
Natural Fatty Acid Guards against Brain Endothelial Cell Death and Microvascular Pathology following Ischemic Insult in the Presence of Acute Hyperglycemia

 , ,
, ,  and
and 
Abstract
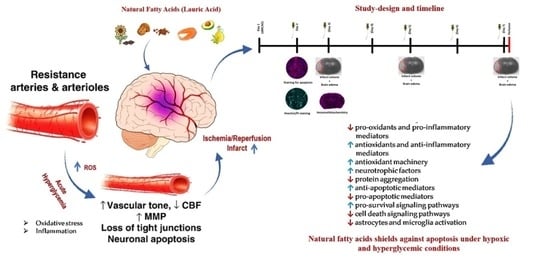
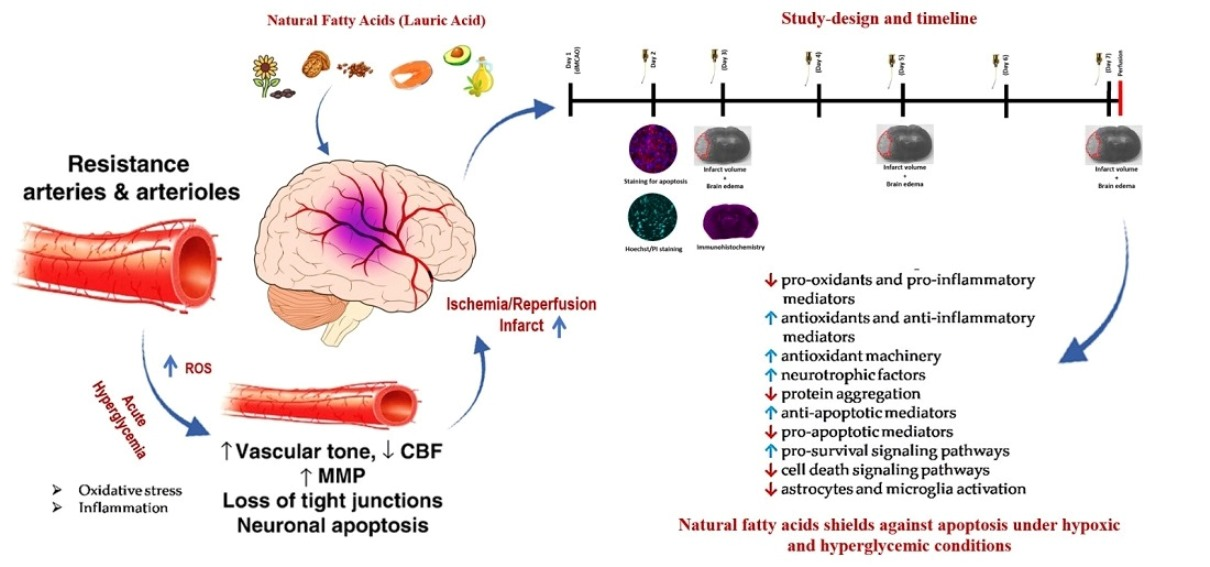
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Ischemic Stroke Model
2.3. Experimental Groups and Dosing
2.4. Immunohistochemistry and Quantification
2.5. Immunostaining for Apoptotic Marker/Cleaved Caspase-3
2.6. Hoechst/Propidium Iodide (PI) Staining
2.7. Lactate Dehydrogenase (LDH) Colorimetric Assay
2.8. Western Blotting
2.9. Food Intake and Body Weight
2.10. Statistical Analysis
3. Results
3.1. LA Reduces Infarct Volumes and Brain Edema in Ischemic Stroke Brain, Challenged by Acute Hyperglycemia
3.2. LA Improved Structural and Functional Characters of Microvasculature in Peri-Infarct Region
3.3. LA Reduces Lipid Peroxidation Production in Brain Microvasculature
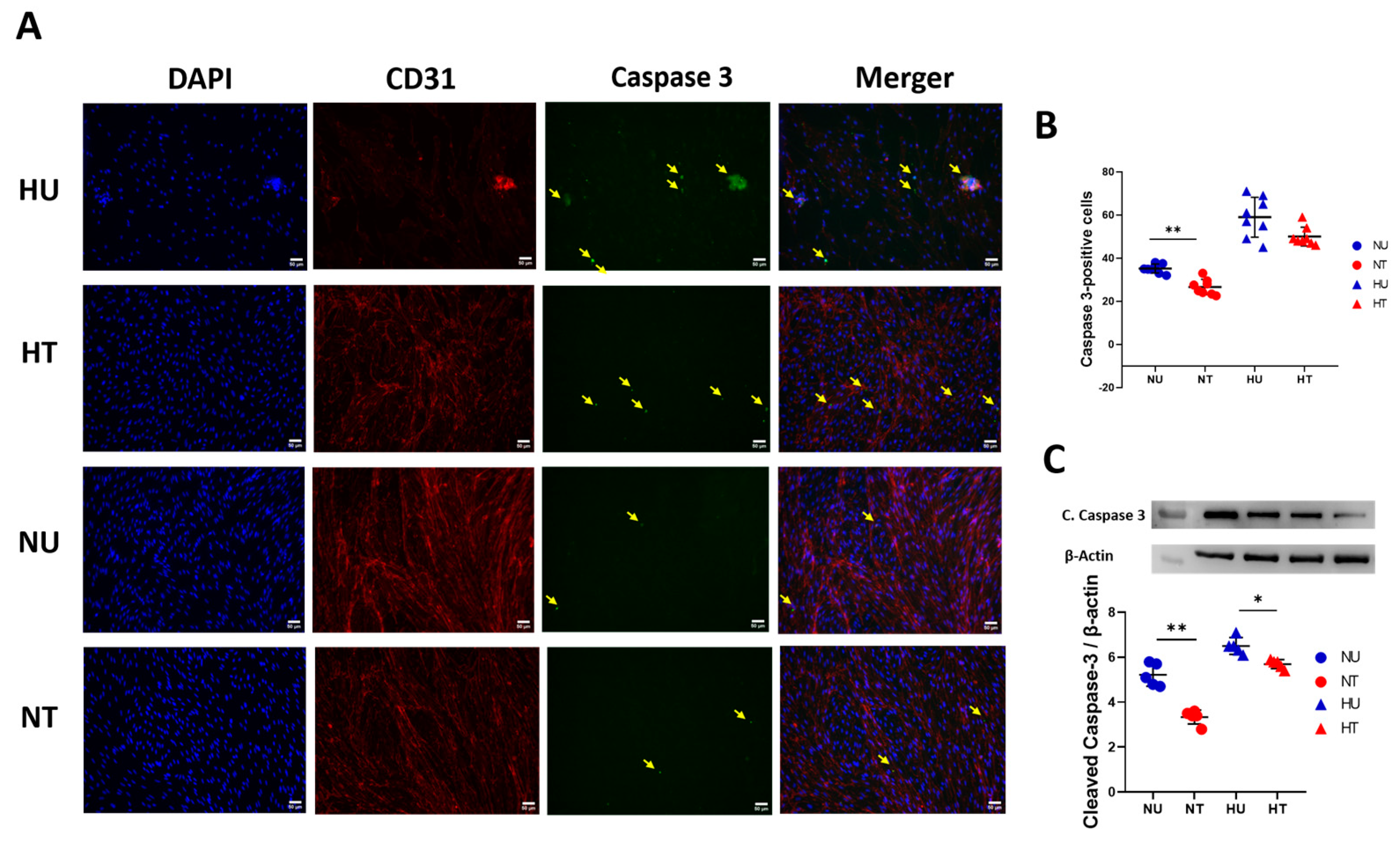
3.4. LA Administration Shields against Endothelial Cells Apoptosis under Hyperglycemic Ischemic Conditions in PBECs
3.5. LA Rescues Cells Death in PBECs under Hyperglycemic Ischemic Conditions
4. Discussion
5. Conclusions
6. Scope and Limitation of the Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Denorme, F.; Portier, I.; Kosaka, Y.; Campbell, R.A. Hyperglycemia exacerbates ischemic stroke outcome independent of platelet glucose uptake. J. Thromb. Haemost. 2021, 19, 536–546. [Google Scholar] [CrossRef]
- Tziomalos, K.; Dimitriou, P.; Bouziana, S.D.; Spanou, M.; Kostaki, S.; Angelopoulou, S.-M.; Papadopoulou, M.; Giampatzis, V.; Savopoulos, C.; Hatzitolios, A.I. Stress hyperglycemia and acute ischemic stroke in-hospital outcome. Metabolism 2017, 67, 99–105. [Google Scholar] [CrossRef]
- Khan, M.A.; Schultz, S.; Othman, A.; Fleming, T.; Lebrón-Galán, R.; Rades, D.; Clemente, D.; Nawroth, P.P.; Schwaninger, M. Hyperglycemia in stroke impairs polarization of monocytes/macrophages to a protective noninflammatory cell type. J. Neurosci. 2016, 36, 9313–9325. [Google Scholar] [CrossRef]
- Gliem, M.; Schwaninger, M.; Jander, S. Protective features of peripheral monocytes/macrophages in stroke. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 329–338. [Google Scholar] [CrossRef]
- Haque, A.U. Cerebrovascular Accidents in Diabetic Patients Can HBA1c Predict Early Outcome and Mortality. Int. J. Pathol. 2019, 17, 72–77. [Google Scholar]
- Palaiodimou, L.; Lioutas, V.-A.; Lambadiari, V.; Paraskevas, G.P.; Voumvourakis, K.; Tsivgoulis, G. Glycemia management in acute ischemic stroke: Current concepts and novel therapeutic targets. Postgrad. Med. 2019, 131, 423–437. [Google Scholar] [CrossRef]
