
Urticaria: A Narrative Overview of Differential Diagnosis


Abstract
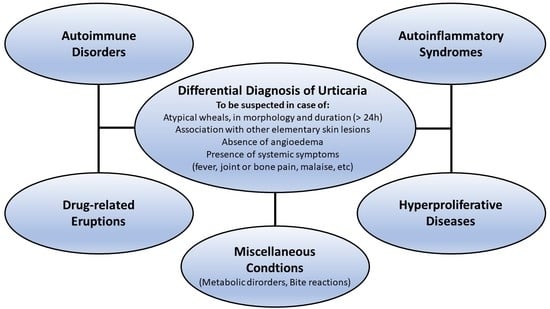
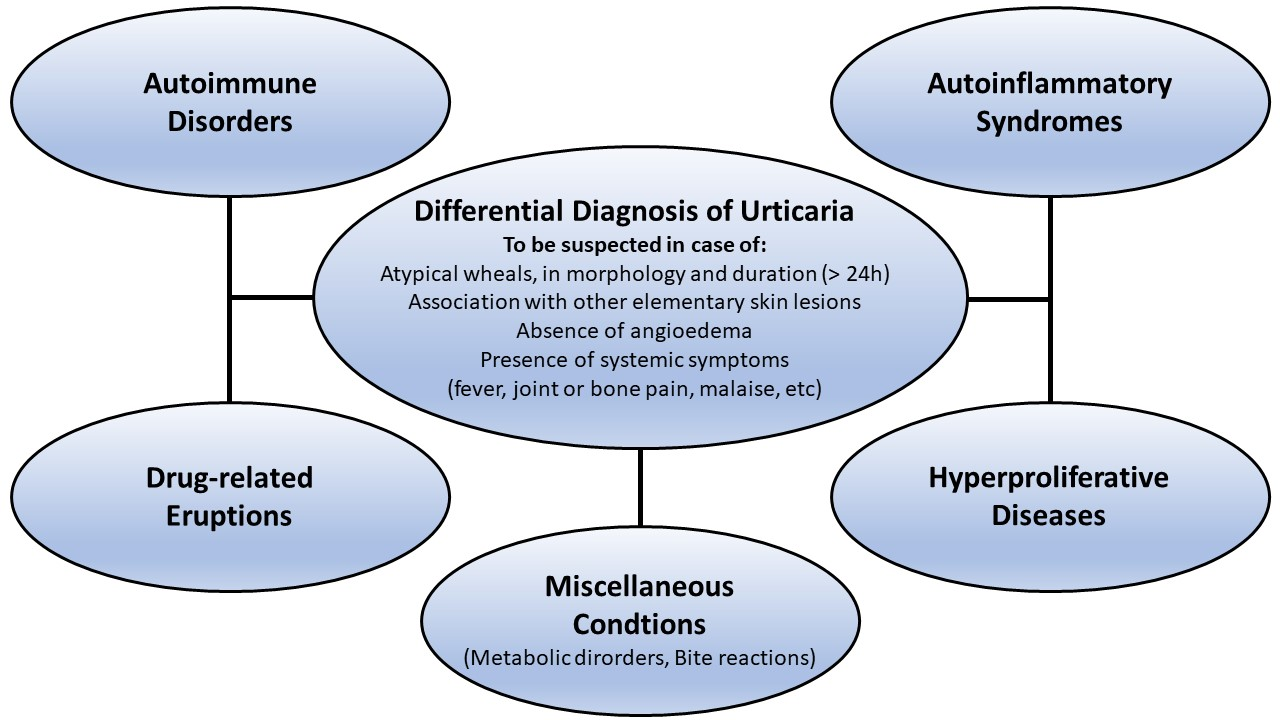
1. Introduction
1.1. Definition, Classification and Pathogenesis of Classic Urticaria
1.2. Epidemiology
1.3. Clinical and Histologic Aspects
1.4. Therapy
1.5. Differential Diagnosis
2. Autoinflammatory Urticarial Syndromes
2.1. Neutrophilic Urticarial Dermatosis
2.2. Cryopyrin-Associated Periodic Syndrome
2.3. Schnitzler’s Syndrome
2.4. Adult-Onset Still’s Disease
2.5. Systemic-Onset Juvenile Idiopathic Arthritis
2.6. Mevalonate Kinase Deficienc
2.7. TNF-Receptor-Associated Periodic Syndrome
3. Immune-Mediated Conditions
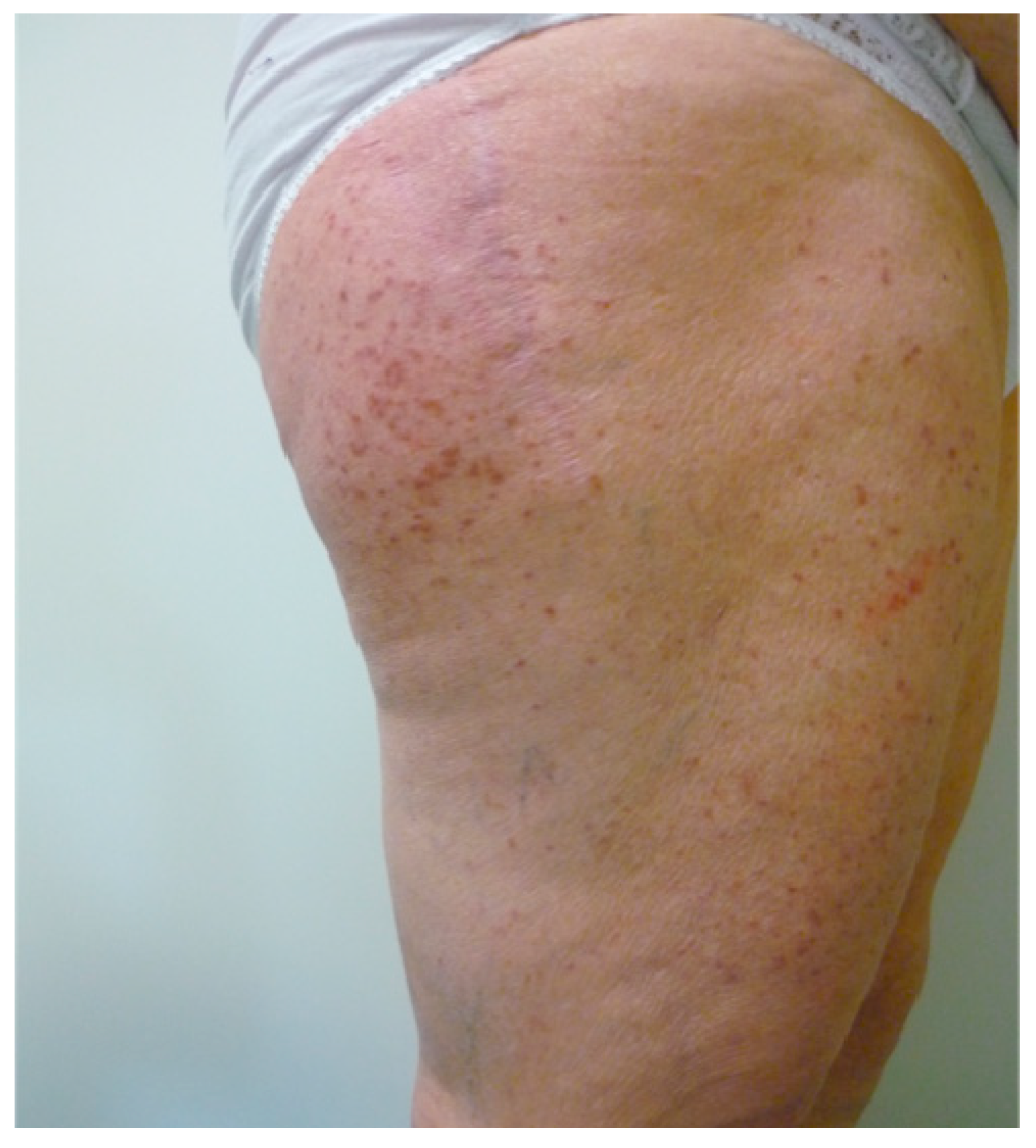
3.1. Urticarial Dermatitis
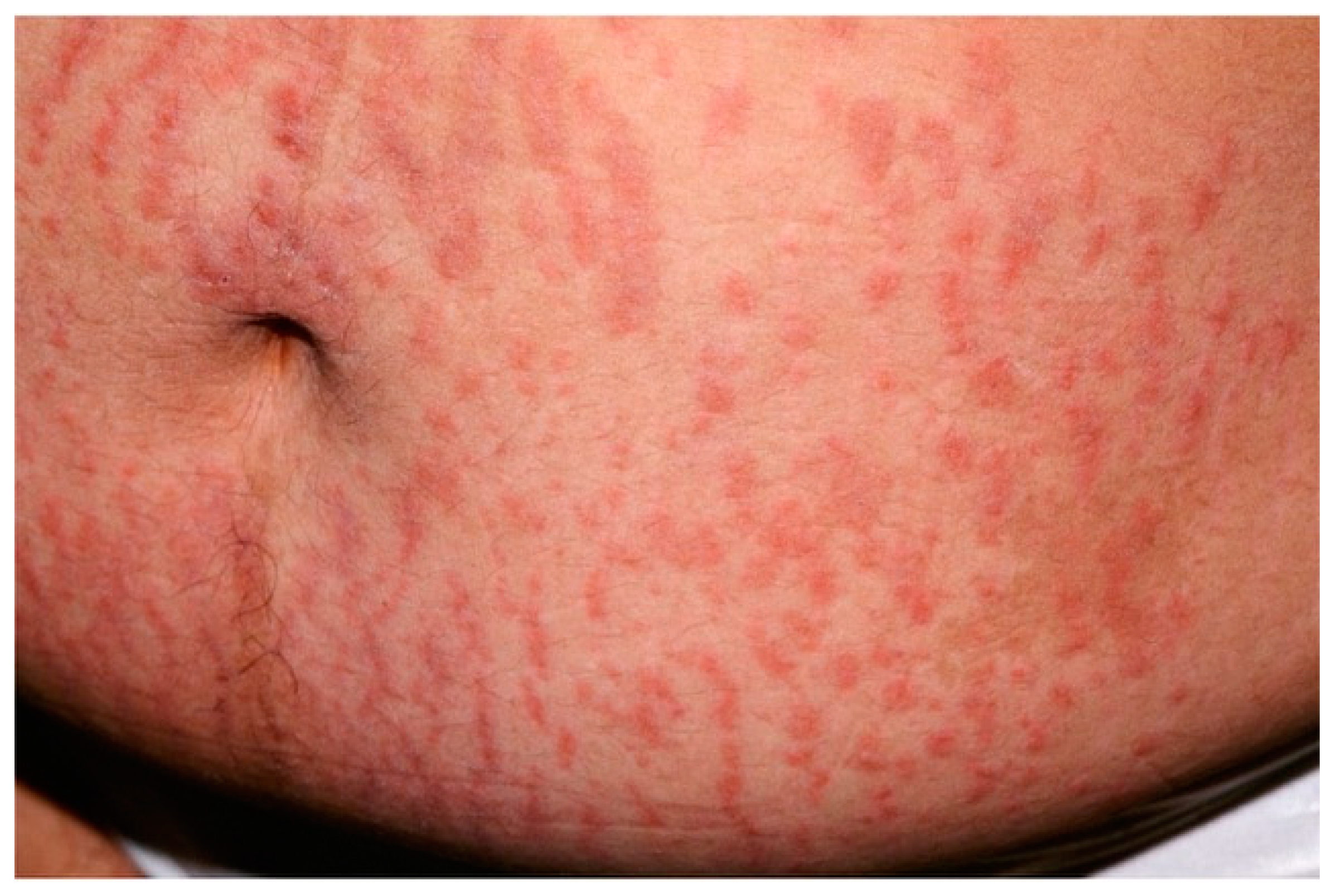
3.2. Urticarial Vasculitis
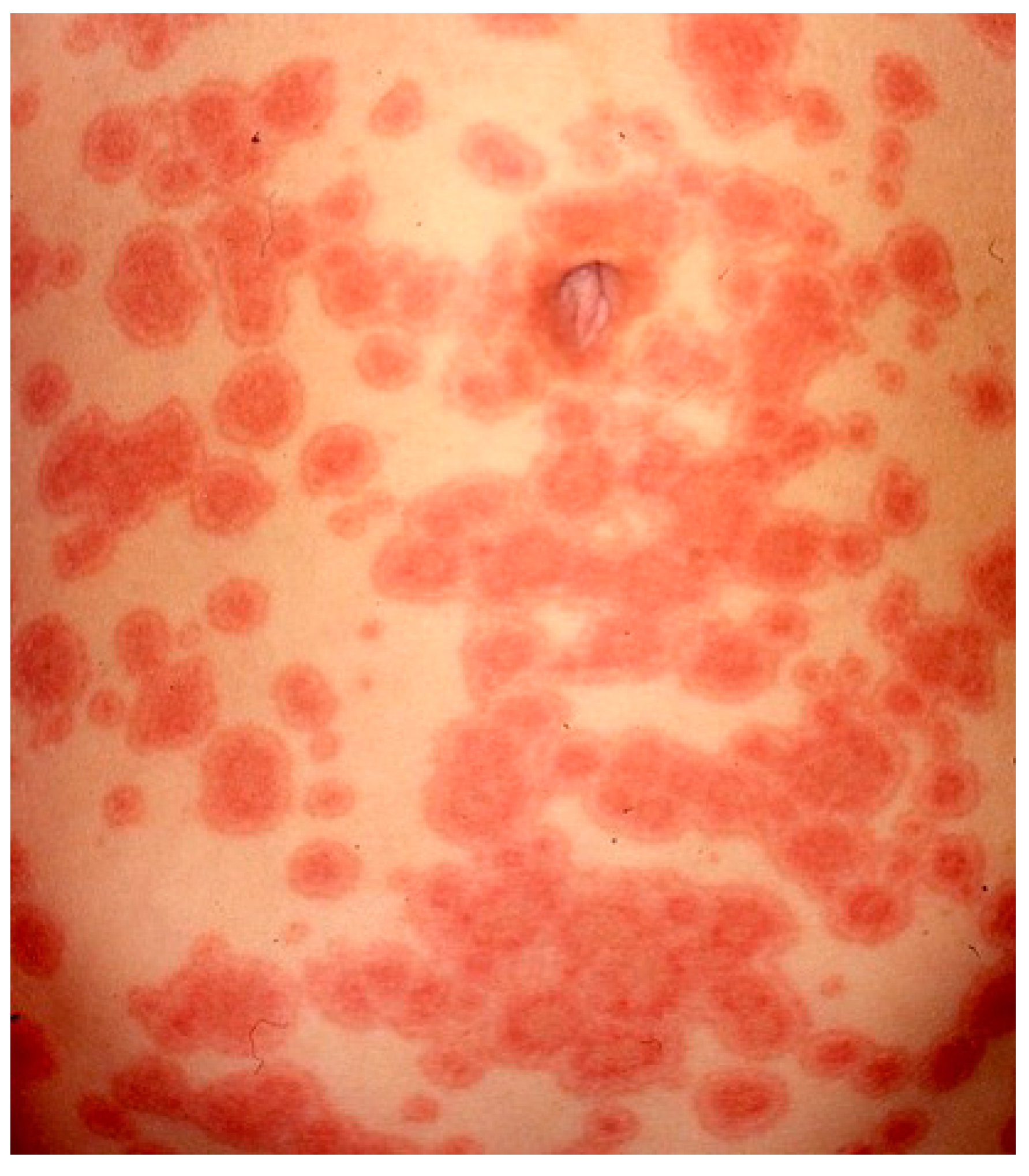
3.3. Cutaneous Lupus Erythematosus
4. Autoimmune Disorders
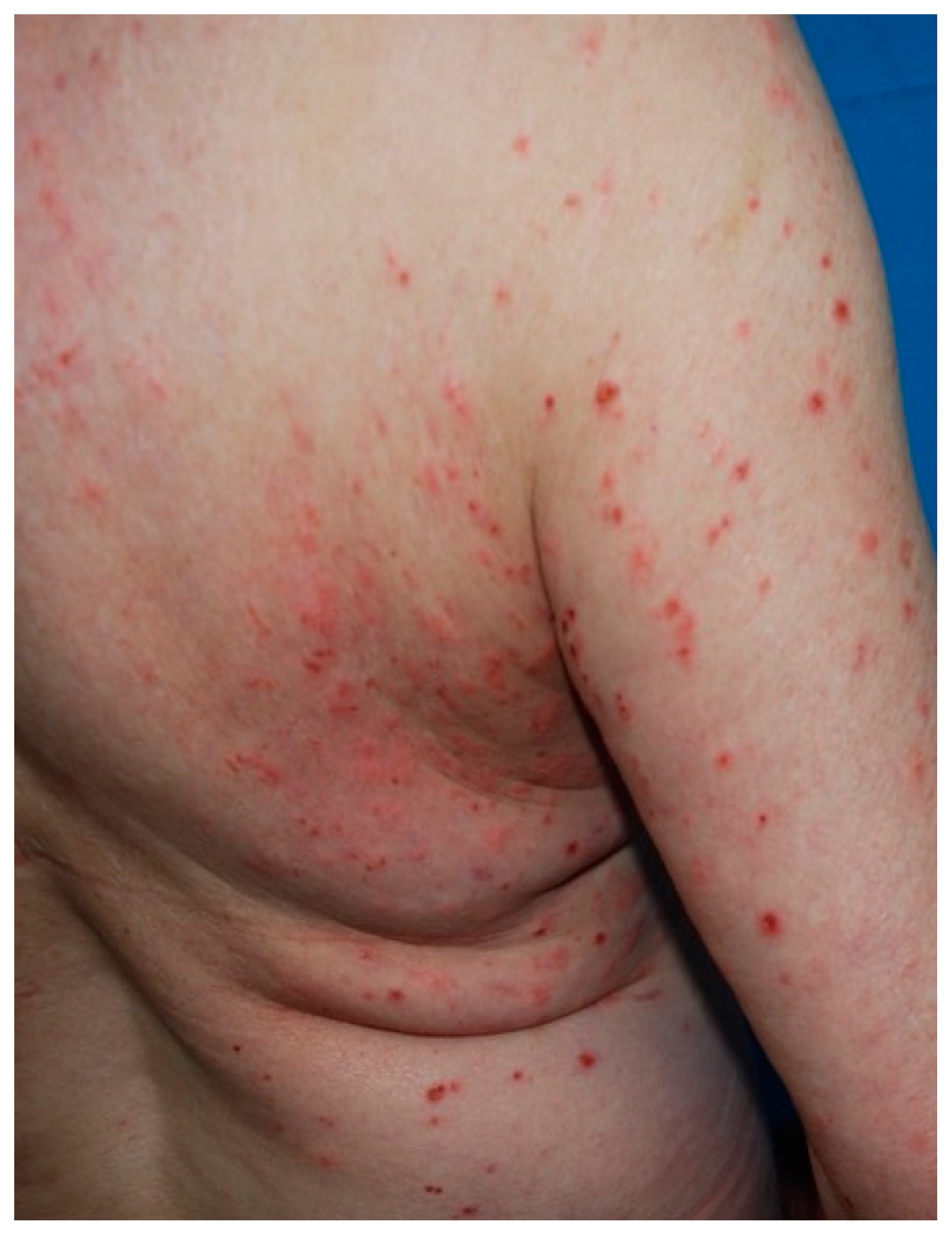
4.1. Bullous Pemphigoid
4.2. Pemphigoid Gestationis
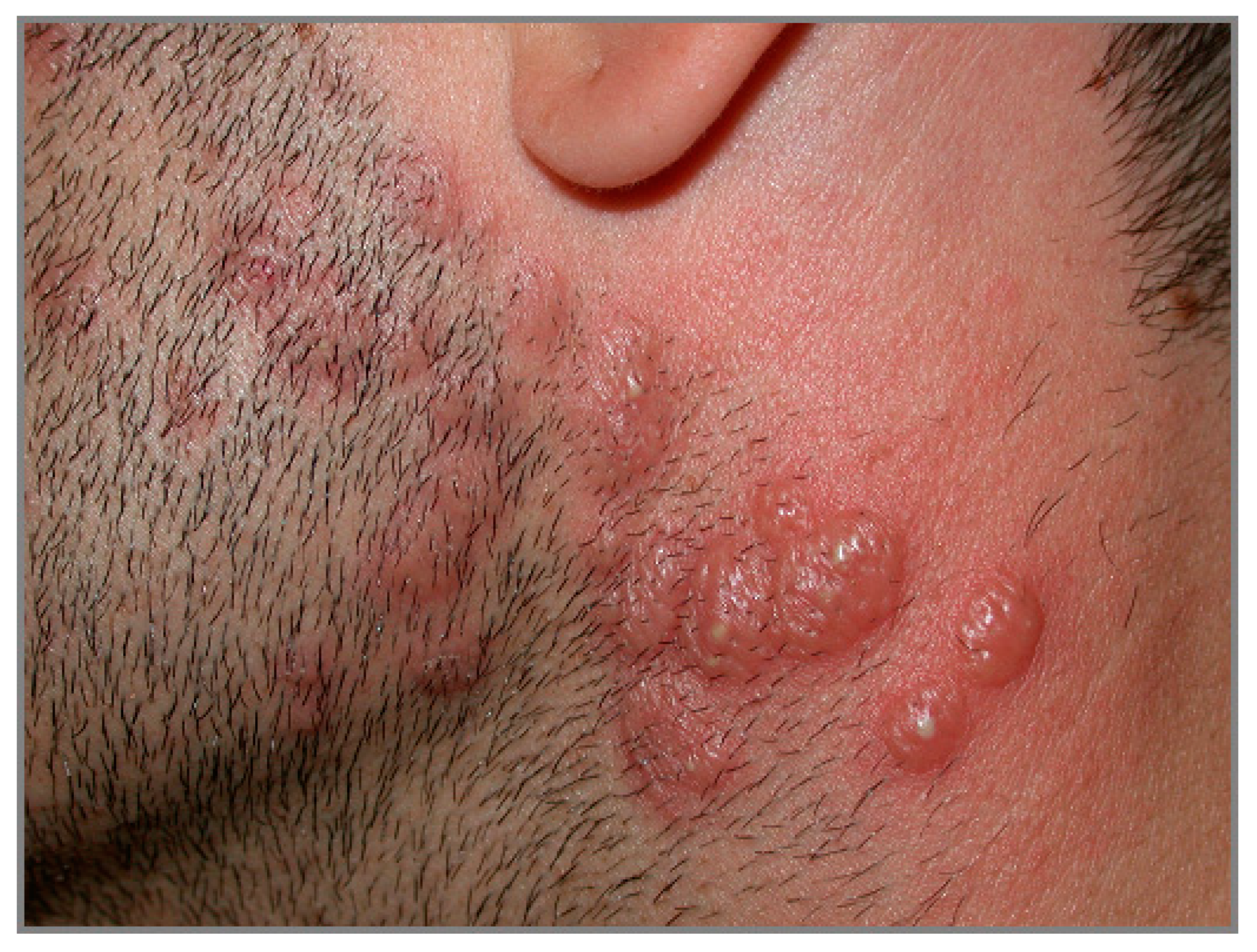
4.3. Dermatitis Herpetiformis
4.4. Autoimmune Progesterone Dermatitis
5. Hyperproliferative Diseases
5.1. Mastocytosis
5.2. Hypereosinophilic Syndrome
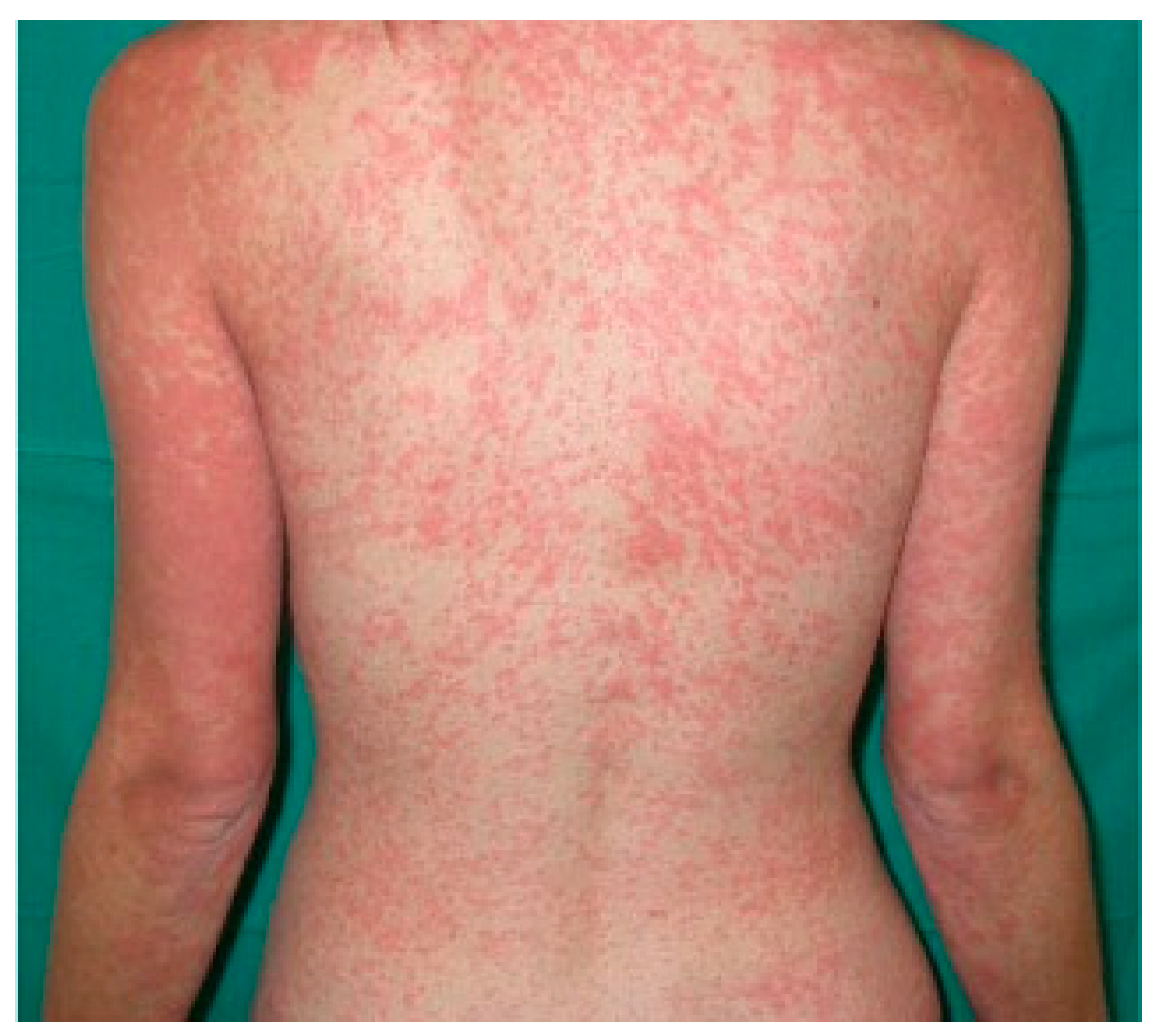
6. Drug-Related Eruptions
6.1. Iatrogenic Rash
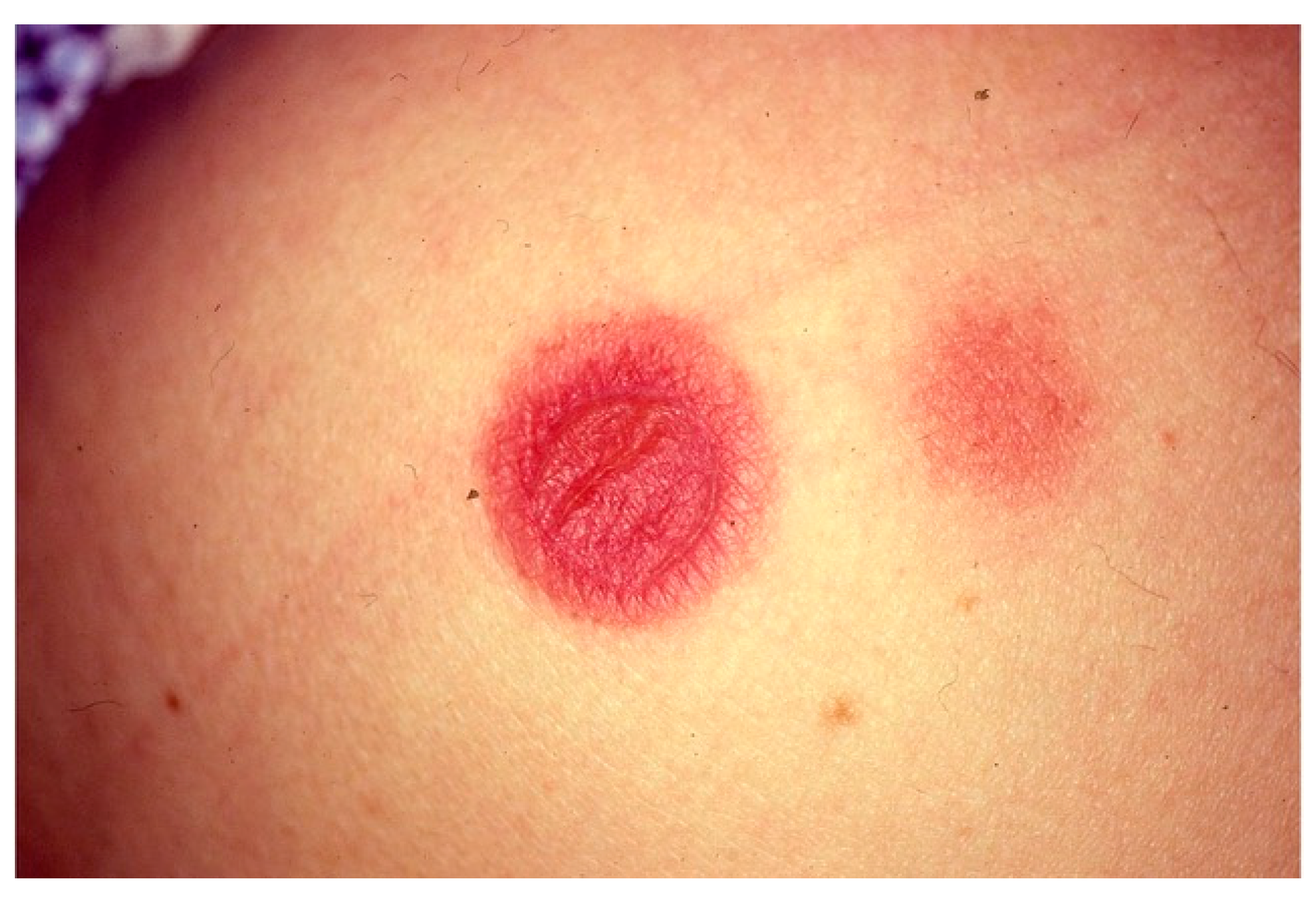
6.2. Fixed Drug Eruption
7. Other Inflammatory Conditions
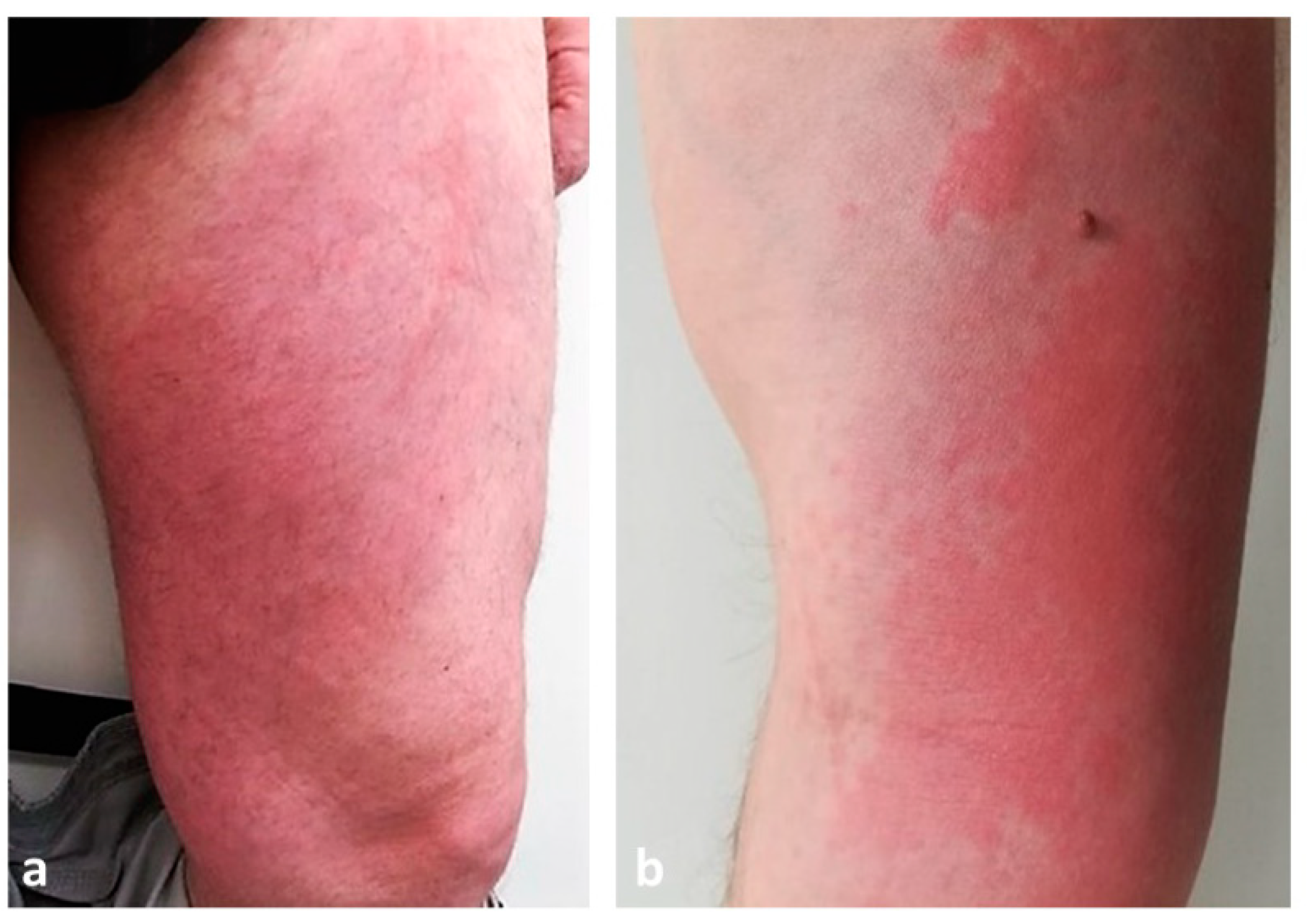
7.1. Wells Syndrome
7.2. Sweet Syndrome
7.3. Polymorphic Eruption of Pregnancy
7.4. Erythema Multiforme
7.5. Hyper IgE Syndromes
7.6. Insect Bite Reactions
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Krause, K.; Grattan, C.E.; Bindslev-Jensen, C.; Gattorno, M.; Kallinich, T.; de Koning, H.D.; Lachmann, H.J.; Lipsker, D.; Navarini, A.A.; Simon, A.; et al. How not to miss autoinflammatory diseases masquerading as urticaria. Allergy 2012, 67, 1465–1474. [Google Scholar] [PubMed]
- Radonjic-Hoesli, S.; Hofmeier, K.S.; Micaletto, S.; Schmid-Grendelmeier, P.; Bircher, A.; Simon, D. Urticaria and Angioedema: An Update on Classification and Pathogenesis. Clin. Rev. Allergy Immunol. 2018, 54, 88–101. [Google Scholar]
- Kolkhir, P.; Church, M.K.; Weller, K.; Metz, M.; Schmetzer, O.; Maurer, M. Autoimmune chronic spontaneous urticaria: What we know and what we do not know. J. Allergy Clin. Immunol. 2017, 139, 1772–1781.e1. [Google Scholar]
