
Likely Pathogenic Variants of Cav1.3 and Nav1.1 Encoding Genes in Amyotrophic Lateral Sclerosis Could Elucidate the Dysregulated Pain Pathways

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
2.1. Analysis of the Cav1.3 and Cav1.2 Channels Encoding Gene
2.2. Analysis of the Piezo, Nav Channel, NMDA, GABA and Glycine Receptor Encoding Genes
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| ALS | Amyotrophic lateral sclerosis |
| CACNA1D | Voltage-gated channel subunit alpha1 D |
| CPG | Central pattern generators |
| GABA | Gamma-aminobutyric acid |
| NMDA | N-methyl-d-aspartate |
| PASNA | Primary aldosteronism, seizures and neurological abnormalities |
| PIC | Persistent inward currents |
| VGLUT1 | Vesicular glutamate transporter 1 |
| WDR | Wide dynamic range |
References
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Charcot, J.-M.J.A. Deux Cas d’atrophie Musculaire Progressive: Avec lÈsions de la Substance Grise et des Faisceaux antÈrolatÈraux de la Moelle ÈpiniËre; Masson: Paris, France, 1869. [Google Scholar]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef]
- Kurland, L.T.; Mulder, D.W. Epidemiologic investigations of amyotrophic lateral sclerosis. 2. Familial aggregations indicative of dominant inheritance. I. Neurology 1955, 5, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Kurland, L.T.; Mulder, D.W. Epidemiologic investigations of amyotrophic lateral sclerosis. 2. Familial aggregations indicative of dominant inheritance. II. Neurology 1955, 5, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.; Heverin, M.; McLaughlin, R.L.; Hardiman, O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019, 76, 1367–1374. [Google Scholar] [CrossRef]
- Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.; Vosa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e1266. [Google Scholar] [CrossRef]
- Zhang, S.; Cooper-Knock, J.; Weimer, A.K.; Shi, M.; Moll, T.; Marshall, J.N.G.; Harvey, C.; Nezhad, H.G.; Franklin, J.; Souza, C.D.S.; et al. Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 2022, 110, 992–1008.e11. [Google Scholar] [CrossRef]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef]
- Vaughan, S.K.; Kemp, Z.; Hatzipetros, T.; Vieira, F.; Valdez, G. Degeneration of proprioceptive sensory nerve endings in mice harboring amyotrophic lateral sclerosis-causing mutations. J. Comp. Neurol. 2015, 523, 2477–2494. [Google Scholar] [CrossRef]
- Held, A.; Major, P.; Sahin, A.; Reenan, R.A.; Lipscombe, D.; Wharton, K.A. Circuit Dysfunction in SOD1-ALS Model First Detected in Sensory Feedback Prior to Motor Neuron Degeneration Is Alleviated by BMP Signaling. J. Neurosci. 2019, 39, 2347–2364. [Google Scholar] [CrossRef] [PubMed]
