
The Role of Glutamate Receptors in Epilepsy


Abstract
1. Introduction
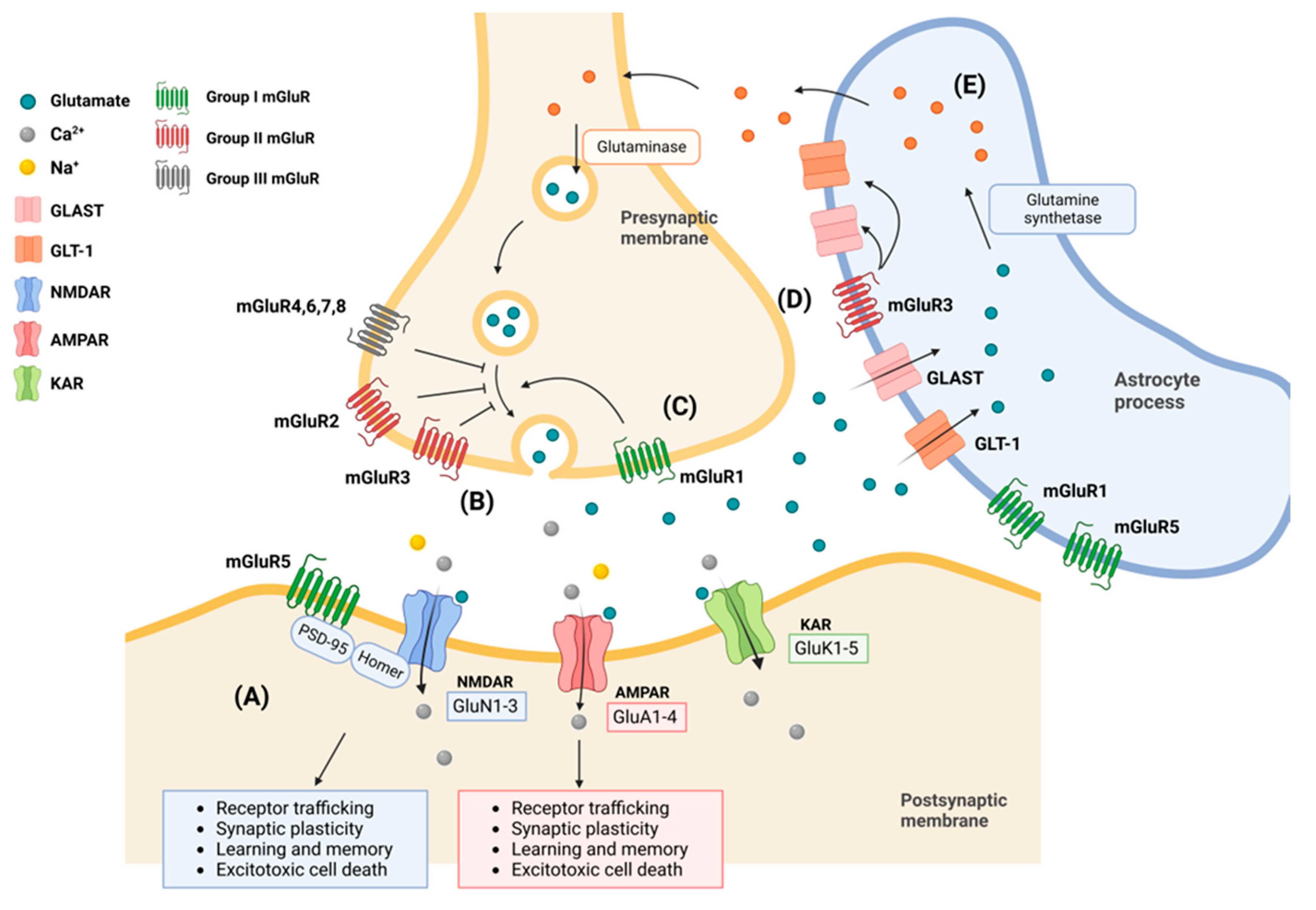
2. Subcellular Structure and Physiology of Glutamate Receptors
2.1. AMPA Receptors (AMPARs)
2.2. N-Methyl-D-Aspartate Receptors (NMDARs)
2.3. Kainate Receptors
2.4. Metabotropic Receptors
2.5. Astrocytes in Glutamate Uptake and Release
2.6. Cannabidiol (CBD) and Glutamate Signaling
3. Main Mechanism of Glutamate Receptor in Epilepsy
3.1. NMDA Receptors Mutation
3.2. AMPA Receptors Mutation
3.3. Anti-NMDA Antibody Encephalitis
4. Antiseizure Medications Acting on Glutamate Receptors
5. Treatment of SE by Glutamate Receptor Antagonists
6. Future Perspective of ASM
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scharfman, H.E. The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 2007, 7, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Wo, Z.G.; Oswald, R.E. Unraveling the modular design of glutamate-gated ion channels. Trends Neurosci. 1995, 18, 161–168. [Google Scholar] [PubMed]
- Sobolevsky, A.I. Structure and gating of tetrameric glutamate receptors. J. Physiol. 2015, 593, 29–38. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Lehre, K.P.; Danbolt, N.C. The number of glutamate transporter subtype molecules at glutamatergic synapses: Chemical and stereological quantification in young adult brain. J. Neurosci. 1998, 18, 8751–8757. [Google Scholar] [CrossRef]
- Krugler, P.; Schleyear, V. Developmental expression of glutamate transporter and glutamate dehydrogenase in astrocytes of the postnatal rat hippocampus. Hippocampus 2004, 14, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Njapo, S.A.; Rastogi, V.; Hedna, V.S. Taming glutamate excitotoxicity: Strategic pathway modulation for neuroprotection. CNS Drugs 2015, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Eid, T.; Gruenbaum, S.E.; Dhaher, R.; Lee, T.S.W.; Zhou, Y.; Danbolt, N.C. The glutamate-glutamine cycle in epilepsy. Adv. Neurobiol. 2016, 13, 351–400. [Google Scholar]
- Willard, S.S.; Koochekpour, S. Glutamate, glutamate receptors and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948–959. [Google Scholar] [CrossRef]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef]
- Rodríguez-Moreno, A.; Herreras, O.; Lerma, J. Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron 1997, 19, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Moreno, A.; Sihra, T.S. Presynaptic kainate receptor facilitation of glutamate release involves protein kinase A in the rat hippocampus. J. Physiol. 2004, 557, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T. The biochemistry, ultrastructure, and subunit assembly mechanism of AMPA receptors. Mol. Neurobiol. 2010, 42, 161–184. [Google Scholar] [CrossRef]
- Hollmann, M.; Hartley, M.; Heinemann, S. Ca2+ permeability of KA-AMPA—Gated glutamate receptor channels depends on subunit composition. Science 1991, 252, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Sans, N.; Vissel, B.; Petralia, R.S.; Wang, Y.X.; Chang, K.; Royle, G.A.; Wang, C.Y.; O’Gorman, S.; Heinemann, S.F.; Wenthold, R.J. Aberrant formation of glutamate receptor complexes in hippocampal neurons of mice lacking the GluR2 AMPA receptor subunit. J. Neurosci. 2003, 23, 9367–9373. [Google Scholar] [CrossRef]
- Hume, R.I.; Dingledine, R.; Heinemann, S.F. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science 1991, 253, 1028–1031. [Google Scholar] [CrossRef]
- Sommer, B.; Köhler, M.; Sprengel, R.; Seeburg, P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef]
- Malinow, R.; Malenka, R.C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef]
- Cull-Candy, S.; Kelly, L.; Farrant, M. Regulation of Ca2+- permeable AMPA receptors: Synaptic plasticity and beyond. Curr. Opin. Neurobiol. 2006, 16, 288–297. [Google Scholar] [CrossRef]
- Anggono, V.; Huganir, R.L. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 2012, 22, 461–469. [Google Scholar] [CrossRef]
- Liu, S.J.; Zukin, R.S. Ca2+- permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007, 30, 126–134. [Google Scholar] [CrossRef]
- Wiltgen, B.J.; Royle, G.A.; Gray, E.E.; Abdipranoto, A.; Thangthaeng, N.; Jacobs, N.; Saab, F.; Tonegawa, S.; Heinemann, S.F.; O’Dell, T.J.; et al. A role for calcium-permeable AMPA receptors in synaptic plasticity and learning. PLoS ONE 2010, 5, e12818. [Google Scholar] [CrossRef]
- Pellegrini-Giampietro, D.E.; Gorter, J.A.; Bennet, M.V.; Zukin, R.S. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997, 20, 464–470. [Google Scholar] [CrossRef]
- Kwak, S.; Weiss, J.H. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr. Opin. Neurobiol. 2006, 16, 281–287. [Google Scholar] [CrossRef]
- Russo, I.; Bonini, D.; Via, L.L.; Barlati, S.; Barbon, A. AMPA receptor properties are modulated in the early stages following pilocarpine-induced status epilepticus. Neuromolecular Med. 2013, 15, 324–338. [Google Scholar] [CrossRef]
- Hestrin, S.; Nicoll, R.A.; Perkel, D.J.; Sah, P. Analysis of excitatory synaptic action in pyramidal cells using whole-cell recording from rat hippocampal slices. J. Physiol. 1990, 422, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Trussell, L.O.; Zhang, S.; Raman, I.M. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron 1993, 10, 1185–1196. [Google Scholar] [CrossRef]
- Geiger, J.R.; Lübke, J.; Roth, A.; Frotscher, M.; Jonas, P. Submillisecond AMPA receptor-mediated signaling at a principal neuron-interneuron synapse. Neuron 1997, 18, 1009–1023. [Google Scholar] [CrossRef]
- Paoletti, P. Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 2011, 33, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Skrenkova, K.; Hemelikova, K.; Kolcheva, M.; Kortus, S.; Kaniakova, M.; Krausova, B.; Horak, M. Structural features in the glycine-binding sites of the GluN1 and GluN3A subunits regulate the surface delivery of NMDA receptors. Sci. Rep. 2019, 9, 12303. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, C.; Shigemoto, R.; Bessho, Y.; Nakanishi, S.; Mizuno, N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994, 347, 150–160. [Google Scholar] [CrossRef]
- Ogden, K.K.; Traynelis, S.F. New advances in NMDA receptor pharmacology. Trends Pharmacol. Sci. 2011, 32, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar] [PubMed]
