
The Role of COX-2 and PGE2 in the Regulation of Immunomodulation and Other Functions of Mesenchymal Stromal Cells

Abstract
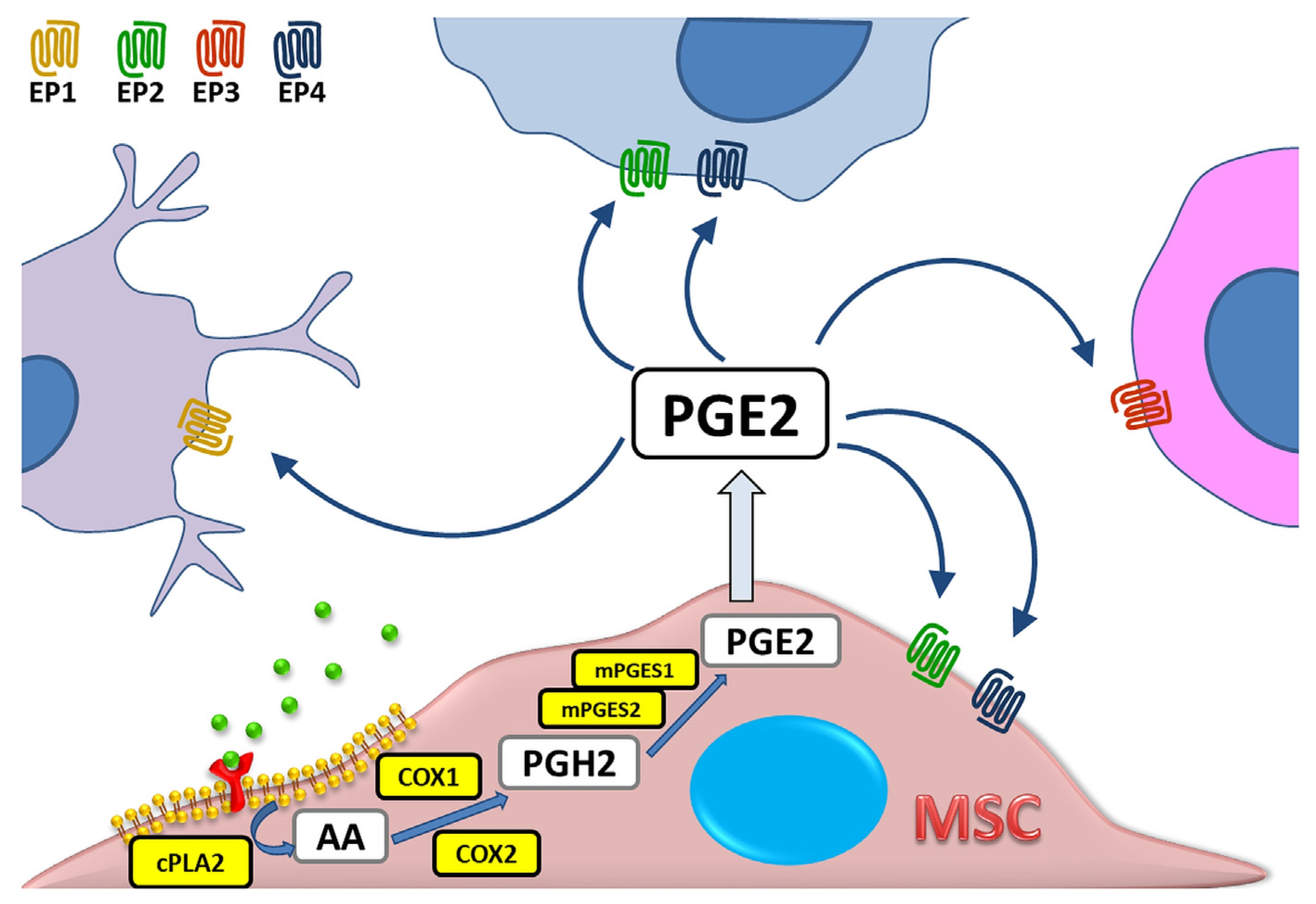
1. Introduction
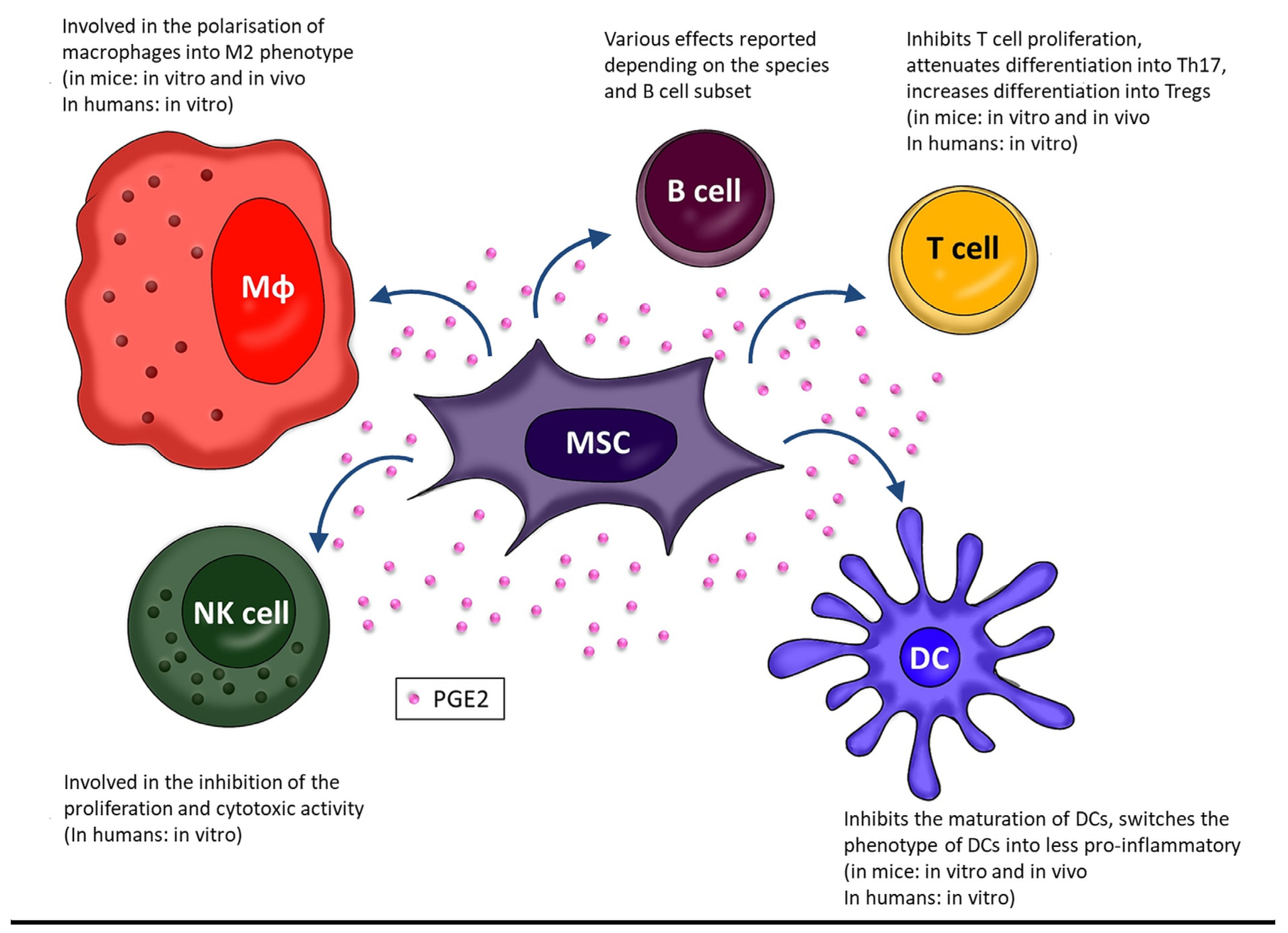
2. PGE2 as Part of Immunomodulatory Activity of MSCs
2.1. Macrophages (Mɸ)
2.2. Dendritic Cells
2.3. Natural Killer Cells
2.4. B Cells
2.5. T Cells

{kind=link}
{kind=link}
| Target Cell Type | Identified COX2/PGE2-Dependent Mechanism of MSC’ Activity on Target Cells | Species Used | MSC Type | Type of the Study | Refs |
|---|---|---|---|---|---|
| Macrophages (Mɸ) | ↑ secretion of IL-10 | Mice | BM-MSCs | In vivo | [33] |
| ↑ secretion of IL-10; ↓ secretion of TNF, IL-6, IL-10p70 | Mice | BM-MSCs | In vitro | [34] | |
| ↓ activation of NLRP3 inflammasome in macrophages from the liver with induced acute failure | Mice | BM-MSCs | In vivo | [38] | |
| ↑ number of M2 macrophages in injured myocardium of diabetic rats | Rats | AT-MSCs | In vivo | [39] | |
| ↓secretion of TNF in M1 macrophages | Human | BM-MSCs | In vitro | [29,40] | |
| Dendritic cells (DCs) | ↓ differentiation of monocytes into immature DCs (CD14neg, CD1apos) and mature DCs (CD80pos, CD86pos, CD83pos) | Human | BM-MSCs | In vitro | [43] |
| ↓ secretion of TNF and IL-12 | Human | BM-MSCs | In vitro | [44] | |
| ↓ production of osteopontin | Human | AT-MSCs | In vitro | [45] | |
| Induction of regulatory DCs with CD11cpos, MHCIIhigh, CD80low, CD86low, DEC205low phenotype | Mice | ND | In vitro, In vivo | [46] | |
| ↓ expression of CD40, ↓ secretion of TNF | Mice | AT-MSCs | In vitro | [47] | |
| Natural killer cells (NK cells) | ↓ proliferation and cytotoxic activity (synergistic action with IDO) | Human | BM-MSCs | In vitro | [50] |
| B cells | ↑antibody production (IgM, IgA, IgG); ↑ proliferation | Human | UC-MSCs | In vitro | [57] |
| ↓ production of IgE by LPS/IL-4 stimulated B cells isolated form mice with induced allergic conjunctivitis | Mice | BM-MSCs | In vivo | [58] | |
| ↓ maturation (% CD27pos/CD19pos) | Human | AT-MSCs | In vitro | [59] | |
| ↓ secretion of IL-10 by LPS-stimulated mice B cells | Mice | BM-MSCs | In vitro | [60] | |
| ↑ secretion of IL-10 by human CD23posCD43pos B regulatory subset | Human | BM-MSCs | In vitro | [61] | |
| T cells | ↓ activation and proliferation of T cells | Human | AT-MSCs WJ-MSCs | In vitro | [70] |
| ↓ T cells proliferation | Human | BM-MSCs | In vitro | [72] | |
| ↓ T cells proliferation by MSC exposed to apoptotic cells | Human | UC-MSCs | In vitro | [74] | |
| ↓ T cells proliferation by MSC spheroids | Human | BM-MSCs | In vitro | [75] | |
| induction of human CD4pos CD25High FoxP3pos T cells | Human | BM-MSCs | In vitro | [77] | |
| induction of IL-10 posIFN-γposCD4 pos regulatory T type 1 (T(R)1)-like cells | Pigs | BM-MSCs | In vitro | [79] | |
| ↓ inflammation by increasing FOXP3 Tregs in mice model of colitis | Mice | AT-MSCs | In vivo | [81] | |
| ↑ percentage of Treg in spleen and kidney in mice with ischemia-reperfusion acute kidney injury | Mice | BM-MSCs | In vitro | [82] | |
| ↑ secretion of CCL12 and CCL5 (↑chemoattraction of T cells towards MSCs), ↓ proliferation of Tc, ↑ production of Treg | Rats | ND | In vitro | [83] | |
| inhibition of follicular helper-like T cells | Human | UC-MSCs | In vitro | [85] | |
| ↓ Th17 differentiation | Mice | BM-MSCs | In vitro | [86] | |
| ↓ Th17 differentiation; ↓production of IL-17, IL-22, IFN-gamma, and TNF-alpha by fully differentiated Th17 | Human | BM-MSCs | In vitro | [87] |
3. The Role of PGE2 on Other Physiological Properties of MSCs
3.1. Supporting Hematopoiesis
3.2. Proliferation
3.3. Migration
3.4. Differentiation
4. Cancerogenesis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, W.L. The eicosanoids and their biochemical mechanisms of action. Biochem. J. 1989, 259, 315–324. [Google Scholar] [CrossRef]
- Leslie, C.C. Regulation of arachidonic acid availability for eicosanoid production. Biochem. Cell Biol. 2004, 82, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Huang, H.; Guo, Z.; Chang, Y.; Li, Z. Role of prostaglandin E2 in tissue repair and regeneration. Theranostics 2021, 11, 8836–8854. [Google Scholar] [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef]
- Berenbaum, F. Proinflammatory cytokines, prostaglandins, and the chondrocyte: Mechanisms of intracellular activation. Jt. Bone Spine 2000, 67, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Xiao, C.Y.; Yuhki, K.; Hara, A.; Fujino, T.; Kuriyama, S.; Yamada, T.; Takayama, K.; Takahata, O.; Karibe, H.; Taniguchi, T.; et al. Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation 2004, 109, 2462–2468. [Google Scholar] [CrossRef]
