
Reactive Oxygen Species Regulation of Chemoresistance and Metastatic Capacity of Melanoma: Role of the Cancer Stem Cell Marker CD271

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
- low-cell density, then maintained in complete medium containing 1 µM of vemurafenib (R1),
- normal-cell density, initially treated with 20 μM vemurafenib and then cultured in a complete medium containing 5 µM of vemurafenib (R5) for at least 4 weeks before being used for subsequent studies.
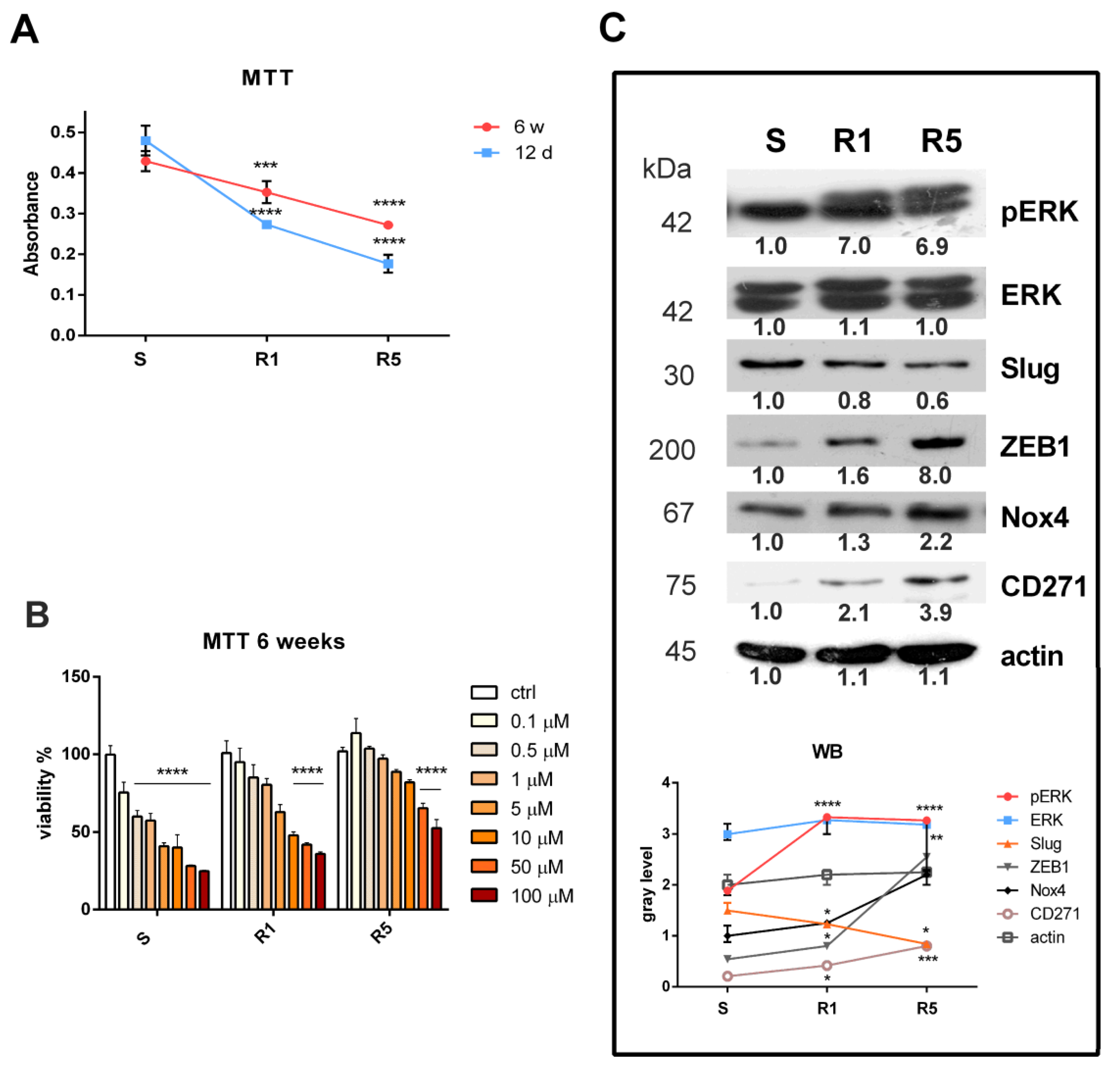
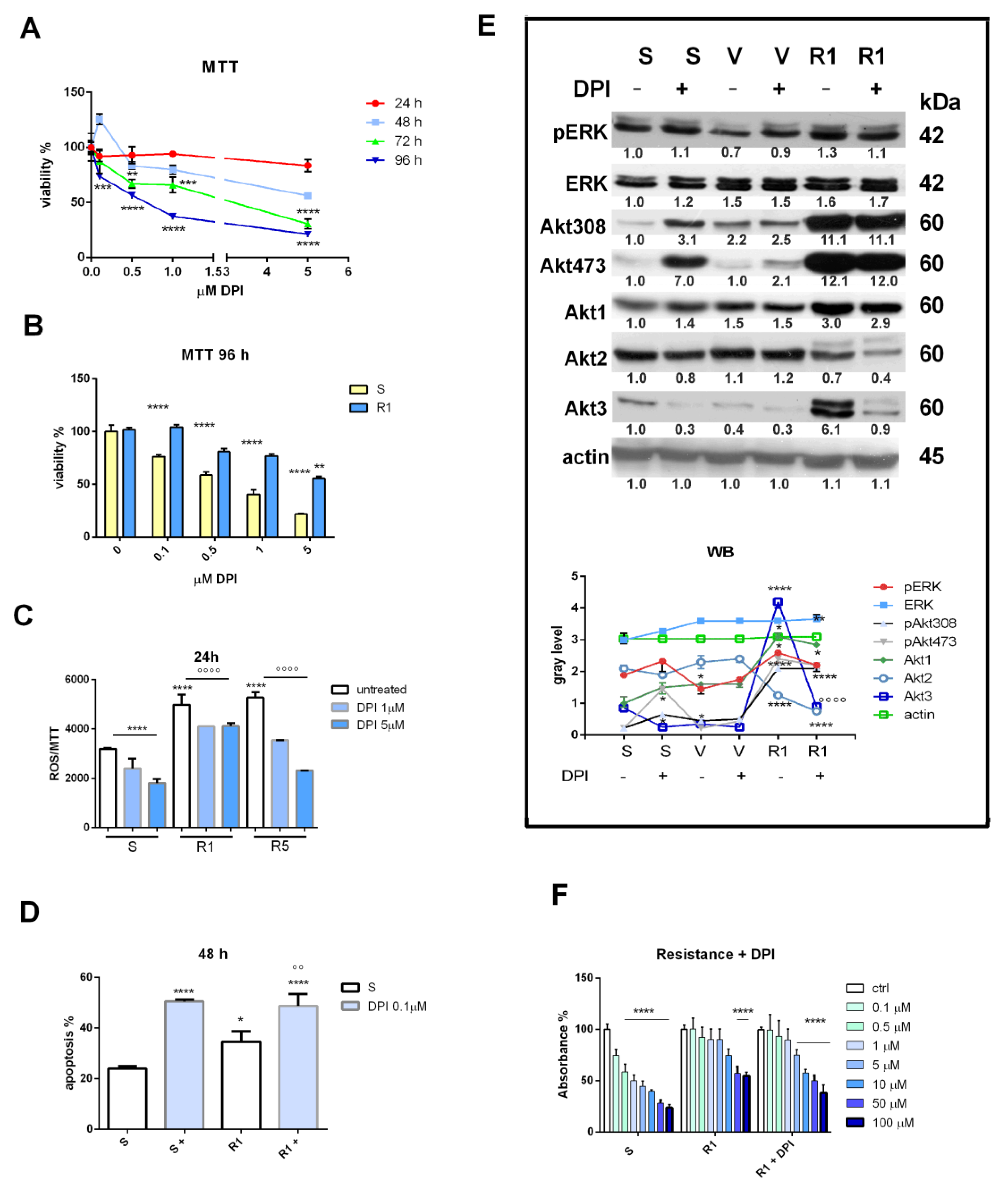
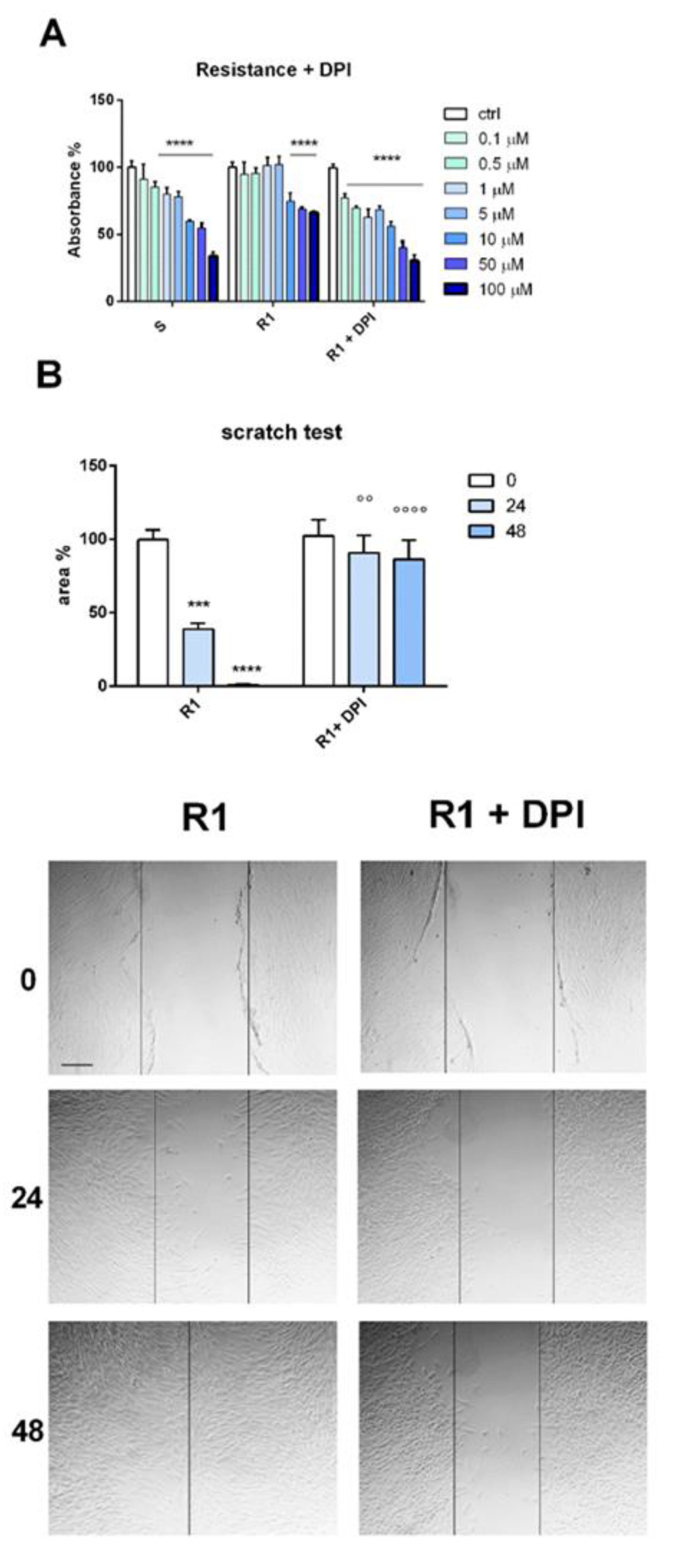
2.2. MTT Assay
2.3. ROS Detection
2.4. Cellular Extract Preparation
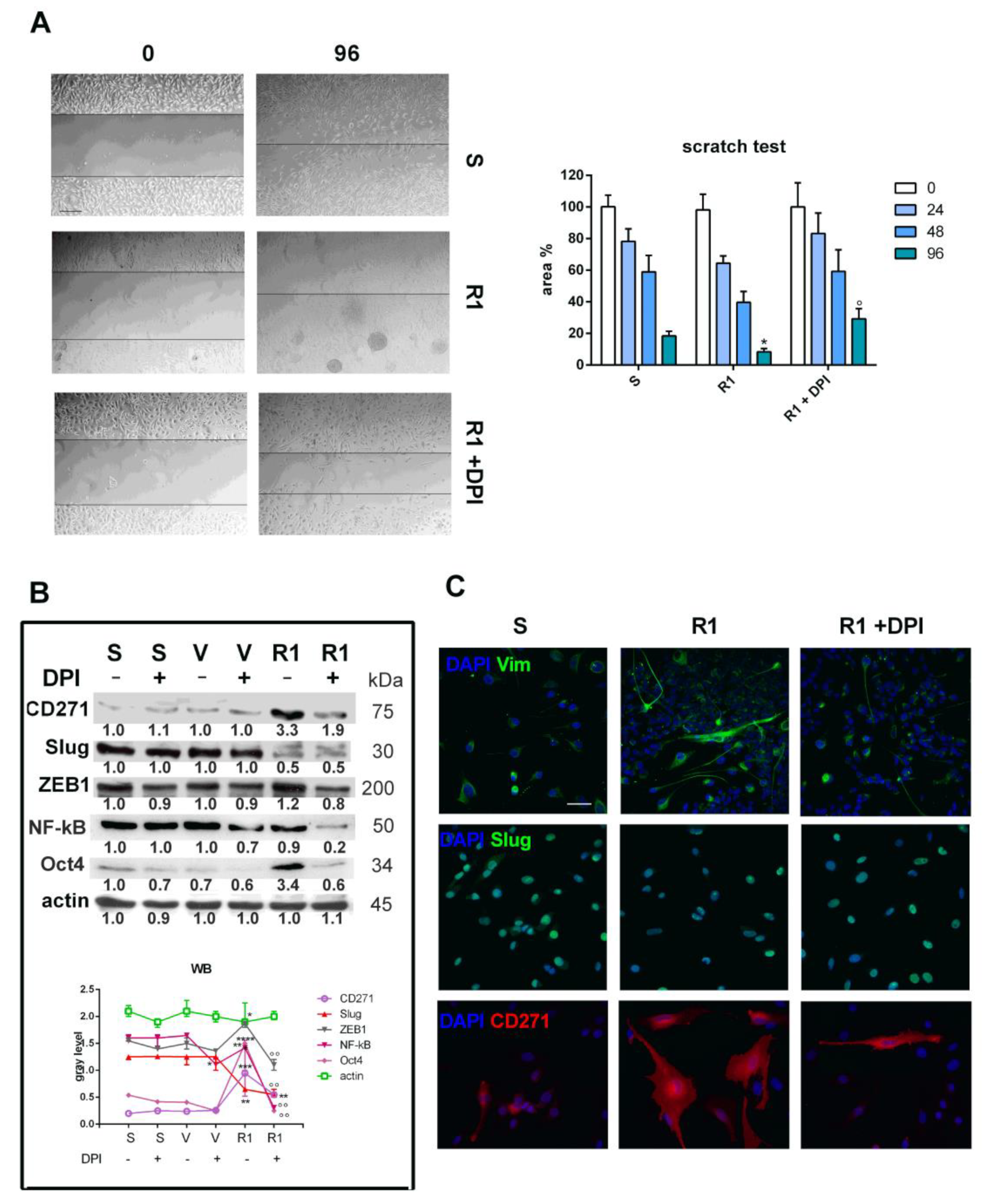
2.5. SDS PAGE and Western Blot
2.6. Immunofluorescence and Confocal Microscopy
2.7. Migration Assay
2.8. Apoptosis Analysis
2.9. Statistical Analysis
3. Results
3.1. Induction of Resistance in the SK-MEL-28 Cell Line
3.2. Nox Inhibition and Drug Resistance
3.3. Effect of Nox Inhibition on Cell Migration and the EMT Process
3.4. Drug-Resistant Primary Melanoma Cells: Effect of Nox Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Verykiou, S.; Alexander, M.; Edwards, N.; Plummer, R.; Chaudhry, B.; Lovat, P.E.; Hill, D.S. Harnessing autophagy to overcome mitogen-activated protein kinase kinase inhibitor-induced resistance in metastatic melanoma. Br. J. Dermatol. 2019, 180, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Rizos, H.; Scolyer, R.A.; Long, G.V. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur. J. Cancer 2016, 62, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Long, G.V. Systemic treatment for BRAF-mutant melanoma: Where do we go next? Lancet Oncol. 2014, 15, e371–e381. [Google Scholar] [CrossRef] [PubMed]