- Magerl, M.; Altrichter, S.; Borzova, E.; Giménez-Arnau, A.; Grattan, C.E.H.; Lawlor, F.; Mathelier-Fusade, P.; Meshkova, R.Y.; Zuberbier, T.; Metz, M.; et al. The definition, diagnostic testing, and management of chronic inducible urticarias—The EAACI/GA(2) LEN/EDF/UNEV consensus recommendations 2016 update and revision. Allergy 2016, 71, 780–802. [Google Scholar]
- Balp, M.M.; Lopes da Silva, N.; Vietri, J.; Tian, H.; Ensina, L.F. The Burden of Chronic Urticaria from Brazilian Patients’ Perspective. Dermatol. Ther. 2017, 7, 535–545. [Google Scholar]
- Zuberbier, T.; Balke, M.; Worm, M.; Edenharter, G.; Maurer, M. Epidemiology of urticaria: A representative cross-sectional population survey. Clin. Exp. Dermatol. 2010, 35, 869–873. [Google Scholar] [CrossRef]
- Kim, B.R.; Yang, S.; Choi, J.W.; Choi, C.W.; Youn, S.W. Epidemiology and comorbidities of patients with chronic urticaria in Korea: A nationwide population-based study. J. Dermatol. 2018, 45, 10–16. [Google Scholar]
- Wertenteil, S.; Strunk, A.; Garg, A. Prevalence estimates for chronic urticaria in the United States: A sex- and age-adjusted population analysis. J. Am. Acad. Dermatol. 2019, 81, 152–156. [Google Scholar] [PubMed]
- Balp, M.M.; Vietri, J.; Tian, H.; Isherwood, G. The Impact of Chronic Urticaria from the Patient’s Perspective: A Survey in Five European Countries. Patient-Patient-Cent. Outcomes Res. 2015, 8, 551–558. [Google Scholar]
- Lee, S.J.; Ha, E.K.; Jee, H.M.; Lee, K.S.; Lee, S.W.; Kim, M.A.; Kim, D.H.; Jung, Y.-H.; Sheen, Y.H.; Sung, M.S.; et al. Prevalence and Risk Factors of Urticaria With a Focus on Chronic Urticaria in Children. Allergy Asthma Immunol. Res. 2017, 9, 212–219. [Google Scholar] [PubMed]
- Balp, M.-M.; Weller, K.; Carboni, V.; Chirilov, A.; Papavassilis, C.; Severin, T.; Tian, H.; Zuberbier, T.; Maurer, M. Prevalence and clinical characteristics of chronic spontaneous urticaria in pediatric patients. Pediatr. Allergy Immunol. 2018, 29, 630–636. [Google Scholar] [PubMed]
- Nettis, E.; Foti, C.; Ambrifi, M.; Baiardini, I.; Bianchi, L.; Borghi, A.; Caminati, M.; Canonica, G.W.; Casciaro, M.; Colli, L.; et al. Urticaria: Recommendations from the Italian Society of Allergology, Asthma and Clinical Immunology and the Italian Society of Allergological, Occupational and Environmental Dermatology. Clin. Mol. Allergy 2020, 18, 8. [Google Scholar] [PubMed]
- Hon, K.L.; Leung, A.K.C.; Ng, W.G.G.; Loo, S.K. Chronic Urticaria: An Overview of Treatment and Recent Patents. Recent. Patents Inflamm. Allergy Drug Discov. 2019, 13, 27–37. [Google Scholar]
