
MyBrain-Seq: A Pipeline for MiRNA-Seq Data Analysis in Neuropsychiatric Disorders
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
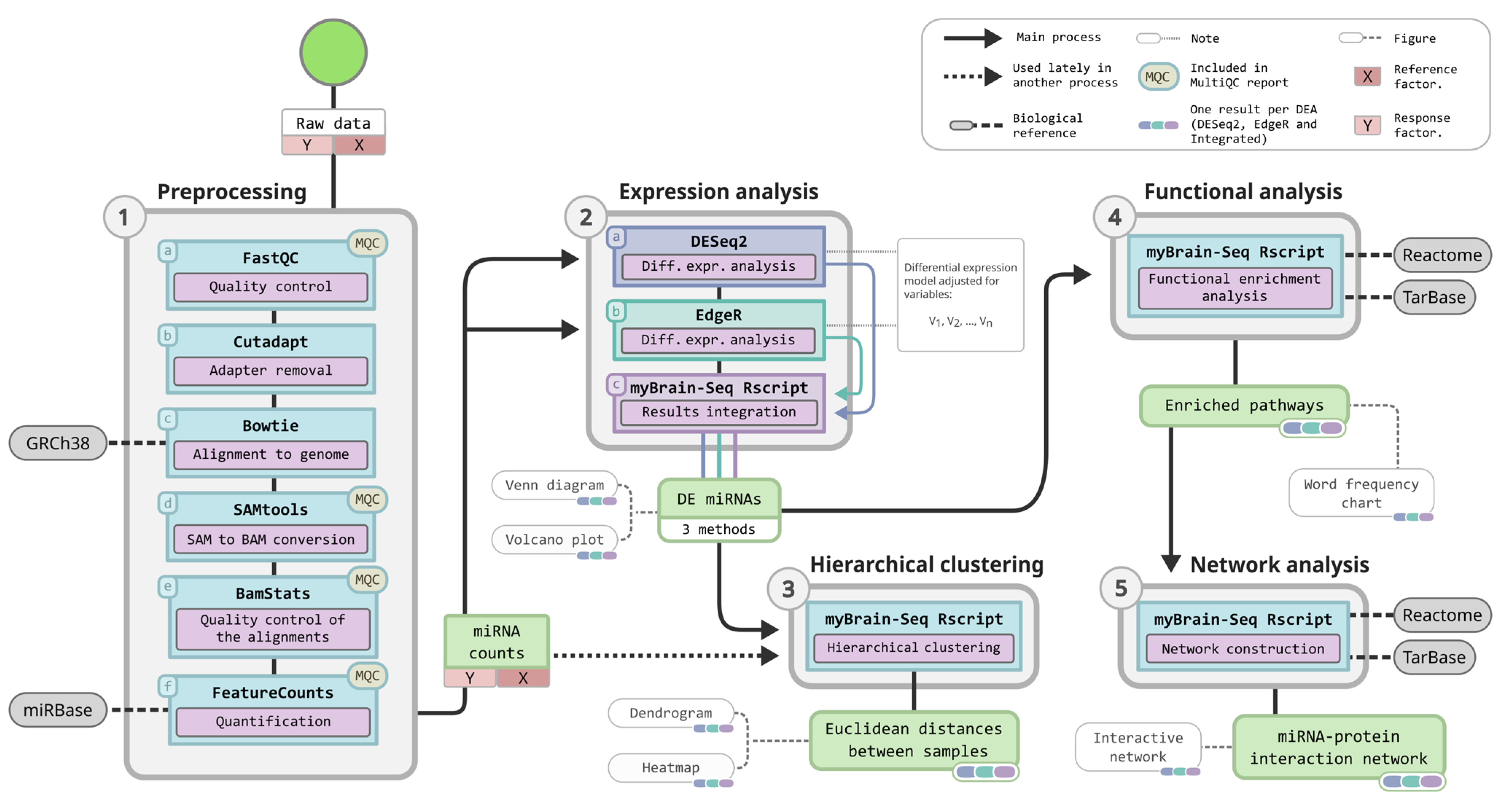
2.1. MyBrain-Seq, a Pipeline for miRNA-Seq Analysis
2.2. Quality Control and Adapter Removal
2.3. Alignment to the Reference Genome
2.4. Transcripts Annotation and Quantification
2.5. Differential Expression Analysis
2.6. Hierarchical Clustering
2.7. Functional Analysis
2.8. MiRNA–Protein Interaction Network
2.9. Summarization of the Quality Controls
2.10. MyBrain-Seq Implementation
2.11. Case Study Dataset: Treatment Resistant Schizophrenia
3. Results and Discussion
3.1. MyBrain-Seq Execution
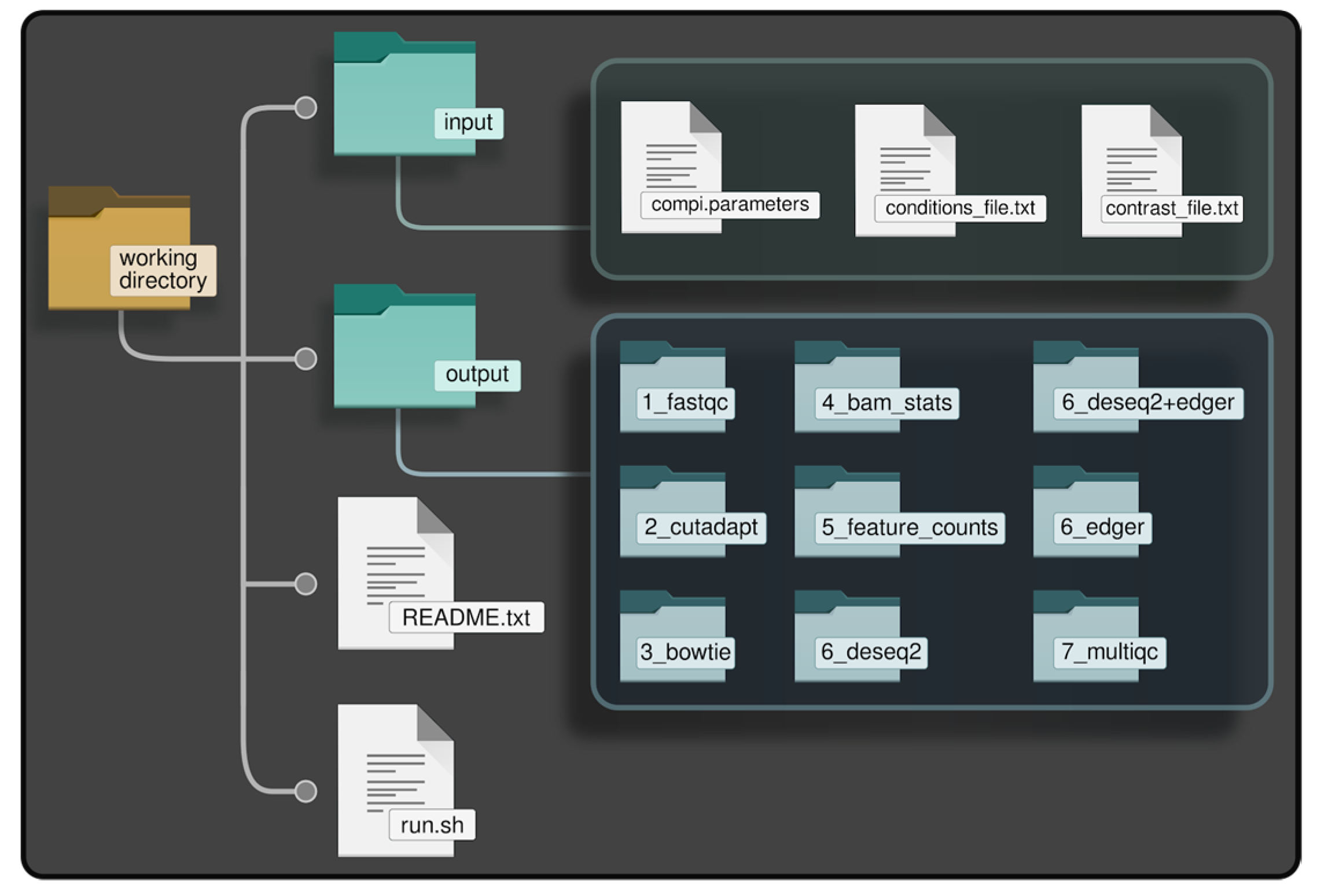
- Creation of the directory tree in the local file system, referred to as “working directory”, shown in Figure 2. The working directory consists of a main directory with two subdirectories: “/input” and “/output”. The input subdirectory is where the parameter files of myBrain-Seq should be placed; the output subdirectory will contain the results after myBrain-Seq execution. This working directory can be initialized using the utilities included in the myBrain-Seq Docker image. This initialization creates a “run.sh” file, used to run the pipeline and templates of the other files required by myBrain-Seq (those inside “/input”). A “README.txt” file is also created with the instructions to fill the template files and run the pipeline.Figure 2. Working directory of myBrain-Seq.