- Verykiou, S.; Edwards, N.; Hill, D. How breakthroughs in translational research have impacted treatment strategies for melanoma. Br. J. Dermatol. 2018, 178, 5–8. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Vidal, A.; Redmer, T. Decoding the Role of CD271 in Melanoma. Cancers 2020, 12, 2460. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, S.; Wang, J.; Zhang, Y.; Xiong, Y.; Zeng, S.X.; Lu, H. Inactivating p53 is essential for nerve growth factor receptor to promote melanoma-initiating cell-stemmed tumorigenesis. Cell Death Dis. 2020, 11, 550. [Google Scholar] [CrossRef]
- Restivo, G.; Diener, J.; Cheng, P.F.; Kiowski, G.; Bonalli, M.; Biedermann, T.; Reichmann, E.; Levesque, M.P.; Dummer, R.; Sommer, L. The low neurotrophin receptor CD271 regulates phenotype switching in melanoma. Nat. Commun. 2017, 8, 1988. [Google Scholar] [CrossRef]
- Murphy, G.F.; Wilson, B.J.; Girouard, S.D.; Frank, N.Y.; Frank, M.H. Stem cells and targeted approaches to melanoma cure. Mol. Asp. Med. 2014, 39, 33–49. [Google Scholar] [CrossRef]
- Pramanik, A.; Xu, Z.; Ingram, N.; Coletta, P.L.; Millner, P.A.; Tyler, A., II; Hughes, T.A. Hyaluronic-Acid-Tagged Cubosomes Deliver Cytotoxics Specifically to CD44-Positive Cancer Cells. Mol. Pharm. 2022, 19, 4601–4611. [Google Scholar] [CrossRef]
- Beretti, F.; Manni, P.; Longo, C.; Argenziano, G.; Farnetani, F.; Cesinaro, A.M.; Witkowski, A.M.; De Pol, A.; Pellacani, G. CD271 is expressed in melanomas with more aggressive behaviour, with correlation of characteristic morphology by in vivo reflectance confocal microscopy. Br. J. Dermatol. 2015, 172, 662–668. [Google Scholar] [CrossRef]