- Zuberbier, T.; Abdul Latiff, A.H.; Abuzakouk, M.; Aquilina, S.; Asero, R.; Baker, D.; Ballmer-Weber, B.; Bangert, C.; Ben-Shoshan, M.; Bernstein, J.A.; et al. The international EAACI/GA2LEN/EuroGuiDerm/APAAACI guideline for the definition, classification, diagnosis, and management of urticaria. Allergy 2022, 77, 734–766. [Google Scholar]
- Manti, S.; Giallongo, A.; Papale, M.; Parisi, G.F.; Leonardi, S. Monoclonal Antibodies in Treating Chronic Spontaneous Urticaria: New Drugs for an Old Disease. J. Clin. Med. 2022, 11, 4453. [Google Scholar] [PubMed]
- Khan, S.; Chopra, C.; Mitchell, A.; Nakonechna, A.; Yong, P.; Karim, M.Y. Resistant Chronic Spontaneous Urticaria—A Case Series Narrative Review of Treatment Options. Allergy Rhinol. 2022, 13, 21526575221144950. [Google Scholar] [CrossRef] [PubMed]
- Grattan, C.E.; O’Donnell, B.F.; Francis, D.M.; Niimi, N.; Barlow, R.J.; Seed, P.T.; Black, A.K.; Greaves, M.W. Randomized double-blind study of cyclosporin in chronic ‘idiopathic’ urticaria. Br. J. Dermatol. 2000, 143, 365–372. [Google Scholar] [CrossRef]
- Zuberbier, T.; Iffländer, J.; Semmler, C.; Henz, B.M. Acute urticaria: Clinical aspects and therapeutic responsiveness. Acta Derm.-Venereol. 1996, 76, 295–297. [Google Scholar]
- Matos, A.L.; Figueiredo, C.; Gonçalo, M. Differential Diagnosis of Urticarial Lesions. Front. Allergy 2022, 3, 808543. [Google Scholar] [CrossRef]
- Brodell, L.A.; Beck, L.A. Differential diagnosis of chronic urticaria. Ann. Allergy Asthma Immunol. 2008, 100, 181–188; discussion 188–190, 215. [Google Scholar]
- Moltrasio, C.; Romagnuolo, M.; Marzano, A.V. NLRP3 inflammasome and NLRP3-related autoinflammatory diseases: From cryopyrin function to targeted therapies. Front. Immunol. 2022, 13, 1007705. [Google Scholar]
- Wu, D.; Shen, M.; Yao, Q. Cutaneous Manifestations of Autoinflammatory Diseases. Rheumatol. Immunol. Res. 2021, 2, 217–225. [Google Scholar] [PubMed]
- Gusdorf, L.; Lipsker, D. Neutrophilic urticarial dermatosis: A review. Ann. Dermatol. Venereol. 2018, 145, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Gusdorf, L.; Lipsker, D. Neutrophilic urticarial dermatosis: An entity bridging monogenic and polygenic autoinflammatory disorders, and beyond. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 685–690. [Google Scholar] [PubMed]
- Simon, A.; Asli, B.; Braun-Falco, M.; De Koning, H.; Fermand, J.P.; Grattan, C.; Krause, K.; Lachmann, H.; Lenormand, C.; Martinez-Taboada, V.; et al. Schnitzler’s syndrome: Diagnosis, treatment, and follow-up. Allergy 2013, 68, 562–568. [Google Scholar]