- Bakhashab, S.; Ahmed, F.W.; Schulten, H.-J.; Bashir, A.; Karim, S.; Al-Malki, A.L.; Gari, M.A.; Abuzenadah, A.M.; Chaudhary, A.G.; Alqahtani, M.H.; et al. Metformin improves the angiogenic potential of human CD34+ cells co-incident with downregulating CXCL10 and TIMP1 gene expression and increasing VEGFA under hyperglycemia and hypoxia within a therapeutic window for myocardial infarction. Cardiovasc. Diabetol. 2016, 15, 27. [Google Scholar] [CrossRef]
- Jiang, Y.; Müller, K.; Khan, M.A.; Assmann, J.C.; Lampe, J.; Kilau, K.; Richter, M.; Kleint, M.; A Ridder, D.; Hübner, N.; et al. Cerebral angiogenesis ameliorates pathological disorders in Nemo-deficient mice with small-vessel disease. J. Cereb. Blood Flow Metab. 2021, 41, 219–235. [Google Scholar] [CrossRef]
- Alfieri, D.F.; Lehmann, M.F.; Flauzino, T.; de Araújo, M.C.M.; Pivoto, N.; Tirolla, R.M.; Simão, A.N.C.; Maes, M.; Reiche, E.M.V. Immune-inflammatory, metabolic, oxidative, and nitrosative stress biomarkers predict acute ischemic stroke and short-term outcome. Neurotox. Res. 2020, 38, 330–343. [Google Scholar] [CrossRef]
- Zille, M.; Ikhsan, M.; Jiang, Y.; Lampe, J.; Wenzel, J.; Schwaninger, M. The impact of endothelial cell death in the brain and its role after stroke: A systematic review. Cell Stress 2019, 3, 330. [Google Scholar] [CrossRef]
- Korshunova, I.; Rhein, S.; García-González, D.; Stölting, I.; Pfisterer, U.; Barta, A.; Dmytriyeva, O.; Kirkeby, A.; Schwaninger, M.; Khodosevich, K. Genetic modification increases the survival and the neuroregenerative properties of transplanted neural stem cells. JCI Insight 2020, 5, e126268. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Prasad, S.; Cucullo, L. Role and Function of Dehydrogenases in CNS and Blood-Brain Barrier Pathophysiology. In Dehydrogenases; IntechOpen: London, UK, 2012. [Google Scholar]
- Zaidi, A.A.; Khan, M.A.; Shahreyar, Z.A.; Ahmed, H. Lauric acid: Its role in behavioral modulation, neuro-inflammatory and oxidative stress markers in haloperidol induced Parkinson’s disease. Pak. J. Pharm. Sci. 2020, 33, 755–763. [Google Scholar] [PubMed]
- Nishimura, Y.; Moriyama, M.; Kawabe, K.; Satoh, H.; Takano, K.; Azuma, Y.