- Boyle, S.E.; Fedele, C.G.; Corbin, V.; Wybacz, E.; Szeto, P.; Lewin, J.; Young, R.J.; Wong, A.; Fuller, R.; Spillane, J.; et al. CD271 Expression on Patient Melanoma Cells Is Unstable and Unlinked to Tumorigenicity. Cancer Res. 2016, 76, 3965–3977. [Google Scholar] [CrossRef]
- Cheli, Y.; Bonnazi, V.F.; Jacquel, A.; Allegra, M.; De Donatis, G.M.; Bahadoran, P.; Bertolotto, C.; Ballotti, R. CD271 is an imperfect marker for melanoma initiating cells. Oncotarget 2014, 5, 5272–5283. [Google Scholar] [CrossRef]
- Lehraiki, A.; Cerezo, M.; Rouaud, F.; Abbe, P.; Allegra, M.; Kluza, J.; Marchetti, P.; Imbert, V.; Cheli, Y.; Bertolotto, C.; et al. Increased CD271 expression by the NF-kB pathway promotes melanoma cell survival and drives acquired resistance to BRAF inhibitor vemurafenib. Cell Discov. 2015, 1, 15030. [Google Scholar] [CrossRef]
- Filipp, F.V.; Li, C.; Boiko, A.D. CD271 is a molecular switch with divergent roles in melanoma and melanocyte development. Sci. Rep. 2019, 9, 7696. [Google Scholar] [CrossRef]
- Redmer, T.; Walz, I.; Klinger, B.; Khouja, S.; Welte, Y.; Schäfer, R.; Regenbrecht, C. The role of the cancer stem cell marker CD271 in DNA damage response and drug resistance of melanoma cells. Oncogenesis 2017, 6, e291. [Google Scholar] [CrossRef]
- Tang, Y.; Durand, S.; Dalle, S.; Caramel, J. EMT-Inducing Transcription Factors, Drivers of Melanoma Phenotype Switching, and Resistance to Treatment. Cancers 2020, 12, 2154. [Google Scholar] [CrossRef]
- Yuan, L.; Mishra, R.; Patel, H.; Abdulsalam, S.; Greis, K.D.; Kadekaro, A.L.; Merino, E.J.; Garrett, J.T. Utilization of reactive oxygen species targeted therapy to prolong the efficacy of BRAF inhibitors in melanoma. J. Cancer 2018, 9, 4665–4676. [Google Scholar] [CrossRef]