- Dantzer, R. Cytokine, sickness behavior, and depression. Neurol. Clin. 2006, 24, 441–460. [Google Scholar] [CrossRef]
- Vardeh, D.; Wang, D.; Costigan, M.; Lazarus, M.; Saper, C.B.; Woolf, C.J.; Fitzgerald, G.A.; Samad, T.A. COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J. Clin. Investig. 2009, 119, 287–294. [Google Scholar] [CrossRef]
- Giuliano, F.; Warner, T.D. Origins of prostaglandin E2: Involvements of cyclooxygenase (COX)-1 and COX-2 in human and rat systems. J. Pharm. Exp. Ther. 2002, 303, 1001–1006. [Google Scholar] [CrossRef]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef]
- Wu, Z.; Nakanishi, H. Phosphatidylserine-containing liposomes: Potential pharmacological interventions against inflammatory and immune diseases through the production of prostaglandin E(2) after uptake by myeloid derived phagocytes. Arch. Immunol. Ther. Exp. (Warsz) 2011, 59, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Montinari, M.R.; Minelli, S.; De Caterina, R. The first 3500years of aspirin history from its roots—A concise summary. Vasc. Pharm. 2019, 113, 1–8. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Gotoh, T.; Tomisato, W.; Mima, S.; Hoshino, T.; Hwang, H.J.; Takenaka, H.; Tsuchiya, T.; Mori, M.; Mizushima, T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004, 11, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharm. 2020, 180, 114147. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.Y. Role of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in Cancer Prevention and Cancer Promotion. Adv. Pharm. Sci. 2019, 2019, 3418975. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Greenbaum, A.; Hsu, Y.M.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Mallis, P.; Michalopoulos, E.; Chatzistamatiou, T.; Giokas, C.S. Interplay between mesenchymal stromal cells and immune system: Clinical applications in immune-related diseases. Explor. Immunol. 2021, 1, 112–139. [Google Scholar] [CrossRef]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed]
- Hoogduijn, M.J.; Verstegen, M.M.; Engela, A.U.; Korevaar, S.S.; Roemeling-van Rhijn, M.; Merino, A.; Franquesa, M.; de Jonge, J.; Ijzermans, J.N.; Weimar, W.; et al. No evidence for circulating mesenchymal stem cells in patients with organ injury. Stem Cells Dev. 2014, 23, 2328–2335. [Google Scholar] [CrossRef] [PubMed]
- Burdzinska, A.; Gala, K.; Paczek, L. Myogenic stem cells. Folia Histochem. Cytobiol. 2008, 46, 401–412. [Google Scholar] [CrossRef]
- Kota, D.J.; Prabhakara, K.S.; Toledano-Furman, N.; Bhattarai, D.; Chen, Q.; DiCarlo, B.; Smith, P.; Triolo, F.; Wenzel, P.L.; Cox, C.S., Jr.; et al. Prostaglandin E2 Indicates Therapeutic Efficacy of Mesenchymal Stem Cells in Experimental Traumatic Brain Injury. Stem Cells 2017, 35, 1416–1430. [Google Scholar] [CrossRef]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Nauta, A.J.; Fibbe, W.E. Immunomodulatory properties of mesenchymal stromal cells. Blood 2007, 110, 3499–3506. [Google Scholar] [CrossRef]
- Bouffi, C.; Bony, C.; Courties, G.; Jorgensen, C.; Noel, D. IL-6-dependent PGE2 secretion by mesenchymal stem cells inhibits local inflammation in experimental arthritis. PLoS ONE 2010, 5, e14247. [Google Scholar] [CrossRef]
- English, K.; Barry, F.P.; Field-Corbett, C.P.; Mahon, B.P. IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol. Lett. 2007, 110, 91–100. [Google Scholar] [CrossRef]
- Saldana, L.; Bensiamar, F.; Valles, G.; Mancebo, F.J.; Garcia-Rey, E.; Vilaboa, N. Immunoregulatory potential of mesenchymal stem cells following activation by macrophage-derived soluble factors. Stem. Cell Res. Ther. 2019, 10, 58. [Google Scholar] [CrossRef]
- Le Blanc, K.; Mougiakakos, D. Multipotent mesenchymal stromal cells and the innate immune system. Nat. Rev. Immunol. 2012, 12, 383–396. [Google Scholar] [CrossRef]
- Dymowska, M.; Aksamit, A.; Zielniok, K.; Kniotek, M.; Kaleta, B.; Roszczyk, A.; Zych, M.; Dabrowski, F.; Paczek, L.; Burdzinska, A. Interaction between Macrophages and Human Mesenchymal Stromal Cells Derived from Bone Marrow and Wharton’s Jelly-A Comparative Study. Pharmaceutics 2021, 13, 1822. [Google Scholar] [CrossRef] [PubMed]
- Francois, M.; Romieu-Mourez, R.; Li, M.; Galipeau, J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol. Ther. 2012, 20, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M.; et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Maggini, J.; Mirkin, G.; Bognanni, I.; Holmberg, J.; Piazzon, I.M.; Nepomnaschy, I.; Costa, H.; Canones, C.; Raiden, S.; Vermeulen, M.; et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS ONE 2010, 5, e9252. [Google Scholar] [CrossRef]