- The second step is the preparation of the data. In addition to the FASTQ files, myBrain-Seq needs a reference genome (or Bowtie index) and a GFF file with miRNA annotations as biological references to perform the analysis. It is recommended, but not mandatory, to put all these files inside subdirectories under “/input”. Nevertheless, if they are in other locations (e.g., a shared directory to save disk space), the provided “run.sh” will take care of this and create the appropriate Docker volume bindings in a transparent manner for the user.
- The third step is the configuration of the analysis. This comprises the creation of three files, namely: “compi.parameters”, “conditions_file.txt” and “contrast_file.txt”. These files are usually placed into the “input” directory.
- The “compi.parameters” file contains the paths and parameters needed for the analysis, i.e.: path of the working directory, paths to FASTQ files and biological references, paths to “conditions_file.txt” and to “contrast_file.txt” and the adapter sequence. For more information about the optional parameters that can be added, refer to the myBrain-Seq user manual (https://github.com/sing-group/my-brain-seq).
- The “conditions_file.txt” contains the metadata regarding names and conditions of each fastQ file. This file is used by myBrain-Seq to link each sample with a condition and its covariates. Each row of this file contains the name of the FASTQ file, its condition, a user label for that sample and zero or more columns describing the covariates for that sample (e.g., age, sex). All the covariates added in this file will be used in the DEA to adjust the statistical model.
- The “contrast_file.txt” contains the conditions to compare during the analysis and a label for each contrast. Conditions included in this file must be the same as those stated in “contitions_file.txt”. MyBrain-Seq can perform several contrasts in the same pipeline execution if several contrasts are specified in this file, one per line.
- The final step is running myBrain-Seq analysis using the “run.sh” script created during the working directory initialization (step number 1). This script will use “compi.parameters” as reference, mount all the needed Docker volumes (by extracting the path from the Compi parameters file) and create a directory for the log files of the current execution. MyBrain-Seq users do not need to modify this file, as it is ready to use. Thus, users only need to run the script using the path to “compi.parameters” as the unique argument to start the myBrain-Seq analysis.
- Both final and intermediate results are saved in the “/output” directory. Such output files are placed in directories corresponding to the different steps of the workflow, namely: “1_fastqc”, “2_cutadapt”, “3_bowtie”, “4_bam_stats”, “5_feature_counts”, “6_deseq2”, “6_deseq2+edger”, “6_edger” and “7_multiqc”. Results from the hierarchical clustering, functional analysis and network analysis are placed in the directories prefixed with “6_”, according to the data from which they were generated. Files from the same contrast are grouped in subdirectories named with the contrast label.

