
Adipokines in Rheumatoid Arthritis: Emerging Biomarkers and Therapeutic Targets

 ,
,  , , , , and
, , , , and
Abstract
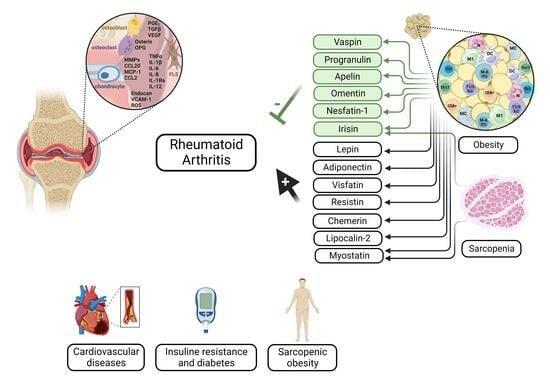
:
1. Introduction
1.1. Overview of Rheumatoid Arthritis
1.2. Role of Adipose Tissue in RA Pathophysiology
1.3. Significance of Adipokines in RA
1.4. Adipose Tissue–Skeletal Muscle Cross-Talk in RA
2. Adipokines in Rheumatoid Arthritis
2.1. Leptin
2.2. Adiponectin
2.3. Visfatin
2.4. Resistin
2.5. Other Adipokines
2.5.1. Vaspin
2.5.2. Chemerin
2.5.3. Omentin
2.5.4. Progranulin
2.5.5. Lipocalin 2
2.5.6. Nesfatin-1
2.5.7. Apelin
2.6. Adipomyokines
2.6.1. Myostatin
2.6.2. Irisin
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Finckh, A.; Gilbert, B.; Hodkinson, B.; Bae, S.C.; Thomas, R.; Deane, K.D.; Alpizar-Rodriguez, D.; Lauper, K. Global epidemiology of rheumatoid arthritis. Nat. Rev. Rheumatol. 2022, 18, 591–602. [Google Scholar] [CrossRef]
- Lee, D.M.; Weinblatt, M.E. Rheumatoid arthritis. Lancet 2001, 358, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Federico, L.E.; Johnson, T.M.; England, B.R.; Wysham, K.D.; George, M.D.; Sauer, B.; Hamilton, B.C.; Hunter, C.D.; Duryee, M.J.; Thiele, G.M.; et al. Circulating Adipokines and Associations with Incident Cardiovascular Disease in Rheumatoid Arthritis. Arthritis Care Res. 2023, 75, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Nikiphorou, E.; de Lusignan, S.; Mallen, C.D.; Khavandi, K.; Bedarida, G.; Buckley, C.D.; Galloway, J.; Raza, K. Cardiovascular risk factors and outcomes in early rheumatoid arthritis: A population-based study. Heart 2020, 106, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Anzaghe, M.; Schulke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. The immunology of rheumatoid arthritis. Nat. Immunol. 2021, 22, 10–18. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewe, R.B.M.; Bergstra, S.A.; Kerschbaumer, A.; Sepriano, A.; Aletaha, D.; Caporali, R.; Edwards, C.J.; Hyrich, K.L.; Pope, J.E.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann. Rheum. Dis. 2023, 82, 3–18. [Google Scholar] [CrossRef]
- Romao, V.C.; Fonseca, J.E. Etiology and Risk Factors for Rheumatoid Arthritis: A State-of-the-Art Review. Front. Med. 2021, 8, 689698. [Google Scholar] [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Kheder, R.K.; Faraj, T.A.; Abdulabbas, H.S.; Esmaeili, S.-A. Impacts of Porphyromonas gingivalis periodontitis on rheumatoid arthritis autoimmunity. Int. Immunopharmacol. 2023, 118, 109936. [Google Scholar]
- Alsalahy, M.M.; Nasser, H.S.; Hashem, M.M.; Elsayed, S.M. Effect of tobacco smoking on tissue protein citrullination and disease progression in patients with rheumatoid arthritis. Saudi Pharm. J. 2010, 18, 75–80. [Google Scholar] [CrossRef] [PubMed]
- George, M.D.; Baker, J.F. The Obesity Epidemic and Consequences for Rheumatoid Arthritis Care. Curr. Rheumatol. Rep. 2016, 18, 6. [Google Scholar] [CrossRef]
- Caminer, A.C.; Haberman, R.; Scher, J.U. Human microbiome, infections, and rheumatic disease. Clin. Rheumatol. 2017, 36, 2645–2653. [Google Scholar] [CrossRef]
- Masdottir, B.; Jonsson, T.; Manfredsdottir, V.; Vikingsson, A.; Brekkan, A.; Valdimarsson, H. Smoking, rheumatoid factor isotypes and severity of rheumatoid arthritis. Rheumatology 2000, 39, 1202–1205. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Joyce Wu, H.J.; Mauro, D.; Schett, G.; Ciccia, F. The gut-joint axis in rheumatoid arthritis. Nat. Rev. Rheumatol. 2021, 17, 224–237. [Google Scholar] [CrossRef]
- Paolino, S.; Pacini, G.; Patane, M.; Alessandri, E.; Cattelan, F.; Goegan, F.; Pizzorni, C.; Gotelli, E.; Cutolo, M. Interactions between microbiota, diet/nutrients and immune/inflammatory response in rheumatic diseases: Focus on rheumatoid arthritis. Reumatologia 2019, 57, 151–157. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, K.; Xiong, Q.; Zhang, J.; Cai, B.; Huang, Z.; Yang, B.; Wei, B.; Chen, J.; Niu, Q. Gut microbiota in pre-clinical rheumatoid arthritis: From pathogenesis to preventing progression. J. Autoimmun. 2023, 103001. [Google Scholar] [CrossRef]
- Dong, Y.; Yao, J.; Deng, Q.; Li, X.; He, Y.; Ren, X.; Zheng, Y.; Song, R.; Zhong, X.; Ma, J. Relationship between gut microbiota and rheumatoid arthritis: A bibliometric analysis. Front. Immunol. 2023, 14, 910. [Google Scholar]
- Moeez, S.; John, P.; Bhatti, A. Anti-citrullinated protein antibodies: Role in pathogenesis of RA and potential as a diagnostic tool. Rheumatol. Int. 2013, 33, 1669–1673. [Google Scholar] [PubMed]
- Taneja, V.; Krco, C.J.; Behrens, M.D.; Luthra, H.S.; Griffiths, M.M.; David, C.S. B cells are important as antigen presenting cells for induction of MHC-restricted arthritis in transgenic mice. Mol. Immunol. 2007, 44, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Alivernini, S.; Firestein, G.S.; McInnes, I.B. The pathogenesis of rheumatoid arthritis. Immunity 2022, 55, 2255–2270. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, H. Regulation of Immune Responses and Chronic Inflammation by Fibroblast-Like Synoviocytes. Front. Immunol. 2019, 10, 1395. [Google Scholar] [CrossRef]
- Robert, M.; Miossec, P. IL-17 in Rheumatoid Arthritis and Precision Medicine: From Synovitis Expression to Circulating Bioactive Levels. Front. Med. 2018, 5, 364. [Google Scholar] [CrossRef]
- Nygaard, G.; Firestein, G.S. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat. Rev. Rheumatol. 2020, 16, 316–333. [Google Scholar] [CrossRef]
- Tsaltskan, V.; Firestein, G.S. Targeting fibroblast-like synoviocytes in rheumatoid arthritis. Curr. Opin. Pharmacol. 2022, 67, 102304. [Google Scholar] [CrossRef]
- Sakthiswary, R.; Uma Veshaaliini, R.; Chin, K.Y.; Das, S.; Sirasanagandla, S.R. Pathomechanisms of bone loss in rheumatoid arthritis. Front. Med. 2022, 9, 962969. [Google Scholar] [CrossRef]
- Paleolog, E.M. Angiogenesis in rheumatoid arthritis. Arthritis Res. 2002, 4 (Suppl. S3), S81–S90. [Google Scholar] [CrossRef]
- Mahmoud, D.E.; Kaabachi, W.; Sassi, N.; Tarhouni, L.; Rekik, S.; Jemmali, S.; Sehli, H.; Kallel-Sellami, M.; Cheour, E.; Laadhar, L. The synovial fluid fibroblast-like synoviocyte: A long-neglected piece in the puzzle of rheumatoid arthritis pathogenesis. Front. Immunol. 2022, 13, 942417. [Google Scholar] [CrossRef]
- Purnell, J.Q. Definitions, Classification, and Epidemiology of Obesity; MDText.com, Inc.: South Dartmouth, MA, USA, 2015. [Google Scholar]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Lanthier, N.; Leclercq, I.A. Adipose tissues as endocrine target organs. Best. Pract. Res. Clin. Gastroenterol. 2014, 28, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Smorlesi, A.; Frontini, A.; Barbatelli, G.; Cinti, S. White, brown and pink adipocytes: The extraordinary plasticity of the adipose organ. Eur. J. Endocrinol. 2014, 170, R159–R171. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M. Reassessing Human Adipose Tissue. N. Engl. J. Med. 2022, 386, 768–779. [Google Scholar] [CrossRef]
- Yang, F.T.; Stanford, K.I. Batokines: Mediators of Inter-Tissue Communication (a Mini-Review). Curr. Obes. Rep. 2022, 11, 1–9. [Google Scholar] [CrossRef]
- Gu, X.; Wang, L.; Liu, S.; Shan, T. Adipose tissue adipokines and lipokines: Functions and regulatory mechanism in skeletal muscle development and homeostasis. Metabolism 2023, 139, 155379. [Google Scholar] [CrossRef]
- Mathis, D. Immunological goings-on in visceral adipose tissue. Cell Metab. 2013, 17, 851–859. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Ren, Y.; Zhao, H.; Yin, C.; Lan, X.; Wu, L.; Du, X.; Griffiths, H.R.; Gao, D. Adipokines, Hepatokines and Myokines: Focus on Their Role and Molecular Mechanisms in Adipose Tissue Inflammation. Front. Endocrinol. 2022, 13, 873699. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khazáai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- James, P.T.; Leach, R.; Kalamara, E.; Shayeghi, M. The worldwide obesity epidemic. Obes. Res. 2001, 9 (Suppl. S4), 228S–233S. [Google Scholar] [CrossRef] [PubMed]
- Bapat, S.P.; Whitty, C.; Mowery, C.T.; Liang, Y.; Yoo, A.; Jiang, Z.; Peters, M.C.; Zhang, L.J.; Vogel, I.; Zhou, C.; et al. Obesity alters pathology and treatment response in inflammatory disease. Nature 2022, 604, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Chartrand, D.J.; Murphy-Despres, A.; Almeras, N.; Lemieux, I.; Larose, E.; Despres, J.P. Overweight, Obesity, and CVD Risk: A Focus on Visceral/Ectopic Fat. Curr. Atheroscler. Rep. 2022, 24, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Kredel, L.I.; Siegmund, B. Adipose-tissue and intestinal inflammation—Visceral obesity and creeping fat. Front. Immunol. 2014, 5, 462. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Thomou, T.; Zhu, Y.; Karagiannides, I.; Pothoulakis, C.; Jensen, M.D.; Kirkland, J.L. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013, 17, 644–656. [Google Scholar] [CrossRef]
- Rana, M.N.; Neeland, I.J. Adipose Tissue Inflammation and Cardiovascular Disease: An Update. Curr. Diab Rep. 2022, 22, 27–37. [Google Scholar] [CrossRef]
- Vecchie, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Fruhbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef]
- Jayedi, A.; Khan, T.A.; Aune, D.; Emadi, A.; Shab-Bidar, S. Body fat and risk of all-cause mortality: A systematic review and dose-response meta-analysis of prospective cohort studies. Int. J. Obes. 2022, 46, 1573–1581. [Google Scholar] [CrossRef]
- Alalwan, T.A. Phenotypes of Sarcopenic Obesity: Exploring the Effects on Peri-Muscular Fat, the Obesity Paradox, Hormone-Related Responses and the Clinical Implications. Geriatrics 2020, 5, 8. [Google Scholar] [CrossRef]
- Shimabukuro, M. Leptin Resistance and Lipolysis of White Adipose Tissue: An Implication to Ectopic Fat Disposition and Its Consequences. J. Atheroscler. Thromb. 2017, 24, 1088–1089. [Google Scholar] [CrossRef]
- Guilak, F.; Fermor, B.; Keefe, F.J.; Kraus, V.B.; Olson, S.A.; Pisetsky, D.S.; Setton, L.A.; Weinberg, J.B. The role of biomechanics and inflammation in cartilage injury and repair. Clin. Orthop. Relat. Res. 2004, 423, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Aune, D.; Heath, A.K. Adiposity and the risk of rheumatoid arthritis: A systematic review and meta-analysis of cohort studies. Sci. Rep. 2020, 10, 16006. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Jacobsen, S.; Klarlund, M.; Pedersen, B.V.; Wiik, A.; Wohlfahrt, J.; Frisch, M. Environmental risk factors differ between rheumatoid arthritis with and without auto-antibodies against cyclic citrullinated peptides. Arthritis Res. Ther. 2006, 8, R133. [Google Scholar] [CrossRef] [PubMed]
- Crowson, C.S.; Matteson, E.L.; Davis, J.M., 3rd; Gabriel, S.E. Contribution of obesity to the rise in incidence of rheumatoid arthritis. Arthritis Care Res. 2013, 65, 71–77. [Google Scholar] [CrossRef]
- Symmons, D.P.; Bankhead, C.R.; Harrison, B.J.; Brennan, P.; Barrett, E.M.; Scott, D.G.; Silman, A.J. Blood transfusion, smoking, and obesity as risk factors for the development of rheumatoid arthritis: Results from a primary care-based incident case-control study in Norfolk, England. Arthritis Rheum. 1997, 40, 1955–1961. [Google Scholar] [CrossRef]
- Uhlig, T.; Hagen, K.B.; Kvien, T.K. Current tobacco smoking, formal education, and the risk of rheumatoid arthritis. J. Rheumatol. 1999, 26, 47–54. [Google Scholar]
- Wesley, A.; Bengtsson, C.; Elkan, A.C.; Klareskog, L.; Alfredsson, L.; Wedren, S.; Epidemiological Investigation of Rheumatoid Arthritis Study Group. Association between body mass index and anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis: Results from a population-based case-control study. Arthritis Care Res. 2013, 65, 107–112. [Google Scholar] [CrossRef]
- Harpsoe, M.C.; Basit, S.; Andersson, M.; Nielsen, N.M.; Frisch, M.; Wohlfahrt, J.; Nohr, E.A.; Linneberg, A.; Jess, T. Body mass index and risk of autoimmune diseases: A study within the Danish National Birth Cohort. Int. J. Epidemiol. 2014, 43, 843–855. [Google Scholar] [CrossRef]
- Linauskas, A.; Overvad, K.; Symmons, D.; Johansen, M.B.; Stengaard-Pedersen, K.; de Thurah, A. Body Fat Percentage, Waist Circumference, and Obesity As Risk Factors for Rheumatoid Arthritis: A Danish Cohort Study. Arthritis Care Res. 2019, 71, 777–786. [Google Scholar] [CrossRef]
- Ljung, L.; Rantapaa-Dahlqvist, S. Abdominal obesity, gender and the risk of rheumatoid arthritis—A nested case-control study. Arthritis Res. Ther. 2016, 18, 277. [Google Scholar] [CrossRef]
- Lu, B.; Hiraki, L.T.; Sparks, J.A.; Malspeis, S.; Chen, C.Y.; Awosogba, J.A.; Arkema, E.V.; Costenbader, K.H.; Karlson, E.W. Being overweight or obese and risk of developing rheumatoid arthritis among women: A prospective cohort study. Ann. Rheum. Dis. 2014, 73, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Pahau, H.; Brown, M.A.; Paul, S.; Thomas, R.; Videm, V. Cardiovascular disease is increased prior to onset of rheumatoid arthritis but not osteoarthritis: The population-based Nord-Trondelag health study (HUNT). Arthritis Res. Ther. 2014, 16, R85. [Google Scholar] [CrossRef]