-T.; Nakamura, Y. Lauric acid alleviates neuroinflammatory responses by activated microglia: Involvement of the GPR40-dependent pathway. Neurochem. Res. 2018, 43, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Lubjuhn, J.; Gastens, A.; von Wilpert, G.; Bargiotas, P.; Herrmann, O.; Murikinati, S.; Rabie, T.; Marti, H.; Amende, I.; Hampton, T.G.; et al. Functional testing in a mouse stroke model induced by occlusion of the distal middle cerebral artery. J. Neurosci. Methods 2009, 184, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Couret, D.; Bourane, S.; Catan, A.; Nativel, B.; Planesse, C.; Dorsemans, A.C.; Ait-Arsa, I.; Cournot, M.; Rondeau, P.; Patche, J.; et al. A hemorrhagic transformation model of mechanical stroke therapy with acute hyperglycemia in mice. J. Comp. Neurol. 2018, 526, 1006–1016. [Google Scholar] [CrossRef]
- Kang, J.-B.; Kim, D.-K.; Park, D.-J.; Shah, M.-A.; Kim, M.-O.; Jung, E.-J.; Lee, H.-S.; Koh, P.-O. Hyperglycemia aggravates decrease in alpha-synuclein expression in a middle cerebral artery occlusion model. Lab. Anim. Res. 2018, 34, 195–202. [Google Scholar] [CrossRef]
- Kravchenko, I.; Golovenko, N.Y.; Larionov, V.B.; Aleksandrova, A.I.; Ovcharenko, N.V. Effect of lauric acid on transdermal penetration of phenazepam in vivo. Bull. Exp. Biol. Med. 2003, 136, 579–581. [Google Scholar] [CrossRef]
- Assmann, J.C.; Müller, K.; Wenzel, J.; Walther, T.; Brands, J.; Thornton, P.; Allan, S.M.; Schwaninger, M. Isolation and cultivation of primary brain endothelial cells from adult mice. Bio. Potoc. 2017, 7, e2294. [Google Scholar] [CrossRef]
- Yousef, H.; Czupalla, C.J.; Lee, D.; Chen, M.B.; Burke, A.N.; Zera, K.A.; Zandstra, J.; Berber, E.; Lehallier, B.; Mathur, V.; et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 2019, 25, 988–1000. [Google Scholar] [CrossRef]
- Di Spiezio, A.; Sandin, E.S.; Dore, R.; Müller-Fielitz, H.; Storck, S.E.; Bernau, M.; Mier, W.; Oster, H.; Jöhren, O.; Pietrzik, C.U.; et al. The LepR-mediated leptin transport across brain barriers controls food reward. Mol. Metab. 2018, 8, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Müller-Fielitz, H.; Pokorná, B.; Vollbrandt, T.; Stölting, I.; Nadrowitz, R.; et al. The β-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef]
- Usatyuk, P.V.; Natarajan, V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J. Biol. Chem. 2004, 279, 11789–11797. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, S.-L.; Qi, S.-H. ALDH2 protects against ischemic stroke in rats by facilitating 4-HNE clearance and AQP4 down-regulation. Neurochem. Res. 2018, 43, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-X.; Huang, Y.; Zeng, J.; Hao, H.; Petroski, G.F.; Lu, H.; Liu, X.; Liu, Z. Admission glucose levels may increase the risk for early neurological deterioration in females with acute ischemic stroke. Front. Neurol. 2020, 11, 548892. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-F.; Chao, A.C.; Hu, H.H.; Lin, R.T.; Chen, C.H.; Chan, L.; Lin, H.J.; Sun, Y.; Lin, Y.Y.; Chen, P.L.; et al. Hyperglycemia predicts unfavorable outcomes in acute ischemic stroke patients treated with intravenous thrombolysis among a Chinese population: A prospective cohort study. J. Neurol. Sci. 2018, 388, 195–202. [Google Scholar] [CrossRef]