- Brownstone, R.M.; Lancelin, C. Escape from homeostasis: Spinal microcircuits and progression of amyotrophic lateral sclerosis. J. Neurophysiol. 2018, 119, 1782–1794. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Mora, G.; Lauria, G. Pain in amyotrophic lateral sclerosis. Lancet Neurol. 2017, 16, 144–157. [Google Scholar] [CrossRef]
- Kwak, S. Pain in amyotrophic lateral sclerosis: A narrative review. J. Yeungnam Med. Sci. 2022, 39, 181–189. [Google Scholar] [CrossRef]
- Miller, T.M.; Layzer, R.B. Muscle cramps. Muscle Nerve 2005, 32, 431–442. [Google Scholar] [CrossRef]
- Smart, K.M.; Blake, C.; Staines, A.; Doody, C. Clinical indicators of ‘nociceptive’, ‘peripheral neuropathic’ and ‘central’ mechanisms of musculoskeletal pain. A Delphi survey of expert clinicians. Man. Ther. 2010, 15, 80–87. [Google Scholar] [CrossRef]
- Sonkodi, B. Delayed Onset Muscle Soreness (DOMS): The Repeated Bout Effect and Chemotherapy-Induced Axonopathy May Help Explain the Dying-Back Mechanism in Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases. Brain Sci. 2021, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Sonkodi, B. Miswired Proprioception in Amyotrophic Lateral Sclerosis in Relation to Pain Sensation (and in Delayed Onset Muscle Soreness)—Is Piezo2 Channelopathy a Principal Transcription Activator in Proprioceptive Terminals Besides Being the Potential Primary Damage? Life 2023, 13, 657. [Google Scholar]
- Sonkodi, B.; Hortobágyi, T. Amyotrophic lateral sclerosis and delayed onset muscle soreness in light of the impaired blink and stretch reflexes – watch out for Piezo2. Open Med. 2022, 17, 397–402. [Google Scholar] [CrossRef]
- Szczot, M.; Liljencrantz, J.; Ghitani, N.; Barik, A.; Lam, R.; Thompson, J.H.; Bharucha-Goebel, D.; Saade, D.; Necaise, A.; Donkervoort, S.; et al. PIEZO2 mediates injury-induced tactile pain in mice and humans. Sci. Transl. Med. 2018, 10, eaat9892. [Google Scholar] [CrossRef]
- Aguiar, P.; Sousa, M.; Lima, D. NMDA channels together with L-type calcium currents and calcium-activated nonspecific cationic currents are sufficient to generate windup in WDR neurons. J. Neurophysiol. 2010, 104, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.M.; Surmeier, D.J.; Lee, K.H.; Sorkin, L.S.; Honda, C.N.; Tsong, Y.; Willis, W.D. Classification of primate spinothalamic and somatosensory thalamic neurons based on cluster analysis. J. Neurophysiol. 1986, 56, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Price, D.D.; Dubner, R. Mechanisms of first and second pain in the peripheral and central nervous systems. J. Investig. Derm. 1977, 69, 167–171. [Google Scholar] [CrossRef]