- Rodriguez, L.A.; Tolosa, L.B.; Ruigomez, A.; Johansson, S.; Wallander, M.A. Rheumatoid arthritis in UK primary care: Incidence and prior morbidity. Scand. J. Rheumatol. 2009, 38, 173–177. [Google Scholar] [CrossRef]
- Voigt, L.F.; Koepsell, T.D.; Nelson, J.L.; Dugowson, C.E.; Daling, J.R. Smoking, obesity, alcohol consumption, and the risk of rheumatoid arthritis. Epidemiology 1994, 5, 525–532. [Google Scholar]
- Feng, J.; Chen, Q.; Yu, F.; Wang, Z.; Chen, S.; Jin, Z.; Cai, Q.; Liu, Y.; He, J. Body Mass Index and Risk of Rheumatoid Arthritis: A Meta-Analysis of Observational Studies. Medicine 2016, 95, e2859. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Yang, M.; Fu, H.; Ma, N.; Wei, T.; Tang, Q.; Hu, Z.; Liang, Y.; Yang, Z.; Zhong, R. Body mass index and the risk of rheumatoid arthritis: A systematic review and dose-response meta-analysis. Arthritis Res. Ther. 2015, 17, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zou, W.; Brestoff, J.R.; Rohatgi, N.; Wu, X.; Atkinson, J.P.; Harris, C.A.; Teitelbaum, S.L. Fat-Produced Adipsin Regulates Inflammatory Arthritis. Cell Rep. 2019, 27, 2809–2816.e2803. [Google Scholar] [CrossRef]
- Moon, J.; Kim, D.; Kim, E.K.; Lee, S.Y.; Na, H.S.; Kim, G.N.; Lee, A.; Jung, K.; Choi, J.W.; Park, S.H.; et al. Brown adipose tissue ameliorates autoimmune arthritis via inhibition of Th17 cells. Sci. Rep. 2020, 10, 12374. [Google Scholar] [CrossRef]
- Sime, K.; Choy, E.H.; Williams, A.S. Alterations to adipose tissue morphology during inflammatory arthritis is indicative of vasculopathology in DBA/1 mice. Adipocyte 2017, 6, 87–101. [Google Scholar] [CrossRef]
- Shi, H.; Wu, H.; Winkler, M.A.; Belin de Chantemele, E.J.; Lee, R.; Kim, H.W.; Weintraub, N.L. Perivascular adipose tissue in autoimmune rheumatic diseases. Pharmacol. Res. 2022, 182, 106354. [Google Scholar] [CrossRef]
- Heliovaara, M.; Aho, K.; Aromaa, A.; Knekt, P.; Reunanen, A. Smoking and risk of rheumatoid arthritis. J. Rheumatol. 1993, 20, 1830–1835. [Google Scholar] [PubMed]
- Turesson, C.; Bergstrom, U.; Pikwer, M.; Nilsson, J.A.; Jacobsson, L.T. A high body mass index is associated with reduced risk of rheumatoid arthritis in men, but not in women. Rheumatology 2016, 55, 307–314. [Google Scholar] [CrossRef] [PubMed]
- van der Helm-van Mil, A.H.; van der Kooij, S.M.; Allaart, C.F.; Toes, R.E.; Huizinga, T.W. A high body mass index has a protective effect on the amount of joint destruction in small joints in early rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Son, K.M.; Kang, S.H.; Seo, Y.I.; Kim, H.A. Association of body composition with disease activity and disability in rheumatoid arthritis. Korean J. Intern. Med. 2021, 36, 214–222. [Google Scholar] [CrossRef]
- Letarouilly, J.G.; Flipo, R.M.; Cortet, B.; Tournadre, A.; Paccou, J. Body composition in patients with rheumatoid arthritis: A narrative literature review. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X211015006. [Google Scholar] [CrossRef]
- Bosy-Westphal, A.; Muller, M.J. Diagnosis of obesity based on body composition-associated health risks-Time for a change in paradigm. Obes. Rev. 2021, 22 (Suppl. S2), e13190. [Google Scholar] [CrossRef]
- Patsalos, O.; Dalton, B.; Leppanen, J.; Ibrahim, M.A.A.; Himmerich, H. Impact of TNF-alpha Inhibitors on Body Weight and BMI: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2020, 11, 481. [Google Scholar] [CrossRef]
- Fatel, E.C.S.; Rosa, F.T.; Simao, A.N.C.; Dichi, I. Adipokines in rheumatoid arthritis. Adv. Rheumatol. 2018, 58, 25. [Google Scholar] [CrossRef]
- Baker, J.F.; Katz, P.; Weber, D.R.; Gould, P.; George, M.D.; Long, J.; Zemel, B.S.; Giles, J.T. Adipocytokines and Associations With Abnormal Body Composition in Rheumatoid Arthritis. Arthritis Care Res. 2023, 75, 616–624. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.C. Circulating leptin level in rheumatoid arthritis and its correlation with disease activity: A meta-analysis. Z. Rheumatol. 2016, 75, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bae, S.C. Circulating adiponectin and visfatin levels in rheumatoid arthritis and their correlation with disease activity: A meta-analysis. Int. J. Rheum. Dis. 2018, 21, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.; Hasseli, R.; Ohl, S.; Lange, U.; Frommer, K.W.; Muller-Ladner, U. Adipokines and Autoimmunity in Inflammatory Arthritis. Cells 2021, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.; Frommer, K.; Vasile, M.; Müller-Ladner, U. Adipocytokines as driving forces in rheumatoid arthritis and related inflammatory diseases? Arthritis Rheum. 2011, 63, 1159–1169. [Google Scholar] [CrossRef]
- Azamar-Llamas, D.; Hernandez-Molina, G.; Ramos-Avalos, B.; Furuzawa-Carballeda, J. Adipokine Contribution to the Pathogenesis of Osteoarthritis. Mediators Inflamm. 2017, 2017, 5468023. [Google Scholar] [CrossRef]
- Carrion, M.; Frommer, K.W.; Perez-Garcia, S.; Muller-Ladner, U.; Gomariz, R.P.; Neumann, E. The Adipokine Network in Rheumatic Joint Diseases. Int. J. Mol. Sci. 2019, 20, 4091. [Google Scholar] [CrossRef]
- Giles, J.T.; Ferrante, A.W.; Broderick, R.; Zartoshti, A.; Rose, J.; Downer, K.; Zhang, H.Z.; Winchester, R.J. Adipose Tissue Macrophages in Rheumatoid Arthritis: Prevalence, Disease-Related Indicators, and Associations With Cardiometabolic Risk Factors. Arthritis Care Res. 2018, 70, 175–184. [Google Scholar] [CrossRef]
- Bennett, J.L.; Pratt, A.G.; Dodds, R.; Sayer, A.A.; Isaacs, J.D. Rheumatoid sarcopenia: Loss of skeletal muscle strength and mass in rheumatoid arthritis. Nat. Rev. Rheumatol. 2023, 19, 239–251. [Google Scholar] [CrossRef]
- Laurindo, L.F.; de Maio, M.C.; Barbalho, S.M.; Guiguer, E.L.; Araujo, A.C.; de Alvares Goulart, R.; Flato, U.A.P.; Junior, E.B.; Detregiachi, C.R.P.; Dos Santos Haber, J.F.; et al. Organokines in Rheumatoid Arthritis: A Critical Review. Int. J. Mol. Sci. 2022, 23, 6193. [Google Scholar] [CrossRef]
- Rome, S. Muscle and Adipose Tissue Communicate with Extracellular Vesicles. Int. J. Mol. Sci. 2022, 23, 7052. [Google Scholar] [CrossRef]
- Sudol-Szopinska, I.; Kontny, E.; Zaniewicz-Kaniewska, K.; Prohorec-Sobieszek, M.; Saied, F.; Maslinski, W. Role of inflammatory factors and adipose tissue in pathogenesis of rheumatoid arthritis and osteoarthritis. Part I: Rheumatoid adipose tissue. J. Ultrason. 2013, 13, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Ruiz-Fernandez, C.; Pino, J.; Mera, A.; Gonzalez-Gay, M.A.; Gomez, R.; Lago, F.; Mobasheri, A.; Gualillo, O. Adipokines: Linking metabolic syndrome, the immune system, and arthritic diseases. Biochem. Pharmacol. 2019, 165, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Andonian, B.J.; Huffman, K.M. Skeletal muscle disease in rheumatoid arthritis: The center of cardiometabolic comorbidities? Curr. Opin. Rheumatol. 2020, 32, 297–306. [Google Scholar] [CrossRef]
- Wu, D.; Luo, Y.; Li, T.; Zhao, X.; Lv, T.; Fang, G.; Ou, P.; Li, H.; Luo, X.; Huang, A.; et al. Systemic complications of rheumatoid arthritis: Focus on pathogenesis and treatment. Front. Immunol. 2022, 13, 1051082. [Google Scholar] [CrossRef]
- Andonian, B.J.; Johannemann, A.; Hubal, M.J.; Pober, D.M.; Koss, A.; Kraus, W.E.; Bartlett, D.B.; Huffman, K.M. Altered skeletal muscle metabolic pathways, age, systemic inflammation, and low cardiorespiratory fitness associate with improvements in disease activity following high-intensity interval training in persons with rheumatoid arthritis. Arthritis Res. Ther. 2021, 23, 187. [Google Scholar] [CrossRef] [PubMed]
- An, H.J.; Tizaoui, K.; Terrazzino, S.; Cargnin, S.; Lee, K.H.; Nam, S.W.; Kim, J.S.; Yang, J.W.; Lee, J.Y.; Smith, L.; et al. Sarcopenia in Autoimmune and Rheumatic Diseases: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 5678. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, M.; Ruiz-Fernandez, C.; Cordero-Barreal, A.; Ait Eldjoudi, D.; Pino, J.; Farrag, Y.; Gualillo, O. Adipokines as targets in musculoskeletal immune and inflammatory diseases. Drug Discov. Today 2022, 27, 103352. [Google Scholar] [CrossRef] [PubMed]
- Little, R.D.; Prieto-Potin, I.; Perez-Baos, S.; Villalvilla, A.; Gratal, P.; Cicuttini, F.; Largo, R.; Herrero-Beaumont, G. Compensatory anabolic signaling in the sarcopenia of experimental chronic arthritis. Sci. Rep. 2017, 7, 6311. [Google Scholar] [CrossRef]
- Huffman, K.M.; Jessee, R.; Andonian, B.; Davis, B.N.; Narowski, R.; Huebner, J.L.; Kraus, V.B.; McCracken, J.; Gilmore, B.F.; Tune, K.N.; et al. Molecular alterations in skeletal muscle in rheumatoid arthritis are related to disease activity, physical inactivity, and disability. Arthritis Res. Ther. 2017, 19, 12. [Google Scholar] [CrossRef]
- Baker, J.F.; Giles, J.T.; Weber, D.; George, M.D.; Leonard, M.B.; Zemel, B.S.; Long, J.; Katz, P. Sarcopenic obesity in rheumatoid arthritis: Prevalence and impact on physical functioning. Rheumatology 2022, 61, 2285–2294. [Google Scholar] [CrossRef]
- Khoja, S.S.; Patterson, C.G.; Goodpaster, B.H.; Delitto, A.; Piva, S.R. Skeletal muscle fat in individuals with rheumatoid arthritis compared to healthy adults. Exp. Gerontol. 2020, 129, 110768. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.R.; Wolff, C.A.; Suer, M.K.; Harber, M.P. Relationship between intermuscular adipose tissue infiltration and myostatin before and after aerobic exercise training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R461–R468. [Google Scholar] [CrossRef] [PubMed]
- Moratal, C.; Raffort, J.; Arrighi, N.; Rekima, S.; Schaub, S.; Dechesne, C.; Chinetti, G.; Dani, C. IL-1β-and IL-4-polarized macrophages have opposite effects on adipogenesis of intramuscular fibro-adipogenic progenitors in humans. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Kalinkovich, A.; Livshits, G. Sarcopenic obesity or obese sarcopenia: A cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res. Rev. 2017, 35, 200–221. [Google Scholar] [CrossRef] [PubMed]
- Al Saedi, A.; Hassan, E.B.; Duque, G. The diagnostic role of fat in osteosarcopenia. J. Lab. Precis. Med. 2019, 4, 1–8. [Google Scholar] [CrossRef]
- Moschou, D.; Krikelis, M.; Georgakopoulos, C.; Mole, E.; Chronopoulos, E.; Tournis, S.; Mavragani, C.; Makris, K.; Dontas, I.; Gazi, S. Sarcopenia in Rheumatoid arthritis. A narrative review. J. Frailty Sarcopenia Falls 2023, 8, 44–52. [Google Scholar] [CrossRef]
- Lee, M.K.; Jeong, H.H.; Kim, M.J.; Ryu, H.; Baek, J.; Lee, B. Nutrients against Glucocorticoid-Induced Muscle Atrophy. Foods 2022, 11, 687. [Google Scholar] [CrossRef]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef]
- Gilson, H.; Schakman, O.; Combaret, L.; Lause, P.; Grobet, L.; Attaix, D.; Ketelslegers, J.M.; Thissen, J.P. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology 2007, 148, 452–460. [Google Scholar] [CrossRef]
- Pereira, R.M.; Freire de Carvalho, J. Glucocorticoid-induced myopathy. Jt. Bone Spine 2011, 78, 41–44. [Google Scholar] [CrossRef]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Dao, T.; Kirk, B.; Phu, S.; Vogrin, S.; Duque, G. Prevalence of Sarcopenia and its Association with Antirheumatic Drugs in Middle-Aged and Older Adults with Rheumatoid Arthritis: A Systematic Review and Meta-analysis. Calcif. Tissue Int. 2021, 109, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.; Wong-Pack, M.; Liu, T.; Adachi, J.; Lau, A.; Ma, J.; Papaioannou, A.; Rodrigues, I.B. Risk Factors and Clinical Outcomes Associated with Sarcopenia in Rheumatoid Arthritis: A Systematic Review and Meta-analysis. J. Clin. Rheumatol. 2023, 10-1097. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.W.; Kim, C.J.; Kim, J.W.; Kim, H.A.; Suh, C.H.; Jung, J.Y. The Assessment of Muscle Mass and Function in Patients with Long-Standing Rheumatoid Arthritis. J. Clin. Med. 2021, 10, 3458. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.; Wang, J.X.; Xu, Y.C.; Zong, H.X.; Teng, Y.Z.; Xu, S.Q. Sarcopenia May Be a Risk Factor for Osteoporosis in Chinese Patients with Rheumatoid Arthritis. Int. J. Gen. Med. 2022, 15, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Brance, M.L.; Di Gregorio, S.; Pons-Estel, B.A.; Quagliato, N.J.; Jorfen, M.; Berbotto, G.; Cortese, N.; Raggio, J.C.; Palatnik, M.; Chavero, I.; et al. Prevalence of Sarcopenia and Whole-Body Composition in Rheumatoid Arthritis. J. Clin. Rheumatol. 2021, 27, S153–S160. [Google Scholar] [CrossRef]
- Yamada, Y.; Tada, M.; Mandai, K.; Hidaka, N.; Inui, K.; Nakamura, H. Glucocorticoid use is an independent risk factor for developing sarcopenia in patients with rheumatoid arthritis: From the CHIKARA study. Clin. Rheumatol. 2020, 39, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Fenton, C.; Webster, J.; Martin, C.; Fareed, S.; Wehmeyer, C.; Mackie, H.; Jones, R.; Seabright, A.; Lewis, J.; Lai, Y.-C. Therapeutic glucocorticoids prevent bone loss but drive muscle wasting when administered in chronic polyarthritis. Arthritis Res. Ther. 2019, 21, 1–12. [Google Scholar] [CrossRef]
- Mochizuki, T.; Yano, K.; Ikari, K.; Okazaki, K. Sarcopenia in Japanese younger patients with rheumatoid arthritis: A cross-sectional study. Mod. Rheumatol. 2021, 31, 504–505. [Google Scholar] [CrossRef]