- Mody, I.; Lambert, J.D.; Heinemann, U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J. Neurophysiol. 1987, 57, 869–888. [Google Scholar] [CrossRef]
- Gnegy, M.E. Ca2+/calmodulin signaling in NMDA-induced synaptic plasticity. Crit. Rev. Neurobiol. 2000, 14, 91–129. [Google Scholar] [CrossRef] [PubMed]
- Husi, H.; Ward, M.A.; Choudhary, J.S.; Blackstock, W.P.; Grant, S.G. Proteomic analysis of NMDAR-adhesion protein signaling complexes. Nat. Neurosci. 2000, 3, 661–669. [Google Scholar] [CrossRef]
- Massey, P.V.; Johnson, B.E.; Moult, P.R.; Auberson, Y.P.; Brown, M.W.; Molnar, E.; Collingridge, G.L.; Bashir, Z.I. Differential roles of NR2A and NR2B-containing NMDARs in cortical long-term potentiation and long-term depression. J. Neurosci. 2004, 24, 7821–7828. [Google Scholar] [CrossRef]
- Pinheiro, P.; Mulle, C. Kainate receptors. Cell Tissue Res. 2006, 326, 457–482. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, M.A.; Gryder, D.; Castaneda, D.; Yonekawa, W.; Banks, M.K.; Lia, H. GluR5 kainate receptors, seizures, and the amygdala. Ann. N. Y. Acad. Sci. 2003, 985, 150–162. [Google Scholar] [CrossRef]
- Patel, S.; Meldrum, B.S.; Collins, J.F. Distribution of [3H]kainic acid and binding sites in the rat brain: In vivo and in vitro receptor autoradiography. Neurosci. Lett. 1986, 70, 301–307. [Google Scholar] [CrossRef]
- Bloss, E.B.; Hunter, R.G. Hippocampal kainate receptors. Vitam. Horm. 2010, 82, 167–184. [Google Scholar]
- Pressey, J.C.; Woodin, M.A. Kainate receptor regulation of synaptic inhibition in the hippocampus. J. Physiol. 2021, 599, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, S.; Lerma, J. Non-canonical Signaling, the Hidden Life of Ligand-Gated Ion Channels. Neuron 2016, 92, 316–329. [Google Scholar] [CrossRef]
- Mulle, C.; Crépel, V. Regulation and dysregulation of neuronal circuits by KARs. Neuropharmacology 2021, 197, 108699. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Lagowska, J. Epileptogenic action of intra-amygdaloid injection of kainic acid. Comptes Rendus Hebd. Séances Acad. Sci. Série. Sci. Nat. 1978, 287, 813–816. [Google Scholar]
- Ben-Ari, Y.; Tremblay, E.; Ottersen, O.P. Primary and secondary cerebral lesions produced by kainic acid injections in the rat. Comptes Rendus Hebd. Séances Acad. Sci. Série. Sci. Nat. 1979, 288, 991–994. [Google Scholar]
- Ben-Ari, Y.; Tremblay, E.; Ottersen, O.P.; Meldrum, B.S. The role of epileptic activity in hippocampal and “remote” cerebral lesions induced by kainic acid. Brain Res. 1980, 191, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Bahn, S.; Volk, B.; Wisden, W. Kainate receptor gene expression in the developing rat brain. J. Neurosci. Off. J. Soc. Neurosci. 1994, 14, 5525–5547. [Google Scholar] [CrossRef]
- Werner, P.; Voigt, M.; Keinänen, K.; Wisden, W.; Seeburg, P.H. Cloning of a putative high-affinity kainate receptor expressed predominantly in hippocampal CA3 cells. Nature 1991, 351, 742–744. [Google Scholar] [CrossRef]
- Wisden, W.; Seeburg, P.H. A complex mosaic of high-affinity kainate receptors in rat brain. J. Neurosci. Off. J. Soc. Neurosci. 1993, 13, 3582–3598. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Cossart, R. Kainate, a double agent that generates seizures: Two decades of progress. Trends Neurosci. 2000, 23, 580–587. [Google Scholar] [CrossRef]
- Lévesque, M.; Langlois, J.M.P.; Lema, P.; Courtemanche, R.; Bilodeau, G.A.; Carmant, L. Synchronized gamma oscillations (30–50 Hz) in the amygdalo-hippocampal network in relation with seizure propagation and severity. Neurobiol. Dis. 2009, 35, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.; Fièvre, S.; Gorlewicz, A.; Mulle, C. Kainate receptors in the hippocampus. Eur. J. Neurosci. 2014, 39, 1835–1844. [Google Scholar] [CrossRef]
- Pahl, S.; Tapken, D.; Haering, S.C.; Hollmann, M. Traffiking of kaintae receptors. Membranes 2014, 4, 565–595. [Google Scholar] [CrossRef]
- Scholefield, C.L.; Atlason, P.T.; Jane, D.E.; Molnár, E. Assembly and trafficking of homomeric and heteromeric kainate receptors with impaired ligand binding sites. Neurochem. Res. 2019, 44, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.D.; Wilkinson, K.A.; Henley, J.M.; Mellor, J.R. Kainate receptors and synaptic plasticity. Neuropharmacology 2021, 196, 108540. [Google Scholar] [CrossRef]
- Merlin, L.R.; Wong, R.S. Role of Group I metabotropic glutamate receptors in the patterning of epileptiform activities in vitro. J. Neurophysiol. 1997, 78, 539–544. [Google Scholar] [CrossRef]
- Mannaioni, G.; Marino, M.J.; Valenti, O.; Traynelis, S.F.; Conn, P.J. Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J. Neurosci. 2001, 21, 5925–5934. [Google Scholar] [CrossRef]
- Notenboom, R.G.E.; Hampson, D.R.; Jansen, G.H.; van Rijen, P.C.; van Veelen, C.W.M.; van Nieuwenhuizen, O.; de Graan, P.N.E. Up-regulation of hippocampal metabotropic glutamate receptor 5 in temporal lobe epilepsy patients. Brain 2006, 129, 91–107. [Google Scholar] [CrossRef]
- Hermans, E.; Challiss, R.A. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: Prototypic family C G-protein-coupled receptors. Biochem. J. 2001, 359, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.R.; Chia, S.C.; Chen, P.M.; Gao, H.; Lee, W.L.; Yeo, T.S.; Burgunder, J.M.; Probst, A.; Sim, M.K.; Ling, E.A. Metabotropic glutamate receptor 2/3 in the hippocampus of patients with mesial temporal lobe epilepsy, and of rats and mice after pilocarpine-induced status epilepticus. Epilepsy Res. 2004, 59, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Pacheco Otalora, L.F.; Couoh, J.; Shigamoto, R.; Zarei, M.M.; Garrido Sanabria, E.R. Abnormal mGluR2/3 expression in the perforant path termination zones and mossy fibers of chronically epileptic rats. Brain Res. 2006, 1098, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Gorter, J.A.; Ijlst-Keizers, H.; Rozemuller, A.J.; Yankaya, B.; Leenstra, S.; Troost, D. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: Opposite regulation of glutamate transporter proteins. Eur. J. Neurosci. 2003, 17, 2106–2118. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; van Vliet, E.A.; Mayboroda, O.A.; Troost, D.; da Silva, F.H.; Gorter, J.A. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur. J. Neurosci. 2000, 12, 2333–2344. [Google Scholar] [CrossRef]