- Takayama, K.; Garcia-Cardena, G.; Sukhova, G.K.; Comander, J.; Gimbrone, M.A., Jr.; Libby, P. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J. Biol. Chem. 2002, 277, 44147–44154. [Google Scholar] [CrossRef]
- Luque-Campos, N.; Bustamante-Barrientos, F.A.; Pradenas, C.; Garcia, C.; Araya, M.J.; Bohaud, C.; Contreras-Lopez, R.; Elizondo-Vega, R.; Djouad, F.; Luz-Crawford, P.; et al. The Macrophage Response Is Driven by Mesenchymal Stem Cell-Mediated Metabolic Reprogramming. Front. Immunol. 2021, 12, 624746. [Google Scholar] [CrossRef]
- Rogers, L.M.; Anders, A.P.; Doster, R.S.; Gill, E.A.; Gnecco, J.S.; Holley, J.M.; Randis, T.M.; Ratner, A.J.; Gaddy, J.A.; Osteen, K.; et al. Decidual stromal cell-derived PGE(2) regulates macrophage responses to microbial threat. Am. J. Reprod. Immunol. 2018, 80, e13032. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Ding, H.; Shi, X.; Ren, H. Mesenchymal stem cell-secreted prostaglandin E2 ameliorates acute liver failure via attenuation of cell death and regulation of macrophage polarization. Stem. Cell Res. Ther. 2021, 12, 15. [Google Scholar] [CrossRef]
- Jin, L.; Deng, Z.; Zhang, J.; Yang, C.; Liu, J.; Han, W.; Ye, P.; Si, Y.; Chen, G. Mesenchymal stem cells promote type 2 macrophage polarization to ameliorate the myocardial injury caused by diabetic cardiomyopathy. J. Transl. Med. 2019, 17, 251. [Google Scholar] [CrossRef]
- Vasandan, A.B.; Jahnavi, S.; Shashank, C.; Prasad, P.; Kumar, A.; Prasanna, S.J. Human Mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE(2)-dependent mechanism. Sci. Rep. 2016, 6, 38308. [Google Scholar] [CrossRef]
- Cao, X.; Duan, L.; Hou, H.; Liu, Y.; Chen, S.; Zhang, S.; Liu, Y.; Wang, C.; Qi, X.; Liu, N.; et al. IGF-1C hydrogel improves the therapeutic effects of MSCs on colitis in mice through PGE2-mediated M2 macrophage polarization. Theranostics 2020, 10, 7697–7709. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Moretta, L. Interactions between mesenchymal stem cells and dendritic cells. Adv. Biochem. Eng. Biotechnol. 2013, 130, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Abdelrazik, H.; Becchetti, F.; Moretta, L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: Central role of MSC-derived prostaglandin E2. Blood 2009, 113, 6576–6583. [Google Scholar] [CrossRef]
- Wehner, R.; Wehrum, D.; Bornhauser, M.; Zhao, S.; Schakel, K.; Bachmann, M.P.; Platzbecker, U.; Ehninger, G.; Rieber, E.P.; Schmitz, M. Mesenchymal stem cells efficiently inhibit the proinflammatory properties of 6-sulfo LacNAc dendritic cells. Haematologica 2009, 94, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Scutera, S.; Salvi, V.; Lorenzi, L.; Piersigilli, G.; Lonardi, S.; Alotto, D.; Casarin, S.; Castagnoli, C.; Dander, E.; D’Amico, G.; et al. Adaptive Regulation of Osteopontin Production by Dendritic Cells Through the Bidirectional Interaction With Mesenchymal Stromal Cells. Front. Immunol. 2018, 9, 1207. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, W.; Huang, Q.; Gu, Y.; Shi, Y.; Huang, J.; Zhao, F.; Liu, Q.; Wei, X.; Jin, M.; et al. Mesenchymal stem cells alleviate bacteria-induced liver injury in mice by inducing regulatory dendritic cells. Hepatology 2014, 59, 671–682. [Google Scholar] [CrossRef]
- Anderson, P.; Gonzalez-Rey, E.; O’Valle, F.; Martin, F.; Oliver, F.J.; Delgado, M. Allogeneic Adipose-Derived Mesenchymal Stromal Cells Ameliorate Experimental Autoimmune Encephalomyelitis by Regulating Self-Reactive T Cell Responses and Dendritic Cell Function. Stem Cells Int. 2017, 2017, 2389753. [Google Scholar] [CrossRef]
- Spaggiari, G.M.; Capobianco, A.; Becchetti, S.; Mingari, M.C.; Moretta, L. Mesenchymal stem cell-natural killer cell interactions: Evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood 2006, 107, 1484–1490. [Google Scholar] [CrossRef]
- Sotiropoulou, P.A.; Perez, S.A.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 2006, 24, 74–85. [Google Scholar] [CrossRef]
- Spaggiari, G.M.; Capobianco, A.; Abdelrazik, H.; Becchetti, F.; Mingari, M.C.; Moretta, L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: Role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 2008, 111, 1327–1333. [Google Scholar] [CrossRef]
- Braun, D.; Longman, R.S.; Albert, M.L. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood 2005, 106, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.C.; Zhou, X.; Cuchens, M.; Jones, Q. Prostaglandin E2 suppressed IL-15-mediated human NK cell function through down-regulation of common gamma-chain. J. Immunol. 2001, 166, 885–891. [Google Scholar] [CrossRef]