- Mochizuki, T.; Yano, K.; Ikari, K.; Okazaki, K. Sarcopenia-associated factors in Japanese patients with rheumatoid arthritis: A cross-sectional study. Geriatr. Gerontol. Int. 2019, 19, 907–912. [Google Scholar] [CrossRef]
- Tada, M.; Yamada, Y.; Mandai, K.; Hidaka, N. Relationships of the stand-up time to falls and fractures in patients with rheumatoid arthritis: Results from the CHIKARA study. Int. J. Rheum. Dis. 2021, 24, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Yamada, Y.; Mandai, K.; Matsumoto, Y.; Hidaka, N. Osteosarcopenia synergistically increases the risk of falls in patients with rheumatoid arthritis. Osteoporos. Sarcopenia 2021, 7, 140–145. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyere, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Park, D.-J.; Kang, J.-H.; Xu, H.; Lee, K.-E.; Lee, S.-S. Thu0152 sarcopenia is associated with persistent disease activity during follow-up of rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 349. [Google Scholar] [CrossRef]
- Roh, E.; Choi, K.M. Health Consequences of Sarcopenic Obesity: A Narrative Review. Front. Endocrinol. 2020, 11, 332. [Google Scholar] [CrossRef]
- Ji, T.; Li, Y.; Ma, L. Sarcopenic Obesity: An Emerging Public Health Problem. Aging Dis. 2022, 13, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Tutan, D.; Sen Uzeli, U. A scientometric analysis of sarcopenic obesity: Future trends and new perspectives. Medicine 2023, 102, e34244. [Google Scholar] [CrossRef]
- Rolland, Y.; Dray, C.; Vellas, B.; Barreto, P.S. Current and investigational medications for the treatment of sarcopenia. Metabolism 2023, 149, 155597. [Google Scholar] [CrossRef]
- Saitoh, M.; Ishida, J.; Ebner, N.; Anker, S.D.; Springer, J.; von Haehling, S. Myostatin inhibitors as pharmacological treatment for muscle wasting and muscular dystrophy. JCSM Clin. Rep. 2017, 2, 1–10. [Google Scholar] [CrossRef]
- Guimarães, N.S.; Guimarães, M.F.B.d.R.; Souza, V.A.d.; Kakehasi, A.M. Effect of biological disease-modifying antirheumatic drugs on body composition in patients with rheumatoid arthritis: A systematic review and meta-analysis. Adv. Rheumatol. 2022, 62, 16. [Google Scholar]
- Hein, T.R.; Peterson, L.; Bartikoski, B.J.; Portes, J.; Espirito Santo, R.C.; Xavier, R.M. The effect of disease-modifying anti-rheumatic drugs on skeletal muscle mass in rheumatoid arthritis patients: A systematic review with meta-analysis. Arthritis Res. Ther. 2022, 24, 171. [Google Scholar] [CrossRef] [PubMed]
- Ben Tekaya, A.; Mehmli, T.; Ben Sassi, M.; Teyeb, Z.; Bouden, S.; Rouached, L.; Mahmoud, I.; Dziri, C.; Abdelmoula, L. Effects of biologic and target synthetic disease-modifying anti-rheumatic drugs on sarcopenia in spondyloarthritis and rheumatoid arthritis: A systematic review and meta-analysis. Clin. Rheumatol. 2023, 42, 979–997. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G. Sarcopenia and exercise “The State of the Art”. J. Funct. Morphol. Kinesiol. 2017, 2, 40. [Google Scholar] [CrossRef]
- Vlietstra, L.; Hendrickx, W.; Waters, D.L. Exercise interventions in healthy older adults with sarcopenia: A systematic review and meta-analysis. Australas. J. Ageing 2018, 37, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Beckwee, D.; Delaere, A.; Aelbrecht, S.; Baert, V.; Beaudart, C.; Bruyere, O.; de Saint-Hubert, M.; Bautmans, I. Exercise Interventions for the Prevention and Treatment of Sarcopenia. A Systematic Umbrella Review. J. Nutr. Health Aging 2019, 23, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.Z.; No, M.H.; Heo, J.W.; Park, D.H.; Kang, J.H.; Kim, S.H.; Kwak, H.B. Role of exercise in age-related sarcopenia. J. Exerc. Rehabil. 2018, 14, 551–558. [Google Scholar] [CrossRef]
- Bilski, J.; Pierzchalski, P.; Szczepanik, M.; Bonior, J.; Zoladz, J.A. Multifactorial Mechanism of Sarcopenia and Sarcopenic Obesity. Role of Physical Exercise, Microbiota and Myokines. Cells 2022, 11, 160. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.A. Exercise metabolism in 2016: Health benefits of exercise—More than meets the eye! Nat. Rev. Endocrinol. 2017, 13, 72–74. [Google Scholar] [CrossRef]
- Johnston, A.P.; De Lisio, M.; Parise, G. Resistance training, sarcopenia, and the mitochondrial theory of aging. Appl. Physiol. Nutr. Metab. 2008, 33, 191–199. [Google Scholar] [CrossRef]
- Heo, J.-W.; No, M.-H.; Min, D.-H.; Kang, J.-H.; Kwak, H.-B. Aging-induced Sarcopenia and Exercise. Off. J. Korean Acad. Kinesiol. 2017, 19, 43–59. [Google Scholar]
- Ko, I.G.; Jeong, J.W.; Kim, Y.H.; Jee, Y.S.; Kim, S.E.; Kim, S.H.; Jin, J.J.; Kim, C.J.; Chung, K.J. Aerobic exercise affects myostatin expression in aged rat skeletal muscles: A possibility of antiaging effects of aerobic exercise related with pelvic floor muscle and urethral rhabdosphincter. Int. Neurourol. J. 2014, 18, 77–85. [Google Scholar] [CrossRef]
- Bouaziz, W.; Schmitt, E.; Kaltenbach, G.; Geny, B.; Vogel, T. Health benefits of endurance training alone or combined with diet for obese patients over 60: A review. Int. J. Clin. Pract. 2015, 69, 1032–1049. [Google Scholar] [CrossRef]
- Chen, H.T.; Chung, Y.C.; Chen, Y.J.; Ho, S.Y.; Wu, H.J. Effects of Different Types of Exercise on Body Composition, Muscle Strength, and IGF-1 in the Elderly with Sarcopenic Obesity. J. Am. Geriatr. Soc. 2017, 65, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.; Paton, B.; Rosenblatt, B.; Gissane, C.; Patterson, S.D. Blood flow restriction training in clinical musculoskeletal rehabilitation: A systematic review and meta-analysis. Br. J. Sports Med. 2017, 51, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Porcari, J.P.; McLean, K.P.; Foster, C.; Kernozek, T.; Crenshaw, B.; Swenson, C. Effects of electrical muscle stimulation on body composition, muscle strength, and physical appearance. J. Strength. Cond. Res. 2002, 16, 165–172. [Google Scholar] [PubMed]
- Musumeci, G. The use of vibration as physical exercise and therapy. J. Funct. Morphol. Kinesiol. 2017, 2, 17. [Google Scholar] [CrossRef]
- Rausch Osthoff, A.K.; Niedermann, K.; Braun, J.; Adams, J.; Brodin, N.; Dagfinrud, H.; Duruoz, T.; Esbensen, B.A.; Gunther, K.P.; Hurkmans, E.; et al. 2018 EULAR recommendations for physical activity in people with inflammatory arthritis and osteoarthritis. Ann. Rheum. Dis. 2018, 77, 1251–1260. [Google Scholar] [CrossRef]
- Baillet, A.; Vaillant, M.; Guinot, M.; Juvin, R.; Gaudin, P. Efficacy of resistance exercises in rheumatoid arthritis: Meta-analysis of randomized controlled trials. Rheumatology 2012, 51, 519–527. [Google Scholar] [CrossRef]
- Munneke, M.; de Jong, Z.; Zwinderman, A.H.; Ronday, H.K.; van Schaardenburg, D.; Dijkmans, B.A.; Kroon, H.M.; Vliet Vlieland, T.P.; Hazes, J.M. Effect of a high-intensity weight-bearing exercise program on radiologic damage progression of the large joints in subgroups of patients with rheumatoid arthritis. Arthritis Rheum. 2005, 53, 410–417. [Google Scholar] [CrossRef]
- Hurkmans, E.; van der Giesen, F.J.; Vliet Vlieland, T.P.; Schoones, J.; Van den Ende, E.C. Dynamic exercise programs (aerobic capacity and/or muscle strength training) in patients with rheumatoid arthritis. Cochrane Database Syst. Rev. 2009, 2009, CD006853. [Google Scholar] [CrossRef]
- Andersson, S.E.M.; Lange, E.; Kucharski, D.; Svedlund, S.; Onnheim, K.; Bergquist, M.; Josefsson, E.; Lord, J.M.; Martensson, I.L.; Mannerkorpi, K.; et al. Moderate- to high intensity aerobic and resistance exercise reduces peripheral blood regulatory cell populations in older adults with rheumatoid arthritis. Immun. Ageing 2020, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Lourenzi, F.M.; Jones, A.; Pereira, D.F.; Santos, J.; Furtado, R.N.V.; Natour, J. Effectiveness of an overall progressive resistance strength program for improving the functional capacity of patients with rheumatoid arthritis: A randomized controlled trial. Clin. Rehabil. 2017, 31, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Xu, A.; Gao, C.; Wang, Z.; Wu, X. The effect of physical exercise on rheumatoid arthritis: An overview of systematic reviews and meta-analysis. J. Adv. Nurs. 2021, 77, 506–522. [Google Scholar] [CrossRef]
- Strasser, B.; Leeb, G.; Strehblow, C.; Schobersberger, W.; Haber, P.; Cauza, E. The effects of strength and endurance training in patients with rheumatoid arthritis. Clin. Rheumatol. 2011, 30, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Marcora, S.M.; Lemmey, A.B.; Maddison, P.J. Can progressive resistance training reverse cachexia in patients with rheumatoid arthritis? Results of a pilot study. J. Rheumatol. 2005, 32, 1031–1039. [Google Scholar]
- Joo, Y.B.; Lee, K.B.; Sul, B.; Lee, H.S.; Lim, S.H.; Park, Y.J. Effect of resistance exercise on serum leptin levels in a prospective longitudinal study of women patients with rheumatoid arthritis. Arthritis Res. Ther. 2022, 24, 76. [Google Scholar] [CrossRef]
- Lemmey, A.B.; Marcora, S.M.; Chester, K.; Wilson, S.; Casanova, F.; Maddison, P.J. Effects of high-intensity resistance training in patients with rheumatoid arthritis: A randomized controlled trial. Arthritis Rheum. 2009, 61, 1726–1734. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.; Ferraz, R.B.; Kurimori, C.O.; Guedes, L.K.; Lima, F.R.; de Sa-Pinto, A.L.; Gualano, B.; Roschel, H. Low-Load Resistance Training With Blood-Flow Restriction in Relation to Muscle Function, Mass, and Functionality in Women With Rheumatoid Arthritis. Arthritis Care Res. 2020, 72, 787–797. [Google Scholar] [CrossRef]
- Piva, S.R.; Khoja, S.S.; Toledo, F.G.S.; Chester-Wasko, M.; Fitzgerald, G.K.; Goodpaster, B.H.; Smith, C.N.; Delitto, A. Neuromuscular Electrical Stimulation Compared to Volitional Exercise for Improving Muscle Function in Rheumatoid Arthritis: A Randomized Pilot Study. Arthritis Care Res. 2019, 71, 352–361. [Google Scholar] [CrossRef]
- Liao, C.D.; Chen, H.C.; Huang, S.W.; Liou, T.H. Exercise therapy for sarcopenia in rheumatoid arthritis: A meta-analysis and meta-regression of randomized controlled trials. Clin. Rehabil. 2022, 36, 145–157. [Google Scholar] [CrossRef]
- Oliveira, A.A.; Martins, F.M.; Júnior, R.F.; Michelin, M.A.; Sousa, A.P.; Nunes, P.R.P.; Murta, E.F.C.; Chica, J.E.L.; Orsatti, F.L. Rheumatoid arthritis-increased gene expressions in muscle atrophy are restored back to control as a response to acute resistance exercise. Rev. Bras. De Ciência E Mov. 2018, 26, 24–33. [Google Scholar] [CrossRef]
- Cutolo, M.; Nikiphorou, E. Don’t neglect nutrition in rheumatoid arthritis! RMD Open 2018, 4, e000591. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Romero-Yuste, S.; Chamizo Carmona, E.; Nolla, J.M. Sarcopenia, immune-mediated rheumatic diseases, and nutritional interventions. Aging Clin. Exp. Res. 2021, 33, 2929–2939. [Google Scholar] [CrossRef] [PubMed]
- Schaffler, A.; Ehling, A.; Neumann, E.; Herfarth, H.; Tarner, I.; Scholmerich, J.; Muller-Ladner, U.; Gay, S. Adipocytokines in synovial fluid. JAMA 2003, 290, 1709. [Google Scholar] [PubMed]
- Sglunda, O.; Mann, H.; Hulejova, H.; Kuklova, M.; Pecha, O.; Plestilova, L.; Filkova, M.; Pavelka, K.; Vencovsky, J.; Senolt, L. Decreased circulating visfatin is associated with improved disease activity in early rheumatoid arthritis: Data from the PERAC cohort. PLoS ONE 2014, 9, e103495. [Google Scholar] [CrossRef] [PubMed]
- Chihara, K.; Hattori, N.; Ichikawa, N.; Matsuda, T.; Saito, T. Re-evaluation of serum leptin and adiponectin concentrations normalized by body fat mass in patients with rheumatoid arthritis. Sci. Rep. 2020, 10, 15932. [Google Scholar] [CrossRef]
- Presle, N.; Pottie, P.; Dumond, H.; Guillaume, C.; Lapicque, F.; Pallu, S.; Mainard, D.; Netter, P.; Terlain, B. Differential distribution of adipokines between serum and synovial fluid in patients with osteoarthritis. Contribution of joint tissues to their articular production. Osteoarthr. Cartil. 2006, 14, 690–695. [Google Scholar] [CrossRef]
- Recinella, L.; Orlando, G.; Ferrante, C.; Chiavaroli, A.; Brunetti, L.; Leone, S. Adipokines: New Potential Therapeutic Target for Obesity and Metabolic, Rheumatic, and Cardiovascular Diseases. Front. Physiol. 2020, 11, 578966. [Google Scholar] [CrossRef]
- Misch, M.; Puthanveetil, P. The Head-to-Toe Hormone: Leptin as an Extensive Modulator of Physiologic Systems. Int. J. Mol. Sci. 2022, 23, 5439. [Google Scholar] [CrossRef]
- Abella, V.; Scotece, M.; Conde, J.; Pino, J.; Gonzalez-Gay, M.A.; Gomez-Reino, J.J.; Mera, A.; Lago, F.; Gomez, R.; Gualillo, O. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017, 13, 100–109. [Google Scholar] [CrossRef]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Paquet, J.; Goebel, J.C.; Delaunay, C.; Pinzano, A.; Grossin, L.; Cournil-Henrionnet, C.; Gillet, P.; Netter, P.; Jouzeau, J.Y.; Moulin, D. Cytokines profiling by multiplex analysis in experimental arthritis: Which pathophysiological relevance for articular versus systemic mediators? Arthritis Res. Ther. 2012, 14, R60. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L., Jr.; Shao, W.H.; Vanniasinghe, A.S.; Amon, M.A.; Holub, M.C.; Kovalszky, I.; Wade, J.D.; Doll, M.; Cohen, P.L.; Manolios, N.; et al. Toward understanding the role of leptin and leptin receptor antagonism in preclinical models of rheumatoid arthritis. Peptides 2011, 32, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Liu, Y.; Yang, M.; Wang, S.; Zhang, M.; Wang, X.; Ko, K.H.; Hua, Z.; Sun, L.; Cao, X.; et al. Leptin exacerbates collagen-induced arthritis via enhancement of Th17 cell response. Arthritis Rheum. 2012, 64, 3564–3573. [Google Scholar] [CrossRef] [PubMed]