- Pitsch, J.; Schoch, S.; Gueler, N.; Flor, P.J.; van der Putten, H.; Becker, A.J. Functional role of mGluR1 and mGluR4 in pilocarpine-induced temporal lobe epilepsy. Neurobiol. Dis. 2007, 26, 623–633. [Google Scholar] [CrossRef]
- Hamilton, N.B.; Attwell, D. Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 2010, 11, 227–238. [Google Scholar] [CrossRef]
- Parri, H.R.; Gould, T.M.; Crunelli, V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat. Neurosci. 2001, 4, 803–812. [Google Scholar] [CrossRef]
- Fellin, T.; Pascual, O.; Gobbo, S.; Pozzan, T.; Haydon, P.G.; Carmignoto, G. Neuronal Synchrony Mediated by Astrocytic Glutamate through Activation of Extrasynaptic NMDA Receptors. Neuron 2004, 43, 729–743. [Google Scholar] [CrossRef]
- Angulo, M.C.; Kozlov, A.S.; Charpak, S.; Audinat, E. Glutamate Released from Glial Cells Synchronizes Neuronal Activity in the Hippocampus. J. Neurosci. 2004, 24, 6920–6927. [Google Scholar] [CrossRef]
- Perea, G.; Araque, A. Properties of Synaptically Evoked Astrocyte Calcium Signal Reveal Synaptic Information Processing by Astrocytes. J. Neurosci. 2005, 25, 2192–2203. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Sanzgiri, R.P.; Parpura, V.; Haydon, P.G. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J. Neurosci. 1998, 18, 6822–6829. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, P.; Bergersen, L.H.; Bhaukaurally, K.; Bezzi, P.; Santello, M.; Domercq, M.; Matute, C.; Tonello, F.; Gundersen, V.; Volterra, A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 2007, 10, 331–339. [Google Scholar] [CrossRef]
- Perea, G.; Araque, A. Astrocytes Potentiate Transmitter Release at Single Hippocampal Synapses. Science 2007, 317, 1083–1086. [Google Scholar] [CrossRef]
- Alcoreza, O.B.; Patel, D.C.; Tewari, B.P.; Sontheiner, H. Dysregulation of abient glutamate and glutamate receptors in epilepsy: An astrocytic perspective. Front. Neurol. 2021, 12, 652159. [Google Scholar] [CrossRef]
- Losi, G.; Mariotti, L.; Carmignoto, G. GABAergic interneuron to astrocyte signalling: A neglected form of cell communication in the brain. Philos. Trans. R. Soc. Lond. B Bio. Sci. 2014, 369, 20130609. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Jiang, L.; Goldman, S.A.; Nedergaard, M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1998, 1, 683–692. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratu, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces AB-induced neuroinflammation and promotes hippocampal neurogenesis through PPARү involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef] [PubMed]
- Atalay, S.; Jarocka-Karpowicz, I.; Skrzydlewska, E. Antioxidative and Anti-Inflammatory Properties of Cannabidiol. Antioxidants 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Pellati, F.; Borgonetti, V.; Brighenti, V.; Biagi, M.; Benvenuti, S.; Corsi, L. Cannabis sativa L. and nonpsychoacitve cannabinoids: Their chemistry and role against oxidative stress, inflammation and cancer. Biomed Res. Int. 2018, 2018, 1691428. [Google Scholar] [CrossRef]
- Kozela, E.; Pietr, M.; Juknat, A.; Rimmerman, N.; Levy, R.; Vogel, Z. Cannabinoids delta(9)-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-kappaB and interferon-beta/STAT proinflammatory pathways in BV-2 microglial cells. J. Biol. Chem. 2010, 285, 1616–1626. [Google Scholar] [CrossRef]
- Schiavon, A.P.; Soares, L.M.; Bonato, J.M.; Milani, H.; Guimarães, F.S.; Weffort de Oliveira, R.M. Protective effects of cannabidiol against hippocampal cell death and cognitive impairment induced by bilateral common carotid artery occlusion in mice. Neurotox. Res. 2014, 26, 307–316. [Google Scholar] [CrossRef]
- Friedman, L.K.; Wongvravit, J.P. Anticonvulsant and neuroprotective effects of cannabidiol during the juvenile period. J. Neuropathol. Exp. Neurol. 2018, 77, 904–919. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.A.; Whalley, B.J. The proposed mechanism of action of CBD in epilepsy. Epileptic Disord. 2020, 22, 10–15. [Google Scholar]
- Zavala-Tecuapetla, C.; Luna-Munguia, H.; López-Meraz, M.L.; Cuellar-Herrera, M. Advances and Challenges of Cannabidiol as an Anti-Seizure Strategy: Preclinical Evidence. Int. J. Mol. Sci. 2022, 23, 16181. [Google Scholar] [CrossRef] [PubMed]
- Premoli, M.; Aria, F.; Bonini, S.A.; Maccarinelli, G.; Gianoncelli, A.; Pina, S.D.; Tambaro, S.; Memo, M.; Mastinu, A. Cannabidiol: Recent advances and new insights for neuropsychiatric disorders treatment. Life Sci. 2019, 224, 120–127. [Google Scholar] [CrossRef]
- Pretzsch, C.M.; Freyberg, J.; Voinescu, B.; Lythgoe, D.; Horder, J.; Mendez, M.A.; Wichers, R.; Ajram, L.; Ivin, G.; Heasman, M.; et al. Effects of cannabidiol on brain excitation and inhibition systems; a randomised placebo-controlled single dose trial during magnetic resonance spectroscopy in adults with and without autism spectrum disorder. Neuropsychopharmacology 2019, 44, 1398–1405. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjogren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Guggenhuber, S.; Monory, K.; Lutz, B.; Klugmann, M. AAV vector-mediated overexpression of CB1 cannabinoid receptor in pyramidal neurons of the hippocampus protects against seizure-induced excitoxicity. PLoS ONE 2010, 5, e15707. [Google Scholar] [CrossRef]
- Ruehle, S.; Remmers, F.; Romo-Parra, H.; Massa, F.; Wickert, M.; Wörtge, S.; Häring, M.; Kaiser, N.; Marsicano, G.; Pape, H.C.; et al. Cannabinoid CB1 receptor in dorsal telencephalic glutamatergic neurons: Distinctive sufficiency for hippocampus-dependent and amygdala-dependent synaptic and behavioral functions. J. Neurosci. 2013, 33, 10264–10277. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, Y.; Kano, M. Endocannabinoid-Mediated Control of Neural Circuit Excitability and Epileptic Seizures. Front. Neural Circuits 2022, 15, 781113. [Google Scholar] [CrossRef]
- Devinsky, O.; Marsh, E.; Friedman, D.; Thiele, E.; Laux, L.; Sullivan, J.; Miller, I.; Flamini, R.; Wilfong, A.; Filloux, F.; et al. Cannabidiol in patients with treatment-resistant epilepsy: An open-label interventional trial. Lancet Neurol. 2016, 15, 270–278. [Google Scholar] [CrossRef]