- Holt, D.; Ma, X.; Kundu, N.; Fulton, A. Prostaglandin E(2) (PGE (2)) suppresses natural killer cell function primarily through the PGE(2) receptor EP4. Cancer Immunol. Immunother. 2011, 60, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Lee, Y.; Kim, M.S.; Kang, Y.J.; Park, Y.J.; Jung, H.; Kim, T.D.; Lee, H.G.; Choi, I.; Yoon, S.R. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front. Immunol. 2018, 9, 1859. [Google Scholar] [CrossRef]
- Knudsen, N.H.; Manguso, R.T. Tumor-Derived PGE2 Gives NK Cells a Headache. Immunity 2020, 53, 1131–1132. [Google Scholar] [CrossRef] [PubMed]
- Tabera, S.; Perez-Simon, J.A.; Diez-Campelo, M.; Sanchez-Abarca, L.I.; Blanco, B.; Lopez, A.; Benito, A.; Ocio, E.; Sanchez-Guijo, F.M.; Canizo, C.; et al. The effect of mesenchymal stem cells on the viability, proliferation and differentiation of B-lymphocytes. Haematologica 2008, 93, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.R.; Yang, Z.X.; Han, Z.B.; Meng, L.; Liang, L.; Feng, X.M.; Yang, S.G.; Chi, Y.; Chen, D.D.; Wang, Y.W.; et al. Mesenchymal stem cells support proliferation and terminal differentiation of B cells. Cell. Physiol. Biochem. 2012, 30, 1526–1537. [Google Scholar] [CrossRef]
- Su, W.; Wan, Q.; Huang, J.; Han, L.; Chen, X.; Chen, G.; Olsen, N.; Zheng, S.G.; Liang, D. Culture medium from TNF-alpha-stimulated mesenchymal stem cells attenuates allergic conjunctivitis through multiple antiallergic mechanisms. J. Allergy Clin. Immunol. 2015, 136, 423–432 e428. [Google Scholar] [CrossRef] [PubMed]
- Shin, T.H.; Lee, B.C.; Choi, S.W.; Shin, J.H.; Kang, I.; Lee, J.Y.; Kim, J.J.; Lee, H.K.; Jung, J.E.; Choi, Y.W.; et al. Human adipose tissue-derived mesenchymal stem cells alleviate atopic dermatitis via regulation of B lymphocyte maturation. Oncotarget 2017, 8, 512–522. [Google Scholar] [CrossRef]
- Hermankova, B.; Zajicova, A.; Javorkova, E.; Chudickova, M.; Trosan, P.; Hajkova, M.; Krulova, M.; Holan, V. Suppression of IL-10 production by activated B cells via a cell contact-dependent cyclooxygenase-2 pathway upregulated in IFN-gamma-treated mesenchymal stem cells. Immunobiology 2016, 221, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cai, C.; Xu, D.; Liu, Q.; Zheng, S.; Liu, L.; Li, G.; Zhang, X.; Li, X.; Ma, Y.; et al. Human Mesenchymal Stem Cell-Treated Regulatory CD23(+)CD43(+) B Cells Alleviate Intestinal Inflammation. Theranostics 2019, 9, 4633–4647. [Google Scholar] [CrossRef]
- Chen, R.; Cao, Y.; Tian, Y.; Gu, Y.; Lu, H.; Zhang, S.; Xu, H.; Su, Z. PGE2 ameliorated viral myocarditis development and promoted IL-10-producing regulatory B cell expansion via MAPKs/AKT-AP1 axis or AhR signaling. Cell. Immunol. 2020, 347, 104025. [Google Scholar] [CrossRef]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Kickler, K.; Maltby, K.; Ni Choileain, S.; Stephen, J.; Wright, S.; Hafler, D.A.; Jabbour, H.N.; Astier, A.L. Prostaglandin E2 affects T cell responses through modulation of CD46 expression. J. Immunol. 2012, 188, 5303–5310. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Tammik, L.; Sundberg, B.; Haynesworth, S.E.; Ringden, O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand. J. Immunol. 2003, 57, 11–20. [Google Scholar] [CrossRef]
- Glennie, S.; Soeiro, I.; Dyson, P.J.; Lam, E.W.; Dazzi, F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 2005, 105, 2821–2827. [Google Scholar] [CrossRef]
- Keyser, K.A.; Beagles, K.E.; Kiem, H.P. Comparison of mesenchymal stem cells from different tissues to suppress T-cell activation. Cell Transpl. 2007, 16, 555–562. [Google Scholar] [CrossRef]
- Beyth, S.; Borovsky, Z.; Mevorach, D.; Liebergall, M.; Gazit, Z.; Aslan, H.; Galun, E.; Rachmilewitz, J. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood 2005, 105, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Najar, M.; Raicevic, G.; Boufker, H.I.; Fayyad Kazan, H.; De Bruyn, C.; Meuleman, N.; Bron, D.; Toungouz, M.; Lagneaux, L. Mesenchymal stromal cells use PGE2 to modulate activation and proliferation of lymphocyte subsets: Combined comparison of adipose tissue, Wharton’s Jelly and bone marrow sources. Cell. Immunol. 2010, 264, 171–179. [Google Scholar] [CrossRef]
- Burr, S.P.; Dazzi, F.; Garden, O.A. Mesenchymal stromal cells and regulatory T cells: The Yin and Yang of peripheral tolerance? Immunol. Cell Biol. 2013, 91, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Zafranskaya, M.; Nizheharodava, D.; Yurkevich, M.; Ivanchik, G.; Demidchik, Y.; Kozhukh, H.; Fedulov, A. PGE2 contributes to in vitro MSC-mediated inhibition of non-specific and antigen-specific T cell proliferation in MS patients. Scand. J. Immunol. 2013, 78, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, B.; Kudlik, G.; Monostori, E.; Uher, F. Activated T-cells and pro-inflammatory cytokines differentially regulate prostaglandin E2 secretion by mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2012, 419, 215–220. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, S.; Wu, S.; Qi, J.; Li, W.; Liu, S.; Cong, Y.; Chen, H.; Lu, L.; Shi, S.; et al. Clearance of apoptotic cells by mesenchymal stem cells contributes to immunosuppression via PGE2. EBioMedicine 2019, 45, 341–350. [Google Scholar] [CrossRef]