- Busso, N.; So, A.; Chobaz-Peclat, V.; Morard, C.; Martinez-Soria, E.; Talabot-Ayer, D.; Gabay, C. Leptin signaling deficiency impairs humoral and cellular immune responses and attenuates experimental arthritis. J. Immunol. 2002, 168, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Bernotiene, E.; Palmer, G.; Talabot-Ayer, D.; Szalay-Quinodoz, I.; Gabay, C. Delayed resolution of acute inflammation during zymosan-induced arthritis in leptin-deficient mice. Arthritis Res. Ther. 2003, 5, 93. [Google Scholar] [CrossRef]
- Sugioka, Y.; Tada, M.; Okano, T.; Nakamura, H.; Koike, T. Acquired leptin resistance by high-fat feeding reduces inflammation from collagen antibody-induced arthritis in mice. Clin. Exp. Rheumatol. 2012, 30, 707–713. [Google Scholar]
- Tong, K.M.; Shieh, D.C.; Chen, C.P.; Tzeng, C.Y.; Wang, S.P.; Huang, K.C.; Chiu, Y.C.; Fong, Y.C.; Tang, C.H. Leptin induces IL-8 expression via leptin receptor, IRS-1, PI3K, Akt cascade and promotion of NF-kappaB/p300 binding in human synovial fibroblasts. Cell Signal 2008, 20, 1478–1488. [Google Scholar] [CrossRef]
- Wu, J.; Qu, Y.; Zhang, Y.P.; Deng, J.X.; Yu, Q.H. RHAMM induces progression of rheumatoid arthritis by enhancing the functions of fibroblast-like synoviocytes. BMC Musculoskelet. Disord. 2018, 19, 455. [Google Scholar] [CrossRef]
- Sun, X.; Wei, J.; Tang, Y.; Wang, B.; Zhang, Y.; Shi, L.; Guo, J.; Hu, F.; Li, X. Leptin-induced migration and angiogenesis in rheumatoid arthritis is mediated by reactive oxygen species. FEBS Open Bio 2017, 7, 1899–1908. [Google Scholar] [CrossRef]
- Figenschau, Y.; Knutsen, G.; Shahazeydi, S.; Johansen, O.; Sveinbjornsson, B. Human articular chondrocytes express functional leptin receptors. Biochem. Biophys. Res. Commun. 2001, 287, 190–197. [Google Scholar] [CrossRef]
- Cordero-Barreal, A.; Gonzalez-Rodriguez, M.; Ruiz-Fernandez, C.; Eldjoudi, D.A.; AbdElHafez, Y.R.F.; Lago, F.; Conde, J.; Gomez, R.; Gonzalez-Gay, M.A.; Mobasheri, A.; et al. An Update on the Role of Leptin in the Immuno-Metabolism of Cartilage. Int. J. Mol. Sci. 2021, 22, 2411. [Google Scholar] [CrossRef]
- Lee, S.W.; Rho, J.H.; Lee, S.Y.; Kim, J.H.; Cheong, J.H.; Kim, H.Y.; Jeong, N.Y.; Chung, W.T.; Yoo, Y.H. Leptin protects rat articular chondrocytes from cytotoxicity induced by TNF-alpha in the presence of cyclohexamide. Osteoarthr. Cartil. 2015, 23, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Kishida, Y.; Hirao, M.; Tamai, N.; Nampei, A.; Fujimoto, T.; Nakase, T.; Shimizu, N.; Yoshikawa, H.; Myoui, A. Leptin regulates chondrocyte differentiation and matrix maturation during endochondral ossification. Bone 2005, 37, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Simopoulou, T.; Malizos, K.; Iliopoulos, D.; Stefanou, N.; Papatheodorou, L.; Ioannou, M.; Tsezou, A. Differential expression of leptin and leptin’s receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism. Osteoarthr. Cartil. 2007, 15, 872–883. [Google Scholar] [CrossRef]
- Zhao, X.; Dong, Y.; Zhang, J.; Li, D.; Hu, G.; Yao, J.; Li, Y.; Huang, P.; Zhang, M.; Zhang, J.; et al. Leptin changes differentiation fate and induces senescence in chondrogenic progenitor cells. Cell Death Dis. 2016, 7, e2188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, P.; Li, G.; Lv, Z.; Hu, G.; Xu, Q. Activation of the leptin pathway by high expression of the long form of the leptin receptor (Ob-Rb) accelerates chondrocyte senescence in osteoarthritis. Bone Joint Res. 2019, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Du, S.; Huang, L.; Li, J.; Xiao, L.; Tong, P. Leptin promotes apoptosis and inhibits autophagy of chondrocytes through upregulating lysyl oxidase-like 3 during osteoarthritis pathogenesis. Osteoarthr. Cartil. 2016, 24, 1246–1253. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Shen, C.; Li, H.; Fan, Q.; Ding, J.; Jin, F.C.; Sha, L. Leptin induces the apoptosis of chondrocytes in an in vitro model of osteoarthritis via the JAK2-STAT3 signaling pathway. Mol. Med. Rep. 2016, 13, 3684–3690. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Fujio, K. Emerging role of leptin in joint inflammation and destruction. Immunol. Med. 2022, 45, 27–34. [Google Scholar] [CrossRef]
- Otero, M.; Lago, R.; Lago, F.; Reino, J.J.; Gualillo, O. Signalling pathway involved in nitric oxide synthase type II activation in chondrocytes: Synergistic effect of leptin with interleukin-1. Arthritis Res. Ther. 2005, 7, R581–R591. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Amling, M.; Takeda, S.; Priemel, M.; Schilling, A.F.; Beil, F.T.; Shen, J.; Vinson, C.; Rueger, J.M.; Karsenty, G. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell 2000, 100, 197–207. [Google Scholar] [CrossRef]
- Micheletti, C.; Jolic, M.; Grandfield, K.; Shah, F.A.; Palmquist, A. Bone structure and composition in a hyperglycemic, obese, and leptin receptor-deficient rat: Microscale characterization of femur and calvarium. Bone 2023, 172, 116747. [Google Scholar] [CrossRef]
- Vaira, S.; Yang, C.; McCoy, A.; Keys, K.; Xue, S.; Weinstein, E.J.; Novack, D.V.; Cui, X. Creation and preliminary characterization of a leptin knockout rat. Endocrinology 2012, 153, 5622–5628. [Google Scholar] [CrossRef]
- Pogoda, P.; Egermann, M.; Schnell, J.C.; Priemel, M.; Schilling, A.F.; Alini, M.; Schinke, T.; Rueger, J.M.; Schneider, E.; Clarke, I.; et al. Leptin inhibits bone formation not only in rodents, but also in sheep. J. Bone Miner. Res. 2006, 21, 1591–1599. [Google Scholar] [CrossRef]
- Holloway, W.R.; Collier, F.M.; Aitken, C.J.; Myers, D.E.; Hodge, J.M.; Malakellis, M.; Gough, T.J.; Collier, G.R.; Nicholson, G.C. Leptin inhibits osteoclast generation. J. Bone Miner. Res. 2002, 17, 200–209. [Google Scholar] [CrossRef]
- Bokarewa, M.; Bokarew, D.; Hultgren, O.; Tarkowski, A. Leptin consumption in the inflamed joints of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Nguyen, N.U.; Dumoulin, G.; Aubin, F.; Cedoz, J.P.; Wendling, D. Relationship between growth hormone-IGF-I-IGFBP-3 axis and serum leptin levels with bone mass and body composition in patients with rheumatoid arthritis. Rheumatology 2005, 44, 120–125. [Google Scholar] [CrossRef]
- Otero, M.; Lago, R.; Gomez, R.; Lago, F.; Dieguez, C.; Gomez-Reino, J.J.; Gualillo, O. Changes in plasma levels of fat-derived hormones adiponectin, leptin, resistin and visfatin in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1198–1201. [Google Scholar] [CrossRef]
- Xibille-Friedmann, D.; Bustos-Bahena, C.; Hernandez-Gongora, S.; Burgos-Vargas, R.; Montiel-Hernandez, J.L. Two-year follow-up of plasma leptin and other cytokines in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 930–931. [Google Scholar] [CrossRef] [PubMed]
- Rho, Y.H.; Solus, J.; Sokka, T.; Oeser, A.; Chung, C.P.; Gebretsadik, T.; Shintani, A.; Pincus, T.; Stein, C.M. Adipocytokines are associated with radiographic joint damage in rheumatoid arthritis. Arthritis Rheum. 2009, 60, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Olama, S.M.; Senna, M.K.; Elarman, M. Synovial/serum leptin ratio in rheumatoid arthritis: The association with activity and erosion. Rheumatol. Int. 2012, 32, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wei, J.; Li, H.; Ouyang, X.; Sun, X.; Tang, Y.; Chen, H.; Wang, B.; Li, X. Leptin Upregulates Peripheral CD4(+)CXCR5(+)ICOS(+) T Cells via Increased IL-6 in Rheumatoid Arthritis Patients. J. Interferon Cytokine Res. 2018, 38, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Dervisevic, A.; Resic, H.; Sokolovic, S.; Babic, N.; Avdagic, N.; Zaciragic, A.; Beciragic, A.; Fajkic, A.; Lepara, O.; Hadzovic-Dzuvo, A. Leptin is associated with disease activity but not with anthropometric indices in rheumatoid arthritis patients. Arch. Med. Sci. 2018, 14, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Arias-de la Rosa, I.; Escudero-Contreras, A.; Ruiz-Ponce, M.; Cuesta-Lopez, L.; Roman-Rodriguez, C.; Perez-Sanchez, C.; Ruiz-Limon, P.; Ruiz, R.G.; Leiva-Cepas, F.; Alcaide, J.; et al. Pathogenic mechanisms involving the interplay between adipose tissue and auto-antibodies in rheumatoid arthritis. iScience 2022, 25, 104893. [Google Scholar] [CrossRef]
- Tian, G.; Liang, J.N.; Pan, H.F.; Zhou, D. Increased leptin levels in patients with rheumatoid arthritis: A meta-analysis. Ir. J. Med. Sci. 2014, 183, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Scotece, M.; Lopez, V.; Abella, V.; Hermida, M.; Pino, J.; Lago, F.; Gomez-Reino, J.J.; Gualillo, O. Differential expression of adipokines in infrapatellar fat pad (IPFP) and synovium of osteoarthritis patients and healthy individuals. Ann. Rheum. Dis. 2014, 73, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Rihl, M.; Heufelder, A.; Loch, O.; Schattenkirchner, M. Leptin serum levels are not correlated with disease activity in patients with rheumatoid arthritis. Metabolism 1999, 48, 745–748. [Google Scholar] [CrossRef]
- Popa, C.; Netea, M.G.; Radstake, T.R.; van Riel, P.L.; Barrera, P.; van der Meer, J.W. Markers of inflammation are negatively correlated with serum leptin in rheumatoid arthritis. Ann. Rheum. Dis. 2005, 64, 1195–1198. [Google Scholar] [CrossRef]
- Hizmetli, S.; Kisa, M.; Gokalp, N.; Bakici, M.Z. Are plasma and synovial fluid leptin levels correlated with disease activity in rheumatoid arthritis ? Rheumatol. Int. 2007, 27, 335–338. [Google Scholar] [CrossRef]
- Gunaydin, R.; Kaya, T.; Atay, A.; Olmez, N.; Hur, A.; Koseoglu, M. Serum leptin levels in rheumatoid arthritis and relationship with disease activity. South. Med. J. 2006, 99, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Park, M.C.; Park, Y.B.; Lee, S.K. Measurement of the serum leptin level could assist disease activity monitoring in rheumatoid arthritis. Rheumatol. Int. 2007, 27, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Wislowska, M.; Rok, M.; Jaszczyk, B.; Stepien, K.; Cicha, M. Serum leptin in rheumatoid arthritis. Rheumatol. Int. 2007, 27, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Seven, A.; Guzel, S.; Aslan, M.; Hamuryudan, V. Serum and synovial fluid leptin levels and markers of inflammation in rheumatoid arthritis. Rheumatol. Int. 2009, 29, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Canoruç, N.; Kale, E.; Turhanoğlu, A.D.; Özmen, Ş.; Ogün, C.; Kaplan, A. Plasma resistin and leptin levels in overweight and lean patients with rheumatoid arthritis. Turk. J. Med. Sci. 2009, 39, 447–451. [Google Scholar] [CrossRef]
- Targonska-Stepniak, B.; Dryglewska, M.; Majdan, M. Adiponectin and leptin serum concentrations in patients with rheumatoid arthritis. Rheumatol. Int. 2010, 30, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.; Ali, H.A.-h.; Ibrahim, H.M. Possible role of leptin, and tumor necrosis factor-alpha in hypoandrogenicity in patients with early rheumatoid arthritis. Egypt. Rheumatol. 2011, 33, 209–215. [Google Scholar] [CrossRef]
- Yoshino, T.; Kusunoki, N.; Tanaka, N.; Kaneko, K.; Kusunoki, Y.; Endo, H.; Hasunuma, T.; Kawai, S. Elevated serum levels of resistin, leptin, and adiponectin are associated with C-reactive protein and also other clinical conditions in rheumatoid arthritis. Intern. Med. 2011, 50, 269–275. [Google Scholar] [CrossRef]
- Allam, A.; Radwan, A. The relationship of serum leptin levels with disease activity in Egyptian patients with rheumatoid arthritis. Egypt. Rheumatol. 2012, 34, 185–190. [Google Scholar] [CrossRef]
- Mirfeizi, Z.; Noubakht, Z.; Rezaie, A.E.; Jokar, M.H.; Sarabi, Z.S. Plasma levels of leptin and visfatin in rheumatoid arthritis patients; is there any relationship with joint damage? Iran. J. Basic. Med. Sci. 2014, 17, 662–666. [Google Scholar]
- Abdalla, M.; Effat, D.; Sheta, M.; Hamed, W.E. Serum Leptin levels in Rheumatoid arthritis and relationship with disease activity. Egypt. Rheumatol. 2014, 36, 1–5. [Google Scholar] [CrossRef]
- Bustos Rivera-Bahena, C.; Xibille-Friedmann, D.X.; Gonzalez-Christen, J.; Carrillo-Vazquez, S.M.; Montiel-Hernandez, J.L. Peripheral blood leptin and resistin levels as clinical activity biomarkers in Mexican Rheumatoid Arthritis patients. Reumatol. Clin. 2016, 12, 323–326. [Google Scholar] [CrossRef]
- Oner, S.Y.; Volkan, O.; Oner, C.; Mengi, A.; Direskeneli, H.; Tasan, D.A. Serum leptin levels do not correlate with disease activity in rheumatoid arthritis. Acta Reumatol. Port. 2015, 40, 50–54. [Google Scholar] [PubMed]