- Pinggera, A.; Mackenroth, L.; Rump, A.; Schallner, J.; Beleggia, F.; Wollnik, B.; Striessnig, J. New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum. Mol. Genet. 2017, 26, 2923–2932. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Goh, G.; Stolting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Allely, C.S. Pain sensitivity and observer perception of pain in individuals with autistic spectrum disorder. Sci. World J. 2013, 2013, 916178. [Google Scholar] [CrossRef]
- Hoffman, T.; Bar-Shalita, T.; Granovsky, Y.; Gal, E.; Kalingel-Levi, M.; Dori, Y.; Buxbaum, C.; Yarovinsky, N.; Weissman-Fogel, I. Indifference or hypersensitivity? Solving the riddle of the pain profile in individuals with autism. PAIN 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Omata, K.; Anand, S.K.; Hovelson, D.H.; Liu, C.J.; Yamazaki, Y.; Nakamura, Y.; Ito, S.; Satoh, F.; Sasano, H.; Rainey, W.E.; et al. Aldosterone-Producing Cell Clusters Frequently Harbor Somatic Mutations and Accumulate with Age in Normal Adrenals. J. Endocr. Soc. 2017, 1, 787–799. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Seidel, E.; Schewe, J.; Scholl, U.I. Genetic causes of primary aldosteronism. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Tripolszki, K.; Gampawar, P.; Schmidt, H.; Nagy, Z.F.; Nagy, D.; Klivenyi, P.; Engelhardt, J.I.; Szell, M. Comprehensive Genetic Analysis of a Hungarian Amyotrophic Lateral Sclerosis Cohort. Front. Genet. 2019, 10, 732. [Google Scholar] [CrossRef] [PubMed]
- Espino, C.M.; Lewis, C.M.; Ortiz, S.; Dalal, M.S.; Garlapalli, S.; Wells, K.M.; O’Neil, D.A.; Wilkinson, K.A.; Griffith, T.N. NaV1.1 is essential for proprioceptive signaling and motor behaviors. Elife 2022, 11, 79917. [Google Scholar] [CrossRef]
- Than, K.; Kim, E.; Navarro, C.; Chu, S.; Klier, N.; Occiano, A.; Ortiz, S.; Salazar, A.; Valdespino, S.R.; Villegas, N.K.; et al. Vesicle-released glutamate is necessary to maintain muscle spindle afferent excitability but not dynamic sensitivity in adult mice. J. Physiol. 2021, 599, 2953–2967. [Google Scholar] [CrossRef] [PubMed]
- Carranza Rojo, D.; Hamiwka, L.; McMahon, J.M.; Dibbens, L.M.; Arsov, T.; Suls, A.; Stodberg, T.; Kelley, K.; Wirrell, E.; Appleton, B.; et al. De novo SCN1A mutations in migrating partial seizures of infancy. Neurology 2011, 77, 380–383. [Google Scholar] [CrossRef]
- Mantegazza, M.; Gambardella, A.; Rusconi, R.; Schiavon, E.; Annesi, F.; Cassulini, R.R.; Labate, A.; Carrideo, S.; Chifari, R.; Canevini, M.P.; et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc. Natl. Acad. Sci. USA 2005, 102, 18177–18182. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Kim, W.; Cho, Y.W.; Lee, S.K.; Jung, K.Y.; Shin, W.; Kim, D.W.; Kim, W.J.; Lee, H.W.; Kim, W.; et al. Genetic characteristics of non-familial epilepsy. PeerJ 2019, 7, e8278. [Google Scholar] [CrossRef]