- Burand, A.J., Jr.; Di, L.; Boland, L.K.; Boyt, D.T.; Schrodt, M.V.; Santillan, D.A.; Ankrum, J.A. Aggregation of Human Mesenchymal Stromal Cells Eliminates Their Ability to Suppress Human T Cells. Front. Immunol. 2020, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, N.; Miranda, A.; Funes, J.M.; Hevia, G.; Perez, R.; de Leon, J. Oncogenic transformation tunes the cross-talk between mesenchymal stem cells and T lymphocytes. Cell. Immunol. 2014, 289, 174–184. [Google Scholar] [CrossRef]
- English, K.; Ryan, J.M.; Tobin, L.; Murphy, M.J.; Barry, F.P.; Mahon, B.P. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin. Exp. Immunol. 2009, 156, 149–160. [Google Scholar] [CrossRef]
- Soontrapa, K.; Honda, T.; Sakata, D.; Yao, C.; Hirata, T.; Hori, S.; Matsuoka, T.; Kita, Y.; Shimizu, T.; Kabashima, K.; et al. Prostaglandin E2-prostaglandin E receptor subtype 4 (EP4) signaling mediates UV irradiation-induced systemic immunosuppression. Proc. Natl. Acad. Sci. USA 2011, 108, 6668–6673. [Google Scholar] [CrossRef]
- Hsu, W.T.; Lin, C.H.; Chiang, B.L.; Jui, H.Y.; Wu, K.K.; Lee, C.M. Prostaglandin E2 potentiates mesenchymal stem cell-induced IL-10+IFN-gamma+CD4+ regulatory T cells to control transplant arteriosclerosis. J. Immunol. 2013, 190, 2372–2380. [Google Scholar] [CrossRef]
- Tumangelova-Yuzeir, K.; Naydenov, E.; Ivanova-Todorova, E.; Krasimirova, E.; Vasilev, G.; Nachev, S.; Kyurkchiev, D. Mesenchymal Stem Cells Derived and Cultured from Glioblastoma Multiforme Increase Tregs, Downregulate Th17, and Induce the Tolerogenic Phenotype of Monocyte-Derived Cells. Stem Cells Int. 2019, 2019, 6904638. [Google Scholar] [CrossRef]
- An, J.H.; Song, W.J.; Li, Q.; Kim, S.M.; Yang, J.I.; Ryu, M.O.; Nam, A.R.; Bhang, D.H.; Jung, Y.C.; Youn, H.Y. Prostaglandin E2 secreted from feline adipose tissue-derived mesenchymal stem cells alleviate DSS-induced colitis by increasing regulatory T cells in mice. BMC Vet. Res. 2018, 14, 354. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Zhang, L.; Fu, B.; Bai, J.; Zhang, Y.; Cai, G.; Bai, X.; Feng, Z.; Sun, S.; Chen, X. IL-17A improves the efficacy of mesenchymal stem cells in ischemic-reperfusion renal injury by increasing Treg percentages by the COX-2/PGE2 pathway. Kidney Int. 2018, 93, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, S.; Li, P.; Huang, X.P.; Guo, J.; Wu, J.; Mihic, A.; Li, S.H.; Zang, W.F.; Shen, D.; Weisel, R.D.; et al. Preserving prostaglandin E2 level prevents rejection of implanted allogeneic mesenchymal stem cells and restores postinfarction ventricular function. Circulation 2013, 128, S69–S78. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, D.; Liang, J.; Zhang, H.; Feng, X.; Wang, H.; Hua, B.; Liu, B.; Ye, S.; Hu, X.; et al. Umbilical cord mesenchymal stem cell transplantation in severe and refractory systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 2467–2475. [Google Scholar] [CrossRef]
- Wei, J.; Ouyang, X.; Tang, Y.; Li, H.; Wang, B.; Ye, Y.; Jin, M.; Al Azab, M.; Li, W.; Li, X. ER-stressed MSC displayed more effective immunomodulation in RA CD4(+)CXCR5(+)ICOS(+) follicular helper-like T cells through higher PGE2 binding with EP2/EP4. Mod. Rheumatol. 2020, 30, 509–516. [Google Scholar] [CrossRef]
- Tatara, R.; Ozaki, K.; Kikuchi, Y.; Hatanaka, K.; Oh, I.; Meguro, A.; Matsu, H.; Sato, K.; Ozawa, K. Mesenchymal stromal cells inhibit Th17 but not regulatory T-cell differentiation. Cytotherapy 2011, 13, 686–694. [Google Scholar] [CrossRef]
- Ghannam, S.; Pene, J.; Moquet-Torcy, G.; Jorgensen, C.; Yssel, H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J. Immunol. 2010, 185, 302–312. [Google Scholar] [CrossRef]
- Yao, C.; Sakata, D.; Esaki, Y.; Li, Y.; Matsuoka, T.; Kuroiwa, K.; Sugimoto, Y.; Narumiya, S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat. Med. 2009, 15, 633–640. [Google Scholar] [CrossRef]
- Boniface, K.; Bak-Jensen, K.S.; Li, Y.; Blumenschein, W.M.; McGeachy, M.J.; McClanahan, T.K.; McKenzie, B.S.; Kastelein, R.A.; Cua, D.J.; de Waal Malefyt, R. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J. Exp. Med. 2009, 206, 535–548. [Google Scholar] [CrossRef]