- Rodriguez, J.; Lafaurie, G.I.; Bautista-Molano, W.; Chila-Moreno, L.; Bello-Gualtero, J.M.; Romero-Sanchez, C. Adipokines and periodontal markers as risk indicators of early rheumatoid arthritis: A cross-sectional study. Clin. Oral. Investig. 2021, 25, 1685–1695. [Google Scholar] [CrossRef]
- Targonska-Stepniak, B.; Grzechnik, K. Adiponectin and Leptin as Biomarkers of Disease Activity and Metabolic Disorders in Rheumatoid Arthritis Patients. J. Inflamm. Res. 2022, 15, 5845–5855. [Google Scholar] [CrossRef]
- Taylan, A.; Akinci, B.; Toprak, B.; Birlik, M.; Arslan, F.D.; Ekerbicer, H.; Gundogdu, B.; Colak, A.; Engin, B. Association of Leptin Levels and Disease Activity in Patients with Early Rheumatoid Arthritis. Arch. Med. Res. 2021, 52, 544–553. [Google Scholar] [CrossRef]
- Magali Chamorro-Melo, Y.; Calixto, O.J.; Bello-Gualtero, J.M.; Bautista-Molano, W.; Beltran-Ostos, A.; Romero-Sanchez, C. Evaluation of the adipokine profile (adiponectin, resistin, adipsin, vaspin, and leptin) in patients with early rheumatoid arthritis and its correlation with disease activity. Reumatologia 2022, 60, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Romero-Sanchez, C.; De Avila, J.; Ramos-Casallas, A.; Chila-Moreno, L.; Delgadillo, N.A.; Chalem-Choueka, P.; Pacheco-Tena, C.; Bello-Gualtero, J.M.; Bautista-Molano, W. High Levels of Leptin and Adipsin Are Associated with Clinical Activity in Early Rheumatoid Arthritis Patients with Overweight and Periodontal Infection. Diagnostics 2023, 13, 1126. [Google Scholar] [CrossRef] [PubMed]
- Chaparro-Sanabria, J.A.; Bautista-Molano, W.; Bello-Gualtero, J.M.; Chila-Moreno, L.; Castillo, D.M.; Valle-Onate, R.; Chalem, P.; Romero-Sanchez, C. Association of adipokines with rheumatic disease activity indexes and periodontal disease in patients with early rheumatoid arthritis and their first-degree relatives. Int. J. Rheum. Dis. 2019, 22, 1990–2000. [Google Scholar] [CrossRef]
- Kim, K.S.; Choi, H.M.; Ji, H.I.; Song, R.; Yang, H.I.; Lee, S.K.; Yoo, M.C.; Park, Y.B. Serum adipokine levels in rheumatoid arthritis patients and their contributions to the resistance to treatment. Mol. Med. Rep. 2014, 9, 255–260. [Google Scholar] [CrossRef]
- Vasileiadis, G.K.; Lundell, A.C.; Zhang, Y.; Andersson, K.; Gjertsson, I.; Rudin, A.; Maglio, C. Adipocytokines in Untreated Newly Diagnosed Rheumatoid Arthritis: Association with Circulating Chemokines and Markers of Inflammation. Biomolecules 2021, 11, 325. [Google Scholar] [CrossRef]
- Baker, J.F.; England, B.R.; George, M.D.; Wysham, K.; Johnson, T.; Lenert, A.; Kunkel, G.; Sauer, B.; Duryee, M.J.; Monach, P.; et al. Adipocytokines and achievement of low disease activity in rheumatoid arthritis. Semin. Arthritis Rheum. 2022, 55, 152003. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Flake, D.D.; Weinblatt, M.E.; Shadick, N.A.; Ostergaard, M.; Hetland, M.L.; Brahe, C.H.; Hwang, Y.G.; Furst, D.E.; Strand, V.; et al. Adjustment of the multi-biomarker disease activity score to account for age, sex and adiposity in patients with rheumatoid arthritis. Rheumatology 2019, 58, 874–883. [Google Scholar] [CrossRef]
- Curtis, J.R.; Weinblatt, M.E.; Shadick, N.A.; Brahe, C.H.; Ostergaard, M.; Hetland, M.L.; Saevarsdottir, S.; Horton, M.; Mabey, B.; Flake, D.D., 2nd; et al. Validation of the adjusted multi-biomarker disease activity score as a prognostic test for radiographic progression in rheumatoid arthritis: A combined analysis of multiple studies. Arthritis Res. Ther. 2021, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, M.; Zarkesh-Esfahani, H.; Wu, Z.; Maamra, M.; Bidlingmaier, M.; Pockley, A.G.; Watson, P.; Matarese, G.; Strasburger, C.J.; Ross, R.J. Identification of a monoclonal antibody against the leptin receptor that acts as an antagonist and blocks human monocyte and T cell activation. J. Immunol. Methods 2006, 312, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.; De Santo, M.; Comandè, A.; Belsito, E.L.; Andò, S.; Liguori, A.; Leggio, A. Leptin-Activity Modulators and Their Potential Pharmaceutical Applications. Biomolecules 2021, 11, 1045. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, X.; Ye, X.; Li, X.; Wei, J. Roles of leptin on the key effector cells of rheumatoid arthritis. Immunol. Lett. 2021, 233, 92–96. [Google Scholar] [CrossRef]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef]
- Straub, L.G.; Scherer, P.E. Metabolic Messengers: Adiponectin. Nat. Metab. 2019, 1, 334–339. [Google Scholar] [CrossRef]
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2018, 8, 1031–1063. [Google Scholar] [CrossRef]
- Lei, X.; Qiu, S.; Yang, G.; Wu, Q. Adiponectin and metabolic cardiovascular diseases: Therapeutic opportunities and challenges. Genes. Dis. 2023, 10, 1525–1536. [Google Scholar] [CrossRef]
- Huang, C.C.; Law, Y.Y.; Liu, S.C.; Hu, S.L.; Lin, J.A.; Chen, C.J.; Wang, S.W.; Tang, C.H. Adiponectin Promotes VEGF Expression in Rheumatoid Arthritis Synovial Fibroblasts and Induces Endothelial Progenitor Cell Angiogenesis by Inhibiting miR-106a-5p. Cells 2021, 10, 2627. [Google Scholar] [CrossRef]
- Sun, X.; Feng, X.; Tan, W.; Lin, N.; Hua, M.; Wei, Y.; Wang, F.; Li, N.; Zhang, M. Adiponectin exacerbates collagen-induced arthritis via enhancing Th17 response and prompting RANKL expression. Sci. Rep. 2015, 5, 11296. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.M.; Lee, Y.A.; Lee, S.H.; Hong, S.J.; Hahm, D.H.; Choi, S.Y.; Yang, H.I.; Yoo, M.C.; Kim, K.S. Adiponectin may contribute to synovitis and joint destruction in rheumatoid arthritis by stimulating vascular endothelial growth factor, matrix metalloproteinase-1, and matrix metalloproteinase-13 expression in fibroblast-like synoviocytes more than proinflammatory mediators. Arthritis Res. Ther. 2009, 11, R161. [Google Scholar] [CrossRef] [PubMed]
- Skalska, U.; Kontny, E. Adiponectin Isoforms and Leptin Impact on Rheumatoid Adipose Mesenchymal Stem Cells Function. Stem Cells Int. 2016, 2016, 6532860. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Luo, S.; Li, Z. Multifaceted roles of adiponectin in rheumatoid arthritis. Int. Immunopharmacol. 2015, 28, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Lee, Y.A.; Ji, H.I.; Song, R.; Kim, J.Y.; Lee, S.H.; Hong, S.J.; Yoo, M.C.; Yang, H.I. Increased expression of endocan in arthritic synovial tissues: Effects of adiponectin on the expression of endocan in fibroblast-like synoviocytes. Mol. Med. Rep. 2015, 11, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.A.; Ji, H.I.; Lee, S.H.; Hong, S.J.; Yang, H.I.; Chul Yoo, M.; Kim, K.S. The role of adiponectin in the production of IL-6, IL-8, VEGF and MMPs in human endothelial cells and osteoblasts: Implications for arthritic joints. Exp. Mol. Med. 2014, 46, e72. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-A.; Choi, H.M.; Lee, S.-H.; Yang, H.-I.; Yoo, M.C.; Hong, S.-J.; Kim, K.S. Synergy between adiponectin and interleukin-1β on the expression of interleukin-6, interleukin-8, and cyclooxygenase-2 in fibroblast-like synoviocytes. Exp. Mol. Med. 2012, 44, 440–447. [Google Scholar] [CrossRef]
- Krumbholz, G.; Junker, S.; Meier, F.M.P.; Rickert, M.; Steinmeyer, J.; Rehart, S.; Lange, U.; Frommer, K.W.; Schett, G.; Muller-Ladner, U.; et al. Response of human rheumatoid arthritis osteoblasts and osteoclasts to adiponectin. Clin. Exp. Rheumatol. 2017, 35, 406–414. [Google Scholar]
- Qian, J.; Xu, L.; Sun, X.; Wang, Y.; Xuan, W.; Zhang, Q.; Zhao, P.; Wu, Q.; Liu, R.; Che, N.; et al. Adiponectin aggravates bone erosion by promoting osteopontin production in synovial tissue of rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 26. [Google Scholar] [CrossRef]
- Liu, H.; Liu, S.; Ji, H.; Zhao, Q.; Liu, Y.; Hu, P.; Luo, E. An adiponectin receptor agonist promote osteogenesis via regulating bone-fat balance. Cell Prolif. 2021, 54, e13035. [Google Scholar] [CrossRef]
- Yang, J.; Park, O.J.; Kim, J.; Han, S.; Yang, Y.; Yun, C.H.; Han, S.H. Adiponectin Deficiency Triggers Bone Loss by Up-Regulation of Osteoclastogenesis and Down-Regulation of Osteoblastogenesis. Front. Endocrinol. 2019, 10, 815. [Google Scholar] [CrossRef]
- Lewis, J.W.; Edwards, J.R.; Naylor, A.J.; McGettrick, H.M. Adiponectin signalling in bone homeostasis, with age and in disease. Bone Res. 2021, 9, 1–11. [Google Scholar] [CrossRef]
- Lee, Y.A.; Hahm, D.H.; Kim, J.Y.; Sur, B.; Lee, H.M.; Ryu, C.J.; Yang, H.I.; Kim, K.S. Potential therapeutic antibodies targeting specific adiponectin isoforms in rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 245. [Google Scholar] [CrossRef]
- Liu, R.; Zhao, P.; Zhang, Q.; Che, N.; Xu, L.; Qian, J.; Tan, W.; Zhang, M. Adiponectin promotes fibroblast-like synoviocytes producing IL-6 to enhance T follicular helper cells response in rheumatoid arthritis. Clin. Exp. Rheumatol. 2020, 38, 11–18. [Google Scholar]
- Senolt, L.; Pavelka, K.; Housa, D.; Haluzik, M. Increased adiponectin is negatively linked to the local inflammatory process in patients with rheumatoid arthritis. Cytokine 2006, 35, 247–252. [Google Scholar] [CrossRef]
- Giles, J.T.; Allison, M.; Bingham, C.O., 3rd; Scott, W.M., Jr.; Bathon, J.M. Adiponectin is a mediator of the inverse association of adiposity with radiographic damage in rheumatoid arthritis. Arthritis Rheum. 2009, 61, 1248–1256. [Google Scholar] [CrossRef]
- Alkady, E.A.; Ahmed, H.M.; Tag, L.; Abdou, M.A. Serum and synovial adiponectin, resistin, and visfatin levels in rheumatoid arthritis patients. Relation to disease activity. Z. Rheumatol. 2011, 70, 602–608. [Google Scholar] [CrossRef]
- Khajoei, S.; Hassaninevisi, M.; Kianmehr, N.; Seif, F.; Khoshmirsafa, M.; Shekarabi, M.; Samei, A.; Haghighi, A. Serum levels of adiponectin and vitamin D correlate with activity of Rheumatoid Arthritis. Mol. Biol. Rep. 2019, 46, 2505–2512. [Google Scholar] [CrossRef]
- Ozgen, M.; Koca, S.S.; Dagli, N.; Balin, M.; Ustundag, B.; Isik, A. Serum adiponectin and vaspin levels in rheumatoid arthritis. Arch. Med. Res. 2010, 41, 457–463. [Google Scholar] [CrossRef]
- Laurberg, T.B.; Frystyk, J.; Ellingsen, T.; Hansen, I.T.; Jorgensen, A.; Tarp, U.; Hetland, M.L.; Horslev-Petersen, K.; Hornung, N.; Poulsen, J.H.; et al. Plasma adiponectin in patients with active, early, and chronic rheumatoid arthritis who are steroid- and disease-modifying antirheumatic drug-naive compared with patients with osteoarthritis and controls. J. Rheumatol. 2009, 36, 1885–1891. [Google Scholar] [CrossRef]
- Popa, C.; Netea, M.G.; de Graaf, J.; van den Hoogen, F.H.; Radstake, T.R.; Toenhake-Dijkstra, H.; van Riel, P.L.; van der Meer, J.W.; Stalenhoef, A.F.; Barrera, P. Circulating leptin and adiponectin concentrations during tumor necrosis factor blockade in patients with active rheumatoid arthritis. J. Rheumatol. 2009, 36, 724–730. [Google Scholar] [CrossRef]
- Ebina, K.; Fukuhara, A.; Ando, W.; Hirao, M.; Koga, T.; Oshima, K.; Matsuda, M.; Maeda, K.; Nakamura, T.; Ochi, T.; et al. Serum adiponectin concentrations correlate with severity of rheumatoid arthritis evaluated by extent of joint destruction. Clin. Rheumatol. 2009, 28, 445–451. [Google Scholar] [CrossRef]
- Ibrahim, S.M.; Hamdy, M.S.; Amer, N. Plasma and synovial fluid adipocytokines in patients with rheumatoid arthritis and osteoarthritis. Egypt. J. Immunol. 2008, 15, 159–170. [Google Scholar]
- Kondrat’eva, L.V.; Gorbunova Iu, N.; Popkova, T.V.; Nasonov, E.L. The role of adipose tissue in rheumatoid arthritis. Klin. Med. 2014, 92, 62–67. [Google Scholar]
- Chennareddy, S.; Babu, K.V.K.; Kommireddy, S.; Varaprasad, R.; Rajasekhar, L. Serum adiponectin and its impact on disease activity and radiographic joint damage in early rheumatoid arthritis–a cross-sectional study. Indian J. Rheumatol. 2016, 11, 82–85. [Google Scholar] [CrossRef]
- Zhang, Y.; Peltonen, M.; Andersson-Assarsson, J.C.; Svensson, P.A.; Herder, C.; Rudin, A.; Carlsson, L.; Maglio, C. Elevated adiponectin predicts the development of rheumatoid arthritis in subjects with obesity. Scand. J. Rheumatol. 2020, 49, 452–460. [Google Scholar] [CrossRef]
- Tan, W.; Wang, F.; Zhang, M.; Guo, D.; Zhang, Q.; He, S. High adiponectin and adiponectin receptor 1 expression in synovial fluids and synovial tissues of patients with rheumatoid arthritis. Semin. Arthritis Rheum. 2009, 38, 420–427. [Google Scholar] [CrossRef]
- Klein-Wieringa, I.R.; van der Linden, M.P.; Knevel, R.; Kwekkeboom, J.C.; van Beelen, E.; Huizinga, T.W.; van der Helm-van Mil, A.; Kloppenburg, M.; Toes, R.E.; Ioan-Facsinay, A. Baseline serum adipokine levels predict radiographic progression in early rheumatoid arthritis. Arthritis Rheum. 2011, 63, 2567–2574. [Google Scholar] [CrossRef]
- Giles, J.T.; van der Heijde, D.M.; Bathon, J.M. Association of circulating adiponectin levels with progression of radiographic joint destruction in rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Park, H.J.; Kang, M.I.; Lee, H.S.; Lee, S.W.; Lee, S.K.; Park, Y.B. Adipokines, inflammation, insulin resistance, and carotid atherosclerosis in patients with rheumatoid arthritis. Arthritis Res. Ther. 2013, 15, R194. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Sellam, J.; Fellahi, S.; Kotti, S.; Bastard, J.P.; Meyer, O.; Liote, F.; Simon, T.; Capeau, J.; Berenbaum, F. Serum level of adiponectin is a surrogate independent biomarker of radiographic disease progression in early rheumatoid arthritis: Results from the ESPOIR cohort. Arthritis Res. Ther. 2013, 15, R210. [Google Scholar] [CrossRef]
