
Anterograde and Retrograde Propagation of Inoculated Human Tau Fibrils and Tau Oligomers in a Non-Transgenic Rat Tauopathy Model

 , ,
, , 
{kind=link}
{kind=link}
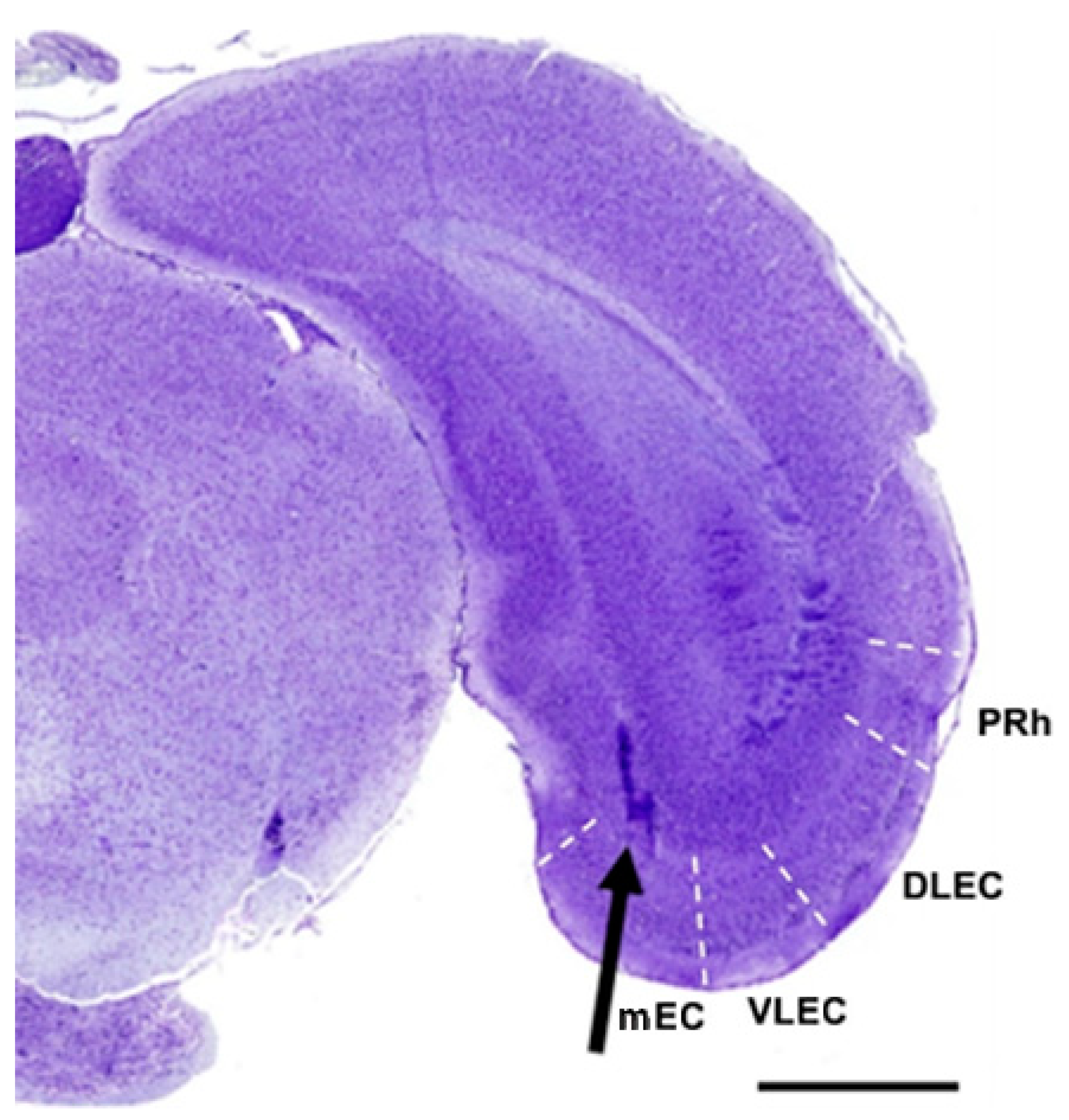
{kind=link}
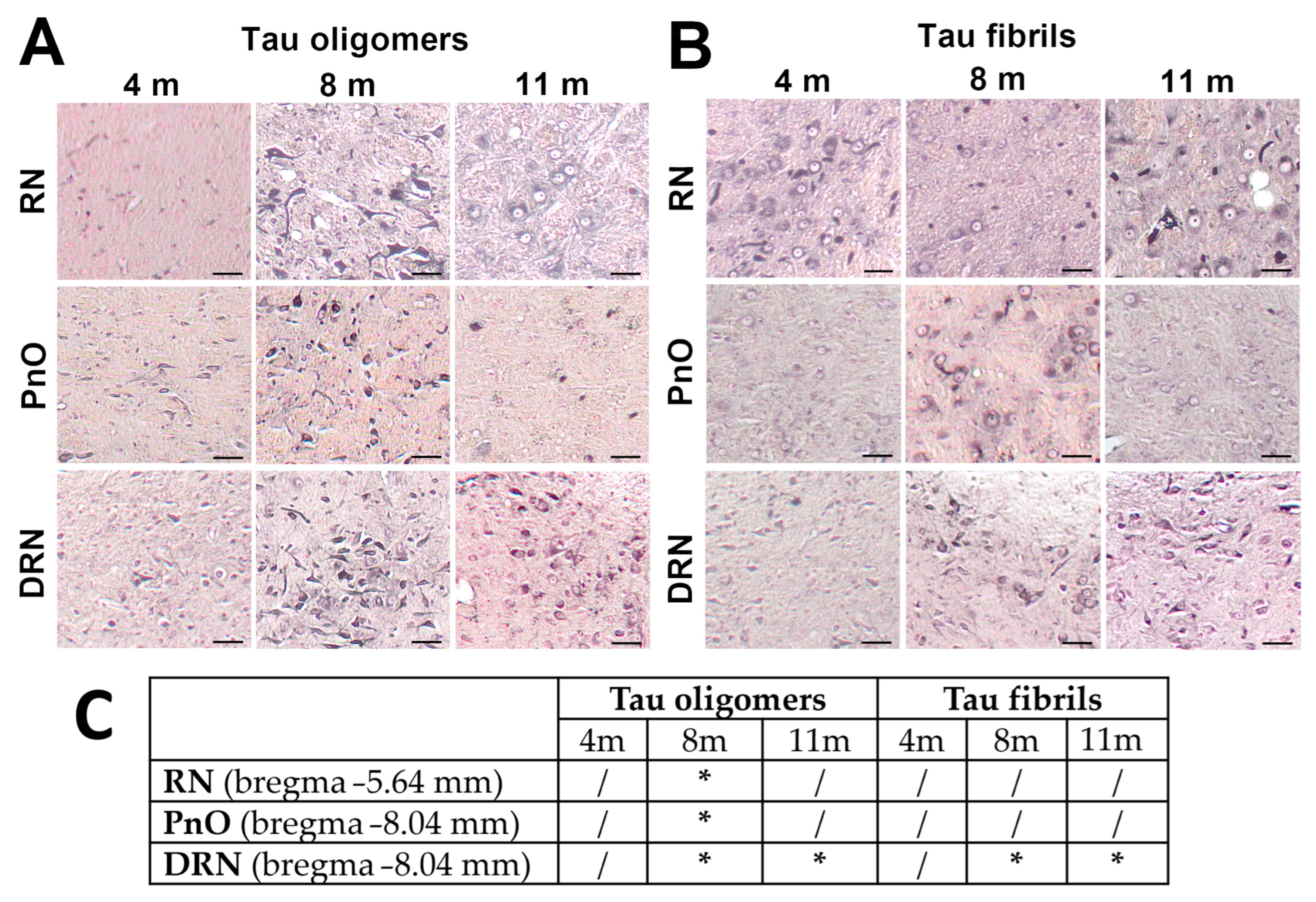
{kind=link}
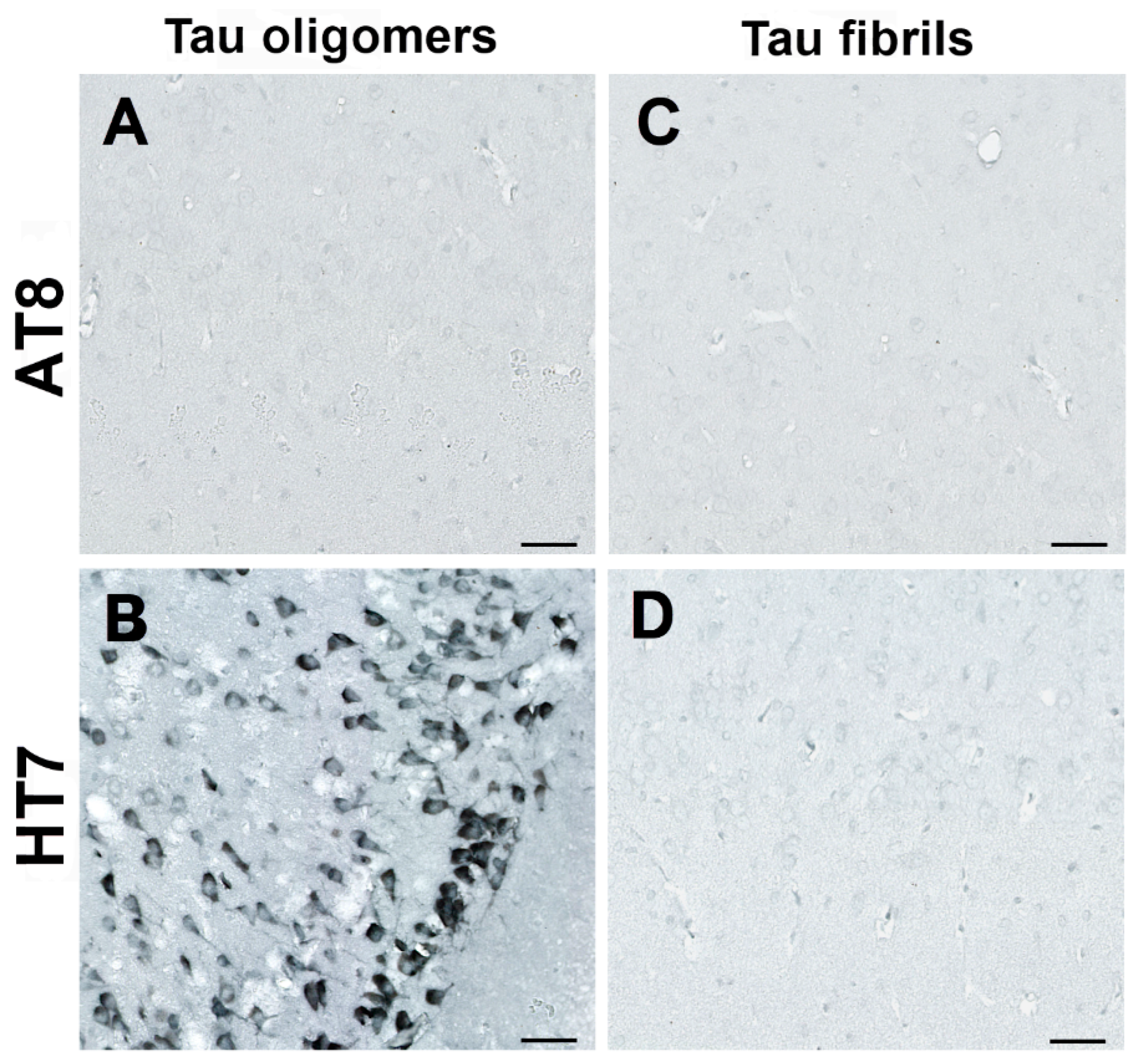
{kind=link}
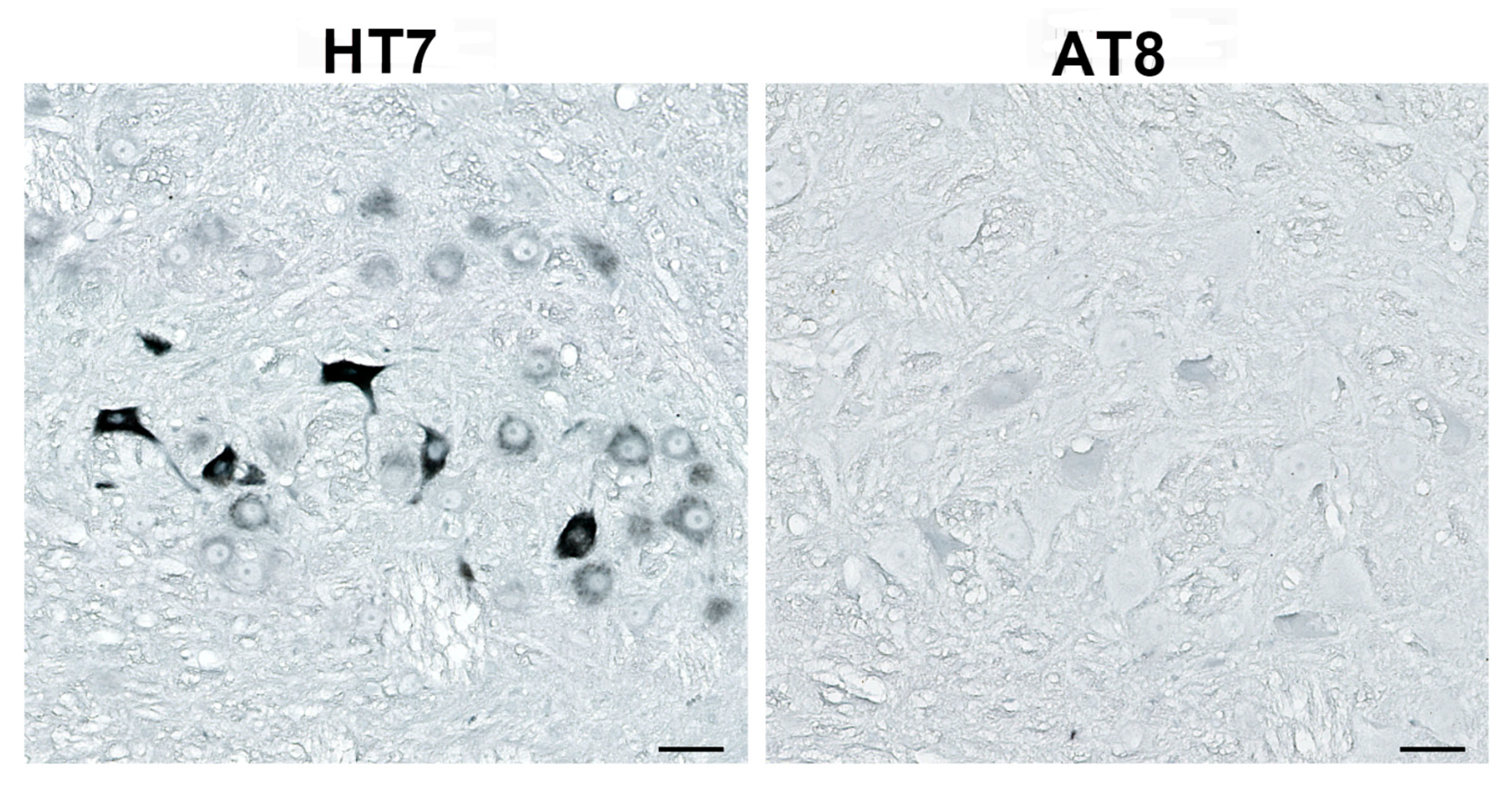
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Human Tau Oligomers and Tau Fibrils
2.3. Experimental Groups and Design
2.4. Stereotaxic Injections
2.5. Histology, Immunohistochemistry, and Staging
2.6. Behavioral Testing
2.7. Statistical Methods
3. Results
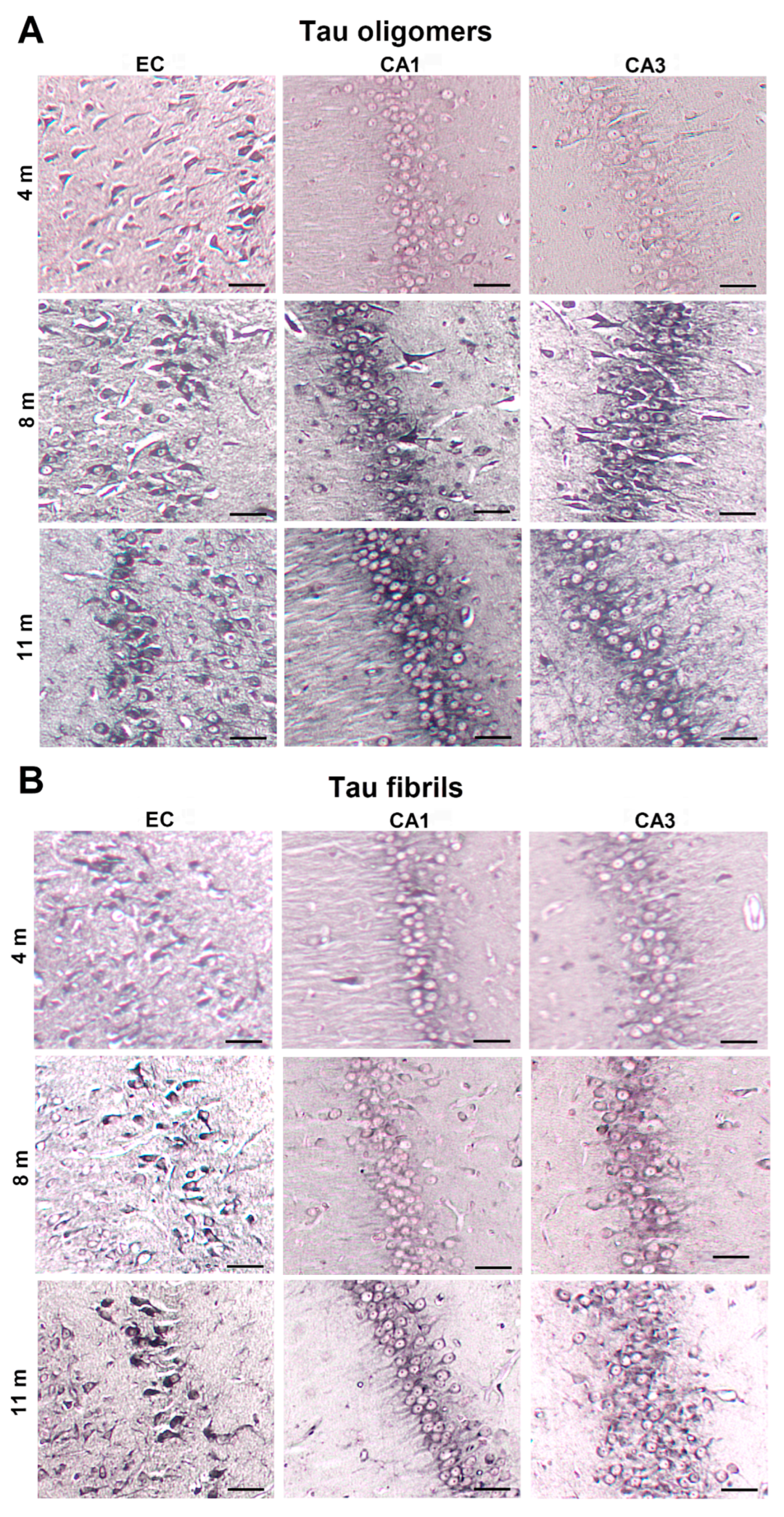
3.1. Injected Human Tau Oligomers and Tau Fibrils Lead to the Appearance of Gallyas-Positive Inclusions in Wild-Type Rats
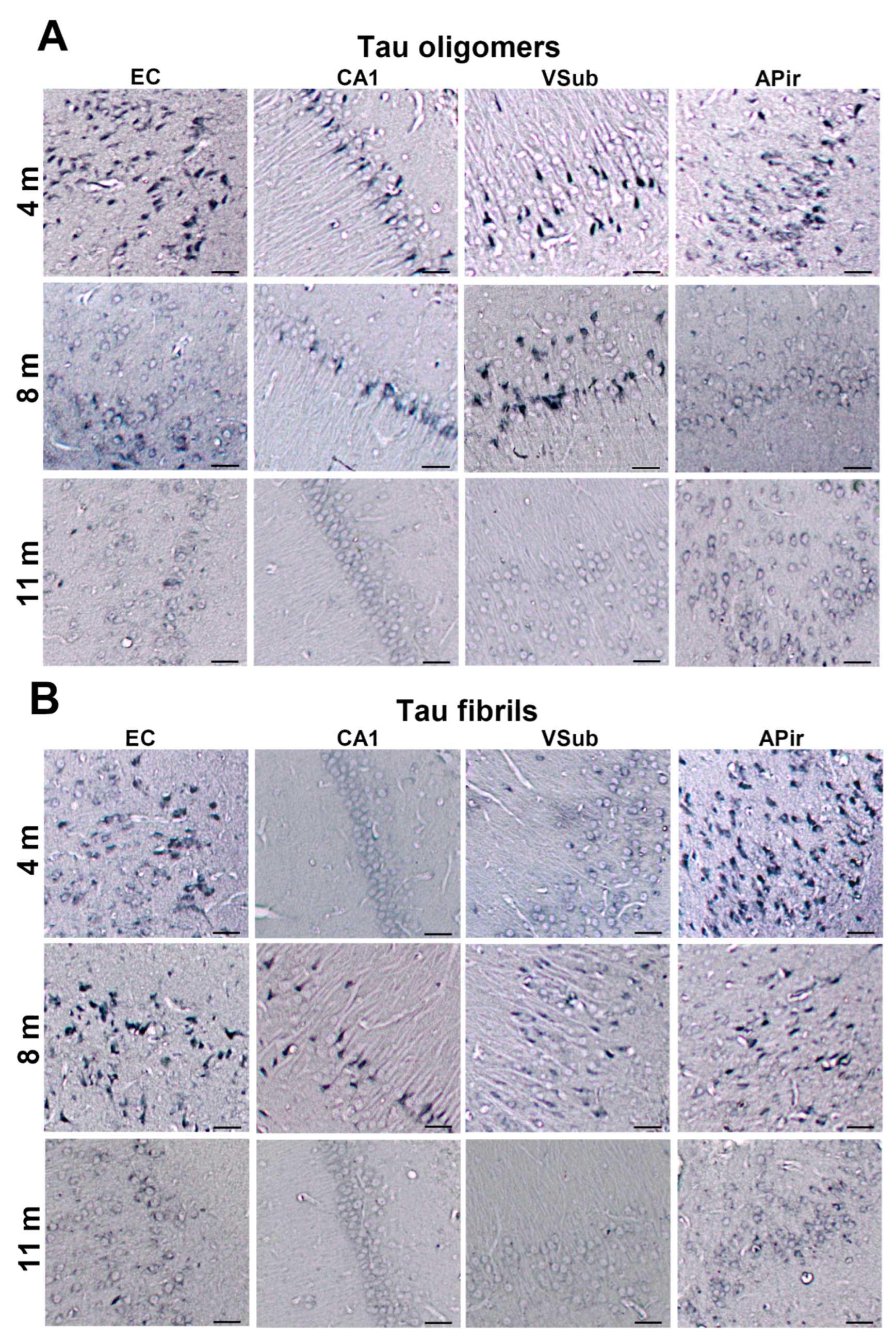
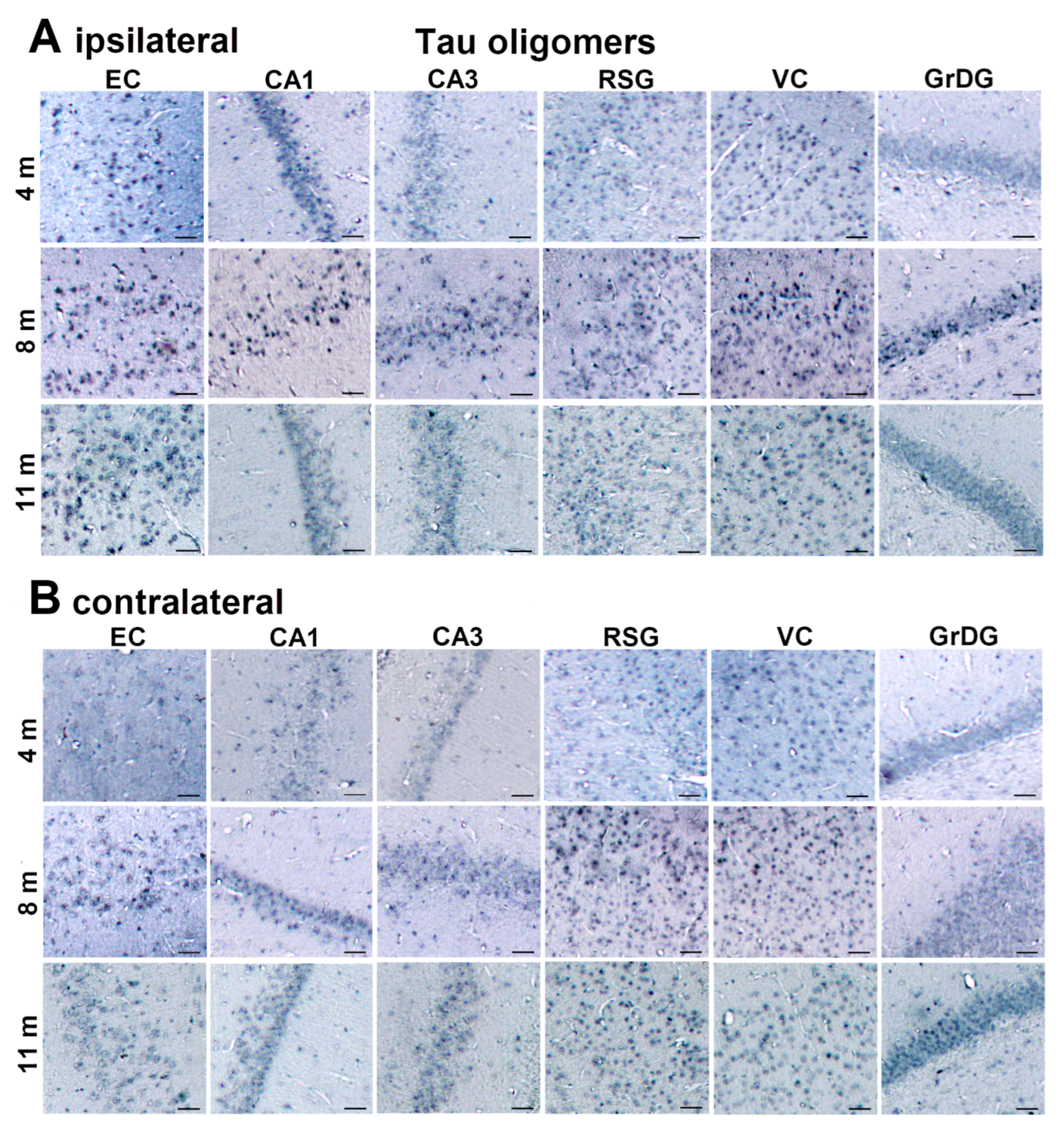
3.2. Injected Human Tau Oligomers and Human Tau Fibrils Initiate and Propagate Tau Pathology in Wild-Type Rats
3.3. Propagation of Tau-Induced Changes
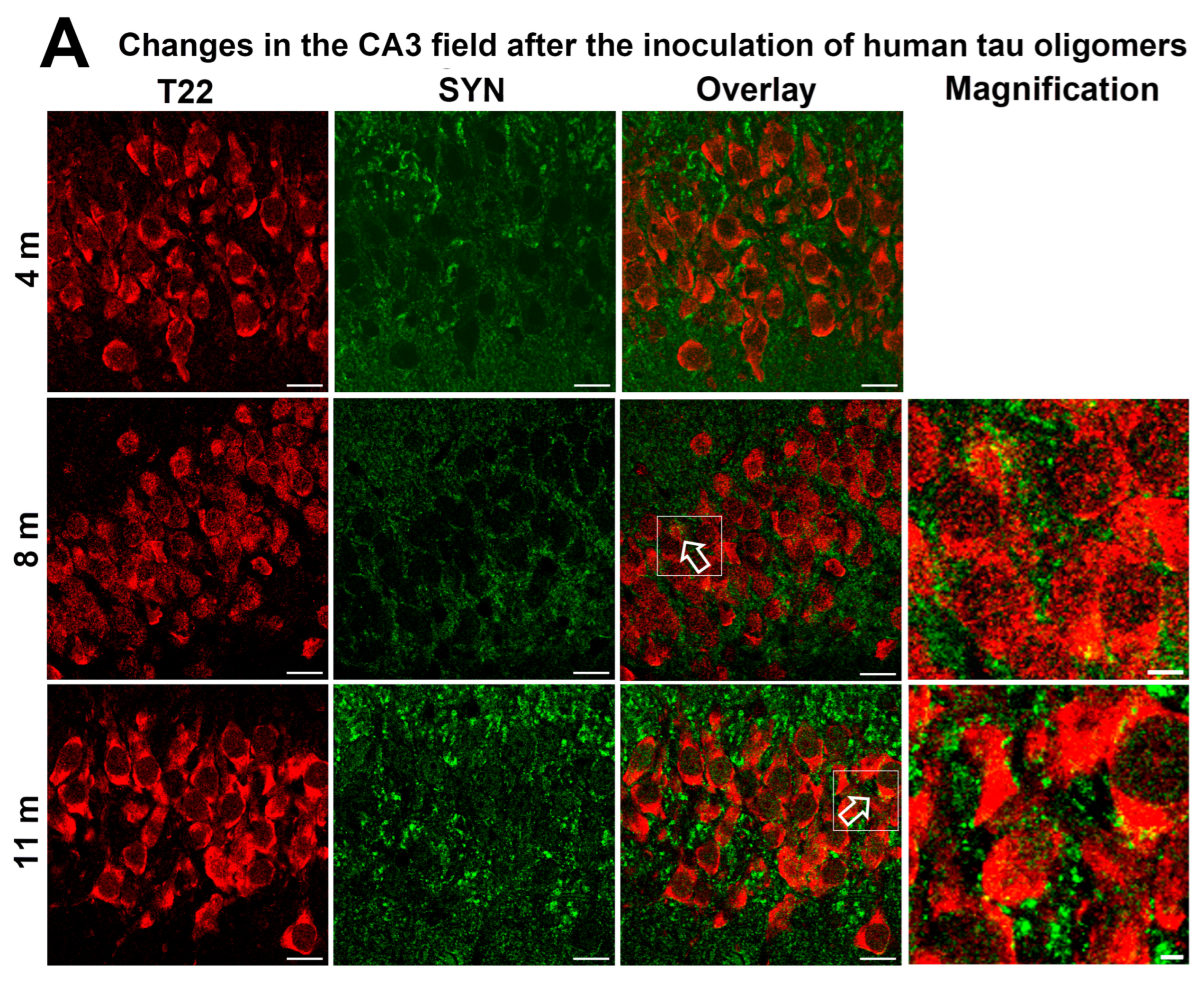
3.4. Human Tau Oligomers and Tau Fibrils Induce the Formation of Conformationally Altered Murine Tau
3.5. Detection of Neurofibrillary Changes and Aβ with ThS
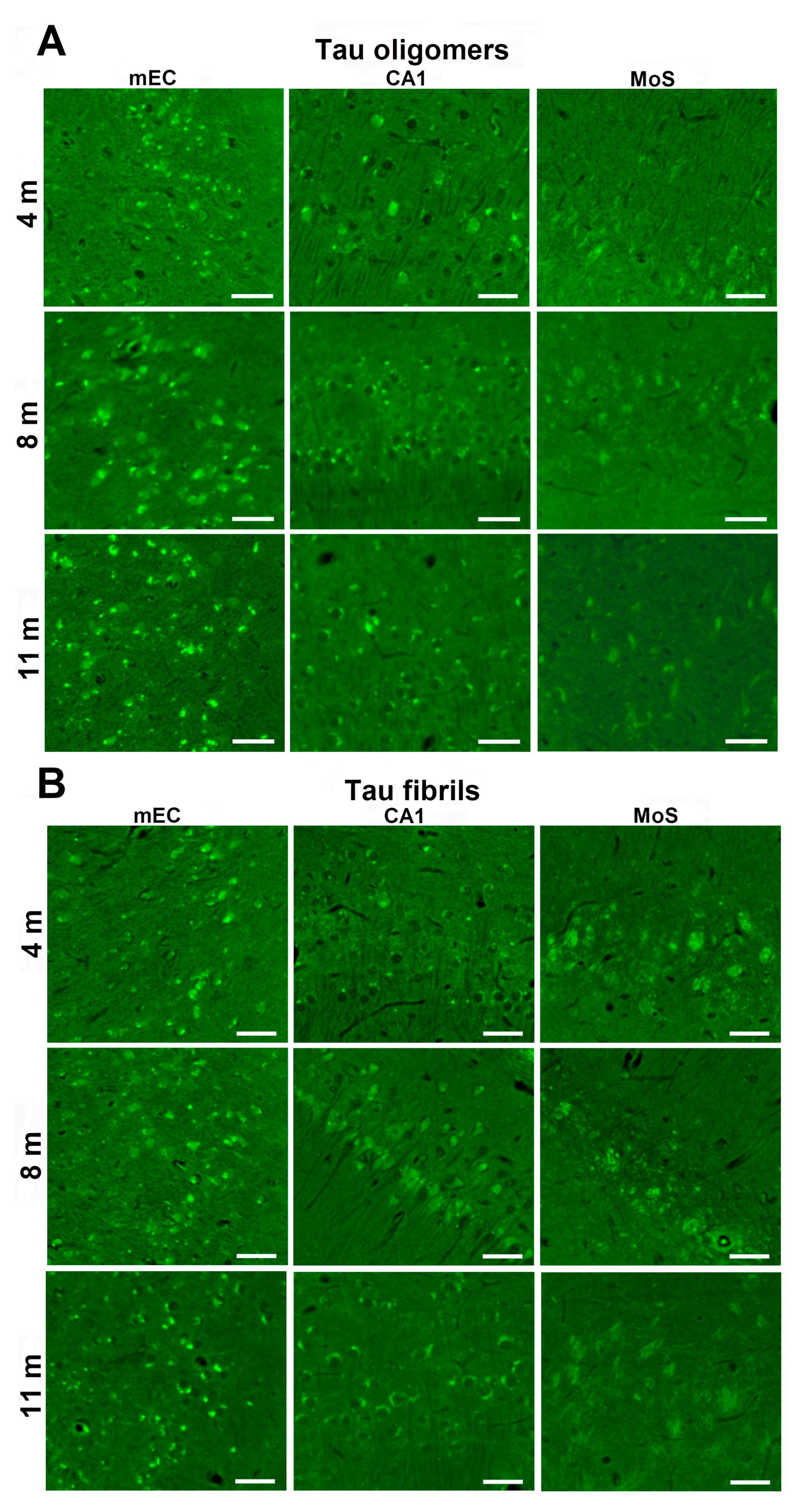
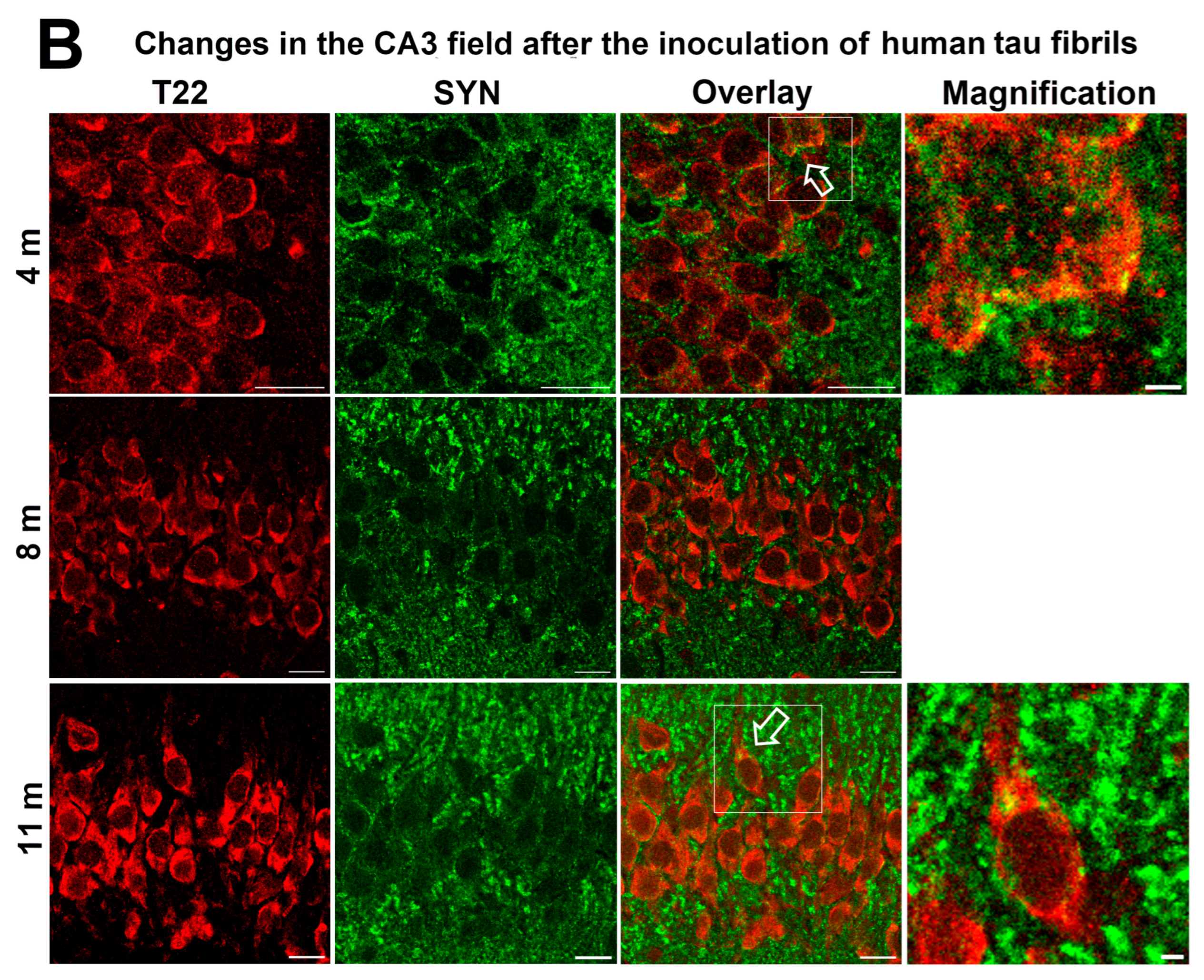
3.6. Synapse Loss in Rats Inoculated with Human Tau Oligomers and Human Tau Fibrils in the mEC
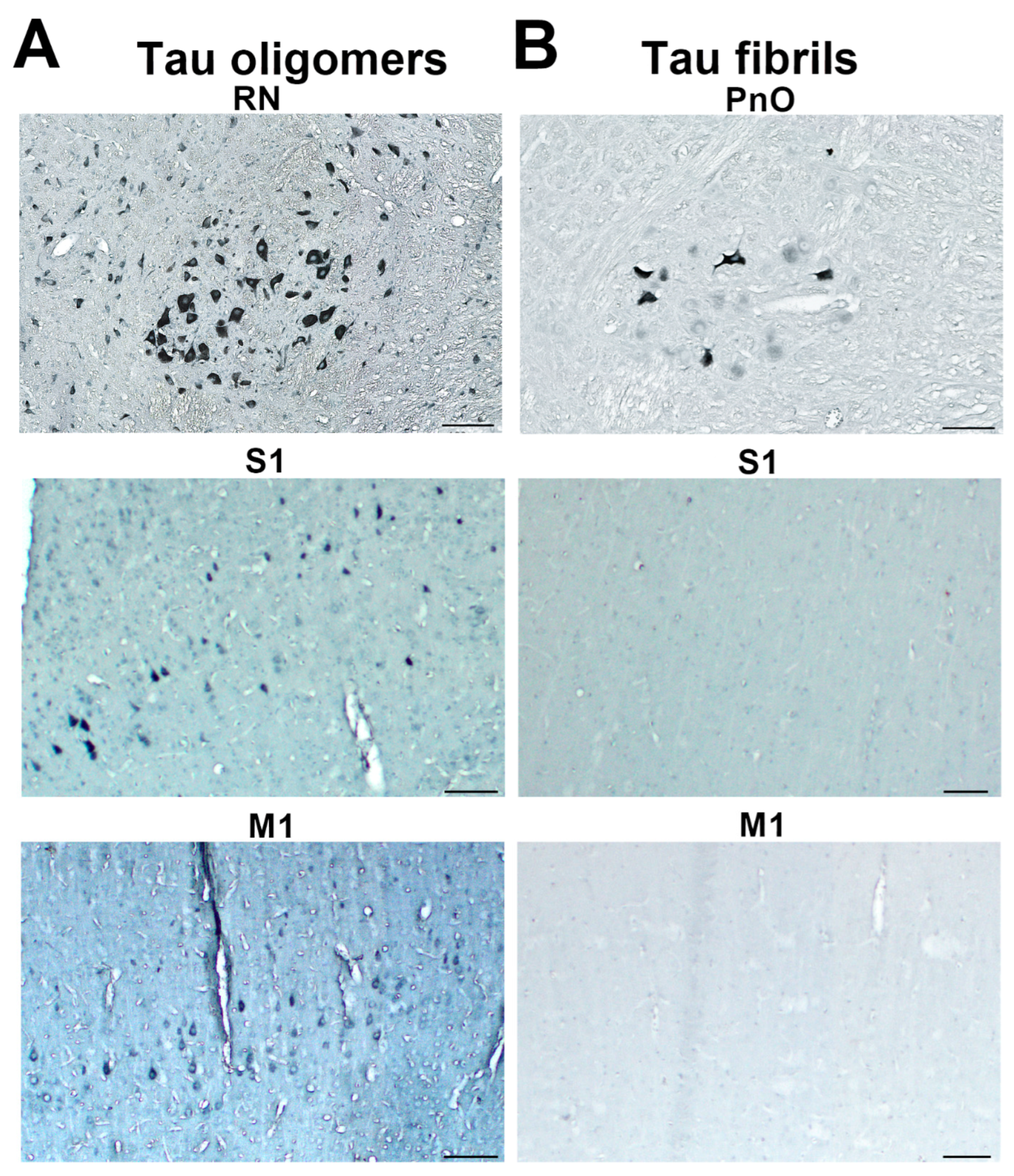
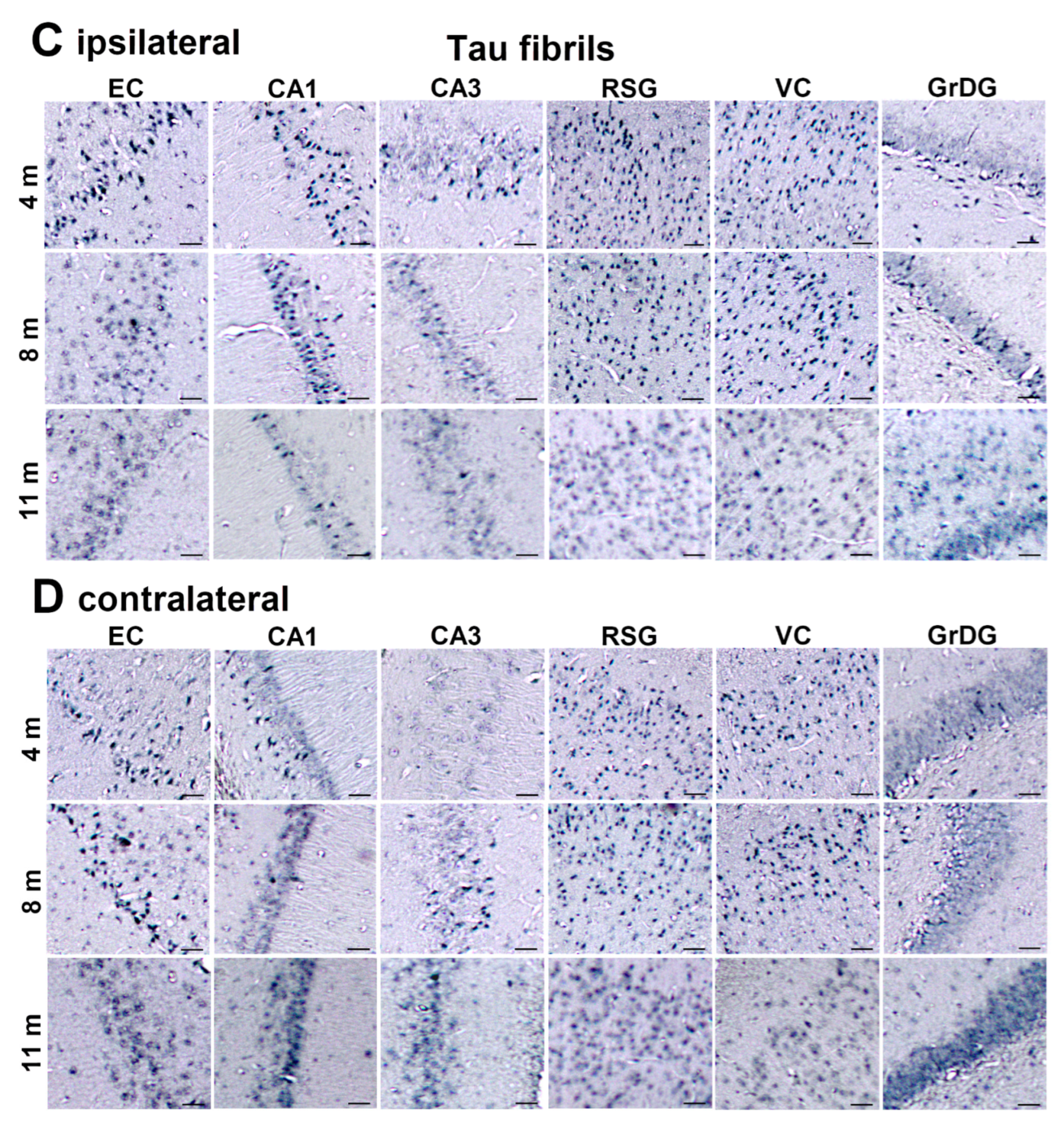
3.7. Wild-Type Rats Injected with Human Tau Fibrils Display Rapid Propagation of Tau Protein-Related Changes Compared with Wild-Type Rats Injected with Human Tau Oligomers
3.8. Progressive Spread of Tau Protein-Related Changes to CA1 and CA3 Fields of the Hippocampus of Wild-Type Rats Are Identified by Gallyas-Braak Silver Impregnation
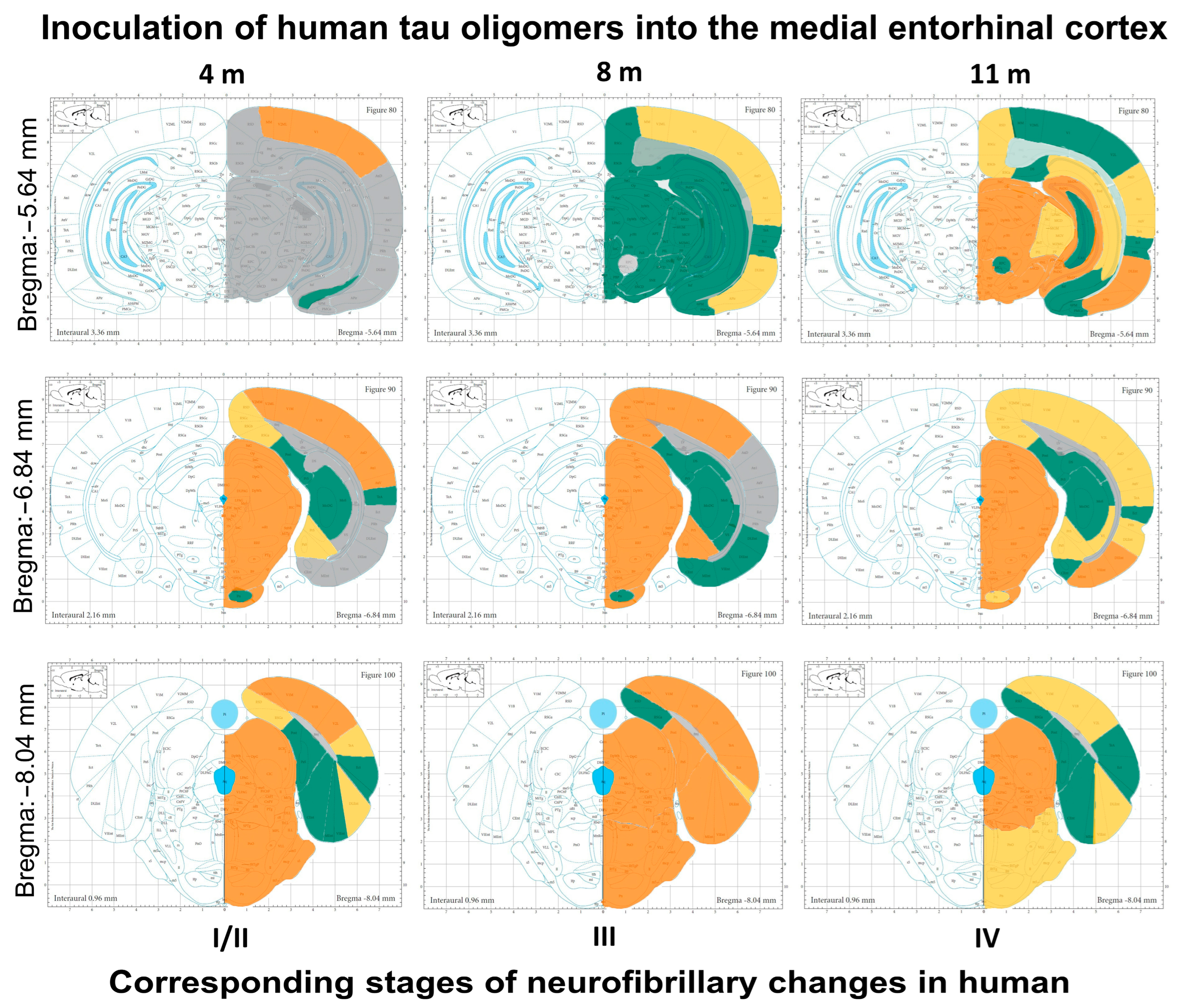
3.9. Summary of Neurofibrillary Changes after Inoculation of Human Tau Oligomers
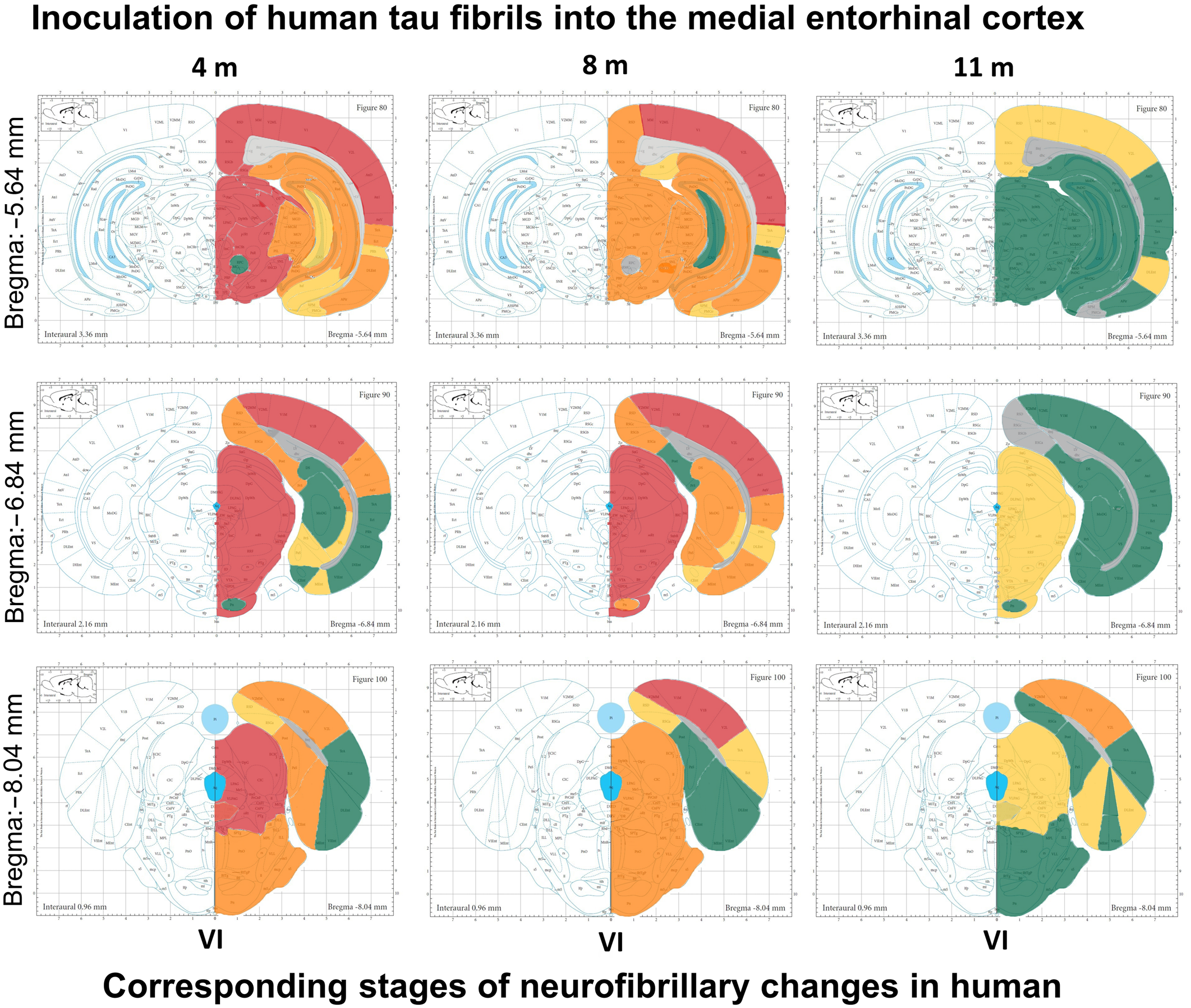
3.10. Summary of Neurofibrillary Changes after Inoculation of Human Tau Fibrils
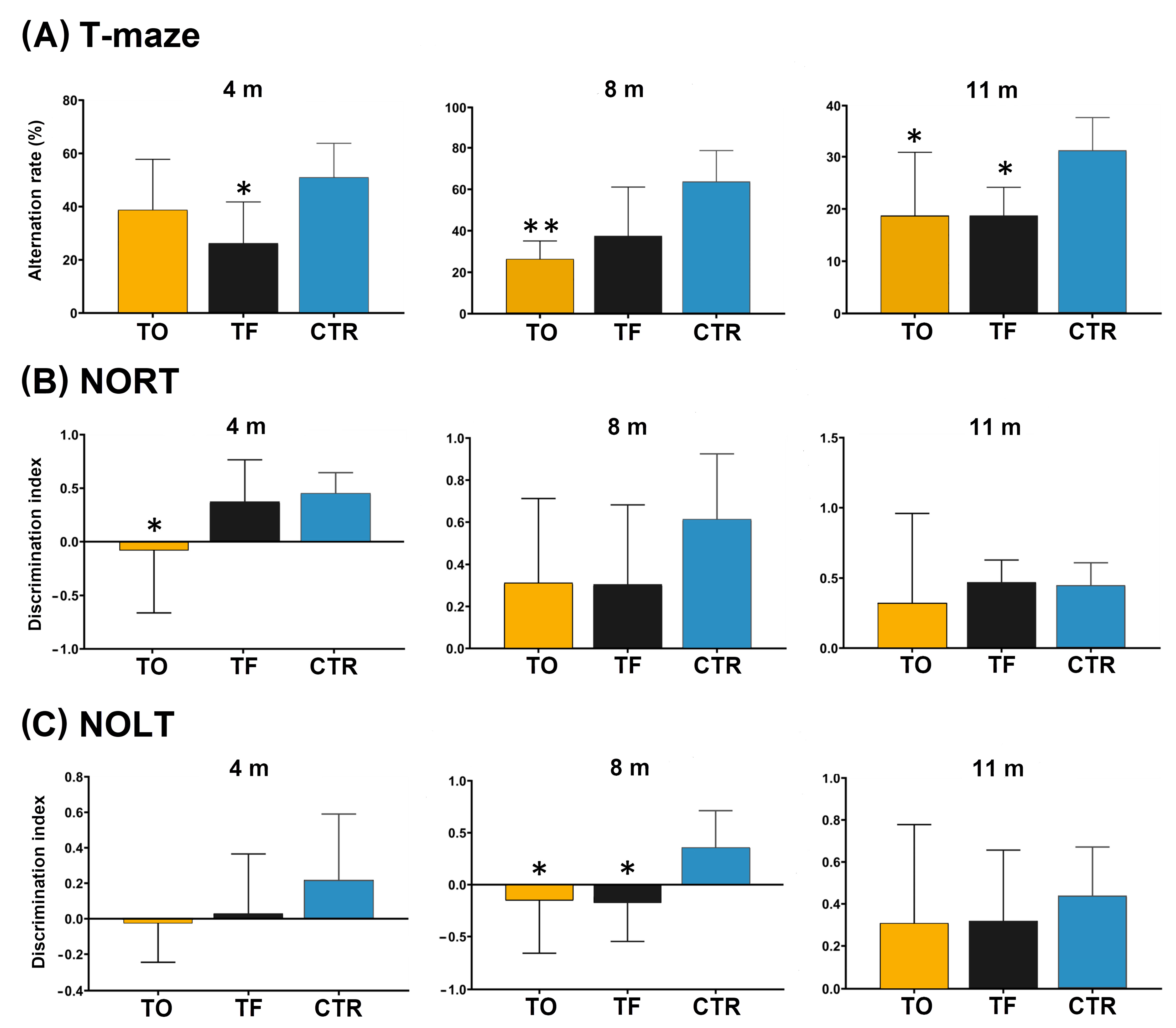
3.11. Behavioral Testing
4. Discussion
4.1. Limitations of the Study
4.2. Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| Aβ | amyloid β |
| AD | Alzheimer’s disease |
| APir | amygdalopiriform transition cortex |
| APP | amyloid precursor protein |
| AT8 | antibody specific for tau phosphorylated at serine 202, threonine 205, and serine 208 |
| CA | Cornu Ammonis (Ammon’s horn) |
| CTR | control group |
| DG | dentate gyrus |
| DI | discrimination index |
| DLEC | dorsolateral entorhinal cortex |
| DRN | dorsal raphe nucleus |
| EC | entorhinal cortex |
| FTDP-17 | frontotemporal dementia with parkinsonism linked to chromosome 17 |
| GrDG | granular layer of the dentate gyrus |
| HT7 | human tau-specific antibody |
| M1 | primary motor cortex |
| MAPT | microtubule-associated protein tau |
| MC1 | antibody specific for a pathological conformation of tau, binds to epitopes between amino acids 7–9 and 313–322 |
| mEC | medial entorhinal cortex |
| MoS | molecular layer of the subiculum |
| MRI | magnetic resonance imaging |
| NIA-AA | National Institute on Aging and Alzheimer’s Association |
| NFT | neurofibrillary tangles |
| NOLT | novel object location test |
| NORT | novel object recognition test |
| PBS | phosphate-buffered saline |
| PET | positron emission tomography |
| PHF | paired helical filaments |
| PnO | pontine reticular nucleus, oral part (nucleus reticularis pontis oralis) |
| PRh | perirhinal cortex |
| RN | red nucleus |
| RSG | retrosplenial granular cortex |
| S1 | primary somatosensory cortex |
| SSS | sterile saline solution |
| SYN | synaptophysin T22 antibody that specifically recognizes oligomeric tau |
| TF | group of animals that received tau fibrils |
| ThS | thioflavin-S |
| TO | group of animals that received tau oligomers |
| VC | visual cortex |
| VLEC | ventrolateral entorhinal cortex |