- Roberts, J.; De Hoog, L.; Bix, G.J. Mice deficient in endothelial α5 integrin are profoundly resistant to experimental ischemic stroke. J. Cereb. Blood Flow Metab. 2017, 37, 85–96. [Google Scholar] [CrossRef]
- Wang, P.; Ren, Q.; Shi, M.; Liu, Y.; Bai, H.; Chang, Y.-Z. Overexpression of Mitochondrial Ferritin Enhances Blood–Brain Barrier Integrity following Ischemic Stroke in Mice by Maintaining Iron Homeostasis in Endothelial Cells. Antioxidants 2022, 11, 1257. [Google Scholar] [CrossRef]
- Yen, T.-L.; Chen, R.-J.; Jayakumar, T.; Lu, W.-J.; Hsieh, C.-Y.; Hsu, M.-J.; Yang, C.-H.; Chang, C.-C.; Lin, Y.-K.; Lin, K.-H.; et al. Andrographolide stimulates p38 mitogen-activated protein kinase–nuclear factor erythroid-2-related factor 2–heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl. Res. 2016, 170, 57–72. [Google Scholar] [CrossRef]
- Pires, P.W.; Earley, S. Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. Elife 2018, 7, e35316. [Google Scholar] [CrossRef]
- Ding, J.; Zhang, Q.; Luo, Q.; Ying, Y.; Liu, Y.; Li, Y.; Wei, W.; Yan, F.; Zhang, H. Alda-1 attenuates lung ischemia-reperfusion injury by reducing 4-hydroxy-2-nonenal in alveolar epithelial cells. Crit. Care Med. 2016, 44, e544–e552. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Liu, M.; Chen, M.; Luo, Y.; Wang, C.; Xu, T.; Jiang, Y.; Guo, Y.; Zhang, J.H. Natural medicine in neuroprotection for ischemic stroke: Challenges and prospective. Pharmacol. Ther. 2020, 216, 107695. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, X.; Han, X.; Yao, L.; Lan, W. A new therapeutic trend: Natural medicine for ameliorating ischemic stroke via PI3K/Akt signaling pathway. Molecules 2022, 27, 7963. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Yin, F.-T.; Zhou, X.-H.; Zhang, A.-H.; Sun, H.; Yan, G.-L.; Wang, X.-J. The signaling pathways and targets of natural compounds from traditional chinese medicine in treating ischemic stroke. Molecules 2022, 27, 3099. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yin, H.; Chen, L.; Liu, H.; Zhao, M.; Zhang, X. Neuroprotective effect of ginkgolide K against acute ischemic stroke on middle cerebral ischemia occlusion in rats. J. Nat. Med. 2012, 66, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yuan, R.; Wang, Y.; Wei, C.; Zhang, S.; Yang, X.; Wu, B.; Liu, M. Early prediction of malignant brain edema after ischemic stroke: A systematic review and meta-analysis. Stroke 2018, 49, 2918–2927. [Google Scholar] [CrossRef] [PubMed]