- Feist, E.; Mitrovic, S.; Fautrel, B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat. Rev. Rheumatol. 2018, 14, 603–618. [Google Scholar] [PubMed]
- Rao, S.; Tsang, L.S.L.; Zhao, M.; Shi, W.; Lu, Q. Adult-onset Still’s disease: A disease at the crossroad of innate immunity and autoimmunity. Front. Med. 2022, 9, 881431. [Google Scholar]
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T. Preliminary criteria for classification of adult Still’s disease. J. Rheumatol. 1992, 19, 424–430. [Google Scholar]
- Cozzi, A.; Papagrigoraki, A.; Biasi, D.; Colato, C.; Girolomoni, G. Cutaneous manifestations of adult-onset Still’s disease: A case report and review of literature. Clin. Rheumatol. 2016, 35, 1377–1382. [Google Scholar]
- Ambler, W.G.; Nanda, K.; Onel, K.B.; Shenoi, S. Refractory systemic onset juvenile idiopathic arthritis: Current challenges and future perspectives. Ann. Med. 2022, 54, 1839–1850. [Google Scholar]
- Prakken, B.; Albani, S.; Martini, A. Juvenile idiopathic arthritis. Lancet 2011, 377, 2138–2149. [Google Scholar] [PubMed]
- Simon, A.; Kremer, H.P.H.; Wevers, R.A.; Scheffer, H.; De Jong, J.G.; Van Der Meer, J.W.M.; Drenth, J.P.H. Mevalonate kinase deficiency: Evidence for a phenotypic continuum. Neurology 2004, 62, 994–997. [Google Scholar] [CrossRef] [PubMed]
- van der Burgh, R.; Ter Haar, N.M.; Boes, M.L.; Frenkel, J. Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Clin. Immunol. 2013, 147, 197–206. [Google Scholar]
- Stoffels, M.; Simon, A. Hyper-IgD syndrome or mevalonate kinase deficiency. Curr. Opin. Rheumatol. 2011, 23, 419–423. [Google Scholar] [CrossRef]
- Menon, S.G.; Efthimiou, P. Tumor necrosis factor-associated periodic syndrome in adults. Rheumatol. Int. 2018, 38, 3–11. [Google Scholar]
- Schmaltz, R.; Vogt, T.; Reichrath, J. Skin manifestations in tumor necrosis factor receptor-associated periodic syndrome (TRAPS). Dermato-endocrinology 2010, 2, 26–29. [Google Scholar] [CrossRef]
- Moreira, A.; Torres, B.; Peruzzo, J.; Mota, A.; Eyerich, K.; Ring, J. Skin symptoms as diagnostic clue for autoinflammatory diseases. An. Bras. Dermatol. 2017, 92, 72–80. [Google Scholar]
- Hannon, G.R.; Wetter, D.A.; Gibson, L.E. Urticarial dermatitis: Clinical features, diagnostic evaluation, and etiologic associations in a series of 146 patients at Mayo Clinic (2006-2012). J. Am. Acad. Dermatol. 2014, 70, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Ben Kridis, W.; Khanfir, A. An uncommon case of pancreatic adenocarcinoma revealed by dermatitis. JGH Open Open Access J. Gastroenterol. Hepatol. 2022, 6, 280–281. [Google Scholar]