| VSub | ventral subiculum |
References
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation Affects the Ability of Tau Protein to Promote Microtubule Assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef] [PubMed]
- Bancher, C.; Brunner, C.; Lassmann, H.; Budka, H.; Jellinger, K.; Wiche, G.; Seitelberger, F.; Grundke-Iqbal, I.; Iqbal, K.; Wisniewski, H.M. Accumulation of Abnormally Phosphorylated τ Precedes the Formation of Neurofibrillary Tangles in Alzheimer’s Disease. Brain Res. 1989, 477, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.M.; Hyman, B.T. Specific Tau Phosphorylation Sites Correlate with Severity of Neuronal Cytopathology in Alzheimer’s Disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar] [CrossRef]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple Isoforms of Human Microtubule-Associated Protein Tau: Sequences and Localization in Neurofibrillary Tangles of Alzheimer’s Disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Hanes, J.; Zilka, N.; Bartkova, M.; Caletkova, M.; Dobrota, D.; Novak, M. Rat Tau Proteome Consists of Six Tau Isoforms: Implication for Animal Models of Human Tauopathies. J. Neurochem. 2009, 108, 1167–1176. [Google Scholar] [CrossRef]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-Synaptic Spread of Tau Pathology In Vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.P.; Buée, L. From the Prion-like Propagation Hypothesis to Therapeutic Strategies of Anti-Tau Immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain Homogenates from Human Tauopathies Induce Tau Inclusions in Mouse Brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and Seed-Competent Tau Monomers Suggest Structural Origins of Aggregation. eLife 2018, 7, e36584. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer Brain-Derived Tau Oligomers Propagate Pathology from Endogenous Tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal Uptake and Propagation of a Rare Phosphorylated High-Molecular-Weight Tau Derived from Alzheimer’s Disease Brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef]
- Maeda, S.; Takashima, A. Tau Oligomers. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1184, pp. 373–380. [Google Scholar]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Guerrero-Muñoz, M.J.; Jackson, G.R.; Kayed, R. Preparation and Characterization of Neurotoxic Tau Oligomers. Biochemistry 2010, 49, 10039–10041. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau Oligomers Impair Memory and Induce Synaptic and Mitochondrial Dysfunction in Wild-Type Mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease: A Practical Approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease. Alzheimers Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Ferreira, D.; Nordberg, A.; Westman, E. Biological Subtypes of Alzheimer Disease: A Systematic Review and Meta-Analysis. Neurology 2020, 94, 436–448. [Google Scholar] [CrossRef]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically Defined Subtypes of Alzheimer’s Disease with Distinct Clinical Characteristics: A Retrospective Study. Lancet Neurol. 2011, 10, 785. [Google Scholar] [CrossRef] [PubMed]
- Risacher, S.L.; Anderson, W.H.; Charil, A.; Castelluccio, P.F.; Shcherbinin, S.; Saykin, A.J.; Schwarz, A.J. Alzheimer Disease Brain Atrophy Subtypes Are Associated with Cognition and Rate of Decline. Neurology 2017, 89, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- Sintini, I.; Graff-Radford, J.; Senjem, M.L.; Schwarz, C.G.; Machulda, M.M.; Martin, P.R.; Jones, D.T.; Boeve, B.F.; Knopman, D.S.; Kantarci, K.; et al. Longitudinal Neuroimaging Biomarkers Differ across Alzheimer’s Disease Phenotypes. Brain 2020, 143, 2281. [Google Scholar] [CrossRef]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau Molecular Diversity Contributes to Clinical Heterogeneity in Alzheimer’s Disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.W.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; Iturria-Medina, Y.; et al. Four Distinct Trajectories of Tau Deposition Identified in Alzheimer’s Disease. Nat. Med. 2021, 27, 871–881. [Google Scholar] [CrossRef]