- Devinsky, O.; Cross, J.H.; Laux, L.; Marsh, E.; Miller, I.; Nabbout, R.; Scheffer, I.E.; Thiele, E.A.; Stephen Wright, S.; Cannabidiol in Dravet Syndrome Study Group. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N. Engl. J. Med. 2017, 376, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Patel, A.D.; Cross, J.H.; Villanueva, V.; Wirrell, E.C.; Privitera, M.; Greenwood, S.M.; Roberts, C.; Checketts, D.; VanLandingham, K.E.; et al. Effect of cannabidiol on drop seizure in the Lennox-Gastaut syndrome. N. Engl. J. Med. 2018, 378, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.; Scheffer, I.E.; Gunning, B.; Sanchez-Carpintero, R.; Gil-Nagel, A.; Perry, M.S.; Saneto, R.P.; Checketts, D.; Dunayevich, E.; Knappertz, V.; et al. Dose-ranging effect of adjunctive oral cannabidiol vs placebo on convulsive seizure frequency in Dravet syndrome. JAMA Neurol. 2020, 77, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Thiele, E.A.; Bebin, E.M.; Bhathal, H.; Jansen, F.E.; Kotulska, K.; Lawson, J.A.; O’Callaghan, F.J.; Wong, M.; Sahebkar, F.; Checketts, D.; et al. Add-on cannabidiol treatment for drug-resistant seizures in tuberous sclerosis complex: A placebo-controlled randomized clinical trial. JAMA Neurol. 2021, 78, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Grunelli, V.; Forda, S.; Kelly, J.S. The reversal potential of excitatory amino acid action on granule cells of the rat dentate gyrus. J. Physiol. 1984, 351, 327–342. [Google Scholar] [CrossRef]
- Duchen, M.R.; Burton, N.R.; Biscore, T.J. An intracellular study of the interaction of N-methyl-D-aspartate with ketamine in the mouse hippocampal slice. Brain Res. 1985, 342, 149–153. [Google Scholar] [CrossRef]
- Peet, M.J.; Gregersen, H.; Mclennan, H. 2-Amino-5-phosphonovalerate and CO2+ selectively block depolarization and burst firing of rat hippocampal CA1 pyramidal neurons by N-methyl-D-aspartate. Neuroscience 1986, 17, 635–641. [Google Scholar] [CrossRef]
- McDonough, J.H., Jr.; Shih, T.M. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci. Biobehav. Rev. 1997, 21, 559–579. [Google Scholar] [CrossRef]
- Dorandeu, F.; Barbier, L.; Dhote, F.; Testylier, G.; Carpentier, P. Ketamine combinations for the field treatment of soman-induced self-sustaining status epilepticus. Review of current data and perspectives. Chem. Biol. Interact. 2013, 203, 154–159. [Google Scholar] [CrossRef]
- Dubey, V.; Dey, S.; Dixit, A.B.; Tripathi, M.; Chandra, P.S.; Banerjee, J. Differential glutamate receptor expression and function in the hippocampus, anterior temporal lobe and neocortex in a pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 2022, 347, 113916. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.; Yong, X.L.H.; Roche, K.W.; Anggono, V. Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J. Neurochem. 2020, 154, 121–143. [Google Scholar] [CrossRef] [PubMed]
- Martel, M.A.; Ryan, T.J.; Bell, K.F.S.; Fowler, J.H.; McMahon, A.; Al-Mubarak, B.; Komiyama, N.H.; Horsburgh, K.; Kind, P.C.; Grant, S.G.N.; et al. The subtype of GluN2 C-terminal domain determines the response to excito_toxic insults. Neuron 2012, 74, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Bellone, C.; Nicoll, R.A. Rapid bidirectional switching of synap_tic NMDA receptors. Neuron 2007, 55, 779–785. [Google Scholar] [CrossRef]
- Ryan, T.J.; Kopanitsa, M.V.; Indersmitten, T.; Nithianantharajah, J.; Afinowi, N.O.; Pettit, C.; Stanford, L.E.; Sprengel, R.; Saksida, L.M.; Bussey, T.J.; et al. Evolution of GluN2A/B cytoplasmic domains diversified vertebrate synaptic plasticity and behavior. Nat. Neurosci. 2013, 16, 25–32. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by per_turbing NMDA receptor- PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau–a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef]
- Ballarin, B.; Tymianski, M. Discovery and development of NA-1 for the treatment of acute ischemic stroke. Acta Pharmacol. Sin. 2018, 39, 661–668. [Google Scholar] [CrossRef]
- Hill, M.D.; Martin, R.H.; Mikulis, D.; Wong, J.H.; Silver, F.L.; terBrugge, K.G.; Milot, G.; Clark, W.M.; Macdonald, R.L.; Kelly, M.E.; et al. Safety and efficacy of NA-1 in pa_tients with iatrogenic stroke after endovascular aneurysm repair (ENACT): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012, 11, 942–950. [Google Scholar] [CrossRef]
- Pagano, J.; Giona, F.; Beretta, S.; Verpelli, C.; Sala, C. N-methyl-d-aspartate receptor function in neuronal and synaptic development and signaling. Curr. Opin. Pharmacol. 2021, 56, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Wasterlain, C.G.; Chen, J.W. Mechanistic and pharmacologic aspects of status epilepticus and its treatment with new antiepileptic drugs. Epilepsia 2008, 49, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.E.; Liu, H.; Niquet, J.; Wasterlain, C.G. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol. Dis. 2013, 54, 225–238. [Google Scholar] [CrossRef]
- Kärkkäinen, O.; Kupila, J.; Häkkinen, M.; Laukkanen, V.; Tupala, E.; Kautiainen, H.; Tiihonen, J.; Storvik, M. AMPA receptors in post-mortem brains of Cloninger type 1 and 2 alcoholics: A whole-hemisphere autoradiography study. Psychiatry Res. 2013, 214, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Fukaya, M.; Hashimoto, K.; Yamasaki, M.; Tsujita, M.; Itakura, M.; Abe, M.; Natsume, R.; Takahashi, M.; Kano, M.; et al. TARPs gamma-2 and gamma-7 are essential for AMPA receptor expression in the cerebellum. Eur. J. Neurosci. 2010, 31, 2204–2220. [Google Scholar] [CrossRef]
- Kimura, M.; Sawada, K.; Miyagawa, T.; Kuwada, M.; Katayama, K.; Nishizawa, Y. Role of glutamate receptors and voltage-dependent calcium and sodium channels in the extracellular glutamate/aspartate accumulation and subsequent neuronal injury induced by oxygen/glucose deprivation in cultured hippocampal neurons. J. Pharmacol. Exp. Ther. 1998, 285, 178–185. [Google Scholar]
- Lamanauskas, N.; Nistril, A. Riluzole blockes persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motorneurons in vitro. Eur. J. Neurosci. 2008, 27, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Falcon-Moya, R.; Sihra, T.S.; Rodríguez-Moreno, A. Kainate receptors: Role in epilepsy. Front. Mol. Neurosci. 2018, 11, 217. [Google Scholar] [CrossRef]
- Negrete-Díaz, J.V.; Falcón-Moya, R.; Rodríguez-Moreno, A. Kainate receptors: From synaptic activity to disease. FEBS J. 2022, 289, 5074–5088. [Google Scholar] [CrossRef]