- Azouzi, N.; Cailloux, J.; Cazarin, J.M.; Knauf, J.A.; Cracchiolo, J.; Al Ghuzlan, A.; Hartl, D.; Polak, M.; Carré, A.; El Mzibri, M.; et al. NADPH Oxidase NOX4 Is a Critical Mediator of BRAF V600E-Induced Downregulation of the Sodium/Iodide Symporter in Papillary Thyroid Carcinomas. Antioxid. Redox Signal. 2017, 26, 864–877. [Google Scholar] [CrossRef]
- Beretti, F.; Farnetani, F.; Bonetti, L.R.; Fabbiani, L.; Zavatti, M.; Maiorana, A.; Pellacani, G.; Maraldi, T. The Interplay between HGF/c-met Axis and Nox4 in BRAF Mutated Melanoma. Int. J. Mol. Sci. 2021, 22, 761. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; Van Den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109. [Google Scholar] [CrossRef] [PubMed]
- Casciaro, F.; Beretti, F.; Zavatti, M.; McCubrey, J.A.; Ratti, S.; Marmiroli, S.; Follo, M.Y.; Maraldi, T. Nuclear Nox4 interaction with prelamin A is associated with nuclear redox control of stem cell aging. Aging 2018, 10, 2911–2934. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liu, S.; Zhang, G.; Kwong, L.N.; Zhu, Y.; Miller, J.P.; Hu, Y.; Zhong, W.; Zeng, J.; Wu, L.; et al. Oncogenic BRAF-Mediated Melanoma Cell Invasion. Cell Rep. 2016, 15, 2012–2024. [Google Scholar] [CrossRef]
- Beretti, F.; Bertoni, L.; Farnetani, F.; Pellegrini, C.; Gorelli, G.; Cesinaro, A.M.; Reggiani Bonetti, L.; Di Nardo, L.; Kaleci, S.; Chester, J.; et al. Melanoma types by in vivo reflectance confocal microscopy correlated with protein and molecular genetic alterations: A pilot study. Exp. Dermatol. 2019, 28, 254–260. [Google Scholar] [CrossRef]