- Rhoads, A.R.; Friedberg, F. Sequence motifs for calmodulin recognition. FASEB J. 1997, 11, 331–340. [Google Scholar] [CrossRef]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Pinggera, A.; Striessnig, J. Ca(v) 1.3 (CACNA1D) L-type Ca(2+) channel dysfunction in CNS disorders. J. Physiol. 2016, 594, 5839–5849. [Google Scholar] [CrossRef]
- Wang, J.; Hamill, O.P. Piezo2-peripheral baroreceptor channel expressed in select neurons of the mouse brain: A putative mechanism for synchronizing neural networks by transducing intracranial pressure pulses. J. Integr. Neurosci. 2021, 20, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.; Ghulam-Jhelani, Z.; Ahn, I.S.; Yang, X.; Wiedau, M.; Simmons, D.; Chandler, S.H. Early deficits in GABA inhibition parallels an increase in L-type Ca(2+) currents in the jaw motor neurons of SOD1(G93A) mouse model for ALS. Neurobiol. Dis. 2023, 177, 105992. [Google Scholar] [CrossRef] [PubMed]
- Liss, B.; Striessnig, J. The Potential of L-Type Calcium Channels as a Drug Target for Neuroprotective Therapy in Parkinson’s Disease. Annu. Rev. Pharm. Toxicol. 2019, 59, 263–289. [Google Scholar] [CrossRef]
- Kang, S.; Cooper, G.; Dunne, S.F.; Dusel, B.; Luan, C.H.; Surmeier, D.J.; Silverman, R.B. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nat. Commun. 2012, 3, 1146. [Google Scholar] [CrossRef] [PubMed]
- Lamanauskas, N.; Nistri, A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur. J. Neurosci. 2008, 27, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Urbani, A.; Belluzzi, O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur. J. Neurosci. 2000, 12, 3567–3574. [Google Scholar] [CrossRef] [PubMed]
- Sonkodi, B. Delayed Onset Muscle Soreness and Critical Neural Microdamage-Derived Neuroinflammation. Biomolecules 2022, 12, 1207. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012, 2012, CD001447. [Google Scholar] [CrossRef]
- Tanaka, Y.; Yamada, M.; Koumura, A.; Sakurai, T.; Hayashi, Y.; Kimura, A.; Hozumi, I.; Inuzuka, T. Cardiac sympathetic function in the patients with amyotrophic lateral sclerosis: Analysis using cardiac [123I] MIBG scintigraphy. J. Neurol. 2013, 260, 2380–2386. [Google Scholar] [CrossRef]
- Pinto, S.; Pinto, A.; De Carvalho, M. Decreased heart rate variability predicts death in amyotrophic lateral sclerosis. Muscle Nerve 2012, 46, 341–345. [Google Scholar] [CrossRef]