- Pelus, L.M.; Hoggatt, J. Pleiotropic effects of prostaglandin E2 in hematopoiesis; prostaglandin E2 and other eicosanoids regulate hematopoietic stem and progenitor cell function. Prostaglandins Other Lipid Mediat. 2011, 96, 3–9. [Google Scholar] [CrossRef]
- DeGowin, R.L.; Gibson, D.P. Prostaglandin-mediated enhancement of erythroid colonies by marrow stromal cells (MSC). Exp. Hematol. 1981, 9, 274–280. [Google Scholar]
- Singh; Pratibha; Orschell, C.M.; Pelus, L.M. Prostaglandin E2 Signaling through EP4 Receptor Promotes Hematopoietic Stem Cell Niche Regeneration and Enhances Hematopoietic Recovery. Blood 2015, 126, 784. [Google Scholar] [CrossRef]
- Lee, B.C.; Kim, H.S.; Shin, T.H.; Kang, I.; Lee, J.Y.; Kim, J.J.; Kang, H.K.; Seo, Y.; Lee, S.; Yu, K.R.; et al. PGE2 maintains self-renewal of human adult stem cells via EP2-mediated autocrine signaling and its production is regulated by cell-to-cell contact. Sci. Rep. 2016, 6, 26298. [Google Scholar] [CrossRef]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim Biophys. Sin (Shanghai) 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.W.; Yun, S.P.; Park, J.H.; Ryu, J.M.; Lee, J.H.; Han, H.J. Cooperation of Epac1/Rap1/Akt and PKA in prostaglandin E(2) -induced proliferation of human umbilical cord blood derived mesenchymal stem cells: Involvement of c-Myc and VEGF expression. J. Cell. Physiol. 2012, 227, 3756–3767. [Google Scholar] [CrossRef]
- Baek, S.H.; Kioussi, C.; Briata, P.; Wang, D.; Nguyen, H.D.; Ohgi, K.A.; Glass, C.K.; Wynshaw-Boris, A.; Rose, D.W.; Rosenfeld, M.G. Regulated subset of G1 growth-control genes in response to derepression by the Wnt pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 3245–3250. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Lee, M.Y.; Ryu, J.M.; Song, C.H.; Han, H.J. Role of HIF-1alpha and VEGF in human mesenchymal stem cell proliferation by 17beta-estradiol: Involvement of PKC, PI3K/Akt, and MAPKs. Am. J. Physiol. Cell. Physiol. 2009, 296, C317–C326. [Google Scholar] [CrossRef]
- Kleiveland, C.R.; Kassem, M.; Lea, T. Human mesenchymal stem cell proliferation is regulated by PGE2 through differential activation of cAMP-dependent protein kinase isoforms. Exp. Cell Res. 2008, 314, 1831–1838. [Google Scholar] [CrossRef]
- Meinkoth, J.L.; Ji, Y.; Taylor, S.S.; Feramisco, J.R. Dynamics of the distribution of cyclic AMP-dependent protein kinase in living cells. Proc. Natl. Acad. Sci. USA 1990, 87, 9595–9599. [Google Scholar] [CrossRef]
- Beene, D.L.; Scott, J.D. A-kinase anchoring proteins take shape. Curr. Opin. Cell Biol. 2007, 19, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Raabe, O.; Addicks, K.; Wenisch, S.; Arnhold, S. Effects of non-steroidal anti-inflammatory drugs on proliferation, differentiation and migration in equine mesenchymal stem cells. Cell Biol. Int. 2011, 35, 235–248. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Zhu, W.; Zhang, H.; Hu, S.; Cong, X. Growth inhibition of mesenchymal stem cells by aspirin: Involvement of the WNT/beta-catenin signal pathway. Clin. Exp. Pharm. Physiol. 2006, 33, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Rennert, R.C.; Sorkin, M.; Garg, R.K.; Gurtner, G.C. Stem cell recruitment after injury: Lessons for regenerative medicine. Regen. Med. 2012, 7, 833–850. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Xu, J.; Woods, C.R.; Mora, A.L.; Spears, W.; Roman, J.; Brigham, K.L. Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am. J. Respir. Cell Mol. Biol. 2005, 33, 145–152. [Google Scholar] [CrossRef]
- Liu, H.; Liu, S.; Li, Y.; Wang, X.; Xue, W.; Ge, G.; Luo, X. The role of SDF-1-CXCR4/CXCR7 axis in the therapeutic effects of hypoxia-preconditioned mesenchymal stem cells for renal ischemia/reperfusion injury. PLoS ONE 2012, 7, e34608. [Google Scholar] [CrossRef]
- Ponte, A.L.; Marais, E.; Gallay, N.; Langonne, A.; Delorme, B.; Herault, O.; Charbord, P.; Domenech, J. The in vitro migration capacity of human bone marrow mesenchymal stem cells: Comparison of chemokine and growth factor chemotactic activities. Stem Cells 2007, 25, 1737–1745. [Google Scholar] [CrossRef]
- Carrero, R.; Cerrada, I.; Lledo, E.; Dopazo, J.; Garcia-Garcia, F.; Rubio, M.P.; Trigueros, C.; Dorronsoro, A.; Ruiz-Sauri, A.; Montero, J.A.; et al. IL1beta induces mesenchymal stem cells migration and leucocyte chemotaxis through NF-kappaB. Stem Cell Rev. Rep. 2012, 8, 905–916. [Google Scholar] [CrossRef]
- Legler, D.F.; Krause, P.; Scandella, E.; Singer, E.; Groettrup, M. Prostaglandin E2 is generally required for human dendritic cell migration and exerts its effect via EP2 and EP4 receptors. J. Immunol. 2006, 176, 966–973. [Google Scholar] [CrossRef]
- Yen, J.H.; Khayrullina, T.; Ganea, D. PGE2-induced metalloproteinase-9 is essential for dendritic cell migration. Blood 2008, 111, 260–270. [Google Scholar] [CrossRef]