- Lehmann, M.; Koedam, E.L.; Barnes, J.; Bartlett, J.W.; Barkhof, F.; Wattjes, M.P.; Schott, J.M.; Scheltens, P.; Fox, N.C. Visual Ratings of Atrophy in MCI: Prediction of Conversion and Relationship with CSF Biomarkers. Neurobiol. Aging 2013, 34, 73–82. [Google Scholar] [CrossRef]
- Lau, H.H.C.; Ingelsson, M.; Watts, J.C. The Existence of Aβ Strains and Their Potential for Driving Phenotypic Heterogeneity in Alzheimer’s Disease. Acta Neuropathol. 2021, 142, 17–39. [Google Scholar] [CrossRef]
- Grinberg, L.T.; Rüb, U.; Ferretti, R.E.L.; Nitrini, R.; Farfel, J.M.; Polichiso, L.; Gierga, K.; Jacob-Filho, W.; Heinsen, H. The Dorsal Raphe Nucleus Shows Phospho-Tau Neurofibrillary Changes before the Transentorhinal Region in Alzheimer’s Disease. A Precocious Onset? Neuropathol. Appl. Neurobiol. 2009, 35, 406–416. [Google Scholar] [CrossRef]
- Šimić, G.; Stanić, G.; Mladinov, M.; Jovanov-Milošević, N.; Kostović, I.; Hof, P.R. Does Alzheimer’s Disease Begin in the Brainstem? Neuropathol. Appl. Neurobiol. 2009, 35, 532–554. [Google Scholar] [CrossRef]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Monoaminergic Neuropathology in Alzheimer’s Disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary Tangles but Not Senile Plaques Parallel Duration and Severity of Alzheimer’s Disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and Neuron Numbers, but Not Amyloid Load, Predict Cognitive Status in Alzheimer’s Disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Tredici, K.D.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Šimić, G.; Gnjidić, M.; Kostović, I. Cytoskeletal Changes as an Alternative View on Pathogenesis of Alzheimer’s Disease. Period. Biol. 1998, 100, 165–173. [Google Scholar]
- Saito, Y.; Ruberu, N.N.; Sawabe, M.; Arai, T.; Tanaka, N.; Kakuta, Y.; Yamanouchi, H.; Murayama, S. Staging of Argyrophilic Grains: An Age-Associated Tauopathy. J. Neuropathol. Exp. Neurol. 2004, 63, 911–918. [Google Scholar] [CrossRef]
- Verny, M.; Jellinger, K.A.; Hauw, J.J.; Bancher, C.; Litvan, I.; Agid, Y. Progressive Supranuclear Palsy: A Clinicopathological Study of 21 Cases. Acta Neuropathol. 1996, 91, 427–431. [Google Scholar] [CrossRef]
- Irwin, D.J.; Brettschneider, J.; McMillan, C.T.; Cooper, F.; Olm, C.; Arnold, S.E.; Van Deerlin, V.M.; Seeley, W.W.; Miller, B.L.; Lee, E.B.; et al. Deep Clinical and Neuropathological Phenotyping of Pick Disease. Ann. Neurol. 2016, 79, 272–287. [Google Scholar] [CrossRef]
- Ahmed, Z.; Cooper, J.; Murray, T.K.; Garn, K.; McNaughton, E.; Clarke, H.; Parhizkar, S.; Ward, M.A.; Cavallini, A.; Jackson, S.; et al. A Novel in Vivo Model of Tau Propagation with Rapid and Progressive Neurofibrillary Tangle Pathology: The Pattern of Spread Is Determined by Connectivity, Not Proximity. Acta Neuropathol. 2014, 127, 667–683. [Google Scholar] [CrossRef]
- Dujardin, S.; Lecolle, K.; Caillierez, R.; Begard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Auregan, G.; Winderickx, J.; et al. Neuron-to-Neuron Wild-Type Tau Protein Transfer through a Trans-Synaptic Mechanism: Relevance to Sporadic Tauopathies. Acta Neuropathol. Commun. 2014, 2, 14. [Google Scholar] [CrossRef]
- Iba, M.; McBride, J.D.; Guo, J.L.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.Y. Tau Pathology Spread in PS19 Tau Transgenic Mice Following Locus Coeruleus (LC) Injections of Synthetic Tau Fibrils Is Determined by the LC’s Afferent and Efferent Connections. Acta Neuropathol. 2015, 130, 349–362. [Google Scholar] [CrossRef]