- Kossard, S.; Hamann, I.; Wilkinson, B. Defining urticarial dermatitis: A subset of dermal hypersensitivity reaction pattern. Arch. Dermatol. 2006, 142, 29–34. [Google Scholar] [CrossRef]
- Mata, D.; Lian, J.; Krakowski, A.; Agarwala, S.; Hafeez, F. Histopathologic and immunophenotypic features of idiopathic dermal hypersensitivity reaction/urticarial dermatitis: A clinicopathologic study. J. Cutan. Pathol. 2021, 48, 592–595. [Google Scholar] [PubMed]
- Banan, P.; Butler, G.; Wu, J. Retrospective chart review in a cohort of patients with urticarial dermatitis. Australas. J. Dermatol. 2014, 55, 137–139. [Google Scholar] [CrossRef]
- García-García, B.; Aubán-Pariente, J.; Munguía-Calzada, P.; Vivanco, B.; Argenziano, G.; Vázquez-López, F. Development of a clinical-dermoscopic model for the diagnosis of urticarial vasculitis. Sci. Rep. 2020, 10, 6092. [Google Scholar]
- Gu, S.L.; Jorizzo, J.L. Urticarial vasculitis. Int. J. Womens Dermatol. 2021, 7, 290–297. [Google Scholar] [PubMed]
- Puhl, V.; Bonnekoh, H.; Scheffel, J.; Hawro, T.; Weller, K.; von den Driesch, P.; Röwert-Huber, H.; Cardoso, J.; Gonçalo, M.; Maurer, M.; et al. A novel histopathological scoring system to distinguish urticarial vasculitis from chronic spontaneous urticaria. Clin. Transl. Allergy 2021, 11, e12031. [Google Scholar] [PubMed]
- Jarrett, P.; Werth, V.P. A review of cutaneous lupus erythematosus: Improving outcomes with a multidisciplinary approach. J. Multidiscip. Healthc. 2019, 12, 419–428. [Google Scholar] [PubMed]
- Jatwani, S.; Holmes, M.P.H. Subacute Cutaneous Lupus Erythematosus [Internet]. StatPearls [Internet]. StatPearls Publishing. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554554/ (accessed on 15 December 2022).
- McDaniel, B.; Sukumaran, S.; Koritala, T.; Tanner, L.S. Discoid Lupus Erythematosus [Internet]. StatPearls [Internet]. StatPearls Publishing. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK493145/ (accessed on 16 February 2023).
- Kole, A.K.; Ghosh, A. Cutaneous manifestations of systemic lupus erythematosus in a tertiary referral center. Indian J. Dermatol. 2009, 54, 132–136. [Google Scholar]
- Spadoni, M.; Jacob, C.; Aikawa, N.; Jesus, A.; Fomin, A.; Silva, C. Chronic autoimmune urticaria as the first manifestation of juvenile systemic lupus erythematosus. Lupus 2011, 20, 763–766. [Google Scholar]