3.2. MyBrain-Seq Results
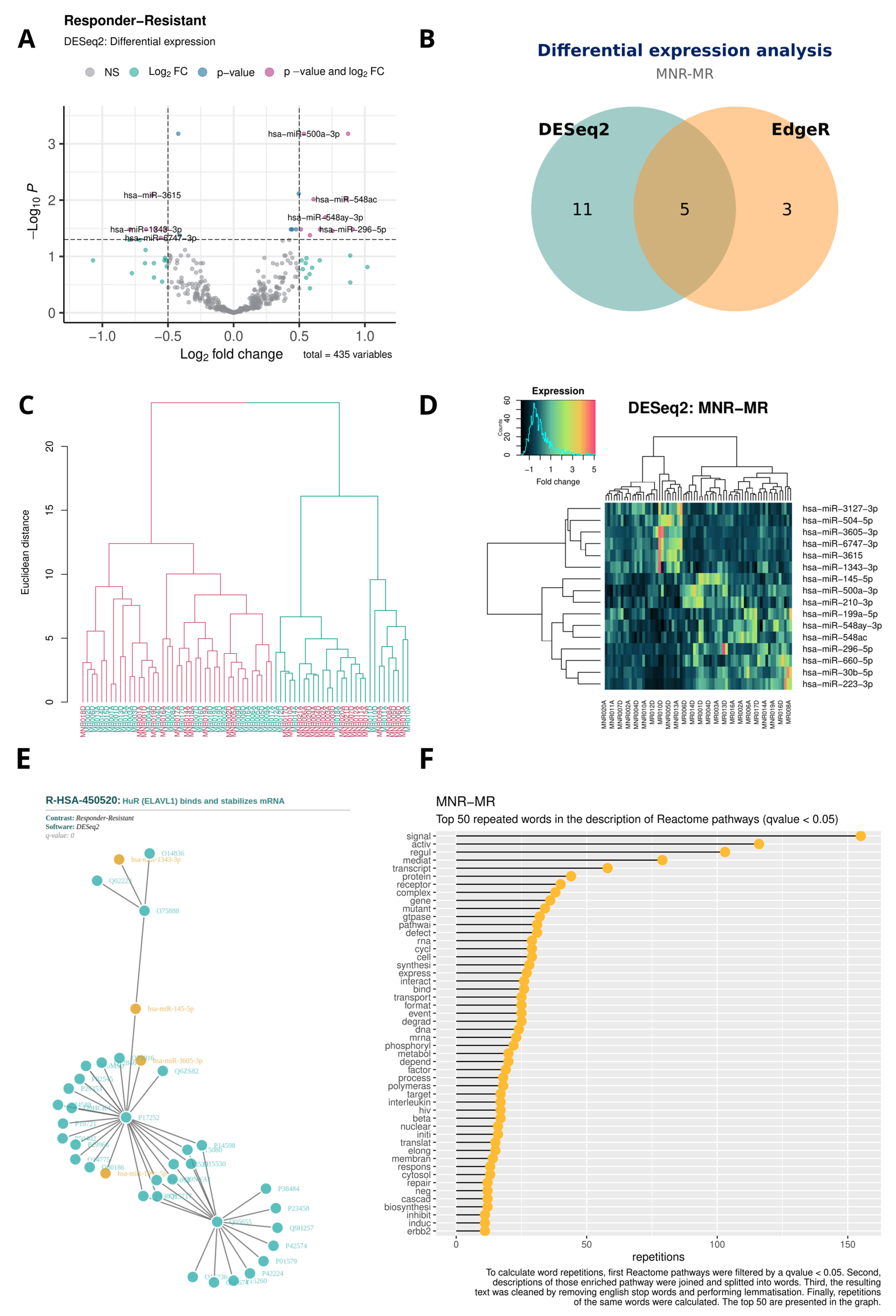
- Volcano plot with the results of each DEA; Figure 3A.
- Venn diagram with the DE miRNA coincidences between DESeq2 and EdgeR; Figure 3B.
- Dendrogram with the result of the hierarchical clustering; Figure 3C.
- Heatmap with the result of the hierarchical clustering; Figure 3D.
- HTML file with a miRNA–protein interaction network of the most enriched pathway; Figure 3E.
- Lollipop chart with the word frequency of the enriched terms; Figure 3F.
- Results of the DEA; Figure 4A. Full table in supplementary Table S1.
- List of DE miRNAs; Figure 4B.
- Enriched pathways; Figure 4C. Full table in supplementary Table S2.
- miRNA–protein interaction network; Figure 4D. Full table in supplementary Table S3.
- Adapter-trimmed FASTQ files.
- BAM and SAM files resulting from the alignment.
- A TXT file with the counts of miRNA per sample.
- A summary of the quantification results.
- A file per contrast with a subset of counts for that contrast.
- A TSV file with the expression per sample of each DE miRNA, used for the hierarchical clustering.