- Min, S.; Chang, R.B.; Prescott, S.L.; Beeler, B.; Joshi, N.R.; Strochlic, D.E.; Liberles, S.D. Arterial Baroreceptors Sense Blood Pressure through Decorated Aortic Claws. Cell Rep. 2019, 29, 2192–2201.e3. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Bentho, O.; Park, M.Y.; Sharabi, Y. Low-frequency power of heart rate variability is not a measure of cardiac sympathetic tone but may be a measure of modulation of cardiac autonomic outflows by baroreflexes. Exp. Physiol. 2011, 96, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Louradour, J.; Bortolotti, O.; Torre, E.; Bidaud, I.; Lamb, N.; Fernandez, A.; Le Guennec, J.Y.; Mangoni, M.E.; Mesirca, P. L-Type Ca(v)1.3 Calcium Channels Are Required for Beta-Adrenergic Triggered Automaticity in Dormant Mouse Sinoatrial Pacemaker Cells. Cells 2022, 11, 1114. [Google Scholar] [CrossRef] [PubMed]
- Oey, P.L.; Vos, P.E.; Wieneke, G.H.; Wokke, J.H.; Blankestijn, P.J.; Karemaker, J.M. Subtle involvement of the sympathetic nervous system in amyotrophic lateral sclerosis. Muscle Nerve 2002, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sonkodi, B.; Hegedűs, Á.; Kopper, B.; Berkes, I. Significantly Delayed Medium-Latency Response of the Stretch Reflex in Delayed-Onset Muscle Soreness of the Quadriceps Femoris Muscles Is Indicative of Sensory Neuronal Microdamage. J. Funct. Morphol. Kinesiol. 2022, 7, 43. [Google Scholar] [CrossRef]
- Fang, Y.; Li, Q.; Li, X.; Luo, G.H.; Kuang, S.J.; Luo, X.S.; Li, Q.Q.; Yang, H.; Liu, Y.; Deng, C.Y.; et al. Piezo1 Participated in Decreased L-Type Calcium Current Induced by High Hydrostatic Pressure via. CaM/Src/Pitx2 Activation in Atrial Myocytes. Front. Cardiovasc. Med. 2022, 9, 842885. [Google Scholar] [CrossRef]
- Radwani, H.; Lopez-Gonzalez, M.J.; Cattaert, D.; Roca-Lapirot, O.; Dobremez, E.; Bouali-Benazzouz, R.; Eiriksdottir, E.; Langel, U.; Favereaux, A.; Errami, M.; et al. Cav1.2 and Cav1.3 L-type calcium channels independently control short- and long-term sensitization to pain. J. Physiol. 2016, 594, 6607–6626. [Google Scholar] [CrossRef]
- Puja, G.; Sonkodi, B.; Bardoni, R. Mechanisms of Peripheral and Central Pain Sensitization: Focus on Ocular Pain. Front. Pharm. 2021, 12, 764396. [Google Scholar] [CrossRef]
- Sonkodi, B.; Bardoni, R.; Hangody, L.; Radak, Z.; Berkes, I. Does Compression Sensory Axonopathy in the Proximal Tibia Contribute to Noncontact Anterior Cruciate Ligament Injury in a Causative Way? A New Theory for the Injury Mechanism. Life 2021, 11, 443. [Google Scholar] [CrossRef]
- Rice, D.A.; McNair, P.J.; Lewis, G.N.; Dalbeth, N. Quadriceps arthrogenic muscle inhibition: The effects of experimental knee joint effusion on motor cortex excitability. Arthritis Res. 2014, 16, 502. [Google Scholar] [CrossRef]
- Comitato, A.; Bardoni, R. Presynaptic Inhibition of Pain and Touch in the Spinal Cord: From Receptors to Circuits. Int. J. Mol. Sci. 2021, 22, 414. [Google Scholar] [CrossRef]