- Yell, J.A.; Mbuagbaw, J.; Burge, S.M. Cutaneous manifestations of systemic lupus erythematosus. Br. J. Dermatol. 1996, 135, 355–362. [Google Scholar] [CrossRef]
- Baigrie, D.; Nookala, V. Bullous Pemphigoid [Internet]. StatPearls [Internet]’ StatPearls Publishing. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK535374/ (accessed on 15 December 2022).
- Bernard, P.; Antonicelli, F. Bullous Pemphigoid: A Review of its Diagnosis, Associations and Treatment. Am. J. Clin. Dermatol. 2017, 18, 513–528. [Google Scholar]
- Fong, M.; Gandhi, G.R.; Gharbi, A.; Hafsi, W. Pemphigoid Gestationis [Internet]. StatPearls [Internet]. StatPearls Publishing. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470287/ (accessed on 21 March 2023).
- Lobato-Berezo, A.; Fernández Figueras, M.T.; Moreno Romero, J.A.; Pujol, R.M. Pemphigoid Gestationis Mimicking Erythema Multiforme With Mucosal Involvement. Actas Dermosifiliogr. 2019, 110, 696–697. [Google Scholar] [CrossRef] [PubMed]
- Kukkamalla, R.M.; Bayless, P. Pemphigoid Gestationis. Clin. Pract. Cases Emerg. Med. 2019, 3, 79–80. [Google Scholar] [CrossRef]
- Daniel, B.S.; Murrell, D.F. Review of autoimmune blistering diseases: The Pemphigoid diseases. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1685–1694. [Google Scholar] [CrossRef]
- Maglie, R.; Quintarelli, L.; Verdelli, A.; Fabbri, P.; Antiga, E.; Caproni, M. Specific dermatoses of pregnancy other than pemphigoid gestationis. G. Ital. Dermatol. Venereol. Organo. Uff Soc. Ital. Dermatol. Sifilogr. 2019, 154, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Salmi, T.; Hervonen, K. Current Concepts of Dermatitis Herpetiformis. Acta Derm.-Venereol. 2020, 100, adv00056. [Google Scholar] [CrossRef]
- Nguyen, C.N.; Kim, S.J. Dermatitis Herpetiformis: An Update on Diagnosis, Disease Monitoring, and Management. Medicina 2021, 57, 843. [Google Scholar] [CrossRef]
- Görög, A.; Antiga, E.; Caproni, M.; Cianchini, G.; De, D.; Dmochowski, M.; Dolinsek, J.; Drenovska, K.; Feliciani, C.; Hervonen, K.; et al. S2k guidelines (consensus statement) for diagnosis and therapy of dermatitis herpetiformis initiated by the European Academy of Dermatology and Venereology (EADV). J. Eur. Acad. Dermatol. Venereol. 2021, 35, 1251–1277. [Google Scholar] [CrossRef]
- Mirza, H.A.; Gharbi, A.; Bhutta, B.S. Dermatitis Herpetiformis [Internet]. StatPearls [Internet]. StatPearls Publishing. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK493163/ (accessed on 19 December 2022).