- Rodrıguez-Moreno, A.; López-García, J.C.; Lerma, J. Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc. Natl. Acad. Sci. USA 2000, 97, 1293–1298. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Crepel, V.; Represa, A. Seizures beget seizures in temporal lobe epilepsies: The boomerang effects of newly formed aberrant kainatergic synapses. Epilepsy Curr. 2008, 8, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.F.; Aroniadou-Anderjaska, V.; Li, H. The physiological role of kainate receptors in the amygdala. Mol. Neurobiol. 2004, 30, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Mazarati, A.M.; Wasterlain, C.G. N-methyl-D-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neurosci. Lett. 1999, 265, 187–190. [Google Scholar] [CrossRef]
- Borris, D.J.; Bertram, E.H.; Kapur, J. Ketamine controls prolonged status epilepticus. Epilepsy Res. 2000, 42, 117–122. [Google Scholar] [CrossRef]
- Dorandeu, F.; Dhote, F.; Barbier, L.; Baccus, B.; Testylier, G. Treatment of status epilepticus with ketamine, are we there yet? CNS Neurosci. Ther. 2013, 19, 411–427. [Google Scholar] [CrossRef]
- Ryley Parrish, R.; Albertson, A.J.; Buckingham, S.C.; Hablitz, J.J.; Mascia, K.L.; Davis Haselden, W.; Lubin, F.D. Status epilepticus triggers early and late alterations in brain-derived neurotrophic factor and NMDA glutamate receptor Grin2b DNA methylation levels in the hippocampus. Neuroscience 2013, 248, 602–619. [Google Scholar] [CrossRef]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific micro-RNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef]
- Jimenez-Mateos, E.M.; Engel, T.; Merino-Serrais, P.; McKiernan, R.C.; Tanaka, K.; Mouri, G.; Sano, T.; O’Tuathaigh, C.; Waddington, J.L.; Prenter, S.; et al. Silencing microRNA-134 produces neuroprotective and prolonged seizure suppressive effects. Nat. Med. 2012, 18, 1087–1094. [Google Scholar] [CrossRef]
- Ohba, C.; Shiina, M.; Tohyama, J.; Haginoya, K.; Lerman-Sagie, T.; Okamoto, N.; Blumkin, L.; Lev, D.; Mukaida, S.; Nozaki, F.; et al. GRIN1 mutations cause encephalopathy with infantile-onset epilepsy, and hyperkinetic and stereotyped movement disorders. Epilepsia 2015, 56, 841–848. [Google Scholar] [CrossRef]
- Lemke, J.R.; Hendricks, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; Pepler, A.; Steiner, I.; Hörtnagel, K.; et al. GRIN2B mutation in West syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef] [PubMed]
- XiangWei, W.S.; Kannan, V.; Xu, Y.C.; Kosobucki, G.J.; Anthony, J.; Schulien, A.J.; Kusumoto, H.; Moufawad El Achkar, C.; Bhattacharya, S.; Lesca, G.; et al. Heterogeneous clinical and functional features of GRIN2D-related developmental and epileptic encephalopathy. Brain 2019, 142, 3009–3027. [Google Scholar] [CrossRef]
- Lemke, J.R.; Lal, D.; Reinthaler, E.M.; Steiner, I.; Nothnagel, M.; Alber, M.; Geider, K.; Laube, B.; Schwake, M.; Finsterwalder, K.; et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 2013, 45, 1067–1072. [Google Scholar] [CrossRef]
- Xu, X.X.; Liu, X.R.; Fan, C.Y.; Jin-Xing Lai, J.X.; Shi, Y.W.; Yang, W.; Su, T.; Xu, J.Y.; Luo, J.H.; Liao, W.P. Functional Investigation of a GRIN2A Variant Associated with Rolandic Epilepsy. Neurosci. Bull. 2018, 34, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhou, Y.; Hu, X.; Gu, Q.; Wu, X.; Cao, M.; Ke, K.; Liu, C. Reduced numbers of cortical GABA-immunoreactive neurons in the chronic D-galactose treatment model of brain aging. Neurosci. Lett. 2013, 549, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Megías, M.; Emri, Z.; Freund, T.F.; Gulyás, A.I. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 2001, 102, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Gataullina, S.; Bienvenu, T.; Nabbout, R.; Huberfeld, G.; Dulac, O. Gene mutations in paediatric epilepsies cause NMDA-pathy, and phasic and tonic GABA-pathy. Dev. Med. Child Neurol. 2019, 61, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory and brain aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [CrossRef]
- Soto, D.; Altafaj, X.; Sindreu, C.; Bayés, A. Glutamate receptor mutations in psychiatric and neurodevelopmental disorders. Commun. Integr. Biol. 2014, 7, e27887. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; Valence, S.; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019, 10, 3094. [Google Scholar] [CrossRef]
- Jia, Z.; Agopyan, N.; Miu, P.; Xiong, Z.; Henderson, J.; Gerlai, R.; Taverna, F.A.; Velumian, A.; MacDonald, J.; Carlen, P.; et al. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 1996, 17, 945–956. [Google Scholar] [CrossRef]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.S.; Lai, M.C.; Huang, H.I.; Wu, S.N.; Huang, C.W. Immunity, Ion Channels and Epilepsy. Int. J. Mol. Sci. 2022, 23, 6446. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S.; Sander, J.W.; Upton, D.; Thompson, P.J.; Patsalos, P.N.; Hirt, D.; Emre, M.; Lowe, D.; Duncan, J.S. The excitatory amino acid antagonist D-CPP-ene (SDZ EAA-494) in patients with epilepsy. Epilepsy Res. 1993, 16, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Manto, M.; Laute, M.A.; Aguera, M.; Rogemond, V.; Pandolfo, M.; Honnorat, J. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases. Ann. Neurol. 2007, 61, 544–551. [Google Scholar] [CrossRef]
- Manto, M.; Dalmau, J.; Didelot, A.; Rogemond, V.; Honnorat, J. In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: Further evidence of synaptic glutamatergic dysfunction. Orphanet J. Rare Dis. 2010, 5, 31. [Google Scholar] [CrossRef]
- Wolf, J.A.; Moyer, J.T.; Lazarewicz, M.T.; Contreras, D.; Benoit-Marand, M.; O’Donnell, P.; Finkel, L.H. NMDA/AMPA ratio impacts state transitions and entrainment to oscillations in a computational model of the nucleus accumbens medium spiny projection neuron. J. Neurosci. 2005, 25, 9080–9095. [Google Scholar] [CrossRef]
- Corona, J.C.; Tapia, R. AMPA receptor activation, but not the accumulation of endogeneous extracellular glutamate, induces paralysis and motor neuron death in rat spinal cord in vivo. J. Neurochem. 2004, 89, 988–997. [Google Scholar] [CrossRef]
- Seki, M.; Suzuki, S.; Iizuka, T.; Shimizu, T.; Nihei, Y.; Suzuki, N.; Dalmau, J. Neurological response to early removal of ovarian teratoma in antiNMDA-R encephalitis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 324–326. [Google Scholar] [CrossRef]