- Gregori-Pla, C.; Blanco, I.; Camps-Renom, P.; Zirak, P.; Serra, I.; Cotta, G.; Maruccia, F.; Prats-Sánchez, L.; Martínez-Domeño, A.; Busch, D.R.; et al. Early microvascular cerebral blood flow response to head-of-bed elevation is related to outcome in acute ischemic stroke. J. Neurol. 2019, 266, 990–997. [Google Scholar] [CrossRef]
- Liu, L.; Yang, C.; Lavayen, B.P.; Tishko, R.J.; Larochelle, J.; Candelario-Jalil, E. Targeted BRD4 protein degradation by dBET1 ameliorates acute ischemic brain injury and improves functional outcomes associated with reduced neuroinflammation and oxidative stress and preservation of blood–brain barrier integrity. J. Neuroinflammation 2022, 19, 168. [Google Scholar] [CrossRef]
- Shadman, J.; Sadeghian, N.; Moradi, A.; Bohlooli, S.; Panahpour, H. Magnesium sulfate protects blood–brain barrier integrity and reduces brain edema after acute ischemic stroke in rats. Metab. Brain Dis. 2019, 34, 1221–1229. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaheryar, Z.A.; Khan, M.A.; Hameed, H.; Mushtaq, M.N.; Muhammad, S.; Shazly, G.A.; Irfan, A.; Jardan, Y.A.B. Natural Fatty Acid Guards against Brain Endothelial Cell Death and Microvascular Pathology following Ischemic Insult in the Presence of Acute Hyperglycemia. Biomedicines 2023, 11, 3342. https://doi.org/10.3390/biomedicines11123342
Shaheryar ZA, Khan MA, Hameed H, Mushtaq MN, Muhammad S, Shazly GA, Irfan A, Jardan YAB. Natural Fatty Acid Guards against Brain Endothelial Cell Death and Microvascular Pathology following Ischemic Insult in the Presence of Acute Hyperglycemia. Biomedicines. 2023; 11(12):3342. https://doi.org/10.3390/biomedicines11123342
Chicago/Turabian StyleShaheryar, Zaib Ali, Mahtab Ahmad Khan, Huma Hameed, Muhammad Naveed Mushtaq, Sajjad Muhammad, Gamal A. Shazly, Ali Irfan, and Yousef A. Bin Jardan. 2023. "Natural Fatty Acid Guards against Brain Endothelial Cell Death and Microvascular Pathology following Ischemic Insult in the Presence of Acute Hyperglycemia" Biomedicines 11, no. 12: 3342. https://doi.org/10.3390/biomedicines11123342
APA StyleShaheryar, Z. A., Khan, M. A., Hameed, H., Mushtaq, M. N., Muhammad, S., Shazly, G. A., Irfan, A., & Jardan, Y. A. B. (2023). Natural Fatty Acid Guards against Brain Endothelial Cell Death and Microvascular Pathology following Ischemic Insult in the Presence of Acute Hyperglycemia. Biomedicines, 11(12), 3342. https://doi.org/10.3390/biomedicines11123342