- Irshad, S.; Haider, M.S.; Master, M.F.; Asif, N.; Khalil, A. Autoimmune Progesterone Dermatitis. Cureus 2021, 13, e19217. [Google Scholar] [CrossRef]
- Nguyen, T.; Razzaque Ahmed, A. Autoimmune progesterone dermatitis: Update and insights. Autoimmun. Rev. 2016, 15, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.L.; Carr, T.F. Iatrogenic autoimmune progesterone dermatitis treated with a novel intramuscular progesterone desensitization protocol. J. Allergy Clin. Immunol. Pract. 2013, 1, 537–538. [Google Scholar] [CrossRef]
- Hartmann, K.; Escribano, L.; Grattan, C.; Brockow, K.; Carter, M.C.; Alvarez-Twose, I.; Matito, A.; Broesby-Olsen, S.; Siebenhaar, F.; Lange, M.; et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J. Allergy Clin. Immunol. 2016, 137, 35–45. [Google Scholar]
- Schaffer, J.V. Pediatric Mastocytosis: Recognition and Management. Am. J. Clin. Dermatol. 2021, 22, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Di Raimondo, C.; Del Duca, E.; Silvaggio, D.; Di Prete, M.; Lombardo, P.; Mazzeo, M.; Spallone, G.; Campione, E.; Botti, E.; Bianchi, L. Cutaneous mastocytosis: A dermatological perspective. Australas. J. Dermatol. 2021, 62, e1–e7. [Google Scholar] [CrossRef]
- Nemat, K.; Abraham, S. Cutaneous mastocytosis in childhood. Allergol. Sel. 2022, 6, 1–10. [Google Scholar] [CrossRef]
- Friedman, A.J. JAAD game changers: Telangiectasia macularis eruptiva perstans (TMEP): A form of cutaneous mastocytosis with potential systemic involvement. J. Am. Acad. Dermatol. 2020, 82, 530. [Google Scholar] [CrossRef] [PubMed]
- Hosking, A.M.; Makdisi, J.; Ortenzio, F.; de Feraudy, S.; Smith, J.; Linden, K. Diffuse cutaneous mastocytosis: Case report and literature review. Pediatr. Dermatol. 2018, 35, e348–e352. [Google Scholar] [CrossRef]
- Giannetti, A.; Filice, E.; Caffarelli, C.; Ricci, G.; Pession, A. Mast Cell Activation Disorders. Medicina 2021, 57, 124. [Google Scholar] [CrossRef]
- Castells, M.; Metcalfe, D.D.; Escribano, L. Diagnosis and treatment of cutaneous mastocytosis in children: Practical recommendations. Am. J. Clin. Dermatol. 2011, 12, 259–270. [Google Scholar] [CrossRef]
- Hardy, W.R.; Anderson, R.E. The hypereosinophilic syndromes. Ann. Intern. Med. 1968, 68, 1220–1229. [Google Scholar] [CrossRef]
- Inayat, F.; O’Neill, S.S.; Zafar, F.; Marupudi, S.; Vasim, I. Idiopathic hypereosinophilic syndrome with cutaneous involvement: A comparative review of 32 cases. BMJ Case Rep. 2018, 11, bcr2018227137. [Google Scholar] [CrossRef]
- Yu, Z.; Dee, E.C.; Menge, T.; Ward, A.; Song, P. Skin-limited idiopathic hypereosinophilic syndrome presenting with retiform purpura. JAAD Case Rep. 2019, 5, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Sharma, A.; Agarwal, S.; Gupta, V. Hypereosinophilic dermatitis: Generalised lichenification and gyrate erythema as the sole manifestation of idiopathic hypereosinophilic syndrome. BMJ Case Rep. 2019, 12, e232142. [Google Scholar] [CrossRef]
- Bigby, M. Rates of cutaneous reactions to drugs. Arch. Dermatol. 2001, 137, 765–770. [Google Scholar]
- Marzano, A.V.; Borghi, A.; Cugno, M. Adverse drug reactions and organ damage: The skin. Eur. J. Intern. Med. 2016, 28, 17–24. [Google Scholar] [CrossRef]
- Ernst, M.; Giubellino, A. Histopathologic Features of Maculopapular Drug Eruption. Dermatopathology 2022, 9, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Shipley, D.; Ormerod, A.D. Drug-induced urticaria. Recognition and treatment. Am. J. Clin. Dermatol. 2001, 2, 151–158. [Google Scholar] [CrossRef]
- Flowers, H.; Brodell, R.; Brents, M.; Wyatt, J.P. Fixed drug eruptions: Presentation, diagnosis, and management. South. Med. J. 2014, 107, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Lellig, E.; Mouton-Faivre, C.; Abs, D.; Bursztejn, A.C. Fixed drug eruption after Pfizer-BioNTech COVID-19 vaccine: A case report. J. Allergy Clin. Immunol. Pract. 2022, 10, 1922–1923. [Google Scholar] [CrossRef]
- Mintoff, D.; Pisani, D.; Betts, A.; Scerri, L. SARS-CoV-2 mRNA vaccine-associated fixed drug eruption. J. Eur. Acad. Dermatol. Venereol. 2021, 35, e560–e563. [Google Scholar] [CrossRef] [PubMed]
- Rekabi, M.; Sadati, E.; Mirzaei, J.; Pourdowlat, G.; Velayati, A.A.; Honarpisheh, P. Fixed drug eruption after the Sinopharm COVID-19 vaccine. JEADV Clin. Pract. 2022, 1, 412–415. [Google Scholar] [CrossRef]
- Anderson, H.J.; Lee, J.B. A Review of Fixed Drug Eruption with a Special Focus on Generalized Bullous Fixed Drug Eruption. Medicina 2021, 57, 925. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.C. Recurrent granulomatous dermatitis with eosinophilia. Trans. St. Johns. Hosp. Dermatol. Soc. 1971, 57, 46–56. [Google Scholar] [PubMed]
- Yu, A.M.; Ito, S.; Leibson, T.; Lavi, S.; Fu, L.W.; Weinstein, M.; Skotnicki, S.M. Pediatric Wells syndrome (eosinophilic cellulitis) after vaccination: A case report and review of the literature. Pediatr. Dermatol. 2018, 35, e262–e264. [Google Scholar] [CrossRef]
- Long, H.; Zhang, G.; Wang, L.; Lu, Q. Eosinophilic Skin Diseases: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 189–213. [Google Scholar] [CrossRef]
- Weins, A.B.; Biedermann, T.; Weiss, T.; Weiss, J.M. Wells syndrome. J. Dtsch. Dermatol. Ges. J. Ger. Soc. Dermatol. 2016, 14, 989–993. [Google Scholar] [CrossRef]
- Toumi, A.; Yarrarapu, S.N.S.; Litaiem, N. Wells Syndrome [Internet]. StatPearls [Internet]. StatPearls Publishing. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532294/ (accessed on 15 February 2023).
- Caputo, R.; Marzano, A.V.; Vezzoli, P.; Lunardon, L. Wells syndrome in adults and children: A report of 19 cases. Arch. Dermatol. 2006, 142, 1157–1161. [Google Scholar] [CrossRef]
- Joshi, T.P.; Friske, S.K.; Hsiou, D.A.; Duvic, M. New Practical Aspects of Sweet Syndrome. Am. J. Clin. Dermatol. 2022, 23, 301–318. [Google Scholar] [CrossRef]
- Marzano, A.V.; Borghi, A.; Wallach, D.; Cugno, M. A Comprehensive Review of Neutrophilic Diseases. Clin. Rev. Allergy Immunol. 2018, 54, 114–130. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.A.; Noe, M.H.; McMahon, C.M.; Gowda, A.; Wu, B.; Ashchyan, H.J.; Perl, A.E.; James, W.D.; Micheletti, R.G.; Rosenbach, M. Sweet syndrome in patients with and without malignancy: A retrospective analysis of 83 patients from a tertiary academic referral center. J. Am. Acad. Dermatol. 2018, 78, 303–309.e4. [Google Scholar] [CrossRef]
- Wallach, D.; Vignon-Pennamen, M.D. Pyoderma gangrenosum and Sweet syndrome: The prototypic neutrophilic dermatoses. Br. J. Dermatol. 2018, 178, 595–602. [Google Scholar] [CrossRef]
- Walker, D.C.; Cohen, P.R. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: Case report and review of drug-induced Sweet’s syndrome. J. Am. Acad. Dermatol. 1996, 34 Pt 2, 918–923. [Google Scholar] [CrossRef]
- Corazza, M.; Lauriola, M.M.; Borghi, A.; Marzola, A.; Virgili, A. Sweet’s syndrome: A retrospective clinical, histopathological and immunohistochemical analysis of 11 cases. Acta Derm.-Venereol. 2008, 88, 601–606. [Google Scholar]
- Sävervall, C.; Sand, F.L.; Thomsen, S.F. Dermatological Diseases Associated with Pregnancy: Pemphigoid Gestationis, Polymorphic Eruption of Pregnancy, Intrahepatic Cholestasis of Pregnancy, and Atopic Eruption of Pregnancy. Dermatol. Res. Pract. 2015, 2015, 979635. [Google Scholar] [CrossRef]
- Lawley, T.J.; Hertz, K.C.; Wade, T.R.; Ackerman, A.B.; Katz, S.I. Pruritic urticarial papules and plaques of pregnancy. JAMA 1979, 241, 1696–1699. [Google Scholar] [CrossRef] [PubMed]
- Brandão, P.; Sousa-Faria, B.; Marinho, C.; Vieira-Enes, P.; Melo, A.; Mota, L. Polymorphic eruption of pregnancy: Review of literature. J. Obs. Gynaecol. J. Inst. Obs. Gynaecol. 2017, 37, 137–140. [Google Scholar] [CrossRef]
- Matz, H.; Orion, E.; Wolf, R. Pruritic urticarial papules and plaques of pregnancy: Polymorphic eruption of pregnancy (PUPPP). Clin. Dermatol. 2006, 24, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Poddighe, D.; Marseglia, G.L. Is There any Relationship Between Extra-Pulmonary Manifestations of Mycoplasma Pneumoniae Infection and Atopy/Respiratory Allergy in Children? Pediatr. Rep. 2016, 8, 6395. [Google Scholar] [CrossRef] [PubMed]
- Siedner-Weintraub, Y.; Gross, I.; David, A.; Reif, S.; Molho-Pessach, V. Paediatric Erythema Multiforme: Epidemiological, Clinical and Laboratory Characteristics. Acta Derm.-Venereol. 2017, 97, 489–492. [Google Scholar] [CrossRef]
- Goldman, R.D. Erythema multiforme in children. Can. Fam. Physician 2022, 68, 507–508. [Google Scholar] [CrossRef]
- Soares, A.; Sokumbi, O. Recent Updates in the Treatment of Erythema Multiforme. Medicina 2021, 57, 921. [Google Scholar] [CrossRef]
- Trayes, K.P.; Love, G.; Studdiford, J.S. Erythema Multiforme: Recognition and Management. Am. Fam. Physician 2019, 100, 82–88. [Google Scholar]
- Lamoreux, M.R.; Sternbach, M.R.; Hsu, W.T. Erythema Multiforme. Am. Fam. Physician 2006, 74, 1883–1888. [Google Scholar]
- Bennardo, L.; Nisticò, S.P.; Dastoli, S.; Provenzano, E.; Napolitano, M.; Silvestri, M.; Passante, M.; Patruno, C. Erythema Multiforme and COVID-19: What Do We Know? Medicina 2021, 57, 828. [Google Scholar] [CrossRef] [PubMed]
- Al-Shaikhly, T.; Ochs, H.D. Hyper IgE syndromes: Clinical and molecular characteristics. Immunol. Cell Biol. 2019, 97, 368–379. [Google Scholar] [CrossRef]
- Hafsi, W.; Yarrarapu, S.N.S. Job Syndrome [Internet]. StatPearls [Internet]. StatPearls Publishing. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK525947/ (accessed on 15 December 2022).