- Peeraer, E.; Bottelbergs, A.; Van Kolen, K.; Stancu, I.-C.; Vasconcelos, B.; Mahieu, M.; Duytschaever, H.; Ver Donck, L.; Torremans, A.; Sluydts, E.; et al. Intracerebral Injection of Preformed Synthetic Tau Fibrils Initiates Widespread Tauopathy and Neuronal Loss in the Brains of Tau Transgenic Mice. Neurobiol. Dis. 2015, 73, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Synthetic Tau Fibrils Mediate Transmission of Neurofibrillary Tangles in a Transgenic Mouse Model of Alzheimer’s-like Tauopathy. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Friedhoff, P.; Von Bergen, M.; Mandelkow, E.M.; Davies, P.; Mandelkow, E. A Nucleated Assembly Mechanism of Alzheimer Paired Helical Filaments. Proc. Natl. Acad. Sci. USA 1998, 95, 15712–15717. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M.Y. Seeding of Normal Tau by Pathological Tau Conformers Drives Pathogenesis of Alzheimer-like Tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and Spreading of Tauopathy in Transgenic Mouse Brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Boluda, S.; Iba, M.; Zhang, B.; Raible, K.M.; Lee, V.M.Y.; Trojanowski, J.Q. Differential Induction and Spread of Tau Pathology in Young PS19 Tau Transgenic Mice Following Intracerebral Injections of Pathological Tau from Alzheimer???S Disease or Corticobasal Degeneration Brains. Acta Neuropathol. 2015, 129, 221–237. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Vasconcelos, B.; Ris, L.; Wang, P.; Villers, A.; Peeraer, E.; Buist, A.; Terwel, D.; Baatsen, P.; Oyelami, T.; et al. Templated Misfolding of Tau by Prion-like Seeding along Neuronal Connections Impairs Neuronal Network Function and Associated Behavioral Outcomes in Tau Transgenic Mice. Acta Neuropathol. 2015, 129, 875–894. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Smolek, T.; Jadhav, S.; Brezovakova, V.; Cubinkova, V.; Valachova, B.; Novak, P.; Zilka, N. First-in-Rat Study of Human Alzheimer’s Disease Tau Propagation. Mol. Neurobiol. 2019, 56, 621–631. [Google Scholar] [CrossRef]
- Levarska, L.; Zilka, N.; Jadhav, S.; Neradil, P.; Novak, M. Of Rodents and Men: The Mysterious Interneuronal Pilgrimage of Misfolded Protein Tau in Alzheimer’s Disease. J. Alzheimers Dis. 2013, 37, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, T.; Dewachter, I.; Lechat, B.; Croes, S.; Termont, A.; Demedts, D.; Borghgraef, P.; Devijver, H.; Filipkowski, R.K.; Kaczmarek, L.; et al. AAV-Tau Mediates Pyramidal Neurodegeneration by Cell-Cycle Re-Entry without Neurofibrillary Tangle Formation in Wild-Type Mice. PLoS ONE 2009, 4, e7280. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Alzheimer’s Disease: Pathogenesis and Prevention. Alzheimers Dement. 2012, 8, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Varo, R.; Mejias-Ortega, M.; Fernandez-Valenzuela, J.J.; Nuñez-Diaz, C.; Caceres-Palomo, L.; Vegas-Gomez, L.; Sanchez-Mejias, E.; Trujillo-Estrada, L.; Garcia-Leon, J.A.; Moreno-Gonzalez, I.; et al. Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis. Int. J. Mol. Sci. 2022, 23, 5404. [Google Scholar] [CrossRef]
- Chang, E.; Kim, S.; Yin, H.; Nagaraja, H.N.; Kuret, J. Pathogenic Missense MAPT Mutations Differentially Modulate Tau Aggregation Propensity at Nucleation and Extension Steps. J. Neurochem. 2008, 107, 1113–1123. [Google Scholar] [CrossRef]
- Guo, J.L.; Narasimhan, S.; Changolkar, L.; He, Z.; Stieber, A.; Zhang, B.; Gathagan, R.J.; Iba, M.; McBride, J.D.; Trojanowski, J.Q.; et al. Unique Pathological Tau Conformers from Alzheimer’s Brains Transmit Tau Pathology in Nontransgenic Mice. J. Exp. Med. 2016, 213, 2635–2654. [Google Scholar] [CrossRef]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological Tau Strains from Human Brains Recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. J. Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef]
- Smolek, T.; Jadhav, S.; Valachova, B.; Vogels, T.; Legath, J.; Novak, P.; Zilka, N. Transmission of Tau Pathology from Human to Rodent Brain: How to Humanise Animal Models for Alzheimer’s Disease Research. J. Alzheimer’s Dis. Park. 2017, 7, 400. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau Pathology Is an Initiating Factor in Sporadic Alzheimer’s Disease. Alzheimers Dement. J. Alzheimers Assoc. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Morrison, J.H.; Hof, P.R. Life and Death of Neurons in the Aging Brain. Science 1997, 278, 412–419. [Google Scholar] [CrossRef]