- Diao, G.; Huang, J.; Zheng, X.; Sun, X.; Tian, M.; Han, J.; Guo, J. Prostaglandin E2 serves a dual role in regulating the migration of dendritic cells. Int. J. Mol. Med. 2021, 47, 207–218. [Google Scholar] [CrossRef]
- Wang, Y.; Lai, S.; Tang, J.; Feng, C.; Liu, F.; Su, C.; Zou, W.; Chen, H.; Xu, D. Prostaglandin E2 promotes human CD34+ cells homing through EP2 and EP4 in vitro. Mol. Med. Rep. 2017, 16, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Osma-Garcia, I.C.; Punzon, C.; Fresno, M.; Diaz-Munoz, M.D. Dose-dependent effects of prostaglandin E2 in macrophage adhesion and migration. Eur. J. Immunol. 2016, 46, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yang, T.; Yu, S.; Liu, C.; He, M.; Hu, C. Prostaglandin E2 increases migration and proliferation of human glioblastoma cells by activating transient receptor potential melastatin 7 channels. J. Cell. Mol. Med. 2018, 22, 6327–6337. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Shao, J.; Washington, M.K.; DuBois, R.N. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J. Biol. Chem. 2001, 276, 18075–18081. [Google Scholar] [CrossRef]
- Yun, S.P.; Ryu, J.M.; Jang, M.W.; Han, H.J. Interaction of profilin-1 and F-actin via a beta-arrestin-1/JNK signaling pathway involved in prostaglandin E(2)-induced human mesenchymal stem cells migration and proliferation. J. Cell. Physiol. 2011, 226, 559–571. [Google Scholar] [CrossRef]
- Lu, X.; Han, J.; Xu, X.; Xu, J.; Liu, L.; Huang, Y.; Yang, Y.; Qiu, H. PGE2 Promotes the Migration of Mesenchymal Stem Cells through the Activation of FAK and ERK1/2 Pathway. Stem Cells Int. 2017, 2017, 8178643. [Google Scholar] [CrossRef]
- Kulesza, A.; Zielniok, K.; Hawryluk, J.; Paczek, L.; Burdzinska, A. Ibuprofen in Therapeutic Concentrations Affects the Secretion of Human Bone Marrow Mesenchymal Stromal Cells, but Not Their Proliferative and Migratory Capacity. Biomolecules 2022, 12, 287. [Google Scholar] [CrossRef]
- Ma, Y.F.; Li, X.J.; Jee, W.S.; McOsker, J.; Liang, X.G.; Setterberg, R.; Chow, S.Y. Effects of prostaglandin E2 and F2 alpha on the skeleton of osteopenic ovariectomized rats. Bone 1995, 17, 549–554. [Google Scholar] [CrossRef]
- Weinreb, M.; Suponitzky, I.; Keila, S. Systemic administration of an anabolic dose of PGE2 in young rats increases the osteogenic capacity of bone marrow. Bone 1997, 20, 521–526. [Google Scholar] [CrossRef]
- Suponitzky, I.; Weinreb, M. Differential effects of systemic prostaglandin E2 on bone mass in rat long bones and calvariae. J. Endocrinol. 1998, 156, 51–57. [Google Scholar] [CrossRef]
- Gajraj, N.M. The effect of cyclooxygenase-2 inhibitors on bone healing. Reg. Anesth. Pain Med. 2003, 28, 456–465. [Google Scholar] [CrossRef]
- Beck, A.; Salem, K.; Krischak, G.; Kinzl, L.; Bischoff, M.; Schmelz, A. Nonsteroidal anti-inflammatory drugs (NSAIDs) in the perioperative phase in traumatology and orthopedics effects on bone healing. Oper. Orthop. Traumatol. 2005, 17, 569–578. [Google Scholar] [CrossRef]
- Brown, K.M.; Saunders, M.M.; Kirsch, T.; Donahue, H.J.; Reid, J.S. Effect of COX-2-specific inhibition on fracture-healing in the rat femur. J. Bone Jt. Surg. Am. 2004, 86, 116–123. [Google Scholar] [CrossRef]
- Arikawa, T.; Omura, K.; Morita, I. Regulation of bone morphogenetic protein-2 expression by endogenous prostaglandin E2 in human mesenchymal stem cells. J. Cell. Physiol. 2004, 200, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Choudhary, S.; Okada, Y.; Voznesensky, O.; Alander, C.; Raisz, L.; Pilbeam, C. Cyclooxygenase-2 gene disruption promotes proliferation of murine calvarial osteoblasts in vitro. Bone 2007, 41, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Schwarz, E.M.; Young, D.A.; Puzas, J.E.; Rosier, R.N.; O’Keefe, R.J. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J. Clin. Investig. 2002, 109, 1405–1415. [Google Scholar] [CrossRef]
- Keila, S.; Kelner, A.; Weinreb, M. Systemic prostaglandin E2 increases cancellous bone formation and mass in aging rats and stimulates their bone marrow osteogenic capacity in vivo and in vitro. J. Endocrinol. 2001, 168, 131–139. [Google Scholar] [CrossRef]
- Chang, J.K.; Li, C.J.; Wu, S.C.; Yeh, C.H.; Chen, C.H.; Fu, Y.C.; Wang, G.J.; Ho, M.L. Effects of anti-inflammatory drugs on proliferation, cytotoxicity and osteogenesis in bone marrow mesenchymal stem cells. Biochem. Pharm. 2007, 74, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.A.; Xie, C.; Zuscik, M.J.; Kingsley, P.; Schwarz, E.M.; Awad, H.; Guldberg, R.; Drissi, H.; Puzas, J.E.; Boyce, B.; et al. Reduced COX-2 expression in aged mice is associated with impaired fracture healing. J. Bone Min. Res. 2009, 24, 251–264. [Google Scholar] [CrossRef]