- Saltari, A.; Dzung, A.; Quadri, M.; Tiso, N.; Facchinello, N.; Hernández-Barranco, A.; Garcia-Silva, S.; Nogués, L.; Stoffel, C.I.; Cheng, P.F.; et al. Specific Activation of the CD271 Intracellular Domain in Combination with Chemotherapy or Targeted Therapy Inhibits Melanoma Progression. Cancer Res. 2021, 81, 6044–6057. [Google Scholar] [CrossRef]
- Silva, C.T.X.; Saddi, V.A.; Silva, K.S.F.E.; Sugita, D.M.; Guillo, L.A. Expression of the cancer stem cell marker OCT4 is associated with worse prognosis and survival in cutaneous melanoma. Melanoma Res. 2021, 31, 439–448. [Google Scholar] [CrossRef]
- Richard, G.; Dalle, S.; Monet, M.; Ligier, M.; Boespflug, A.; Pommier, R.M.; Fouchardière, A.; Perier-Muzet, M.; Depaepe, L.; Barnault, R.; et al. ZEB1-mediated melanoma cell plasticity enhances resistance to MAPK inhibitors. EMBO Mol. Med. 2016, 8, 1143–1161. [Google Scholar] [CrossRef]
- Cordaro, F.G.; De Presbiteris, A.L.; Camerlingo, R.; Mozzillo, N.; Pirozzi, G.; Cavalcanti, E.; Manca, A.; Palmieri, G.; Cossu, A.; Ciliberto, G.; et al. Phenotype characterization of human melanoma cells resistant to dabrafenib. Oncol. Rep. 2017, 38, 2741–2751. [Google Scholar] [CrossRef]
- Pearlman, R.L.; Montes de Oca, M.K.; Pal, H.C.; Afaq, F. Potential therapeutic targets of epithelial-mesenchymal transition in melanoma. Cancer Lett. 2017, 391, 125–140. [Google Scholar] [CrossRef]
- Irvine, M.; Stewart, A.; Pedersen, B.; Boyd, S.; Kefford, R.; Rizos, H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 2018, 7, 72. [Google Scholar] [CrossRef]
- Tian, Y.; Guo, W. A Review of the Molecular Pathways Involved in Resistance to BRAF Inhibitors in Patients with Advanced-Stage Melanoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e920957. [Google Scholar] [CrossRef]