- Šimić, G.; Bexheti, S.; Kelović, Z.; Kos, M.; Grbić, K.; Hof, P.R.; Kostović, I. Hemispheric Asymmetry, Modular Variability and Age-Related Changes in the Human Entorhinal Cortex. Neuroscience 2005, 130, 911–925. [Google Scholar] [CrossRef]
- Gerson, J.E.; Sengupta, U.; Kayed, R. Tau Oligomers as Pathogenic Seeds: Preparation and Propagation in Vitro and in Vivo. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1523, pp. 141–157. [Google Scholar]
- Margittai, M.; Langen, R. Template-Assisted Filament Growth by Parallel Stacking of Tau. Proc. Natl. Acad. Sci. USA 2004, 101, 10278–10283. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Elsevier Science, Academic Press: Amsterdam, The Netherlands, 2007; ISBN 9780080475158. [Google Scholar]
- Witter, M.P.; Doan, T.P.; Jacobsen, B.; Nilssen, E.S.; Ohara, S. Architecture of the Entorhinal Cortex a Review of Entorhinal Anatomy in Rodents with Some Comparative Notes. Front. Syst. Neurosci. 2017, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Bažadona, D.; Fabek, I.; Babić Leko, M.; Bobić Rasonja, M.; Kalinić, D.; Bilić, E.; Raguž, J.D.; Mimica, N.; Borovečki, F.; Hof, P.R.; et al. A Non-Invasive Hidden-Goal Test for Spatial Orientation Deficit Detection in Subjects with Suspected Mild Cognitive Impairment. J. Neurosci. Methods 2020, 332, 108547. [Google Scholar] [CrossRef]
- Gage, G.J.; Kipke, D.R.; Shain, W. Whole Animal Perfusion Fixation for Rodents. J. Vis. Exp. JoVE 2012, 65, e3564. [Google Scholar] [CrossRef]
- Hurtado, D.E.; Molina-Porcel, L.; Iba, M.; Aboagye, A.K.; Paul, S.M.; Trojanowski, J.Q.; Lee, V.M.Y. Aβ Accelerates the Spatiotemporal Progression of Tau Pathology and Augments Tau Amyloidosis in an Alzheimer Mouse Model. Am. J. Pathol. 2010, 177, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Gallyas, F. Silver Staining of Alzheimer’s Neurofibrillary Changes by Means of Physical Development. Acta Morphol. Acad. Sci. Hung. 1971, 19, 1–8. [Google Scholar] [PubMed]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E.; Ohm, T.; Bohl, J. Silver Impregnation of Alzheimer’s Neurofibrillary Changes Counterstained for Basophilic Material and Lipofuscin Pigment. Stain Technol. 2009, 63, 197–200. [Google Scholar] [CrossRef]
- Biernat, J.; Mandelkow, E.M.; Schröter, C.; Lichtenberg-Kraag, B.; Steiner, B.; Berling, B.; Meyer, H.; Mercken, M.; Vandermeeren, A.; Goedert, M. The Switch of Tau Protein to an Alzheimer-like State Includes the Phosphorylation of Two Serine-Proline Motifs Upstream of the Microtubule Binding Region. EMBO J. 1992, 11, 1593–1597. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Vanmechelen, E. Monoclonal Antibody AT8 Recognises Tau Protein Phosphorylated at Both Serine 202 and Threonine 205. Neurosci. Lett. 1995, 189, 167–170. [Google Scholar] [CrossRef]
- Jicha, G.A.; Bowser, R.; Kazam, I.G.; Davies, P. Alz-50 and MC-1, a New Monoclonal Antibody Raised to Paired Helical Filaments, Recognize Conformational Epitopes on Recombinant Tau. J. Neurosci. Res. 1997, 48, 128–132. [Google Scholar] [CrossRef]
- Jicha, G.A.; Berenfeld, B.; Davies, P. Sequence Requirements for Formation of Conformational Variants of Tau Similar to Those Found in Alzheimer’s Disease. J. Neurosci. Res. 1999, 55, 713–723. [Google Scholar] [CrossRef]
- Weaver, C.L.; Espinoza, M.; Kress, Y.; Davies, P. Conformational Change as One of the Earliest Alterations of Tau in Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.J.; Rawlins, J.N.P. T-Maze Alternation in the Rodent. Nat. Protoc. 2006, 1, 7–12. [Google Scholar] [CrossRef]
- Zhang, X.H.; Liu, S.S.; Yi, F.; Zhuo, M.; Li, B.M. Delay-Dependent Impairment of Spatial Working Memory with Inhibition of NR2B-Containing NMDA Receptors in Hippocampal CA1 Region of Rats. Mol. Brain 2013, 6, 13. [Google Scholar] [CrossRef]