- Toussirot, E.; Grandclement, E.; Gaugler, B.; Michel, F.; Wendling, D.; Saas, P.; Dumoulin, G.; CBT-506. Serum adipokines and adipose tissue distribution in rheumatoid arthritis and ankylosing spondylitis. A comparative study. Front. Immunol. 2013, 4, 453. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.F.; England, B.R.; George, M.D.; Wysham, K.; Johnson, T.; Kunkel, G.; Sauer, B.; Hamilton, B.C.; Hunter, C.D.; Duryee, M.J.; et al. Elevations in adipocytokines and mortality in rheumatoid arthritis. Rheumatology 2022, 61, 4924–4934. [Google Scholar] [CrossRef] [PubMed]
- Minamino, H.; Katsushima, M.; Yoshida, T.; Hashimoto, M.; Fujita, Y.; Shirakashi, M.; Yamamoto, W.; Murakami, K.; Murata, K.; Nishitani, K.; et al. Increased circulating adiponectin is an independent disease activity marker in patients with rheumatoid arthritis: A cross-sectional study using the KURAMA database. PLoS ONE 2020, 15, e0229998. [Google Scholar] [CrossRef]
- Lei, Y.; Li, X.; Gao, Z.; Liu, Y.; Zhang, B.; Xia, L.; Lu, J.; Shen, H. Association Between Adiponectin and Clinical Manifestations in Rheumatoid Arthritis. J. Interferon Cytokine Res. 2020, 40, 501–508. [Google Scholar] [CrossRef]
- Chen, X.; Wang, K.; Lu, T.; Wang, J.; Zhou, T.; Tian, J.; Zhou, B.; Long, L.; Zhou, Q. Adiponectin is negatively associated with disease activity and Sharp score in treatment-naive Han Chinese rheumatoid arthritis patients. Sci. Rep. 2022, 12, 2092. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef]
- Dakroub, A.; Nasser, S.A.; Kobeissy, F.; Yassine, H.M.; Orekhov, A.; Sharifi-Rad, J.; Iratni, R.; El-Yazbi, A.F.; Eid, A.H. Visfatin: An emerging adipocytokine bridging the gap in the evolution of cardiovascular diseases. J. Cell Physiol. 2021, 236, 6282–6296. [Google Scholar] [CrossRef]
- Sun, Z.; Lei, H.; Zhang, Z. Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions. Cytokine Growth Factor. Rev. 2013, 24, 433–442. [Google Scholar] [CrossRef]
- Lee, W.J.; Wu, C.S.; Lin, H.; Lee, I.T.; Wu, C.M.; Tseng, J.J.; Chou, M.M.; Sheu, W.H. Visfatin-induced expression of inflammatory mediators in human endothelial cells through the NF-kappaB pathway. Int. J. Obes. 2009, 33, 465–472. [Google Scholar] [CrossRef]
- Kim, S.R.; Bae, Y.H.; Bae, S.K.; Choi, K.S.; Yoon, K.H.; Koo, T.H.; Jang, H.O.; Yun, I.; Kim, K.W.; Kwon, Y.G.; et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim. Biophys. Acta 2008, 1783, 886–895. [Google Scholar] [CrossRef]
- Senolt, L.; Krystufkova, O.; Hulejova, H.; Kuklova, M.; Filkova, M.; Cerezo, L.A.; Belacek, J.; Haluzik, M.; Forejtova, S.; Gay, S.; et al. The level of serum visfatin (PBEF) is associated with total number of B cells in patients with rheumatoid arthritis and decreases following B cell depletion therapy. Cytokine 2011, 55, 116–121. [Google Scholar] [CrossRef]
- Matsui, H.; Tsutsumi, A.; Sugihara, M.; Suzuki, T.; Iwanami, K.; Kohno, M.; Goto, D.; Matsumoto, I.; Ito, S.; Sumida, T. Visfatin (pre-B cell colony-enhancing factor) gene expression in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 571–572. [Google Scholar] [CrossRef]
- El-Hini, S.H.; Mohamed, F.I.; Hassan, A.A.; Ali, F.; Mahmoud, A.; Ibraheem, H.M. Visfatin and adiponectin as novel markers for evaluation of metabolic disturbance in recently diagnosed rheumatoid arthritis patients. Rheumatol. Int. 2013, 33, 2283–2289. [Google Scholar] [CrossRef] [PubMed]
- Ozgen, M.; Koca, S.S.; Aksoy, K.; Dagli, N.; Ustundag, B.; Isik, A. Visfatin levels and intima-media thicknesses in rheumatic diseases. Clin. Rheumatol. 2011, 30, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Nowell, M.A.; Richards, P.J.; Fielding, C.A.; Ognjanovic, S.; Topley, N.; Williams, A.S.; Bryant-Greenwood, G.; Jones, S.A. Regulation of pre-B cell colony-enhancing factor by STAT-3-dependent interleukin-6 trans-signaling: Implications in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2084–2095. [Google Scholar] [CrossRef]
- Jurcovicova, J.; Stofkova, A.; Skurlova, M.; Baculikova, M.; Zorad, S.; Stancikova, M. Alterations in adipocyte glucose transporter GLUT4 and circulating adiponectin and visfatin in rat adjuvant induced arthritis. Gen. Physiol. Biophys. 2010, 29, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Busso, N.; Karababa, M.; Nobile, M.; Rolaz, A.; Van Gool, F.; Galli, M.; Leo, O.; So, A.; De Smedt, T. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS ONE 2008, 3, e2267. [Google Scholar] [CrossRef] [PubMed]
- Presumey, J.; Courties, G.; Louis-Plence, P.; Escriou, V.; Scherman, D.; Pers, Y.M.; Yssel, H.; Pene, J.; Kyburz, D.; Gay, S.; et al. Nicotinamide phosphoribosyltransferase/visfatin expression by inflammatory monocytes mediates arthritis pathogenesis. Ann. Rheum. Dis. 2013, 72, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Islam, S.; Xiong, M.; Nsumu, N.N.; Lee, M.W., Jr.; Zhang, L.Q.; Ueki, Y.; Heruth, D.P.; Lei, G.; Ye, S.Q. Epigenetic regulation of NfatC1 transcription and osteoclastogenesis by nicotinamide phosphoribosyl transferase in the pathogenesis of arthritis. Cell Death Discov. 2019, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Brentano, F.; Schorr, O.; Ospelt, C.; Stanczyk, J.; Gay, R.E.; Gay, S.; Kyburz, D. Pre-B cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum. 2007, 56, 2829–2839. [Google Scholar] [CrossRef]
- Meier, F.M.; Frommer, K.W.; Peters, M.A.; Brentano, F.; Lefevre, S.; Schroder, D.; Kyburz, D.; Steinmeyer, J.; Rehart, S.; Gay, S.; et al. Visfatin/pre-B-cell colony-enhancing factor (PBEF), a proinflammatory and cell motility-changing factor in rheumatoid arthritis. J. Biol. Chem. 2012, 287, 28378–28385. [Google Scholar] [CrossRef]
- Hasseli, R.; Frommer, K.W.; Schwarz, M.; Hulser, M.L.; Schreiyack, C.; Arnold, M.; Diller, M.; Tarner, I.H.; Lange, U.; Pons-Kuhnemann, J.; et al. Adipokines and Inflammation Alter the Interaction Between Rheumatoid Arthritis Synovial Fibroblasts and Endothelial Cells. Front. Immunol. 2020, 11, 925. [Google Scholar] [CrossRef]
- Tsiklauri, L.; Werner, J.; Kampschulte, M.; Frommer, K.W.; Berninger, L.; Irrgang, M.; Glenske, K.; Hose, D.; El Khassawna, T.; Pons-Kuhnemann, J.; et al. Visfatin alters the cytokine and matrix-degrading enzyme profile during osteogenic and adipogenic MSC differentiation. Osteoarthr. Cartil. 2018, 26, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Gosset, M.; Berenbaum, F.; Salvat, C.; Sautet, A.; Pigenet, A.; Tahiri, K.; Jacques, C. Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: Possible influence on osteoarthritis. Arthritis Rheum. 2008, 58, 1399–1409. [Google Scholar] [CrossRef]
- Ling, M.; Huang, P.; Islam, S.; Heruth, D.P.; Li, X.; Zhang, L.Q.; Li, D.Y.; Hu, Z.; Ye, S.Q. Epigenetic regulation of Runx2 transcription and osteoblast differentiation by nicotinamide phosphoribosyltransferase. Cell Biosci. 2017, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- He, X.; He, J.; Shi, Y.; Pi, C.; Yang, Y.; Sun, Y.; Ma, C.; Lin, L.; Zhang, L.; Li, Y. Nicotinamide phosphoribosyltransferase (Nampt) may serve as the marker for osteoblast differentiation of bone marrow-derived mesenchymal stem cells. Exp. Cell Res. 2017, 352, 45–52. [Google Scholar] [CrossRef]
- Khalifa, I.A.; Ibrahim, A.; Abdelfattah, A. Relation between serum visfatin and clinical severity in different stages of rheumatoid arthritis. Egypt. Rheumatol. Rehabil. 2014, 40, 1–8. [Google Scholar] [CrossRef]
- Mohammed Ali, D.M.; Al-Fadhel, S.Z.; Al-Ghuraibawi, N.H.A.; Al-Hakeim, H.K. Serum chemerin and visfatin levels and their ratio as possible diagnostic parameters of rheumatoid arthritis. Reumatologia 2020, 58, 67–75. [Google Scholar] [CrossRef]
- Franco-Trepat, E.; Alonso-Perez, A.; Guillan-Fresco, M.; Jorge-Mora, A.; Gualillo, O.; Gomez-Reino, J.J.; Gomez Bahamonde, R. Visfatin as a therapeutic target for rheumatoid arthritis. Expert. Opin. Ther. Targets 2019, 23, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.P.; Chen, W.P.; Wu, L.D. Visfatin: A potential therapeutic target for rheumatoid arthritis. J. Int. Med. Res. 2009, 37, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Polyakova, Y.V.; Zavodovsky, B.V.; Sivordova, L.E.; Akhverdyan, Y.R.; Zborovskaya, I.A. Visfatin and Rheumatoid Arthritis: Pathogenetic Implications and Clinical Utility. Curr. Rheumatol. Rev. 2020, 16, 224–239. [Google Scholar] [CrossRef]
- Nowell, M.; Evans, L.; Williams, A. PBEF/NAMPT/visfatin: A promising drug target for treating rheumatoid arthritis? Future Med. Chem. 2012, 4, 751–769. [Google Scholar] [CrossRef]
- Jebur, M.M.; Al-qaisi, A.H.J.; Harbi, N.S. Evaluation Serum Chemerin and Visfatin Levels with Rheumatoid Arthritis: Possible Diagnostic Biomarkers. Int. J. Curr. Res. Rev. 2022, 14, 42. [Google Scholar] [CrossRef]
- Tripathi, D.; Kant, S.; Pandey, S.; Ehtesham, N.Z. Resistin in metabolism, inflammation, and disease. FEBS J. 2020, 287, 3141–3149. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.S.; Weakley, S.M.; Yao, Q.; Chen, C. Resistin: Functional roles and therapeutic considerations for cardiovascular disease. Br. J. Pharmacol. 2012, 165, 622–632. [Google Scholar] [CrossRef]
- Su, C.M.; Huang, C.Y.; Tang, C.H. Characteristics of resistin in rheumatoid arthritis angiogenesis. Biomark. Med. 2016, 10, 651–660. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, X.; Hensley, G.; Chang, L.W.; Liao, W.; Abu-Amer, Y.; Sandell, L.J. Resistin induces expression of proinflammatory cytokines and chemokines in human articular chondrocytes via transcription and messenger RNA stabilization. Arthritis Rheum. 2010, 62, 1993–2003. [Google Scholar] [CrossRef]
- Bokarewa, M.; Nagaev, I.; Dahlberg, L.; Smith, U.; Tarkowski, A. Resistin, an adipokine with potent proinflammatory properties. J. Immunol. 2005, 174, 5789–5795. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Muraoka, S.; Kusunoki, N.; Masuoka, S.; Yamada, S.; Ogasawara, H.; Imai, T.; Akasaka, Y.; Tochigi, N.; Takahashi, H.; et al. Resistin upregulates chemokine production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 263. [Google Scholar] [CrossRef] [PubMed]
- Fedoce, A.G.; Veras, F.P.; Rosa, M.H.; Silva, J.F.; Paiva, I.M.; Schneider, A.H.; Machado, M.R.; Cunha, F.Q.; Cassia Aleixo Tostes, R. Resistin contributes perivascular adipose tissue dysfunction in a rheumatoid arthritis mouse model. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Kassem, E.; Mahmoud, L.; Salah, W. Study of Resistin and YKL-40 in rheumatoid arthritis. J. Am. Sci. 2010, 6, 1004–1012. [Google Scholar]
- Migita, K.; Maeda, Y.; Miyashita, T.; Kimura, H.; Nakamura, M.; Ishibashi, H.; Eguchi, K. The serum levels of resistin in rheumatoid arthritis patients. Clin. Exp. Rheumatol. 2006, 24, 698–701. [Google Scholar]
- Fadda, S.M.; Gamal, S.M.; Elsaid, N.Y.; Mohy, A.M. Resistin in inflammatory and degenerative rheumatologic diseases. Relationship between resistin and rheumatoid arthritis disease progression. Z. Rheumatol. 2013, 72, 594–600. [Google Scholar] [CrossRef]
- Kontunen, P.; Vuolteenaho, K.; Nieminen, R.; Lehtimaki, L.; Kautiainen, H.; Kesaniemi, Y.; Ukkola, O.; Kauppi, M.; Hakala, M.; Moilanen, E. Resistin is linked to inflammation, and leptin to metabolic syndrome, in women with inflammatory arthritis. Scand. J. Rheumatol. 2011, 40, 256–262. [Google Scholar] [CrossRef]
- Hammad, M.H.; Nasef, S.; Musalam, D.; Ahmed, M.; Osman, I.; Hammad, M. Resistin, an adipokine, its relation to inflammation in Systemic Lupus Erythematosus and Rheumatoid Arthritis. Middle East. J. Intern. Med. 2014, 7, 3–9. [Google Scholar] [CrossRef]
- Senolt, L.; Housa, D.; Vernerova, Z.; Jirasek, T.; Svobodova, R.; Veigl, D.; Anderlova, K.; Muller-Ladner, U.; Pavelka, K.; Haluzik, M. Resistin in rheumatoid arthritis synovial tissue, synovial fluid and serum. Ann. Rheum. Dis. 2007, 66, 458–463. [Google Scholar] [CrossRef]
- Forsblad d’Elia, H.; Pullerits, R.; Carlsten, H.; Bokarewa, M. Resistin in serum is associated with higher levels of IL-1Ra in post-menopausal women with rheumatoid arthritis. Rheumatology 2008, 47, 1082–1087. [Google Scholar] [CrossRef]
- Vuolteenaho, K.; Tuure, L.; Nieminen, R.; Laasonen, L.; Leirisalo-Repo, M.; Moilanen, E.; Group, N.E.-R.S. Pretreatment resistin levels are associated with erosive disease in early rheumatoid arthritis treated with disease-modifying anti-rheumatic drugs and infliximab. Scand. J. Rheumatol. 2022, 51, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Tao, S.S.; Zhang, Y.J.; Zhang, C.; Li, L.J.; Zhao, W.; Zhao, M.Q.; Li, P.; Pan, H.F.; Mao, C.; et al. Serum resistin levels in patients with rheumatoid arthritis and systemic lupus erythematosus: A meta-analysis. Clin. Rheumatol. 2015, 34, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gay, M.A.; Garcia-Unzueta, M.T.; Gonzalez-Juanatey, C.; Miranda-Filloy, J.A.; Vazquez-Rodriguez, T.R.; De Matias, J.M.; Martin, J.; Dessein, P.H.; Llorca, J. Anti-TNF-alpha therapy modulates resistin in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2008, 26, 311–316. [Google Scholar] [PubMed]