- Yuan, L.; Mishra, R.; Patel, H.; Alanazi, S.; Wei, X.; Ma, Z.; Garrett, J.T. BRAF Mutant Melanoma Adjusts to BRAF/MEK Inhibitors via Dependence on Increased Antioxidant SOD2 and Increased Reactive Oxygen Species Levels. Cancers 2020, 12, 1661. [Google Scholar] [CrossRef]
- Liu-Smith, F.; Dellinger, R.; Meyskens, F.L. Updates of reactive oxygen species in melanoma etiology and progression. Arch. Biochem. Biophys. 2014, 563, 51–55. [Google Scholar] [CrossRef]
- Polytarchou, C.; Hatziapostolou, M.; Yau, T.O.; Christodoulou, N.; Hinds, P.W.; Kottakis, F.; Sanidas, I.; Tsichlis, P.N. Akt3 induces oxidative stress and DNA damage by activating the NADPH oxidase via phosphorylation of p47 phox. Proc. Natl. Acad. Sci. USA 2020, 117, 28806–28815. [Google Scholar] [CrossRef]
- Aldieri, E.; Riganti, C.; Polimeni, M.; Gazzano, E.; Lussiana, C.; Campia, I.; Ghigo, D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr. Drug Metab. 2008, 9, 686–696. [Google Scholar] [CrossRef]
- Moody, T.W.; Lee, L.; Iordanskaia, T.; Ramos-Alvarez, I.; Moreno, P.; Boudreau, H.E.; Leto, T.L.; Jensen, R.T. PAC1 regulates receptor tyrosine kinase transactivation in a reactive oxygen species-dependent manner. Peptides 2019, 120, 170017. [Google Scholar] [CrossRef]
- Chen, C.C.; Cheng, Y.Y.; Chen, S.C.; Tuan, Y.F.; Chen, Y.J.; Chen, C.Y.; Chen, L.C. Cyclooxygenase-2 expression is up-regulated by 2-aminobiphenyl in a ROS and MAPK-dependent signaling pathway in a bladder cancer cell line. Chem. Res. Toxicol. 2012, 25, 695–705. [Google Scholar] [CrossRef]