- Dember, W.N.; Richman, C.L. Spontaneous Alternation Behavior; Springer: New York, NY, USA, 1989. [Google Scholar]
- Olton, D.S.; Becker, J.T.; Handelmann, G.E. Hippocampus, Space, and Memory. Behav. Brain Sci. 1979, 2, 313–322. [Google Scholar] [CrossRef]
- Ennaceur, A.; Delacour, J. A New One-Trial Test for Neurobiological Studies of Memory in Rats. 1: Behavioral Data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef]
- Vogel-Ciernia, A.; Wood, M.A. Examining Object Location and Object Recognition Memory in Mice. Curr. Protoc. Neurosci. 2014, 69, 8.31.1–8.31.17. [Google Scholar] [CrossRef]
- Winters, B.D.; Bussey, T.J. Transient Inactivation of Perirhinal Cortex Disrupts Encoding, Retrieval, and Consolidation of Object Recognition Memory. J. Neurosci. 2005, 25, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Aggleton, J.P.; Albasser, M.M.; Aggleton, D.J.; Poirier, G.L.; Pearce, J.M. Lesions of the Rat Perirhinal Cortex Spare the Acquisition of a Complex Configural Visual Discrimination Yet Impair Object Recognition. Behav. Neurosci. 2010, 124, 55–68. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse Loss in Frontal Cortex Biopsies in Alzheimer’s Disease: Correlation with Cognitive Severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Braak, F.; Braak, H.; Mandelkow, E.M. A Sequence of Cytoskeleton Changes Related to the Formation of Neurofibrillary Tangles and Neuropil Threads. Acta Neuropathol. 1994, 87, 554–567. [Google Scholar] [CrossRef]
- Swanson, L.W.; Kohler, C. Anatomical Evidence for Direct Projections from the Entorhinal Area to the Entire Cortical Mantle in the Rat. J. Neurosci. 1986, 6, 3010–3023. [Google Scholar] [CrossRef]
- Šimić, G.; Kostović, I.; Winblad, B.; Bogdanović, N. Volume and Number of Neurons of the Human Hippocampal Formation in Normal Aging and Alzheimer’s Disease. J. Comp. Neurol. 1997, 379, 482–494. [Google Scholar] [CrossRef]
- Šimić, G.; Krsnik, Ž.; Knezović, V.; Kelović, Z.; Mathiasen, M.L.; Junaković, A.; Radoš, M.; Mulc, D.; Španić, E.; Quattrocolo, G.; et al. Prenatal Development of the Human Entorhinal Cortex. J. Comp. Neurol. 2022, 530, 2711–2748. [Google Scholar] [CrossRef] [PubMed]
- Witter, M.P.; Wouterlood, F.G.; Naber, P.A.; Van Haeften, T. Anatomical Organization of the Parahippocampal-Hippocampal Network. Ann. N. Y. Acad. Sci. 2006, 911, 1–24. [Google Scholar] [CrossRef]
- Steward, O. Topographic Organization of the Projections from the Entorhinal Area to the Hippocampal Formation of the Rat. J. Comp. Neurol. 1976, 167, 285–314. [Google Scholar] [CrossRef]
- Hjorth-Simonsen, A.; Jeune, B. Origin and Termination of the Hippocampal Perforant Path in the Rat Studied by Silver Impregnation. J. Comp. Neurol. 1972, 144, 215–231. [Google Scholar] [CrossRef]
- Hyman, B.T.; Van Hoesen, G.W.; Kromer, L.J.; Damasio, A.R. Perforant Pathway Changes and the Memory Impairment of Alzheimer’s Disease. Ann. Neurol. 1986, 20, 472–481. [Google Scholar] [CrossRef]
- Thal, D.R.; Holzer, M.; Rüb, U.; Waldmann, G.; Günzel, S.; Zedlick, D.; Schober, R. Alzheimer-Related τ-Pathology in the Perforant Path Target Zone and in the Hippocampal Stratum Oriens and Radiatum Correlates with Onset and Degree of Dementia. Exp. Neurol. 2000, 163, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Steward, O.; Scoville, S.A. Cells of Origin of Entorhinal Cortical Afferents to the Hippocampus and Fascia Dentata of the Rat. J. Comp. Neurol. 1976, 169, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Goldowitz, D.; White, W.F.; Steward, O.; Lynch, G.; Cotman, C. Anatomical Evidence for a Projection from the Entorhinal Cortex to the Contralateral Dentate Gyrus of the Rat. Exp. Neurol. 1975, 47, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C. Intrinsic Connections of the Retrohippocampal Region in the Rat Brain: III. The Lateral Entorhinal Area. J. Comp. Neurol. 1988, 271, 208–228. [Google Scholar] [CrossRef] [PubMed]