3.3. Case Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saliba, A.-E.; Santos, S.C.; Vogel, J. New RNA-Seq Approaches for the Study of Bacterial Pathogens. Curr. Opin. Microbiol. 2017, 35, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Sudhagar, A.; Kumar, G.; El-Matbouli, M. Transcriptome Analysis Based on RNA-Seq in Understanding Pathogenic Mechanisms of Diseases and the Immune System of Fish: A Comprehensive Review. Int. J. Mol. Sci. 2018, 19, 245. [Google Scholar] [CrossRef] [PubMed]
- Kaartokallio, T.; Cervera, A.; Kyllönen, A.; Laivuori, K.; Kere, J.; Laivuori, H. Gene Expression Profiling of Pre-Eclamptic Placentae by RNA Sequencing. Sci. Rep. 2015, 5, 14107. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Program—NCI. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 13 March 2023).
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The Reactome Pathway Knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, D.; López-Fernández, H.; Agís-Balboa, R.C. Application of MiRNA-Seq in Neuropsychiatry: A Methodological Perspective. Comput. Biol. Med. 2021, 135, 31–42. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, D.; Penedo, M.A.; Rivera-Baltanás, T.; Peña-Centeno, T.; Burkhardt, S.; Fischer, A.; Prieto-González, J.M.; Olivares, J.M.; López-Fernández, H.; Agís-Balboa, R.C. MiRNA Differences Related to Treatment-Resistant Schizophrenia. IJMS 2023, 24, 1891. [Google Scholar] [CrossRef]
- Chang, X.; Liu, Y.; Hahn, C.-G.; Gur, R.E.; Sleiman, P.M.A.; Hakonarson, H. RNA-Seq Analysis of Amygdala Tissue Reveals Characteristic Expression Profiles in Schizophrenia. Transl. Psychiatry 2017, 7, e1203. [Google Scholar] [CrossRef]
- Pantazatos, S.P.; Huang, Y.; Rosoklija, G.B.; Dwork, A.J.; Arango, V.; Mann, J.J. Whole-Transcriptome Brain Expression and Exon-Usage Profiling in Major Depression and Suicide: Evidence for Altered Glial, Endothelial and ATPase Activity. Mol. Psychiatry 2017, 22, 760–773. [Google Scholar] [CrossRef]
- Labonté, B.; Engmann, O.; Purushothaman, I.; Menard, C.; Wang, J.; Tan, C.; Scarpa, J.R.; Moy, G.; Loh, Y.-H.E.; Cahill, M.; et al. Sex-Specific Transcriptional Signatures in Human Depression. Nat. Med. 2017, 23, 1102–1111. [Google Scholar] [CrossRef]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. MicroRNA-34c Is a Novel Target to Treat Dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef] [PubMed]
- Neff, R.A.; Wang, M.; Vatansever, S.; Guo, L.; Ming, C.; Wang, Q.; Wang, E.; Horgusluoglu-Moloch, E.; Song, W.; Li, A.; et al. Molecular Subtyping of Alzheimer’s Disease Using RNA Sequencing Data Reveals Novel Mechanisms and Targets. Sci. Adv. 2021, 7, eabb5398. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A Database for Experimentally Supported Human MicroRNA–Disease Associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [PubMed]
- Pomper, N.; Liu, Y.; Hoye, M.L.; Dougherty, J.D.; Miller, T.M. CNS MicroRNA Profiles: A Database for Cell Type Enriched MicroRNA Expression across the Mouse Central Nervous System. Sci. Rep. 2020, 10, 4921. [Google Scholar] [CrossRef] [PubMed]
- Panwar, B.; Omenn, G.S.; Guan, Y. MiRmine: A Database of Human MiRNA Expression Profiles. Bioinformatics 2017, 33, 1554–1560. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, D.; López-Fernández, H.; Agís-Balboa, R.C. On the Reproducibility of MiRNA-Seq Differential Expression Analyses in Neuropsychiatric Diseases. In Practical Applications of Computational Biology & Bioinformatics, 15th International Conference (PACBB 2021); Rocha, M., Fdez-Riverola, F., Mohamad, M.S., Casado-Vara, R., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 41–51. [Google Scholar]