- Tanaka, M.; Miura, Y.; Numanami, H.; Karnan, S.; Ota, A.; Konishi, H.; Hosokawa, Y.; Hanyuda, M. Inhibition of NADPH oxidase 4 induces apoptosis in malignant mesothelioma: Role of reactive oxygen species. Oncol. Rep. 2015, 34, 1726–1732. [Google Scholar] [CrossRef]
- Piszczatowska, K.; Przybylska, D.; Sikora, E.; Mosieniak, G. Inhibition of NADPH Oxidases Activity by Diphenyleneiodonium Chloride as a Mechanism of Senescence Induction in Human Cancer Cells. Antioxidants 2020, 9, 1248. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beretti, F.; Gatti, M.; Zavatti, M.; Bassoli, S.; Pellacani, G.; Maraldi, T. Reactive Oxygen Species Regulation of Chemoresistance and Metastatic Capacity of Melanoma: Role of the Cancer Stem Cell Marker CD271. Biomedicines 2023, 11, 1229. https://doi.org/10.3390/biomedicines11041229
Beretti F, Gatti M, Zavatti M, Bassoli S, Pellacani G, Maraldi T. Reactive Oxygen Species Regulation of Chemoresistance and Metastatic Capacity of Melanoma: Role of the Cancer Stem Cell Marker CD271. Biomedicines. 2023; 11(4):1229. https://doi.org/10.3390/biomedicines11041229
Chicago/Turabian StyleBeretti, Francesca, Martina Gatti, Manuela Zavatti, Sara Bassoli, Giovanni Pellacani, and Tullia Maraldi. 2023. "Reactive Oxygen Species Regulation of Chemoresistance and Metastatic Capacity of Melanoma: Role of the Cancer Stem Cell Marker CD271" Biomedicines 11, no. 4: 1229. https://doi.org/10.3390/biomedicines11041229
APA StyleBeretti, F., Gatti, M., Zavatti, M., Bassoli, S., Pellacani, G., & Maraldi, T. (2023). Reactive Oxygen Species Regulation of Chemoresistance and Metastatic Capacity of Melanoma: Role of the Cancer Stem Cell Marker CD271. Biomedicines, 11(4), 1229. https://doi.org/10.3390/biomedicines11041229