- Faught, E.; Wilder, B.J.; Ramsay, R.E.; Reife, R.A.; Kramer, L.D.; Pledger, G.W.; Karim, R.M. Topiramate placebo-controlled dose-ranging trial in refractory partial epilepsy using 200-, 400-, 600-mg dosages. Neurology 1996, 46, 1684–1690. [Google Scholar] [CrossRef]
- Privitera, M.; Fincham, R.; Penry, J.; Reife, R.; Kramer, L.; Pledger, G.; Karim, R. Topiramate placebo-controlled dose-ranging trial in refractory partial epilepsy using 600-, 800-, and 1,000-mg daily dosages. Neurology 1996, 46, 1678–1683. [Google Scholar] [CrossRef]
- Severt, L.; Coulter, D.A.; Sombati, S. Topiramate selectively blocks kainite currentts in cultured hippocampal neurons. Epilepsia 1995, 36, s38. [Google Scholar]
- Gibbs, J.W.; Sombati, S.; DeLorenzo, R.I.; Coulter, D.A. Cellular actions of topiramate: Blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia 2000, 41, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Velumian, A.A.; Jones, O.T.; Carlen, P.L. Modulation of high-voltage activated calcium channels in dentate granule cells by topiramate. Epilepsia 2000, 41, 52–60. [Google Scholar] [CrossRef]
- Rho, J.M.; Donevan, S.D.; Rogawski, M.A. Mechanism of action of the anticonvulsant felbamate: Opposing effects on N-methyl-D-aspartate and g-aminobutyric acid A receptors. Ann. Neuro. 1994, 35, 229–234. [Google Scholar] [CrossRef] [PubMed]
- McCabe, R.T.; Wasterlain, C.G.; Kucharczyk, N.; Sofia, R.D.; Vogel, J.R. Evidence for anticonvulsant and neuroprotectant action of felbamate mediated by strychnine-insensitive glycine receptors. J. Pharmacol. Exp. Ther. 1993, 264, 1248–1252. [Google Scholar]
- Dooley, D.J.; Taylor, C.P.; Donevan, S.; Feltner, D. Ca2+ channel α2δ ligands: Novel modulators of neurotransmission. Trends Pharmacol. Sci. 2007, 28, 75–82. [Google Scholar] [CrossRef]
- Wang, S.J.; Huang, C.C.; Hsu, K.S.; Tsai, J.J.; Gean, P.W. Inhibition of N-type calcium currents by lamotrigine in rat amygdalar neurones. Neuroreport 1996, 7, 3037–3040. [Google Scholar] [CrossRef]
- Dibue, M.; Kamp, M.A.; Alpdogan, S.; Tevoufouet, E.E.; Neiss, W.F.; Hescheler, J.; Schneider, T. Cav2.3 (R-type) calcium channels are critical for mediating anticonvulsive and neuroprotective properties of lamotrigine in vivo. Epilepsia 2013, 54, 1542–1550. [Google Scholar] [CrossRef]
- Prakriya, M.; Mennerick, S. Selective Depression of Low–Release Probability Excitatory Synapses by Sodium Channel Blockers. Neuron 2000, 26, 671–682. [Google Scholar] [CrossRef]
- Debono, M.-W.; Le Guern, J.; Canton, T.; Doble, A.; Pradier, L. Inhibition by riluzole of electrophysiological responses mediated by rat kainate and NMDA receptors expressed in Xenopus oocytes. Eur. J. Pharmacol. 1993, 235, 283–289. [Google Scholar] [CrossRef]
- Fumagalli, E.; Funicello, M.; Rauen, T.; Gobbi, M.; Mennini, T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur. J. Pharmacol. 2008, 578, 171–176. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, D.S.; Kwak, S.E.; Choi, H.C.; Song, H.K.; Choi, S.Y.; Kwon, O.S.; Kim, Y.I.; Kang, T.C. Anti-glutamatergic effect of riluzole: Comparison with valproic acid. Neuroscience 2007, 147, 136–145. [Google Scholar] [CrossRef]
- Borowicz, K.K.; Sekowski, A.; Drelewska, E.; Czuczwar, S. Rilozole enhances the antiseizure action of conventional antiepileptic drugs against pentetrazole-induced convulsions in mice. Pol. J. Pharmacol. 2004, 56, 187–193. [Google Scholar] [PubMed]
- Du, J.; Vegh, V.; Reutens, D.C. Persistent sodium current blockers can suppress seizures caused by loss of low-threshold D-type potassium currents: Predictions from an in silico study of Kv 1 channel disorder. Epilepsia Open 2020, 5, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Pellegrini, J.W.; Aggarwal, S.K.; Lei, S.Z.; Warach, S.; Jensen, F.E.; Lipton, S.A. Open-channel block of N-methyl-Daspartate (NMDA) responses by memantine: Therapeutic advantage against NMDA receptor-mediated neurotoxicity. J. Neurosci. 1992, 12, 4427–4436. [Google Scholar] [CrossRef]
- Zaitsev, A.V.; Kim, K.; Vasilev, D.S.; Lukomskaya, N.Y.; Lavrentyeva, V.V.; Tumanova, N.L.; Zhuravin, I.A.; Magazanik, L.G. N-methyl-D-aspartate receptor channel blockers prevent pentylenetetrazole-induced convulsions and morphological changes in rat brain neurons. J. Neurosci. Res. 2015, 93, 454–465. [Google Scholar] [CrossRef]
- Sun, Y.; Dhamne, S.C.; Carretero-Guillén, A.; Salvador, R.; Goldenberg, M.C.; Godlewski, B.R.; Pascual-Leone, A.; Madsen, J.R.; Stone, S.S.D.; Ruffini, G.; et al. Drug-Responsive Inhomogeneous Cortical Modulation by Direct Current Stimulation. Ann. Neurol. 2020, 88, 489–502. [Google Scholar] [CrossRef]
- Wada, Y.; Hasegawa, H.; Nakamura, M.; Yamaguchi, N. The NMDA receptor antagonist MK-801 has a dissociative effect on seizure activity of hippocampal-kindled cats. Pharmacol. Biochem. Behav. 1992, 43, 1269–1272. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Jevtovic-Todorovic, V.; Selke, G.; Melson, A.K.; Hershey, T.; Craft, S.; Olney, J.W. Ketamine-Induced NMDA Receptor Hypofunction as a Model of Memory Impairment and Psychosis. Neuropsychopharmacology 1999, 20, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Goda, Y.; Stevens, C.F. Synaptic plasticity: The basis of particular types of learning. Curr. Biol. 1996, 6, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, M.A. AMPA receptors as a molecular target in epilepsy therapy. Acta Neurol. Scand. Suppl. 2013, 197, 9–18. [Google Scholar] [CrossRef]
- Hunt, D.L.; Castillo, P.E. Synaptic plasticity of NMDA receptors: Mechanisms and functional implications. Curr. Opin. Neurobiol. 2012, 22, 496–508. [Google Scholar] [CrossRef]
- Zarnowski, T.; Kleinrok, Z.; Turski, W.A.; Czuczwar, S.J. The competitive NMDA antagonist, D-CPP-ene, potentiates the anticonvulsant activity of conventional antiepileptics against maximal electroshock-induced mice. Neuropharmacology 1994, 33, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Dong, J.; Shen, K.; Bai, Y.; Zhang, Y.; Lv, X.; Chao, J.; Yao, H. NMDA receptor NR2B subunits contribute to PTZ-kindling-induced hippocampal astrocytosis and oxidative stress. Brain Res. Bull. 2015, 114, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Hönack, D. Anticonvulsant and behavioral effects of two novel competitive N-methyl-D-aspartic acid receptor antagonists, CGP 37849 and CGP 39551, in the kindling model of epilepsy. Comparison with MK-801 and carbamazepine. J. Pharmacol. Exp. Ther. 1991, 256, 432–440. [Google Scholar] [PubMed]