- Peixoto, L.; Risso, D.; Poplawski, S.G.; Wimmer, M.E.; Speed, T.P.; Wood, M.A.; Abel, T. How Data Analysis Affects Power, Reproducibility and Biological Insight of RNA-Seq Studies in Complex Datasets. Nucleic Acids Res. 2015, 43, 7664–7674. [Google Scholar] [CrossRef]
- Simoneau, J.; Dumontier, S.; Gosselin, R.; Scott, M.S. Current RNA-Seq Methodology Reporting Limits Reproducibility. Brief. Bioinform. 2021, 22, 140–145. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, B. A Comprehensive Evaluation of Ensembl, RefSeq, and UCSC Annotations in the Context of RNA-Seq Read Mapping and Gene Quantification. BMC Genom. 2015, 16, 97. [Google Scholar] [CrossRef]
- Godard, P.; van Eyll, J. Pathway Analysis from Lists of MicroRNAs: Common Pitfalls and Alternative Strategy. Nucleic Acids Res. 2015, 43, 3490–3497. [Google Scholar] [CrossRef]
- Hansen, K.D.; Wu, Z.; Irizarry, R.A.; Leek, J.T. Sequencing Technology Does Not Eliminate Biological Variability. Nat. Biotechnol. 2011, 29, 572–573. [Google Scholar] [CrossRef]
- McIntyre, L.M.; Lopiano, K.K.; Morse, A.M.; Amin, V.; Oberg, A.L.; Young, L.J.; Nuzhdin, S.V. RNA-Seq: Technical Variability and Sampling. BMC Genom. 2011, 12, 293. [Google Scholar] [CrossRef] [PubMed]
- Vitsios, D.M.; Kentepozidou, E.; Quintais, L.; Benito-Gutiérrez, E.; van Dongen, S.; Davis, M.P.; Enright, A.J. Mirnovo: Genome-Free Prediction of MicroRNAs from Small RNA Sequencing Data and Single-Cells Using Decision Forests. Nucleic Acids Res. 2017, 45, e177. [Google Scholar] [CrossRef] [PubMed]
- Stocks, M.B.; Moxon, S.; Mapleson, D.; Woolfenden, H.C.; Mohorianu, I.; Folkes, L.; Schwach, F.; Dalmay, T.; Moulton, V. The UEA SRNA Workbench: A Suite of Tools for Analysing and Visualizing next Generation Sequencing MicroRNA and Small RNA Datasets. Bioinformatics 2012, 28, 2059–2061. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Lai, J.; Lehman, M.L.; Nelson, C.C. MiRDeep*: An Integrated Application Tool for MiRNA Identification from RNA Sequencing Data. Nucleic Acids Res. 2013, 41, 727–737. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Argasinska, J.; Quinones-Olvera, N.; Finn, R.D.; Bateman, A.; Petrov, A.I. Non-Coding RNA Analysis Using the Rfam Database. Curr. Protoc. Bioinform. 2018, 62, e51. [Google Scholar] [CrossRef]
- Hinske, L.C.; França, G.S.; Torres, H.A.M.; Ohara, D.T.; Lopes-Ramos, C.M.; Heyn, J.; Reis, L.F.L.; Ohno-Machado, L.; Kreth, S.; Galante, P.A.F. MiRIAD-Integrating MicroRNA Inter- and Intragenic Data. Database 2014, 2014, bau099. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A Decade-Long Collection of Experimentally Supported MiRNA–Gene Interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. StarBase v2.0: Decoding MiRNA-CeRNA, MiRNA-NcRNA and Protein-RNA Interaction Networks from Large-Scale CLIP-Seq Data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Yuan, C.; Meng, X.; Li, X.; Illing, N.; Ingle, R.A.; Wang, J.; Chen, M. PceRBase: A Database of Plant Competing Endogenous RNA. Nucleic Acids Res. 2017, 45, D1009–D1014. [Google Scholar] [CrossRef]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the Development of MiRNA Bioinformatics Tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef] [PubMed]
- MicroRNA-Seq Data Standards and Processing Pipeline—ENCODE. Available online: https://www.encodeproject.org/microrna/microrna-seq/#references (accessed on 13 March 2023).