- Greig, L.C.; Woodworth, M.B.; Galazo, M.J.; Padmanabhan, H.; Macklis, J.D. Molecular Logic of Neocortical Projection Neuron Specification, Development and Diversity. Nat. Rev. Neurosci. 2013, 14, 755–769. [Google Scholar] [CrossRef]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; De Decker, R.; Vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-β Pathology Enhances Pathological Fibrillary Tau Seeding Induced by Alzheimer PHF in Vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef]
- Usenovic, M.; Niroomand, S.; Drolet, R.E.; Yao, L.; Gaspar, R.C.; Hatcher, N.G.; Schachter, J.; Renger, J.J.; Parmentier-Batteur, S. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. J. Neurosci. 2015, 35, 14234–14250. [Google Scholar] [CrossRef]
- Bibow, S.; Mukrasch, M.D.; Chinnathambi, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. The Dynamic Structure of Filamentous Tau. Angew. Chem. Int. Ed. Engl. 2011, 50, 11520–11524. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Suh, Y.H.; Checler, F. Amyloid Precursor Protein, Presenilins, and Alpha-Synuclein: Molecular Pathogenesis and Pharmacological Applications in Alzheimer’s Disease. Pharmacol. Rev. 2002, 54, 469–525. [Google Scholar] [CrossRef] [PubMed]
- Reisel, D.; Bannerman, D.M.; Schmitt, W.B.; Deacon, R.M.J.; Flint, J.; Borchardt, T.; Seeburg, P.H.; Rawlins, J.N.P. Spatial Memory Dissociations in Mice Lacking GluR1. Nat. Neurosci. 2002, 5, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.J.; Rawlins, J.N.P. Hippocampal Lesions, Species-Typical Behaviours and Anxiety in Mice. Behav. Brain Res. 2005, 156, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Assini, F.L.; Duzzioni, M.; Takahashi, R.N. Object Location Memory in Mice: Pharmacological Validation and Further Evidence of Hippocampal CA1 Participation. Behav. Brain Res. 2009, 204, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Mumby, D.G.; Gaskin, S.; Glenn, M.J.; Schramek, T.E.; Lehmann, H. Hippocampal Damage and Exploratory Preferences in Rats: Memory for Objects, Places, and Contexts. Learn. Mem. 2002, 9, 49–57. [Google Scholar] [CrossRef]
- Clarke, J.R.; Cammarota, M.; Gruart, A.; Izquierdo, I.; Delgado-García, J.M. Plastic Modifications Induced by Object Recognition Memory Processing. Proc. Natl. Acad. Sci. USA 2010, 107, 2652–2657. [Google Scholar] [CrossRef]
- Götz, J.; Ittner, L.M. Animal Models of Alzheimer’s Disease and Frontotemporal Dementia. Nat. Rev. Neurosci. 2008, 9, 532–544. [Google Scholar] [CrossRef]
- Götz, J.; Probst, A.; Spillantini, M.G.; Schäfer, T.; Jakes, R.; Bürki, K.; Goedert, M. Somatodendritic Localization and Hyperphosphorylation of Tau Protein in Transgenic Mice Expressing the Longest Human Brain Tau Isoform. EMBO J. 1995, 14, 1304–1313. [Google Scholar] [CrossRef]
- Brion, J.P.; Trempdagger, G.; Octavedagger, J.N. Transgenic Expression of the Shortest Human Tau Affects Its Compartmentalization and Its Phosphorylation as in the Pretangle Stage of Alzheimer’s Disease. Am. J. Pathol. 1999, 154, 255–270. [Google Scholar] [CrossRef]
- Ishihara, T.; Hong, M.; Zhang, B.; Nakagawa, Y.; Lee, M.K.; Trojanowski, J.Q.; Lee, V.M.Y. Age-Dependent Emergence and Progression of a Tauopathy in Transgenic Mice Overexpressing the Shortest Human Tau Isoform. Neuron 1999, 24, 751–762. [Google Scholar] [CrossRef]
- Probst, A.; Tolnay, M.; Mistl, C.; Götz, J.; Wiederhold, K.H.; Jaton, A.L.; Hong, M.; Ishihara, T.; Lee, V.M.Y.; Trojanowski, J.Q.; et al. Axonopathy and Amyotrophy in Mice Transgenic for Human Four-Repeat Tau Protein. Acta Neuropathol. 2000, 99, 469–481. [Google Scholar] [CrossRef]
