcrystalline cellulose (Avicel)/GLY | Solvent: methanol Time drying under vacuum: 4–5 h | Amphotericin B | [40] |
| Multilayer film | CS, PC | Solvent: water Drying time: 25h Temperature: 30–35 °C | Clotrimazole | [41] |
| Film | CS/propylene glycol | Solvent: 0.5% acetic acid solution Drying time/temperature: 50 °C/24 h | Metronidzole/ levofloxacin | [42] |
| Monolayer and bilayer mucoadhesive film | CS, HPMC/acetic acid solution glycerin | Monolayer film solvent casting Bilayer films were prepared using double casting | Cefuroxime axetil | [19] |
| Type of Carrier | Polymers/ Excipients | Processing Parameters | API/ Application/ Indication | Ref. |
|---|---|---|---|---|
| Sponge | SA | Freezing time/temperature: 12 h/−20 °C Pressure/temperature: 0.01 atm/−60 °C | Tranexamic acid | [49] |
| Sponge | GG | Freezing temperature: −20 °C | Dental filling | [50] |
| Matrix | Gelatin | Freezing temperature: −15 °C Pressure: 9 × 10−2 to 1.3 × 10−1 mBar | Metronidazole/intra-pocket application | [51] |
| Wafers | SA, CMC | - | Paracetamol/application to mucosal surfaces, including wounds | [18] |
| Wafers | Karaya gum, guar gum, SA, xanthan gum | - | Chlorhexidine digluconate release against Pseudomonas aeruginosa | [52] |
| Wafers | CS | - | Protein drug delivery via the buccal mucosa | [53] |
| Wafers | CS, HPMC, glycerin, acetic acid solution | Monolayer wafer Freezing temperature: −20 °C overnight before freeze-drying Freeze-dried for 24 h in the following conditions: collector temperature: −80 °C; vacuum: 0.1 mBar Bilayer wafer First layer was frozen at −20 °C and before the second layer, an additional overnight freezing step was applied before the freeze-drying cycle | Cefuroxime axetil | [19] |
| Processing Parameters | Solution Parameters | Ambient Parameters |
|---|---|---|
| Applied voltage Distance from needle tip to collector Needle inner diameter Solution flow rate Collector type | Solution viscosity Polymer concentration Polymer molecular weight Polymer solubility Conductivity Surface tension Solvent volatility | Air humidity Ambient temperature |
| Parameters | Values |
|---|---|
| Temperature | °C |
| Needle gauge | G |
| Distance between needle and collector | cm |
| Flow rate range | mL/h |
| High voltage range | kV |
| Structure/ Carrier/ Morphology | Polymers/ Excipients/ Solvent | Processing Parameters | API and/or Application/ Activity/ Indication | Ref. |
|---|---|---|---|---|
| Nanofibers | Eudragit L, Eudragit S/ethanol | Flow rate: 0.5–2 mL/h | Ketoprofen | [61] |
| Core-sheath nanofibers | PVA, CS/acetic acid | Flow rate: 10 µL min−1/5 µL min−1 Distance: 5 cm Voltage: 25 kV Needle diameter: 1.02 mm/0.65 mm | Tetracycline hydrochloride | [62] |
| Nanofibrous membranes | PCL, ZnO, hexafluoroisopropanol | Needle diameter: 20 G Distance: 20 cm Voltage: 18 kV Flow rate: 2 mL h−1 | Osteogenic and antibacterial/guided tissue regeneration | [63] |
| Nanofibers | PLGA, collagen/hexafluoroisopropanol | - | Lidocaine/ epinephrine | [64] |
| Electrospinning-based nanofibers/mucoadhesive two-layer patch | Rivelin® | - | Mucosal diseases clobetasol propionate | [22] |
| Double-layer mucoadhesive buccal film | CS, PVA, acetone/acetic acid | Distance: 20 cm Flow rate: 0.5–2 mL h−1 Voltage: 15 kV | Neem extract | [33] |
| Nanofibrous scaffolds | Gelatin, CS, SA, acetate buffer, citric acid | Voltage: 28 kV Distance: 15 cm Flow rate: 0.794 mL h−1 | Restoring tissue integrity and treating bacterial infections | [65] |
| Electrospinning bilayer patch | PVP, Eudragit, polyethylene oxide, PCL, DCM, DMF | Voltage: 17 kV Flow rate: 1–5 mL h−1 Distance: 19 cm | Clobetasol propionate | [66] |
| Electrospun dual-layer patch delivery | PVP, Eudragit RS100, PEO 400 kDa, PCL DCM/DMF/ACN, ethanol | Solvent: 97% ethanol Voltage: 19 kV Distance: 14 cm Flow rate: 1.5 mL h−1 | Electrospun patch delivery Lidocaine | [67] |