- Klaasen, R.; Herenius, M.M.; Wijbrandts, C.A.; de Jager, W.; van Tuyl, L.H.; Nurmohamed, M.T.; Prakken, B.J.; Gerlag, D.M.; Tak, P.P. Treatment-specific changes in circulating adipocytokines: A comparison between tumour necrosis factor blockade and glucocorticoid treatment for rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Kurowska, P.; Mlyczynska, E.; Dawid, M.; Jurek, M.; Klimczyk, D.; Dupont, J.; Rak, A. Review: Vaspin (SERPINA12) Expression and Function in Endocrine Cells. Cells 2021, 10, 1710. [Google Scholar] [CrossRef] [PubMed]
- Zieger, K.; Weiner, J.; Krause, K.; Schwarz, M.; Kohn, M.; Stumvoll, M.; Bluher, M.; Heiker, J.T. Vaspin suppresses cytokine-induced inflammation in 3T3-L1 adipocytes via inhibition of NFkappaB pathway. Mol. Cell Endocrinol. 2018, 460, 181–188. [Google Scholar] [CrossRef]
- Zhu, X.; Jiang, Y.; Shan, P.F.; Shen, J.; Liang, Q.H.; Cui, R.R.; Liu, Y.; Liu, G.Y.; Wu, S.S.; Lu, Q.; et al. Vaspin attenuates the apoptosis of human osteoblasts through ERK signaling pathway. Amino Acids 2013, 44, 961–968. [Google Scholar] [CrossRef]
- Kamio, N.; Kawato, T.; Tanabe, N.; Kitami, S.; Morita, T.; Ochiai, K.; Maeno, M. Vaspin attenuates RANKL-induced osteoclast formation in RAW264.7 cells. Connect. Tissue Res. 2013, 54, 147–152. [Google Scholar] [CrossRef]
- Wang, H.; Chen, F.; Li, J.; Wang, Y.; Jiang, C.; Wang, Y.; Zhang, M.; Xu, J. Vaspin antagonizes high fat-induced bone loss in rats and promotes osteoblastic differentiation in primary rat osteoblasts through Smad-Runx2 signaling pathway. Nutr. Metab. 2020, 17, 1–16. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, F.; Pei, H.X.; Zhu, X.; Lin, X.; Song, C.Y.; Liang, Q.H.; Liao, E.Y.; Yuan, L.Q. Vaspin regulates the osteogenic differentiation of MC3T3-E1 through the PI3K-Akt/miR-34c loop. Sci. Rep. 2016, 6, 25578. [Google Scholar] [CrossRef]
- Bao, J.P.; Xu, L.H.; Ran, J.S.; Xiong, Y.; Wu, L.D. Vaspin prevents leptin-induced inflammation and catabolism by inhibiting the activation of nuclear factor-kappaB in rat chondrocytes. Mol. Med. Rep. 2017, 16, 2925–2930. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.P.; Jiang, L.F.; Li, J.; Chen, W.P.; Hu, P.F.; Wu, L.D. Visceral adipose tissue-derived serine protease inhibitor inhibits interleukin-1beta-induced catabolic and inflammatory responses in murine chondrocytes. Mol. Med. Rep. 2014, 10, 2191–2197. [Google Scholar] [CrossRef] [PubMed]
- Senolt, L.; Polanska, M.; Filkova, M.; Cerezo, L.A.; Pavelka, K.; Gay, S.; Haluzik, M.; Vencovsky, J. Vaspin and omentin: New adipokines differentially regulated at the site of inflammation in rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1410–1411. [Google Scholar] [CrossRef]
- Wahba, A.S.; Ibrahim, M.E.; Abo-Elmatty, D.M.; Mehanna, E.T. Association of the Adipokines Chemerin, Apelin, Vaspin and Omentin and Their Functional Genetic Variants with Rheumatoid Arthritis. J. Pers. Med. 2021, 11, 976. [Google Scholar] [CrossRef]
- Ha, Y.-J.; Kang, E.-J.; Song, J.-S.; Park, Y.-B.; Lee, S.-K.; Choi, S.T. Plasma chemerin levels in rheumatoid arthritis are correlated with disease activity rather than obesity. Jt. Bone Spine 2013, 81, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Takemura, M.; Serrero, G.; Hayashi, J.; Yue, B.; Tsuboi, A.; Kubo, H.; Mitsuhashi, T.; Mannami, K.; Sato, M.; et al. Increased serum GP88 (Progranulin) concentrations in rheumatoid arthritis. Inflammation 2014, 37, 1806–1813. [Google Scholar] [CrossRef]
- Cerezo, L.A.; Kuklova, M.; Hulejova, H.; Vernerova, Z.; Kasprikova, N.; Veigl, D.; Pavelka, K.; Vencovsky, J.; Senolt, L. Progranulin Is Associated with Disease Activity in Patients with Rheumatoid Arthritis. Mediators Inflamm. 2015, 2015, 740357. [Google Scholar] [CrossRef]
- Chen, J.; Li, S.; Shi, J.; Zhang, L.; Li, J.; Chen, S.; Wu, C.; Shen, B. Serum progranulin irrelated with Breg cell levels, but elevated in RA patients, reflecting high disease activity. Rheumatol. Int. 2016, 36, 359–364. [Google Scholar] [CrossRef]
- Fouad, N.A.; Nassr, M.H.; Fathi, H.M.; Zaki, O.M.; Negm, A.A.; Senara, S.H. Potential value of serum progranulin as an activity biomarker in rheumatoid arthritis patients: Relation to musculoskeletal ultrasonographic evaluation. Egypt. Rheumatol. 2019, 41, 93–97. [Google Scholar] [CrossRef]
- Kvlividze, T.Z.; Zavodovsky, B.V.; Akhverdyan, Y.R.; Polyakova, Y.V.; Sivordova, L.E.; Yakovlev, A.T.; Zborovskaya, I.A. Serum nesfatin-1 as a marker of systemic inflammation in rheumatoid arthritis. Klin. Lab. Diagn. 2019, 64, 53–56. [Google Scholar] [CrossRef]
- Gamal, R.M.; Mohamed, M.E.; Hammam, N.; El Fetoh, N.A.; Rashed, A.M.; Furst, D.E. Preliminary study of the association of serum irisin levels with poor sleep quality in rheumatoid arthritis patients. Sleep. Med. 2020, 67, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ponce, F.; Gamez-Nava, J.I.; Perez-Guerrero, E.E.; Saldana-Cruz, A.M.; Vazquez-Villegas, M.L.; Ponce-Guarneros, J.M.; Huerta, M.; Trujillo, X.; Contreras-Haro, B.; Rocha-Munoz, A.D.; et al. Serum chemerin levels: A potential biomarker of joint inflammation in women with rheumatoid arthritis. PLoS ONE 2021, 16, e0255854. [Google Scholar] [CrossRef]
- Vazquez-Villegas, M.L.; Gamez-Nava, J.I.; Saldana-Cruz, A.M.; Celis, A.; Sanchez-Rodriguez, E.N.; Perez-Guerrero, E.E.; Ramirez-Villafana, M.; Nava-Valdivia, C.A.; Contreras-Haro, B.; Vasquez-Jimenez, J.C.; et al. Functional disability is related to serum chemerin levels in rheumatoid arthritis. Sci. Rep. 2021, 11, 8360. [Google Scholar] [CrossRef]
- Zhang, S.; Rong, G.; Xu, Y.; Jing, J. Elevated Nesfatin-1 Level in Synovium and Synovial Fluid is Associated with Pro-Inflammatory Cytokines in Patients with Rheumatoid Arthritis. Int. J. Gen. Med. 2021, 14, 5269–5278. [Google Scholar] [CrossRef]
- Murillo-Saich, J.D.; Vazquez-Villegas, M.L.; Ramirez-Villafana, M.; Saldana-Cruz, A.M.; Aceves-Aceves, J.A.; Gonzalez-Lopez, L.; Guma, M.; Gamez-Nava, J.I. Association of myostatin, a cytokine released by muscle, with inflammation in rheumatoid arthritis: A cross-sectional study. Medicine 2021, 100, e24186. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Z.; Ma, J.D.; Yang, L.J.; Zou, Y.W.; Zhang, X.P.; Pan, J.; Li, Q.H.; Li, H.G.; Yang, Z.H.; Wu, T.; et al. Myokine myostatin is a novel predictor of one-year radiographic progression in patients with rheumatoid arthritis: A prospective cohort study. Front. Immunol. 2022, 13, 1005161. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ponce, F.; Gamez-Nava, J.I.; Gomez-Ramirez, E.E.; Ramirez-Villafana, M.; Jacobo-Cuevas, H.; Rodriguez-Jimenez, N.A.; Olivas-Flores, E.M.; Esparza-Guerrero, Y.; Martelli-Garcia, A.; Santiago-Garcia, A.P.; et al. Myostatin Levels and the Risk of Myopenia and Rheumatoid Cachexia in Women with Rheumatoid Arthritis. J. Immunol. Res. 2022, 2022, 7258152. [Google Scholar] [CrossRef]
- Soliman, S.A.; Gad, R.; Senosy, T.; Higazi, A.M.; Elshereef, R. Serum irisin level in rheumatoid arthritis patients: Relationship to disease activity, subclinical atherosclerosis, and cardiovascular risk factors. Egypt. Rheumatol. 2022, 44, 109–114. [Google Scholar] [CrossRef]
- Gamez-Nava, J.I.; Ramirez-Villafana, M.; Cons-Molina, F.; Gomez-Ramirez, E.E.; Esparza-Guerrero, Y.; Saldana-Cruz, A.M.; Sanchez-Rodriguez, E.N.; Jacobo-Cuevas, H.; Totsuka-Sutto, S.E.; Perez-Guerrero, E.E.; et al. Serum irisin concentrations and osteoporotic vertebral fractures in women with rheumatoid arthritis: A cross-sectional study. Medicine 2022, 101, e28799. [Google Scholar] [CrossRef]
- Maijer, K.I.; Neumann, E.; Muller-Ladner, U.; Drop, D.A.; Ramwadhdoebe, T.H.; Choi, I.Y.; Gerlag, D.M.; de Hair, M.J.; Tak, P.P. Serum Vaspin Levels Are Associated with the Development of Clinically Manifest Arthritis in Autoantibody-Positive Individuals. PLoS ONE 2015, 10, e0144932. [Google Scholar] [CrossRef]
- Fischer, T.F.; Beck-Sickinger, A.G. Chemerin—Exploring a versatile adipokine. Biol. Chem. 2022, 403, 625–642. [Google Scholar] [CrossRef]
- Helfer, G.; Wu, Q.F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Miyabe, Y.; Takayasu, A.; Fukuda, S.; Miyabe, C.; Ebisawa, M.; Yokoyama, W.; Watanabe, K.; Imai, T.; Muramoto, K.; et al. Chemerin activates fibroblast-like synoviocytes in patients with rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, R158. [Google Scholar] [CrossRef] [PubMed]
- Berg, V.; Sveinbjornsson, B.; Bendiksen, S.; Brox, J.; Meknas, K.; Figenschau, Y. Human articular chondrocytes express ChemR23 and chemerin; ChemR23 promotes inflammatory signalling upon binding the ligand chemerin(21-157). Arthritis Res. Ther. 2010, 12, R228. [Google Scholar] [CrossRef]
- Eisinger, K.; Bauer, S.; Schaffler, A.; Walter, R.; Neumann, E.; Buechler, C.; Muller-Ladner, U.; Frommer, K.W. Chemerin induces CCL2 and TLR4 in synovial fibroblasts of patients with rheumatoid arthritis and osteoarthritis. Exp. Mol. Pathol. 2012, 92, 90–96. [Google Scholar] [CrossRef]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pray, J.; Wu, H.B.; Hansen, B.C.; Shuldiner, A.R.; Fried, S.K.; McLenithan, J.C.; Gong, D.W. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: Possible role in modulating insulin action. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1253–E1261. [Google Scholar] [CrossRef] [PubMed]
- de Souza Batista, C.M.; Yang, R.Z.; Lee, M.J.; Glynn, N.M.; Yu, D.Z.; Pray, J.; Ndubuizu, K.; Patil, S.; Schwartz, A.; Kligman, M.; et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes 2007, 56, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, G.P.; Anithalekshmi, M.S.; Yasir, M.; Parameswaran, S.; Packirisamy, R.M.; Bobby, Z. Role of omentin 1 and IL-6 in type 2 diabetes mellitus patients with diabetic nephropathy. Diabetes Metab. Syndr. 2018, 12, 23–26. [Google Scholar] [CrossRef]
- Robinson, C.; Tsang, L.; Solomon, A.; Woodiwiss, A.J.; Gunter, S.; Millen, A.M.; Norton, G.R.; Fernandez-Lopez, M.J.; Hollan, I.; Dessein, P.H. Omentin concentrations are independently associated with those of matrix metalloproteinase-3 in patients with mild but not severe rheumatoid arthritis. Rheumatol. Int. 2017, 37, 3–11. [Google Scholar] [CrossRef]
- Lan, Y.J.; Sam, N.B.; Cheng, M.H.; Pan, H.F.; Gao, J. Progranulin as a Potential Therapeutic Target in Immune-Mediated Diseases. J. Inflamm. Res. 2021, 14, 6543–6556. [Google Scholar] [CrossRef]
- Schmid, A.; Hochberg, A.; Kreiss, A.F.; Gehl, J.; Patz, M.; Thomalla, M.; Hanses, F.; Karrasch, T.; Schaffler, A. Role of progranulin in adipose tissue innate immunity. Cytokine 2020, 125, 154796. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, M.; Ait Edjoudi, D.; Cordero Barreal, A.; Ruiz-Fernandez, C.; Farrag, M.; Gonzalez-Rodriguez, B.; Lago, F.; Capuozzo, M.; Gonzalez-Gay, M.A.; Mera Varela, A.; et al. Progranulin in Musculoskeletal Inflammatory and Degenerative Disorders, Focus on Rheumatoid Arthritis, Lupus and Intervertebral Disc Disease: A Systematic Review. Pharmaceuticals 2022, 15, 1544. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Lai, Y.; Tian, Q.; Lin, E.A.; Kong, L.; Liu, C. Granulin-epithelin precursor binds directly to ADAMTS-7 and ADAMTS-12 and inhibits their degradation of cartilage oligomeric matrix protein. Arthritis Rheum. 2010, 62, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Hou, C. miR-138 activates NF-kappaB signaling and PGRN to promote rheumatoid arthritis via regulating HDAC4. Biochem. Biophys. Res. Commun. 2019, 519, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, B.; Liu, C.J. Establishment of a surgically-induced model in mice to investigate the protective role of progranulin in osteoarthritis. J. Vis. Exp. 2014, e50924. [Google Scholar] [CrossRef]
- Wang, M.; Tang, D.; Shu, B.; Wang, B.; Jin, H.; Hao, S.; Dresser, K.A.; Shen, J.; Im, H.J.; Sampson, E.R.; et al. Conditional activation of beta-catenin signaling in mice leads to severe defects in intervertebral disc tissue. Arthritis Rheum. 2012, 64, 2611–2623. [Google Scholar] [CrossRef]
- Abella, V.; Pino, J.; Scotece, M.; Conde, J.; Lago, F.; Gonzalez-Gay, M.A.; Mera, A.; Gomez, R.; Mobasheri, A.; Gualillo, O. Progranulin as a biomarker and potential therapeutic agent. Drug Discov. Today 2017, 22, 1557–1564. [Google Scholar] [CrossRef]
- Xia, Q.; Zhu, S.; Wu, Y.; Wang, J.; Cai, Y.; Chen, P.; Li, J.; Heng, B.C.; Ouyang, H.W.; Lu, P. Intra-articular transplantation of atsttrin-transduced mesenchymal stem cells ameliorate osteoarthritis development. Stem Cells Transl. Med. 2015, 4, 523–531. [Google Scholar] [CrossRef]
- Haynes, K.R.; Pettit, A.R.; Duan, R.; Tseng, H.W.; Glant, T.T.; Brown, M.A.; Thomas, G.P. Excessive bone formation in a mouse model of ankylosing spondylitis is associated with decreases in Wnt pathway inhibitors. Arthritis Res. Ther. 2012, 14, R253. [Google Scholar] [CrossRef]