- Patil, A.H.; Halushka, M.K. MiRge3.0: A Comprehensive MicroRNA and TRF Sequencing Analysis Pipeline. NAR Genom. Bioinform. 2021, 3, lqab068. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Evans, J.; Bhagwate, A.; Middha, S.; Bockol, M.; Yan, H.; Kocher, J.-P. CAP-MiRSeq: A Comprehensive Analysis Pipeline for MicroRNA Sequencing Data. BMC Genom. 2014, 15, 423. [Google Scholar] [CrossRef]
- Andrés-León, E.; Núñez-Torres, R.; Rojas, A.M. MiARma-Seq: A Comprehensive Tool for MiRNA, MRNA and CircRNA Analysis. Sci. Rep. 2016, 6, 25749. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, D.; Pérez-Rodríguez, M.; Agís-Balboa, R.C.; López-Fernández, H. Towards a Flexible and Portable Workflow for Analyzing MiRNA-Seq Neuropsychiatric Data: An Initial Replicability Assessment. In Practical Applications of Computational Biology and Bioinformatics, 16th International Conference (PACBB 2022); Fdez-Riverola, F., Rocha, M., Mohamad, M.S., Caraiman, S., Gil-González, A.B., Eds.; Springer International Publishing: Cham, Switzerland, 2023; pp. 31–42. [Google Scholar]
- López-Fernández, H.; Graña-Castro, O.; Nogueira-Rodríguez, A.; Reboiro-Jato, M.; Glez-Peña, D. Compi: A Framework for Portable and Reproducible Pipelines. PeerJ Comput. Sci. 2021, 7, e593. [Google Scholar] [CrossRef]
- Nogueira-Rodríguez, A.; López-Fernández, H.; Graña-Castro, O.; Reboiro-Jato, M.; Glez-Peña, D. Compi Hub: A Public Repository for Sharing and Discovering Compi Pipelines. In Practical Applications of Computational Biology & Bioinformatics, 14th International Conference (PACBB 2020); Panuccio, G., Rocha, M., Fdez-Riverola, F., Mohamad, M.S., Casado-Vara, R., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 51–59. [Google Scholar]
- Andrews, S. FASTQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://scholar.google.com/scholar?hl=en&q=FASTQC.+A+quality+control+tool+for+high+throughput+sequence+data#d=gs_cit&t=1681893164969&u=%2Fscholar%3Fq%3Dinfo%3A7Au96aB8tVoJ%3Ascholar.google.com%2F%26output%3Dcite%26scirp%3D0%26hl%3Den (accessed on 19 April 2023).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential Expression Analysis for Sequence Count Data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A Package for the Generation of Highly-Customizable Venn and Euler Diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2022. Available online: https://bioconductor.org/packages/devel/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html (accessed on 19 April 2023).
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef]
- Galili, T. Dendextend: An R Package for Visualizing, Adjusting and Comparing Trees of Hierarchical Clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. Gplots: Various R Programming Tools for Plotting Data. 2022. Available online: https://cran.r-project.org/web/packages/gplots/gplots.pdf (accessed on 19 April 2023).
- Silge, J.; Robinson, D. Tidytext: Text Mining and Analysis Using Tidy Data Principles in R. JOSS 2016, 1, 37. [Google Scholar] [CrossRef]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Allaire, J.J.; Ellis, P.; Gandrud, C.; Kuo, K.; Lewis, B.W.; Owen, J.; Russell, K.; Rogers, J.; Sese, C.; Yetman, C.J. NetworkD3: D3 JavaScript Network Graphs from R. 2017. Available online: https://cran.r-project.org/web/packages/networkD3/networkD3.pdf (accessed on 19 April 2023).