| Type of Carrier/API/ 3D Printing Method | Polymers/ Excipients | Processing Parameters | Application/ Activity/Indication | Ref. |
|---|---|---|---|---|
| 3D-printed tooth caps with fluoride/FDM | PCL, PVA or PEG | 1/NaF-loaded (10 wt%) composite filaments 1.75 ± 0.15 mm were produced by hot-melt extrusion (HME) 2/PCL/PVA and PCL/PEG filaments produced using FMD (nozzle temperatures set at 145 and 100 °C, the bed heated at 40 and 30 °C, respectively) | Sustained localized release of fluoride from personalized 3D-printed mouthguards at the device–enamel interface can improve the incorporation of fluoride in the tooth matrix and prevent lesion progression | [77] |
| Catechin-based film/SSE | HPMC/mannitol, glycerol, ethanol/water, Tween 80 | 1/Preparation of hydrogel-based printer material/ink 2/Extrusion parameters: printer syringe—27 G nozzle, air pressure through pump—20–70 kPa, the printed hydrogel was dried at room temperature and stored in a desiccator (air drying—AD); alternatively, the hydrogel was frozen at −80 °C and freeze-dried using an FD1000 freeze-dryer (freeze-drying—FD) | Oral cavity, including aphthous stomatitis and oral mucositis ulcers, catechins are flavonoids, and polyphenols exhibit antioxidant, anti-inflammatory, anti-cancer and anti-hypertensive effects | [78] |
| Multilayer film containing an ibuprofen/lidocaine ionic liquid/pressure-assisted microsyringe (PAM) printing | HPMC supporting layer, mannitol, water/Eudragit polymer layer (L100, EPO or RSPO), acetone, active substances | 1/Preparation of hydrogel-based printer material/ink for Layer 1
| Oral mucositis caused by radiation therapy and chemotherapy | [79] |
| Apigenin-loaded mucoadhesive oral film/SSE | HPMC, carbopol, poloxamer/ethanol | 1/Preparation of hydrogel-based printer material/ink 2/Extrusion parameters: printer syringe—27 G nozzle, air pressure through pump—90 kPa, the printed hydrogel was dried at room temperature | Oral leucoplakia, including on the tongue | [80] |
| Release Exponent (n) | Drug Transport Mechanism | Rate as a Function of Time |
|---|---|---|
| 0.5 | Fickian diffusion | t−0.5 |
| 0.45 < n = 0.89 | Non-Fickian transport | tn−1 |
| 0.89 | Case II transport | Zero-order release |
| Higher than 0.89 | Super case II transport | tn−1 |
| Storage Condition | Stability Test Method | ICH Test Temperature/ Humidity/Period in Months |
|---|---|---|
| Room temperature | Long term | 25 ± 2 °C/60 ± 5% RH/12 |
| Intermediate | 30 ± 2 °C/65 ± 5% RH/6 | |
| Accelerated | 40 ± 2 °C/75 ± 5% RH/6 |
| Technology | Advantages | Disadvantages/Challenges | Potential Solutions |
|---|---|---|---|
| Solvent-casting method (SCM) |
|
|
|
| Freeze casting method (LM) |
|
|
|
| Electrospinning method |
|
|
|
| 3D printing method: fused deposition modelling (FDM) |
|
|
|
| 3D printing method: semi-solid extrusion (SSE) |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kida, D.; Konopka, T.; Jurczyszyn, K.; Karolewicz, B. Technological Aspects and Evaluation Methods for Polymer Matrices as Dental Drug Carriers. Biomedicines 2023, 11, 1274. https://doi.org/10.3390/biomedicines11051274
Kida D, Konopka T, Jurczyszyn K, Karolewicz B. Technological Aspects and Evaluation Methods for Polymer Matrices as Dental Drug Carriers. Biomedicines. 2023; 11(5):1274. https://doi.org/10.3390/biomedicines11051274
Chicago/Turabian StyleKida, Dorota, Tomasz Konopka, Kamil Jurczyszyn, and Bożena Karolewicz. 2023. "Technological Aspects and Evaluation Methods for Polymer Matrices as Dental Drug Carriers" Biomedicines 11, no. 5: 1274. https://doi.org/10.3390/biomedicines11051274
APA StyleKida, D., Konopka, T., Jurczyszyn, K., & Karolewicz, B. (2023). Technological Aspects and Evaluation Methods for Polymer Matrices as Dental Drug Carriers. Biomedicines, 11(5), 1274. https://doi.org/10.3390/biomedicines11051274