- Dziki, M.; Hönack, D.; Löscher, W. Kindled rats are more sensitive than non-kindled rats to the behavioural effects of combined treatment with MK-801 and valproate. Eur. J. Pharmacol. 1992, 222, 273–278. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Kurzman, P.S.; Yamaguchi, S.I.; Li, H. Role of AMPA and GluR5 kainate receptors in the development and expression of amygdala kindling in the mouse. Neuropharmacology 2001, 40, 28–35. [Google Scholar] [CrossRef]
- Löscher, W.; Rundfeldt, C.; Hönack, D. Low doses of NMDA receptor antagonists synergistically increase the anticonvulsant effect of the AMPA receptor antagonist NBQX in the kindling model of epilepsy. Eur. J. Neurosci. 1993, 5, 1545–1550. [Google Scholar] [CrossRef]
- Graebenitz, S.; Kedo, O.; Speckmann, E.J.; Gorji, A.; Panneck, H.; Hans, V.; Palomero-Gallagher, N.; Schleicher, A.; Zilles, K.; Pape, H.C. Interictal-like network activity and receptor expression in the epileptic human lateral amygdala. Brain 2011, 134, 2929–2947. [Google Scholar] [CrossRef]
- Hanada, T.; Ido, K.; Kosasa, T. Effect of perampanel, a novel AMPA antagonist, on benzodiazepine-resistant status epilepticus in a lithium-pilocarpine rat model. Pharmacol. Res. Perspect. 2014, 2, e00063. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, T.H.; Qashu, F.; Apland, J.P.; Aroniadou-Anderjaska, V.; Souza, A.P.; Braga, M.F.M. The GluK1 (GluR5) Kainate/{alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonist LY293558 reduces soman-induced seizures and neuropathology. J. Pharmacol. Exp. Ther. 2011, 336, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Chavda, V. Pre- and post-exposure talampanel (GYKI 53773) against kainic acid seizures in neonatal rats. Pharmacol. Rep. PR. 2016, 68, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, B.; Stott, J.J.; Joelle Donofrio, J.; Rogawski, M.A. Treatment of early and late kainic acid-induced status epilepticus with the noncompetitive AMPA receptor antagonist GYKI 52466. Epilepsia 2010, 51, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.; Sekar, S.; Wei, Z.; Moien-Afshari, F.; Taghibiglou, C. Perampanel but Not Amantadine Prevents Behavioral Alterations and Epileptogenesis in Pilocarpine Rat Model of Status Epilepticus. Mol. Neurobiol. 2019, 56, 2508–2523. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Ido, K.; Osada, Y.; Kotani, S.; Tamaoka, A.; Hanada, T. The neuroprotective effect of perampanel in lithium-pilocarpine rat seizure model. Epilepsy Res. 2017, 137, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Rheims, S.; Ryvlin, P. Profile of perampanel and its potential in the treatment of partial onset seizures. Neuropsychiatr. Dis. Treat. 2013, 9, 629–637. [Google Scholar] [CrossRef]
- Wu, T.; Ido, K.; Ohgoh, M.; Hanada, T. Mode of seizure inhibition by sodium channel blockers, an SV2A ligand, and an AMPA receptor antagonist in a rat amygdala kindling model. Epilepsy Res. 2019, 154, 42–49. [Google Scholar] [CrossRef]
- Lai, M.C.; Tzeng, R.C.; Huang, C.W.; Wu, S.N. The Novel Direct Modulatory Effects of Perampanel, an Antagonist of AMPA Receptors, on Voltage-Gated Sodium and M-type Potassium Currents. Biomolecules 2019, 9, 638. [Google Scholar] [CrossRef]
- Krauss, G.L.; Serratosa, J.M.; Villanueva, V.; Endziniene, M.; Hong, Z.; French, J.; Yang, H.; Squillacote, D.; Edwards, H.B.; Zhu, J.; et al. Randomized phase III study 306: Adjunctive perampanel for refractory partial-onset seizures. Neurology 2012, 78, 1408–1415. [Google Scholar] [CrossRef]
- Nishida, T.; Lee, S.K.; Inoue, Y.; Saeki, K.; Ishikawa, K.; Kaneko, S. Adjunctive perampanel in partial-onset seizures: Asia-Pacific, randomized phase III study. Acta Neurol. Scand. 2018, 137, 392–399. [Google Scholar] [CrossRef]
- French, J.A.; Krauss, G.L.; Wechsler, R.T.; Wang, X.F.; DiVentura, B.; Brandt, C.; Trinka, E.J.; O’Brien, T.; Laurenza, A.; Patten, A.; et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy A randomized trial. Neurology 2015, 85, 950–957. [Google Scholar] [CrossRef]
- Davies, J.; Evans, R.H.; Herrling, P.L.; Jones, A.W.; Olverman, H.J.; Pook, P.; Watkins, J.C. CPP, a new potent and selective NMDA antagonist. Depression of central neuron responses, affinity for [3H]D-AP5 binding sites on brain membranes and anticonvulsant activity. Brain Res. 1986, 382, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, J.D.; Hartvig, P.; Karlsten, R.; Gordh, T.; Halldin, M. CSF and plasma pharmacokinetics of the NMDA receptor antagonist CPP after intrathecal, extradural and i.v. administration in anaesthetized pigs. Br. J. Anaesth. 1995, 74, 193–200. [Google Scholar] [CrossRef]
- Rogawski, M.A. Therapeutic potential of excitatory amino acid antagonists: Channel blockers and 2,3-benzodiazepines. Trends Pharmacol. Sci. 1993, 14, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.; Williamson, J.; Bertram, E.H.; Kapur, J. A comparison of three NMDA receptor antagonists in the treatment of prolonged status epilepticus. Epilepsy Res. 2004, 59, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, H.P.; Yeh, J.L.; Kapur, J. Status epilepticus increases intracellular accumulation of GABAA receptors. J. Neurosci. 2005, 25, 5511–5520. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, N.; Foreman, B.; Judd, L.M.; Brenton, J.N.; Nathan, B.R.; McCoy, B.M.; Al-Otaibi, A.; Kilbride, R.; Fernández, I.S.; Mendoza, L.; et al. Intravenous ketamine for the treatment of refractory status epilepticus: A retrospective multicenter study. Epilepsia 2013, 54, 1498–1503. [Google Scholar] [CrossRef]
- Kramer, A.H. Early ketamine to treat refractory status epilepticus. Neurocrit. Care 2012, 16, 299–305. [Google Scholar] [CrossRef]
- Synowiec, A.S.; Singh, D.S.; Yenugadhati, V.; Valeriano, J.P.; Schramke, C.J.; Kelly, K.M. Ketamine use in the treatment of refractory status epilepticus. Epilepsy Res. 2013, 105, 183–188. [Google Scholar] [CrossRef]