- Minegishi, Y. Hyper-IgE syndrome, 2021 update. Allergol. Int. 2021, 70, 407–414. [Google Scholar] [CrossRef]
- Steen, C.J.; Janniger, C.K.; Schutzer, S.E.; Schwartz, R.A. Insect sting reactions to bees, wasps, and ants. Int. J. Dermatol. 2005, 44, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Stingeni, L.; Bianchi, L.; Hansel, K.; Neve, D.; Foti, C.; Corazza, M.; Bini, V.; Moretta, I.; Principato, M. Dermatitis caused by arthropods in domestic environment: An Italian multicentre study. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1526–1533. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Epidemiology | Clinical Features | Histopathology |
|---|---|---|---|
| Urticaria | All ages | Wheals < 24 h, angioedema | Swelling of superficial dermis, dilation of vessels, perivascular inflammatory infiltrate |
| Urticarial Dermatitis | Mean age: 60 y.o. | Overlapping urticarial and eczematous features—simultaneously or sequentially | Dermal hypersensitivity reaction pattern: dermal and perivascular lymphocytic infiltrate + scattered eosinophils. Epidermal spongiosis. Corneum unaffected. |
| Urticarial vasculitis | Mean age: 30–40 y.o. | Wheals (0.5–5 cm) lasting >24 h, dark-red or brown. Painful and/or burning sensation. | Karyorrhexis + erythrocytes outside vessels + fibrin inside vessels + neutrophilic infiltrate |
| Cutaneous lupus erythematosus | Females 3rd–4th decade | Broad spectrum. LE specific and LE nonspecific skin manifestations. | overlapping features: (i) epidermal vacuolization + apoptosis, (ii) interface dermatitis, (iii) dermal inflammation, (iv) follicular plugging, (v) thick basal membrane (vi) dermal mucin deposits |
| Bullous Pemphigoid | Elderly | Polymorphic Tense bullae → erosions/crusts on erythematous urticarial skin | (i) subepidermal rupture (ii) perivascular infiltrate of eosinophils, (iii) eosinophilic spongiosis. DIF: linear deposition of C3 and IgG at the basal membrane |
| Pemphigoid gestationis | Pregnant women/post-partum | Polymorphic: eczematous, erythema multiforme-like, urticaria-like, papules → tense blisters. Starts on the abdomen | (i) papillary edema, (ii) dermal inflammatory infiltrate, (iii) subepidermal blistering. DIF: linear deposition of C and IgG at the dermal-epidermal junction. |
| Dermatitis herpetiformis | Mean age: 30–40 y.o. | Papulo-vesicular rash at the extensor surfaces of the limbs | Vesicle = sub-epidermal split + neutrophilic infiltrate at dermal papillae. Perivascular polymorphic inflammatory infiltrate |
| Autoimmune progesterone dermatitis | Women of childbearing age | Heterogenous: urticaria-like, vesiculobullous, erythema multiforme, eczema, maculopapular eruption, purpura/petechiae | Nonspecific findings |
| Mastocytosis | Different onset age depending on subtype | MPCM: yellowish/brown lesions. TMEP: multiple erythematous/hyperpigmented macules + telangiectasias. Cutaneous mastocytoma: solitary yellowish/brown macule/nodule/plaque. DCM: generalized erythema and pachydermia. | Multifocal infiltrates of mast cells |
| Hypereosinophilic syndrome | All ages | Pruritic, tender, erythematous papules/plaques. Trunk and/or extremities. | Nonspecific findings |
| Iatrogenic rash | All ages | Erythematous macules/papules → confluent. Start on the trunk → extremities symmetrically. | Nonspecific findings |
| Fixed drug eruption | All ages | Red/purple, well-demarcated round/oval patch | (i) vacuolar interface dermatitis (ii) polymorphic infiltration (iii) necrotic keratinocytes (epidermis) (iv) pigment incontinence |
| Wells syndrome | Adults | Erythematous, edematous, infiltrated plaques. Abrupt onset. | Acute: dermal edema + dense eosinophilic infiltrate without vasculitis. Subacute: flame figures (degeneration of collagen due to eosinophil degranulation) Regressive: no eosinophils + microgranulomas |
| Sweet syndrome | All ages | Painful, elevated, edematous, well-demarked red/purple-red papules/plaques | dense neutrophilic infiltrate in the dermis, without vasculitis |
| Polymorphic eruption of pregnancy | Pregnant women | Erythematous and edematous papules → plaques. Starts on the abdomen. | Nonspecific findings |
| Hyper IgE syndromes | 50% neonatal onset | Eczema/atopic dermatitis | Nonspecific findings |
| Insect bite reactions | Any age | Erythematous urticarial papules (2–10 mm) | Nonspecific findings |
| Neutrophilic urticarial dermatosis | Urticarial rash | Intense neutrophilic infiltrate | |
| Erythema multiforme | All ages | Pink/red papules, → plaques → classic target lesion | Early stages: perivascular mononuclear cell infiltrate. Target lesion: dermal edema (peripherically) + necrotic keratinocytes/epidermal changes (centrally) |
| Syndrome | Age of Onset | Duration of Clinical Manifestation | Skin Lesions | Systemic Symptoms |
|---|---|---|---|---|
| Familial cold auto-inflammatory syndrome | Infancy | <24 h | Cold-induced urticarial eruptions | Fever, arthralgia, nausea, vomiting and conjunctivitis |
| Muckle-Wells syndrome | Infancy-adolescence | 1 to 3 days | Wheal-and-flare macules and papules | Fever, arthralgia, conjunctivitis, progressive sensorineural loss and risk of amyloidosis |
| Chronic infantile neurologic, cutaneous and articular syndrome | Neonatal period | Continuous with flares | Persistent urticaria-like rash | Fever, neurologic disorders (aseptic meningitis), ocular manifestations (progressive visual loss due to papilledema and atrophy of optic nerve) and joint involvement (deforming osteo-arthropaty) |
| Schnitzler’s syndrome | Adulthood (mean age 50 years) | 12 to 36 h | Slightly itchy wheals occasionally with hyperpigmented evolution | Recurrent fever, fatigue, general malaise, arthralgia, myalgia, bone pain, hepato-splenomegaly and lymphadenopaty. IgM or IgG monoclonal gammopathy as obligate criterion for diagnosis. |
| Adult-onset Still’s disease | 16–35 years | 24–36 h | Mildly itchy skin rash with salmon-pink macules and papules | High-spiking fever in the afternoon or early evening, arthralgia or arthritis, sore throat, serositis, hepato-splenomegaly and lymphadenopaty |
| Systemic-onset juvenile idiopathic arthritis | <16 years | 24–36 h | Transient pink macular eruption | High-spiking fever, poly-arthritis, serositis, hepato-splenomegaly and lymphadenopaty |
| Mevalonate kinase deficiency | First year of life (mean age 6 months) in HIDS and at birth or ante-natal in mevalonic aciduria | 3–7 days | Evanescent maculo-papular rashes occasionally with purpuric aspects | Recurrent fever, abdominal pain, vomiting and/or diarrhea, lymphadenopathy, arthralgia and non-erosive arthritis in HIDS. Neurologic alterations and hematologic abnormalities in mevalonic aciduria. Elevated IgD levels as laboratoristic finding. |
| TNF-receptor-associated periodic syndrome | <10 years | 5 to 25 days with recurrences every 4 to 6 weeks | Migrating erythema with centrifugal evolution and less frequently urticarial plaques | High fever, abdominal and chest pain, arthralgia, myalgia, eye involvement (uveitis or conjunctivitis) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schettini, N.; Corazza, M.; Schenetti, C.; Pacetti, L.; Borghi, A. Urticaria: A Narrative Overview of Differential Diagnosis. Biomedicines 2023, 11, 1096. https://doi.org/10.3390/biomedicines11041096
Schettini N, Corazza M, Schenetti C, Pacetti L, Borghi A. Urticaria: A Narrative Overview of Differential Diagnosis. Biomedicines. 2023; 11(4):1096. https://doi.org/10.3390/biomedicines11041096
Chicago/Turabian StyleSchettini, Natale, Monica Corazza, Cecilia Schenetti, Lucrezia Pacetti, and Alessandro Borghi. 2023. "Urticaria: A Narrative Overview of Differential Diagnosis" Biomedicines 11, no. 4: 1096. https://doi.org/10.3390/biomedicines11041096
APA StyleSchettini, N., Corazza, M., Schenetti, C., Pacetti, L., & Borghi, A. (2023). Urticaria: A Narrative Overview of Differential Diagnosis. Biomedicines, 11(4), 1096. https://doi.org/10.3390/biomedicines11041096