- Pountos, I.; Giannoudis, P.V.; Jones, E.; English, A.; Churchman, S.; Field, S.; Ponchel, F.; Bird, H.; Emery, P.; McGonagle, D. NSAIDS inhibit in vitro MSC chondrogenesis but not osteogenesis: Implications for mechanism of bone formation inhibition in man. J. Cell. Mol. Med. 2011, 15, 525–534. [Google Scholar] [CrossRef]
- Salem, O.; Wang, H.T.; Alaseem, A.M.; Ciobanu, O.; Hadjab, I.; Gawri, R.; Antoniou, J.; Mwale, F. Naproxen affects osteogenesis of human mesenchymal stem cells via regulation of Indian hedgehog signaling molecules. Arthritis Res. Ther. 2014, 16, R152. [Google Scholar] [CrossRef]
- Alaseem, A.M.; Madiraju, P.; Aldebeyan, S.A.; Noorwali, H.; Antoniou, J.; Mwale, F. Naproxen induces type X collagen expression in human bone-marrow-derived mesenchymal stem cells through the upregulation of 5-lipoxygenase. Tissue Eng. Part A 2015, 21, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Almaawi, A.; Wang, H.T.; Ciobanu, O.; Rowas, S.A.; Rampersad, S.; Antoniou, J.; Mwale, F. Effect of acetaminophen and nonsteroidal anti-inflammatory drugs on gene expression of mesenchymal stem cells. Tissue Eng. Part A 2013, 19, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.S.; Yoo, J.H.; Kim, Y.H.; Paik, S.; Han, C.D.; Lee, J.W. The effects of COX-2 inhibitor during osteogenic differentiation of bone marrow-derived human mesenchymal stem cells. Stem Cells Dev. 2010, 19, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Moses, H.L. Tumor-stroma interactions. Curr. Opin. Genet. Dev. 2005, 15, 97–101. [Google Scholar] [CrossRef]
- Houghton, J.; Stoicov, C.; Nomura, S.; Rogers, A.B.; Carlson, J.; Li, H.; Cai, X.; Fox, J.G.; Goldenring, J.R.; Wang, T.C. Gastric cancer originating from bone marrow-derived cells. Science 2004, 306, 1568–1571. [Google Scholar] [CrossRef]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef]
- Menter, D.G.; Schilsky, R.L.; DuBois, R.N. Cyclooxygenase-2 and cancer treatment: Understanding the risk should be worth the reward. Clin. Cancer Res. 2010, 16, 1384–1390. [Google Scholar] [CrossRef]
- Chen, H.; Cai, W.; Chu, E.S.H.; Tang, J.; Wong, C.C.; Wong, S.H.; Sun, W.; Liang, Q.; Fang, J.; Sun, Z.; et al. Hepatic cyclooxygenase-2 overexpression induced spontaneous hepatocellular carcinoma formation in mice. Oncogene 2017, 36, 4415–4426. [Google Scholar] [CrossRef] [PubMed]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Reinhardt, F.; Herschman, H.R.; Weinberg, R.A. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012, 2, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ren, H.; Zhou, Y.; Shang, L.; Zhang, Y.; Yang, F.; Shi, X. The hypoxia conditioned mesenchymal stem cells promote hepatocellular carcinoma progression through YAP mediated lipogenesis reprogramming. J. Exp. Clin. Cancer Res. 2019, 38, 228. [Google Scholar] [CrossRef]
- Naderi, E.H.; Skah, S.; Ugland, H.; Myklebost, O.; Sandnes, D.L.; Torgersen, M.L.; Josefsen, D.; Ruud, E.; Naderi, S.; Blomhoff, H.K. Bone marrow stroma-derived PGE2 protects BCP-ALL cells from DNA damage-induced p53 accumulation and cell death. Mol. Cancer 2015, 14, 14. [Google Scholar] [CrossRef]
- Martinet, L.; Fleury-Cappellesso, S.; Gadelorge, M.; Dietrich, G.; Bourin, P.; Fournie, J.J.; Poupot, R. A regulatory cross-talk between Vgamma9Vdelta2 T lymphocytes and mesenchymal stem cells. Eur. J. Immunol. 2009, 39, 752–762. [Google Scholar] [CrossRef]
- Ryan, D.; Paul, B.T.; Koziol, J.; ElShamy, W.M. The pro- and anti-tumor roles of mesenchymal stem cells toward BRCA1-IRIS-overexpressing TNBC cells. Breast Cancer Res. 2019, 21, 53. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulesza, A.; Paczek, L.; Burdzinska, A. The Role of COX-2 and PGE2 in the Regulation of Immunomodulation and Other Functions of Mesenchymal Stromal Cells. Biomedicines 2023, 11, 445. https://doi.org/10.3390/biomedicines11020445
Kulesza A, Paczek L, Burdzinska A. The Role of COX-2 and PGE2 in the Regulation of Immunomodulation and Other Functions of Mesenchymal Stromal Cells. Biomedicines. 2023; 11(2):445. https://doi.org/10.3390/biomedicines11020445
Chicago/Turabian StyleKulesza, Agnieszka, Leszek Paczek, and Anna Burdzinska. 2023. "The Role of COX-2 and PGE2 in the Regulation of Immunomodulation and Other Functions of Mesenchymal Stromal Cells" Biomedicines 11, no. 2: 445. https://doi.org/10.3390/biomedicines11020445
APA StyleKulesza, A., Paczek, L., & Burdzinska, A. (2023). The Role of COX-2 and PGE2 in the Regulation of Immunomodulation and Other Functions of Mesenchymal Stromal Cells. Biomedicines, 11(2), 445. https://doi.org/10.3390/biomedicines11020445