- Sabharwal, V.; Ramsay, E.; Martinez, R.; Shumate, R.; Khan, F.; Dave, H.; Iwuchukwu, I.; McGrade, H. Propofol-ketamine combination therapy for effective control of super-refractory status epilepticus. Epilepsy Behav. 2015, 52, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Rosati, A.; L’Erario, M.; Ilvento, L.; Cecchi, C.; Pisano, T.; Mirabile, L.; Guerrini, R. Efficacy and safety of ketamine in refractory status epilepticus in children. Neurology 2012, 79, 2355–2358. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Rosado, B.M.; de Araujo Furtado, M.; Kundrick, E.R.; Walker, K.A.; Stone, M.F.; Schultz, C.R.; Nguyen, D.A.; Lumley, L.A. Ketamine as adjunct to midazolam treatment following soman-induced status epilepticus reduces seizure severity, epileptogenesis, and brain pathology in plasma carboxylesterase knockout mice. Epilepsy Behav. 2020, 111, 107229. [Google Scholar] [CrossRef]
- Santoro, J.D.; Filippakis, A.; Chitnis, T. Ketamine use in refractory status epilepticus associated with anti-NMDA receptor antibody encephalitis. Epilepsy Behav. Rep. 2019, 12, 100326. [Google Scholar] [CrossRef]
- Alkhachroum, A.; Der-Nigoghossian, C.A.; Mathews, E.; Massad, N.; Letchinger, R.; Doyle, K.; Chiu, W.T.; Kromm, J.; Rubinos, C.; Velazquez, A.; et al. Ketamine to treat super-refractory epilepticus. Neurology 2020, 95, e2286–e2294. [Google Scholar] [CrossRef] [PubMed]
- Liebe, J.; Li, S.; Lord, A.; Colic, L.; Kuause, A.L.; Batra, A.; Kretzschmar, M.A.; Sweeney-Reed, C.M.; Behnisch, G.; Schott, B.H.; et al. Factors influencing the cardiovascular response to subanesthetic ketamine: A randomized, placebo-controlled trial. Int. J. Neuropsychopharmacol. 2017, 20, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Perks, A.; Cheema, S.; Mohanraj, R. Anaesthesia and epilepsy. Br. J. Anaesth. 2012, 108, 562–571. [Google Scholar] [CrossRef]
- Hallak, M. Effect of parenteral magnesium sulfate administration on excitatory amino acid receptors in the rat brain. Magnes. Res. 1998, 11, 117–131. [Google Scholar]
- Visser, N.A.; Braun, K.P.J.; Leijten, F.S.S.; van Nieuwenhuizen, O.; Wokke, J.H.J.; van den Bergh, W.M. Magnesium treatment for patients with refractory status epilepticus due to POLG1-mutations. J. Neurol. 2011, 258, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.P.; Wang, X.; Dong, C.H.; Chen, C.H.; Zhao, W.; Zhao, R.Y. Three-week combination treatment with ACTH + magnesium sulfate versus ACTH monotherapy for infantile spasms: A 24-week, randomized, open-label, follow-up study in China. Clin. Ther. 2010, 32, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Euser, A.G.; Cipolla, M.J. Magnesium sulfate for the treatment of eclampsia: A brief review. Stroke 2009, 40, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.R.; Takahashi, D.K.; Thomson, K.E.; Wilcox, K.S. The expression of kainate receptor subunits in hippocampal astrocytes after experimentally induced status epilepticus. J. Neuropathol. Exp. Neurol. 2013, 72, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, M.B.; Smeal, R.M.; Takahashi, D.K.; Vargas, J.R.; Wilcox, K.S. Contributions of astrocytes to epileptogenesis following status epilepticus: Opportunities for preventive therapy? Neurochem. Int. 2013, 63, 660–669. [Google Scholar] [CrossRef]
- Faught, E. BGG492 (selurampanel), an AMPA/kainate receptor antagonist drug for epilepsy. Expert Opin. Investig. Drugs 2014, 23, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Elger, C.E.; Hong, S.B.; Brandt, C.; Mancione, L.; Han, J.; Strohmaier, C. BGG492 as an adjunctive treatment in patient with partial-onset seizure: A 12-week, randomized, double-blind, placebo-controlled, phase II dose-titration study with an open-label extension. Epilepsia 2017, 7, 1217–1226. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J. Metabolism and metabolomics of ketamine: A toxicological approach. Forensic Sci. Res. 2017, 2, 2–10. [Google Scholar] [CrossRef]
- Li, Y.; Jackson, K.A.; Slon, B.; Hardy, J.R.; Franco, M.; William, L.; Poon, P.; Coller, J.K.; Hutchinson, M.R.; Currow, D.C.; et al. CYP2B6*6 allele and age substantially reduce steady-state ketamine clearance in chronic pain patients: Impact on adverse effects. Br. J. Clin. Pharmacol. 2015, 80, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Mir, A.; Qahtani, M.; Bashir, S. GRIN2A-related severe epileptic encephalopathy treated with memantine: An example of precision medicine. J. Pediatr. Genet. 2020, 9, 252–257. [Google Scholar] [CrossRef]
- Pierson, T.M.; Yuan, H.; Marsh, E.D.; Fuentes-Fajardo, K.; Adams, D.R.; Markello, T.; Golas, G.; Simeonov, D.R.; Holloman, C.; Tankovic, A.; et al. GRAIN2A mutation and early- onset epileptic encephalopathy: Personalized therapy with memantine. Ann. Clin. Transl. Neurol. 2014, 1, 190–198. [Google Scholar] [CrossRef]
- Platzer, K.; Yuan, H.; Schütz, H.; Winschel, A.; Chen, W.; Hu, C.; Kusumoto, H.; Heyne, H.O.; Helbig, K.L.; Tang, S.; et al. GRAIN2B encephalopathy: Novel findings on phontype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 2017, 54, 460–470. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Medication | Mechanism | Efficacy | Remarks |
|---|---|---|---|
| Ketamine | Noncompetitive NMDA antagonism | -Treatment of SRSE in adults and children, with control rate ranging from 50% to as high as 90% | -Does not alter intracranial pressure or cerebral blood flow -Lack of adverse cardiopulmonary depression effect |
| Magnesium sulfate | NMDA antagonism | -Associated with an increased seizure threshold and reduced seizures in animal studies; effective add-on therapy in infantile spasm | -Intravenous magnesium sulphate is the standard management in eclamptic seizures. |
| Dizocilpine (MK-801) | Noncompetitive NMDA antagonism | -In SE animal models, MK-801 is superior to the competitive antagonist CPP and noncompetitive antagonist ifenprodil |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.-S.; Huang, T.-H.; Lai, M.-C.; Huang, C.-W. The Role of Glutamate Receptors in Epilepsy. Biomedicines 2023, 11, 783. https://doi.org/10.3390/biomedicines11030783
Chen T-S, Huang T-H, Lai M-C, Huang C-W. The Role of Glutamate Receptors in Epilepsy. Biomedicines. 2023; 11(3):783. https://doi.org/10.3390/biomedicines11030783
Chicago/Turabian StyleChen, Tsang-Shan, Tzu-Hsin Huang, Ming-Chi Lai, and Chin-Wei Huang. 2023. "The Role of Glutamate Receptors in Epilepsy" Biomedicines 11, no. 3: 783. https://doi.org/10.3390/biomedicines11030783
APA StyleChen, T.-S., Huang, T.-H., Lai, M.-C., & Huang, C.-W. (2023). The Role of Glutamate Receptors in Epilepsy. Biomedicines, 11(3), 783. https://doi.org/10.3390/biomedicines11030783