- Zhang, T.C.; Janik, J.J.; Grill, W.M. Modeling effects of spinal cord stimulation on wide-dynamic range dorsal horn neurons: Influence of stimulation frequency and GABAergic inhibition. J. Neurophysiol. 2014, 112, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Foerster, B.R.; Callaghan, B.C.; Petrou, M.; Edden, R.A.; Chenevert, T.L.; Feldman, E.L. Decreased motor cortex gamma-aminobutyric acid in amyotrophic lateral sclerosis. Neurology 2012, 78, 1596–1600. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Martin, L.J. Glycine receptor channels in spinal motoneurons are abnormal in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2011, 31, 2815–2827. [Google Scholar] [CrossRef] [PubMed]
- Branchereau, P.; Martin, E.; Allain, A.E.; Cazenave, W.; Supiot, L.; Hodeib, F.; Laupenie, A.; Dalvi, U.; Zhu, H.; Cattaert, D. Relaxation of synaptic inhibitory events as a compensatory mechanism in fetal SOD spinal motor networks. Elife 2019, 8, 51402. [Google Scholar] [CrossRef]
- Torok, F.; Tezcan, K.; Filippini, L.; Fernandez-Quintero, M.L.; Zanetti, L.; Liedl, K.R.; Drexel, R.S.; Striessnig, J.; Ortner, N.J. Germline de novo variant F747S extends the phenotypic spectrum of CACNA1D Ca2+ channelopathies. Hum. Mol. Genet. 2022, 32, 847–859. [Google Scholar] [CrossRef]
- Ding, J.; Li, X.; Tian, H.; Wang, L.; Guo, B.; Wang, Y.; Li, W.; Wang, F.; Sun, T. SCN1A Mutation-Beyond Dravet Syndrome: A Systematic Review and Narrative Synthesis. Front. Neurol. 2021, 12, 743726. [Google Scholar] [CrossRef]
- Lossin, C. A catalog of SCN1A variants. Brain Dev. 2009, 31, 114–130. [Google Scholar] [CrossRef]
- Huang, W.; Liu, M.; Yan, S.F.; Yan, N. Structure-based assessment of disease-related mutations in human voltage-gated sodium channels. Protein Cell 2017, 8, 401–438. [Google Scholar] [CrossRef]
- Brunklaus, A.; Brunger, T.; Feng, T.; Fons, C.; Lehikoinen, A.; Panagiotakaki, E.; Vintan, M.A.; Symonds, J.; Andrew, J.; Arzimanoglou, A.; et al. The gain of function SCN1A disorder spectrum: Novel epilepsy phenotypes and therapeutic implications. Brain 2022, 145, 3816–3831. [Google Scholar] [CrossRef]
- Carvalho, M.D.; Swash, M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph. Lateral Scler. 2009, 10, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.; Drory, V.; Hardiman, O.; Nakano, I.; Ravits, J.; Robberecht, W.; Shefner, J.; WFN Research Group on ALS/MND. A revision of the El Escorial criteria—2015. Amyotroph Lateral Scler. Front. Degener. 2015, 16, 291–292. [Google Scholar] [CrossRef] [PubMed]



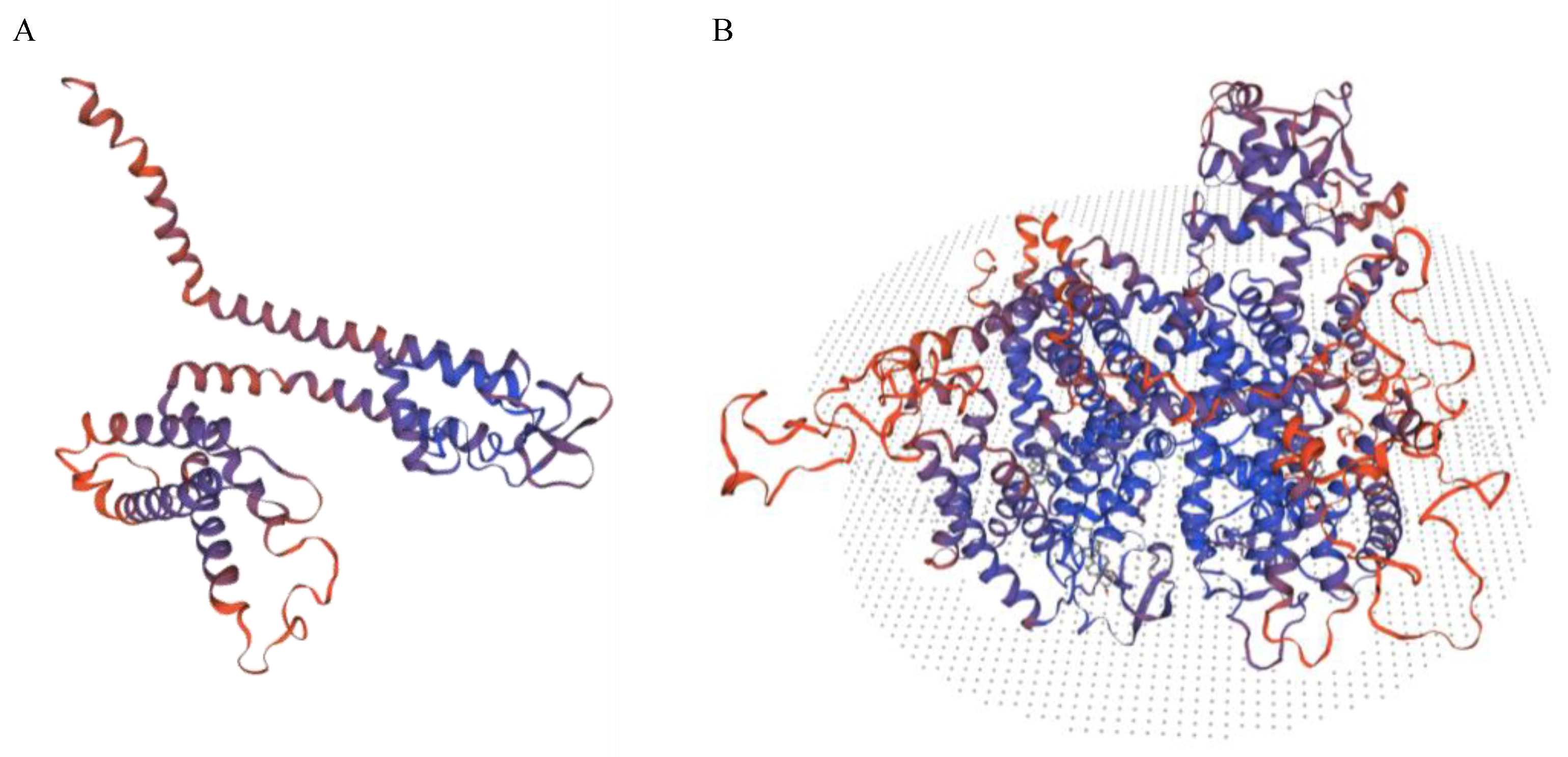{kind=link}
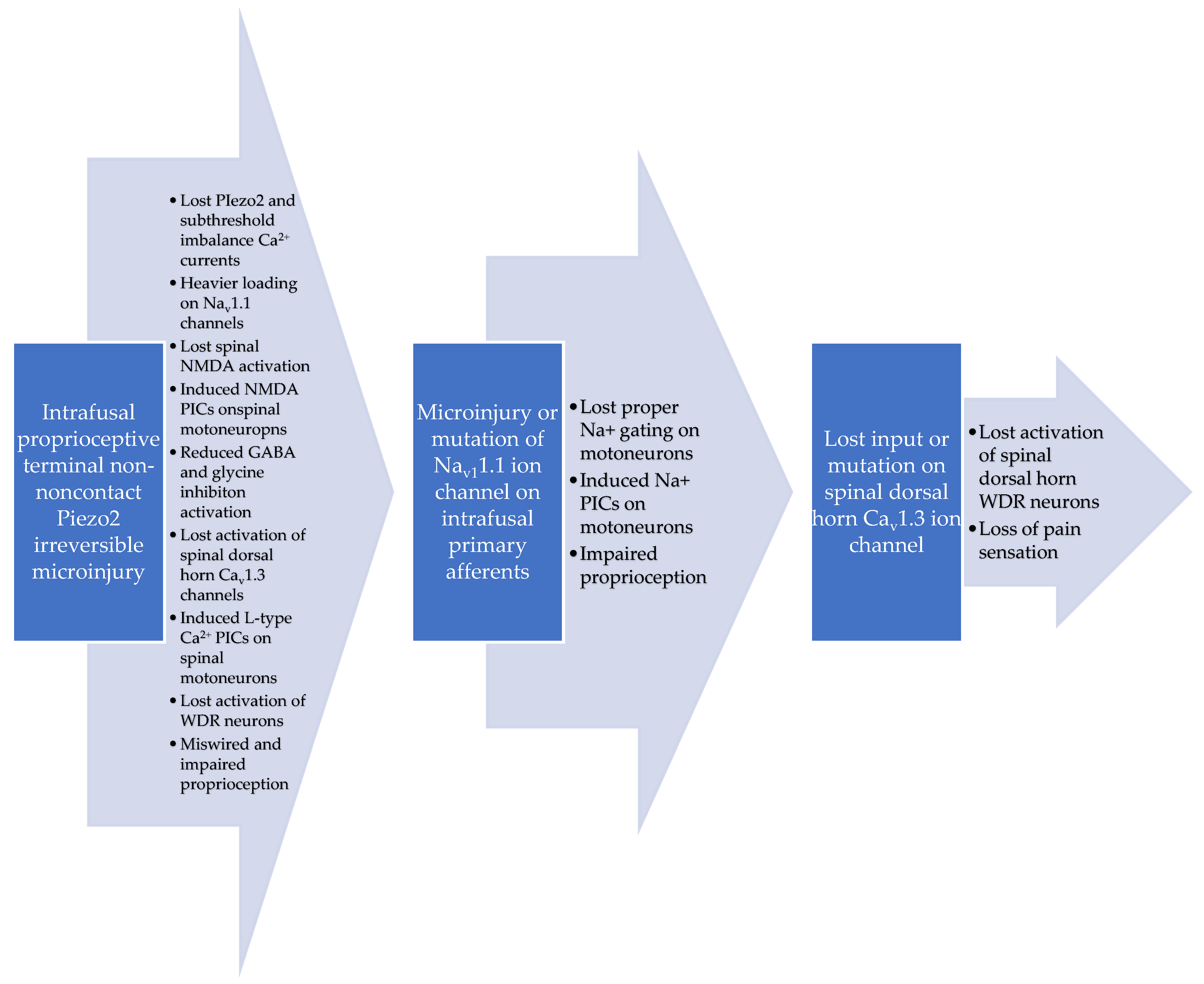{kind=link}