- Bengoetxea, X.; Rodriguez-Perdigon, M.; Ramirez, M.J. Object Recognition Test for Studying Cognitive Impairments in Animal Models of Alzheimer’s Disease. Front. Biosci.-Sch. 2017, 7, 10–29. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Gerson, J.E.; Sengupta, U.; Guerrero-Muñoz, M.J.; Lasagna-Reeves, C.A.; Kayed, R. Specific Targeting of Tau Oligomers in Htau Mice Prevents Cognitive Impairment and Tau Toxicity Following Injection with Brain-Derived Tau Oligomeric Seeds. J. Alzheimers Dis. 2014, 40, S97–S111. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Götz, J. Tau-Based Therapies in Neurodegeneration: Opportunities and Challenges. Nat. Rev. Drug Discov. 2017, 16, 863–883. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Prokop, S.; Gorion, K.-M.M.; Kim, J.D.; Sorrentino, Z.A.; Bell, B.M.; Manaois, A.N.; Chakrabarty, P.; Davies, P.; Giasson, B.I. Tau Ser208 Phosphorylation Promotes Aggregation and Reveals Neuropathological Diversity in Alzheimer’s Disease and Other Tauopathies. Acta Neuropathol. Commun. 2020, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. The Pathological Process Underlying Alzheimer’s Disease in Individuals Under Thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef]
- Audouard, E.; Houben, S.; Masaracchia, C.; Yilmaz, Z.; Suain, V.; Authelet, M.; De Decker, R.; Buée, L.; Boom, A.; Leroy, K.; et al. High-Molecular-Weight Paired Helical Filaments from Alzheimer Brain Induces Seeding of Wild-Type Mouse Tau into an Argyrophilic 4R Tau Pathology in Vivo. Am. J. Pathol. 2016, 186, 2709–2722. [Google Scholar] [CrossRef]
- Caillierez, R.; Bégard, S.; Lécolle, K.; Deramecourt, V.; Zommer, N.; Dujardin, S.; Loyens, A.; Dufour, N.; Aurégan, G.; Winderickx, J.; et al. Lentiviral Delivery of the Human Wild-Type Tau Protein Mediates a Slow and Progressive Neurodegenerative Tau Pathology in the Rat Brain. Mol. Ther. 2013, 21, 1358–1368. [Google Scholar] [CrossRef]
- Jacob, H.J.; Kwitek, A.E. Rat Genetics: Attaching Physiology and Pharmacology to the Genome. Nat. Rev. Genet. 2002, 3, 33–42. [Google Scholar] [CrossRef]
- Whishaw, I.Q.; Metz, G.A.S.; Kolb, B.; Pellis, S.M. Accelerated Nervous System Development Contributes to Behavioral Efficiency in the Laboratory Mouse: A Behavioral Review and Theoretical Proposal. Dev. Psychobiol. 2001, 39, 151–170. [Google Scholar] [CrossRef]
- Do Carmo, S.; Cuello, A.C. Modeling Alzheimer’s Disease in Transgenic Rats. Mol. Neurodegener. 2013, 8, 37. [Google Scholar] [CrossRef]
- Puangmalai, N.; Bhatt, N.; Montalbano, M.; Sengupta, U.; Gaikwad, S.; Ventura, F.; McAllen, S.; Ellsworth, A.; Garcia, S.; Kayed, R. Internalization Mechanisms of Brain-derived Tau Oligomers from Patients with Alzheimer’s Disease, Progressive Supranuclear Palsy, And Dementia with Lewy bodies. Cell Death Dis. 2020, 11, 314. [Google Scholar] [CrossRef] [PubMed]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langer Horvat, L.; Španić Popovački, E.; Babić Leko, M.; Zubčić, K.; Horvat, L.; Mustapić, M.; Hof, P.R.; Šimić, G. Anterograde and Retrograde Propagation of Inoculated Human Tau Fibrils and Tau Oligomers in a Non-Transgenic Rat Tauopathy Model. Biomedicines 2023, 11, 1004. https://doi.org/10.3390/biomedicines11041004
Langer Horvat L, Španić Popovački E, Babić Leko M, Zubčić K, Horvat L, Mustapić M, Hof PR, Šimić G. Anterograde and Retrograde Propagation of Inoculated Human Tau Fibrils and Tau Oligomers in a Non-Transgenic Rat Tauopathy Model. Biomedicines. 2023; 11(4):1004. https://doi.org/10.3390/biomedicines11041004
Chicago/Turabian StyleLanger Horvat, Lea, Ena Španić Popovački, Mirjana Babić Leko, Klara Zubčić, Luka Horvat, Maja Mustapić, Patrick R. Hof, and Goran Šimić. 2023. "Anterograde and Retrograde Propagation of Inoculated Human Tau Fibrils and Tau Oligomers in a Non-Transgenic Rat Tauopathy Model" Biomedicines 11, no. 4: 1004. https://doi.org/10.3390/biomedicines11041004
APA StyleLanger Horvat, L., Španić Popovački, E., Babić Leko, M., Zubčić, K., Horvat, L., Mustapić, M., Hof, P. R., & Šimić, G. (2023). Anterograde and Retrograde Propagation of Inoculated Human Tau Fibrils and Tau Oligomers in a Non-Transgenic Rat Tauopathy Model. Biomedicines, 11(4), 1004. https://doi.org/10.3390/biomedicines11041004