- Vaidyanathan, R.; Xie, Y.; Allaire, J.J.; Cheng, J.; Sievert, C.; Russell, K.; Hughes, E. RStudio Htmlwidgets: HTML Widgets for R. 2023. Available online: https://www.htmlwidgets.org/ (accessed on 19 April 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- López-Fernández, H.; Ferreira, P.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. The Pegi3s Bioinformatics Docker Images Project. In Practical Applications of Computational Biology & Bioinformatics, 15th International Conference (PACBB 2021); Rocha, M., Fdez-Riverola, F., Mohamad, M.S., Casado-Vara, R., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 31–40. [Google Scholar]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dependencies | Version | Dependencies | Version |
|---|---|---|---|
| pegi3s/r_deseq2 | 1.32.0 | pegi3s/samtools_bcftools | 1.10 |
| pegi3s/r_edger | 3.36.0 | pegi3s/r_data-analysis | 4.1.1_v2 |
| pegi3s/r_enhanced-volcano | 1.12.0 | pegi3s/r_venn-diagram | 1.7.0 |
| pegi3s/cutadapt | 1.16 | pegi3s/r_network | 4.1.1_v2_v3 |
| pegi3s/fastqc | 0.11.9 | pegi3s/multiqc | 1.14.0 |
| pegi3s/bowtie1 | 1.2.3 | python3 | 3.8.5 |
| pegi3s/feature-counts | 2.0.0 | DIANA Tarbase annotations | 8 |
| pegi3s/samtools_bcftools | 1.9 | Reactome annotations | 83 |
| Pérez-Rodríguez et al. 2023 [8] | MyBrain-Seq | ||||
|---|---|---|---|---|---|
| Pathway | p-Value | q-Value | Pathway | p-Value | q-Value |
| Metabolism of proteins | 2.32 × 10−55 | 5.03 × 10−52 | HuR (ELAVL1) binds and stabilizes mRNA | 4.22 × 10−8 | 1.22 × 10−5 |
| Gene expression (Transcription) | 1.47 × 10−54 | 1.6 × 10−51 | Activation of anterior HOX genes in hindbrain development during early embryogenesis | 4.80 × 10−8 | 1.22 × 10−5 |
| Cellular responses to stress | 3.18 × 10−47 | 1.73 × 10−44 | Transcriptional regulation by small RNAs | 5.22 × 10−8 | 1.22 × 10−5 |
| Disease | 1.08 × 10−44 | 3.89 × 10−42 | Cyclin E associated events during G1/S transition | 5.31 × 10−8 | 1.22 × 10−5 |
| Metabolism of RNA | 7.31 × 10−43 | 1.99 × 10−40 | MAPK6/MAPK4 signaling | 5.52 × 10−8 | 1.22 × 10−5 |
| Cell Cycle | 1.33 × 10−42 | 3.22 × 10−40 | PPARA activates gene expression | 5.67 × 10−8 | 1.22 × 10−5 |
| Developmental Biology | 8.61 × 10−34 | 1.7 × 10−31 | Cyclin A:Cdk2 associated events at S phase entry | 5.68 × 10−8 | 1.22 × 10−5 |
| Transcriptional Regulation by TP53 | 5.17 × 10−33 | 9.37 × 10−31 | Potential therapeutics for SARS | 5.99 × 10−8 | 1.22 × 10−5 |
| DNA Repair | 2.5 × 10−32 | 4.18 × 10−30 | Assembly of the pre-replicative complex | 7.16 × 10−8 | 1.22 × 10−5 |
| Innate Immune System | 1.94 × 10−31 | 3.01 × 10−29 | SUMOylation of ubiquitinylation proteins | 8.15 × 10−8 | 1.22 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Rodríguez, D.; Agís-Balboa, R.C.; López-Fernández, H. MyBrain-Seq: A Pipeline for MiRNA-Seq Data Analysis in Neuropsychiatric Disorders. Biomedicines 2023, 11, 1230. https://doi.org/10.3390/biomedicines11041230
Pérez-Rodríguez D, Agís-Balboa RC, López-Fernández H. MyBrain-Seq: A Pipeline for MiRNA-Seq Data Analysis in Neuropsychiatric Disorders. Biomedicines. 2023; 11(4):1230. https://doi.org/10.3390/biomedicines11041230
Chicago/Turabian StylePérez-Rodríguez, Daniel, Roberto Carlos Agís-Balboa, and Hugo López-Fernández. 2023. "MyBrain-Seq: A Pipeline for MiRNA-Seq Data Analysis in Neuropsychiatric Disorders" Biomedicines 11, no. 4: 1230. https://doi.org/10.3390/biomedicines11041230
APA StylePérez-Rodríguez, D., Agís-Balboa, R. C., & López-Fernández, H. (2023). MyBrain-Seq: A Pipeline for MiRNA-Seq Data Analysis in Neuropsychiatric Disorders. Biomedicines, 11(4), 1230. https://doi.org/10.3390/biomedicines11041230