| Gene | Transcription Number | Variant | MAF in Non-Finnish European Population in Genome Aggregation Database | American College of Medical Genetics and Genomics (ACMG) Classification | Sample Number |
|---|---|---|---|---|---|
| CACNA1D | NM_001128839 | c.G5864A, p.Arg1955Gln | 0.0053% | Variant of unknown significance (VUS) | 42r |
| CACNA1D | NM_001128839 | c.A485T, p.Glu162Val | 0 | Variant of unknown significance (VUS) | 48r |
| CACNA1D | NM_001128839 | c.C1528T, p.Arg510Ter | 0 | Likely pathogenic | 74r |
| CACNA1D | NM_001128839 | c.C2241A, p.Phe747Leu | 0 | Variant of unknown significance (VUS) | 64r |
| CACNA1D | NM_001128839 | c.G5449A, p.Gly1817Arg | 0 | Variant of unknown significance (VUS) | 54r |
| CACNA1D | NM_001128839 | c.5694_5696del, p.Phe1899delfsTer239 | 0.4556% | Benign | 62r |
| Gene | Transcription Number | Variant | MAF in Non-Finnish European Population in Genome Aggregation Database | American College of Medical Genetics and Genomics (ACMG) Classification | Sample Number |
|---|---|---|---|---|---|
| SCN1A | NM_001165963 | c.A5779G, p.R1927G | 0 | Likely pathogenic | 79r |
| SCN1A | NM_001165963 | c.G2589T, p.L863F | 0 | Pathogenic | 71r |
| SCN1A | NM_001165963 | c.C1193T, p.T398M | 0.0016% | Likely pathogenic | 46r |
| SCN1A | NM_001165963 | c.T682C, p.S228P | 0 | Pathogenic | 55r |
| Minimum age | 40 |
| Maximum age | 73 |
| Average age | 60.0526 |
| Standard deviation | 8.8095 |
| Number of females | 10 |
| Number of males | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, Z.F.; Sonkodi, B.; Pál, M.; Klivényi, P.; Széll, M. Likely Pathogenic Variants of Cav1.3 and Nav1.1 Encoding Genes in Amyotrophic Lateral Sclerosis Could Elucidate the Dysregulated Pain Pathways. Biomedicines 2023, 11, 933. https://doi.org/10.3390/biomedicines11030933
Nagy ZF, Sonkodi B, Pál M, Klivényi P, Széll M. Likely Pathogenic Variants of Cav1.3 and Nav1.1 Encoding Genes in Amyotrophic Lateral Sclerosis Could Elucidate the Dysregulated Pain Pathways. Biomedicines. 2023; 11(3):933. https://doi.org/10.3390/biomedicines11030933
Chicago/Turabian StyleNagy, Zsófia Flóra, Balázs Sonkodi, Margit Pál, Péter Klivényi, and Márta Széll. 2023. "Likely Pathogenic Variants of Cav1.3 and Nav1.1 Encoding Genes in Amyotrophic Lateral Sclerosis Could Elucidate the Dysregulated Pain Pathways" Biomedicines 11, no. 3: 933. https://doi.org/10.3390/biomedicines11030933
APA StyleNagy, Z. F., Sonkodi, B., Pál, M., Klivényi, P., & Széll, M. (2023). Likely Pathogenic Variants of Cav1.3 and Nav1.1 Encoding Genes in Amyotrophic Lateral Sclerosis Could Elucidate the Dysregulated Pain Pathways. Biomedicines, 11(3), 933. https://doi.org/10.3390/biomedicines11030933