- Tang, W.; Lu, Y.; Tian, Q.Y.; Zhang, Y.; Guo, F.J.; Liu, G.Y.; Syed, N.M.; Lai, Y.; Lin, E.A.; Kong, L.; et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 2011, 332, 478–484. [Google Scholar] [CrossRef]
- Abella, V.; Scotece, M.; Conde, J.; Gomez, R.; Lois, A.; Pino, J.; Gomez-Reino, J.J.; Lago, F.; Mobasheri, A.; Gualillo, O. The potential of lipocalin-2/NGAL as biomarker for inflammatory and metabolic diseases. Biomarkers 2015, 20, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Owen, H.C.; Roberts, S.J.; Ahmed, S.F.; Farquharson, C. Dexamethasone-induced expression of the glucocorticoid response gene lipocalin 2 in chondrocytes. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E1023–E1034. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Gomez, R.; Bianco, G.; Scotece, M.; Lear, P.; Dieguez, C.; Gomez-Reino, J.; Lago, F.; Gualillo, O. Expanding the adipokine network in cartilage: Identification and regulation of novel factors in human and murine chondrocytes. Ann. Rheum. Dis. 2011, 70, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Shukla, M.; Cowland, J.B.; Malemud, C.J.; Haqqi, T.M. Neutrophil gelatinase-associated lipocalin is expressed in osteoarthritis and forms a complex with matrix metalloproteinase 9. Arthritis Rheum. 2007, 56, 3326–3335. [Google Scholar] [CrossRef]
- Katano, M.; Okamoto, K.; Arito, M.; Kawakami, Y.; Kurokawa, M.S.; Suematsu, N.; Shimada, S.; Nakamura, H.; Xiang, Y.; Masuko, K.; et al. Implication of granulocyte-macrophage colony-stimulating factor induced neutrophil gelatinase-associated lipocalin in pathogenesis of rheumatoid arthritis revealed by proteome analysis. Arthritis Res. Ther. 2009, 11, R3. [Google Scholar] [CrossRef]
- Wilson, R.; Belluoccio, D.; Little, C.B.; Fosang, A.J.; Bateman, J.F. Proteomic characterization of mouse cartilage degradation in vitro. Arthritis Rheum. 2008, 58, 3120–3131. [Google Scholar] [CrossRef]
- Ayada, C.; Toru, U.; Korkut, Y. Nesfatin-1 and its effects on different systems. Hippokratia 2015, 19, 4–10. [Google Scholar]
- Scotece, M.; Conde, J.; Abella, V.; Lopez, V.; Lago, F.; Pino, J.; Gomez-Reino, J.J.; Gualillo, O. NUCB2/nesfatin-1: A new adipokine expressed in human and murine chondrocytes with pro-inflammatory properties, an in vitro study. J. Orthop. Res. 2014, 32, 653–660. [Google Scholar] [CrossRef]
- Xu, Y.; Zai, Z.; Zhang, T.; Wang, L.; Qian, X.; Xu, D.; Tao, J.; Lu, Z.; Zhang, Z.; Peng, X.; et al. Nesfatin-1 exerts protective effects on acidosis-stimulated chondrocytes and rats with adjuvant-induced arthritis by inhibiting ASIC1a expression. Lab. Investig. 2022, 102, 859–871. [Google Scholar] [CrossRef]
- Chang, J.W.; Lin, Y.Y.; Tsai, C.H.; Liu, S.C.; He, X.Y.; Wu, Y.S.; Huang, C.C.; Tang, C.H. Nesfatin-1 stimulates BMP5 expression and osteoclastogenesis in rheumatoid arthritis. Biochem. Pharmacol. 2023, 215, 115687. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Li, P.; Zheng, Y.; Yang, Y.; Ji, S. Apelin/APJ system in inflammation. Int. Immunopharmacol. 2022, 109, 108822. [Google Scholar] [CrossRef]
- Di Franco, M.; Spinelli, F.R.; Metere, A.; Gerardi, M.C.; Conti, V.; Boccalini, F.; Iannuccelli, C.; Ciciarello, F.; Agati, L.; Valesini, G. Serum levels of asymmetric dimethylarginine and apelin as potential markers of vascular endothelial dysfunction in early rheumatoid arthritis. Mediators Inflamm. 2012, 2012, 347268. [Google Scholar] [CrossRef]
- Fang, P.; She, Y.; Yu, M.; Min, W.; Shang, W.; Zhang, Z. Adipose-Muscle crosstalk in age-related metabolic disorders: The emerging roles of adipo-myokines. Ageing Res. Rev. 2023, 84, 101829. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.-J. Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Consitt, L.A.; Clark, B.C. The Vicious Cycle of Myostatin Signaling in Sarcopenic Obesity: Myostatin Role in Skeletal Muscle Growth, Insulin Signaling and Implications for Clinical Trials. J. Frailty Aging 2018, 7, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Guo, T.; Jou, W.; Chanturiya, T.; Portas, J.; Gavrilova, O.; McPherron, A.C. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE 2009, 4, e4937. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Yao, T.; Zhou, P.; Kazak, L.; Tenen, D.; Lyubetskaya, A.; Dawes, B.A.; Tsai, L.; Kahn, B.B.; Spiegelman, B.M.; et al. Brown Adipose Tissue Controls Skeletal Muscle Function via the Secretion of Myostatin. Cell Metab. 2018, 28, 631–643.e633. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.G.; Lu, M.J.; Wang, B.W.; Sun, H.Y.; Chang, H. Myostatin expression in ventricular myocardium in a rat model of volume-overload heart failure. Eur. J. Clin. Investig. 2006, 36, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; McNally, R.M.; Hoffmann, F.M.; Hornberger, T.A. Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol. Endocrinol. 2013, 27, 1946–1957. [Google Scholar] [CrossRef]
- Haines, M.S.; Dichtel, L.E.; Kimball, A.; Bollinger, B.; Gerweck, A.V.; Bredella, M.A.; Miller, K.K. OR26-03 Lower Serum Myostatin Levels Are Associated with Higher Insulin Sensitivity in Adults with Overweight/Obesity. J. Endocr. Soc. 2020, 4, OR26-03. [Google Scholar] [CrossRef]
- Amor, M.; Itariu, B.K.; Moreno-Viedma, V.; Keindl, M.; Jurets, A.; Prager, G.; Langer, F.; Grablowitz, V.; Zeyda, M.; Stulnig, T.M. Serum Myostatin is Upregulated in Obesity and Correlates with Insulin Resistance in Humans. Exp. Clin. Endocrinol. Diabetes 2019, 127, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.S.; Li, G.; Blumenthal, J.B.; Ortmeyer, H.K. Aerobic exercise + weight loss decreases skeletal muscle myostatin expression and improves insulin sensitivity in older adults. Obesity 2013, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.A.; Sargeant, J.A.; Thackray, A.E.; Yates, T.; Stensel, D.J.; Aithal, G.P.; King, J.A. Effect of exercise intensity on circulating hepatokine concentrations in healthy men. Appl. Physiol. Nutr. Metab. 2019, 44, 1065–1072. [Google Scholar] [CrossRef]
- Su, C.M.; Hu, S.L.; Sun, Y.; Zhao, J.; Dai, C.; Wang, L.; Xu, G.; Tang, C.H. Myostatin induces tumor necrosis factor-alpha expression in rheumatoid arthritis synovial fibroblasts through the PI3K-Akt signaling pathway. J. Cell Physiol. 2019, 234, 9793–9801. [Google Scholar] [CrossRef]
- Wada, Y.; Sudo, M.; Kobayashi, D.; Kuroda, T.; Nakano, M. Serum Myostatin in Patients with Rheumatoid Arthritis and Its Correlations with Body Compositions and the Disease Activity. In Arthritis & Rheumatology; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Fennen, M.; Weinhage, T.; Kracke, V.; Intemann, J.; Varga, G.; Wehmeyer, C.; Foell, D.; Korb-Pap, A.; Pap, T.; Dankbar, B. A myostatin-CCL20-CCR6 axis regulates Th17 cell recruitment to inflamed joints in experimental arthritis. Sci. Rep. 2021, 11, 14145. [Google Scholar] [CrossRef]
- Dankbar, B.; Fennen, M.; Brunert, D.; Hayer, S.; Frank, S.; Wehmeyer, C.; Beckmann, D.; Paruzel, P.; Bertrand, J.; Redlich, K.; et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat. Med. 2015, 21, 1085–1090. [Google Scholar] [CrossRef]
- Hu, S.L.; Chang, A.C.; Huang, C.C.; Tsai, C.H.; Lin, C.C.; Tang, C.H. Myostatin Promotes Interleukin-1beta Expression in Rheumatoid Arthritis Synovial Fibroblasts through Inhibition of miR-21-5p. Front. Immunol. 2017, 8, 1747. [Google Scholar] [CrossRef]
- Furlanetto Junior, R.; Martins, F.M.; Oliveira, A.A.; Nunes, P.R.; Michelin, M.A.; Murta, E.F.; Orsatti, F.L. Loss of Ovarian Function Results in Increased Loss of Skeletal Muscle in Arthritic Rats. Rev. Bras. Ginecol. Obstet. 2016, 38, 56–64. [Google Scholar] [CrossRef]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Rodriguez, A.; Becerril, S.; Ezquerro, S.; Mendez-Gimenez, L.; Fruhbeck, G. Crosstalk between adipokines and myokines in fat browning. Acta Physiol. 2017, 219, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Navarrete, J.M.; Ortega, F.; Serrano, M.; Guerra, E.; Pardo, G.; Tinahones, F.; Ricart, W.; Fernandez-Real, J.M. Irisin is expressed and produced by human muscle and adipose tissue in association with obesity and insulin resistance. J. Clin. Endocrinol. Metab. 2013, 98, E769–E778. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S.; Kuloglu, T.; Aydin, S.; Kalayci, M.; Yilmaz, M.; Cakmak, T.; Albayrak, S.; Gungor, S.; Colakoglu, N.; Ozercan, I.H. A comprehensive immunohistochemical examination of the distribution of the fat-burning protein irisin in biological tissues. Peptides 2014, 61, 130–136. [Google Scholar] [CrossRef]
- Martinez Munoz, I.Y.; Camarillo Romero, E.D.S.; Garduno Garcia, J.J. Irisin a Novel Metabolic Biomarker: Present Knowledge and Future Directions. Int. J. Endocrinol. 2018, 2018, 7816806. [Google Scholar] [CrossRef] [PubMed]
- Colaianni, G.; Cuscito, C.; Mongelli, T.; Pignataro, P.; Buccoliero, C.; Liu, P.; Lu, P.; Sartini, L.; Di Comite, M.; Mori, G.; et al. The myokine irisin increases cortical bone mass. Proc. Natl. Acad. Sci. USA 2015, 112, 12157–12162. [Google Scholar] [CrossRef]
- Kornel, A.; Den Hartogh, D.J.; Klentrou, P.; Tsiani, E. Role of the Myokine Irisin on Bone Homeostasis: Review of the Current Evidence. Int. J. Mol. Sci. 2021, 22, 9136. [Google Scholar] [CrossRef]
- Ning, K.; Wang, Z.; Zhang, X.A. Exercise-induced modulation of myokine irisin in bone and cartilage tissue-Positive effects on osteoarthritis: A narrative review. Front. Aging Neurosci. 2022, 14, 934406. [Google Scholar] [CrossRef]
- Mazur-Bialy, A.; Bilski, J.; Pochec, E.; Brzozowski, T. New insight into the direct anti-inflammatory activity of a myokine irisin against proinflammatory activation of adipocytes. Implication for exercise in obesity. J. Physiol. Pharmacol. 2017, 68, 243–251. [Google Scholar]
- Mazur-Bialy, A.I.; Pochec, E.; Zarawski, M. Anti-Inflammatory Properties of Irisin, Mediator of Physical Activity, Are Connected with TLR4/MyD88 Signaling Pathway Activation. Int. J. Mol. Sci. 2017, 18, 701. [Google Scholar] [CrossRef]
- Kim, H.; Wrann, C.D.; Jedrychowski, M.; Vidoni, S.; Kitase, Y.; Nagano, K.; Zhou, C.; Chou, J.; Parkman, V.A.; Novick, S.J.; et al. Irisin Mediates Effects on Bone and Fat via alphaV Integrin Receptors. Cell 2018, 175, 1756–1768.e17. [Google Scholar] [CrossRef]
- Qiao, X.; Nie, Y.; Ma, Y.; Chen, Y.; Cheng, R.; Yin, W.; Hu, Y.; Xu, W.; Xu, L. Irisin promotes osteoblast proliferation and differentiation via activating the MAP kinase signaling pathways. Sci. Rep. 2016, 6, 18732. [Google Scholar] [CrossRef]
- Storlino, G.; Colaianni, G.; Sanesi, L.; Lippo, L.; Brunetti, G.; Errede, M.; Colucci, S.; Passeri, G.; Grano, M. Irisin Prevents Disuse-Induced Osteocyte Apoptosis. J. Bone Miner. Res. 2020, 35, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Vadala, G.; Di Giacomo, G.; Ambrosio, L.; Cannata, F.; Cicione, C.; Papalia, R.; Denaro, V. Irisin Recovers Osteoarthritic Chondrocytes In Vitro. Cells 2020, 9, 1478. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.S.; Kuo, C.W.; Ko, J.Y.; Chen, Y.S.; Wang, S.Y.; Ke, H.J.; Kuo, P.C.; Lee, C.H.; Wu, J.C.; Lu, W.B.; et al. Irisin Mitigates Oxidative Stress, Chondrocyte Dysfunction and Osteoarthritis Development through Regulating Mitochondrial Integrity and Autophagy. Antioxidants 2020, 9, 810. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Dincer, F.; Mesfum, E.; Mantzoros, C.S. Irisin stimulates muscle growth-related genes and regulates adipocyte differentiation and metabolism in humans. Int. J. Obes. 2014, 38, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Reza, M.M.; Subramaniyam, N.; Sim, C.M.; Ge, X.; Sathiakumar, D.; McFarlane, C.; Sharma, M.; Kambadur, R. Irisin is a pro-myogenic factor that induces skeletal muscle hypertrophy and rescues denervation-induced atrophy. Nat. Commun. 2017, 8, 1104. [Google Scholar] [CrossRef]
- Chang, J.S.; Kong, I.D. Irisin prevents dexamethasone-induced atrophy in C2C12 myotubes. Pflugers Arch. 2020, 472, 495–502. [Google Scholar] [CrossRef]
- Dong, J.; Dong, Y.; Chen, F.; Mitch, W.; Zhang, L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int. J. Obes. 2016, 40, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto-Mikami, E.; Sato, K.; Kurihara, T.; Hasegawa, N.; Fujie, S.; Fujita, S.; Sanada, K.; Hamaoka, T.; Tabata, I.; Iemitsu, M. Endurance training-induced increase in circulating irisin levels is associated with reduction of abdominal visceral fat in middle-aged and older adults. PLoS ONE 2015, 10, e0120354. [Google Scholar] [CrossRef]
- Mahgoub, M.O.; D’Souza, C.; Al Darmaki, R.; Baniyas, M.; Adeghate, E. An update on the role of irisin in the regulation of endocrine and metabolic functions. Peptides 2018, 104, 15–23. [Google Scholar] [CrossRef]
- Raafat Ibrahim, R.; Shafik, N.M.; El-Esawy, R.O.; El-Sakaa, M.H.; Arakeeb, H.M.; El-Sharaby, R.M.; Ali, D.A.; Safwat El-Deeb, O.; Ragab Abd El-Khalik, S. The emerging role of irisin in experimentally induced arthritis: A recent update involving HMGB1/MCP1/Chitotriosidase I-mediated necroptosis. Redox Rep. 2022, 27, 21–31. [Google Scholar] [CrossRef] [PubMed]
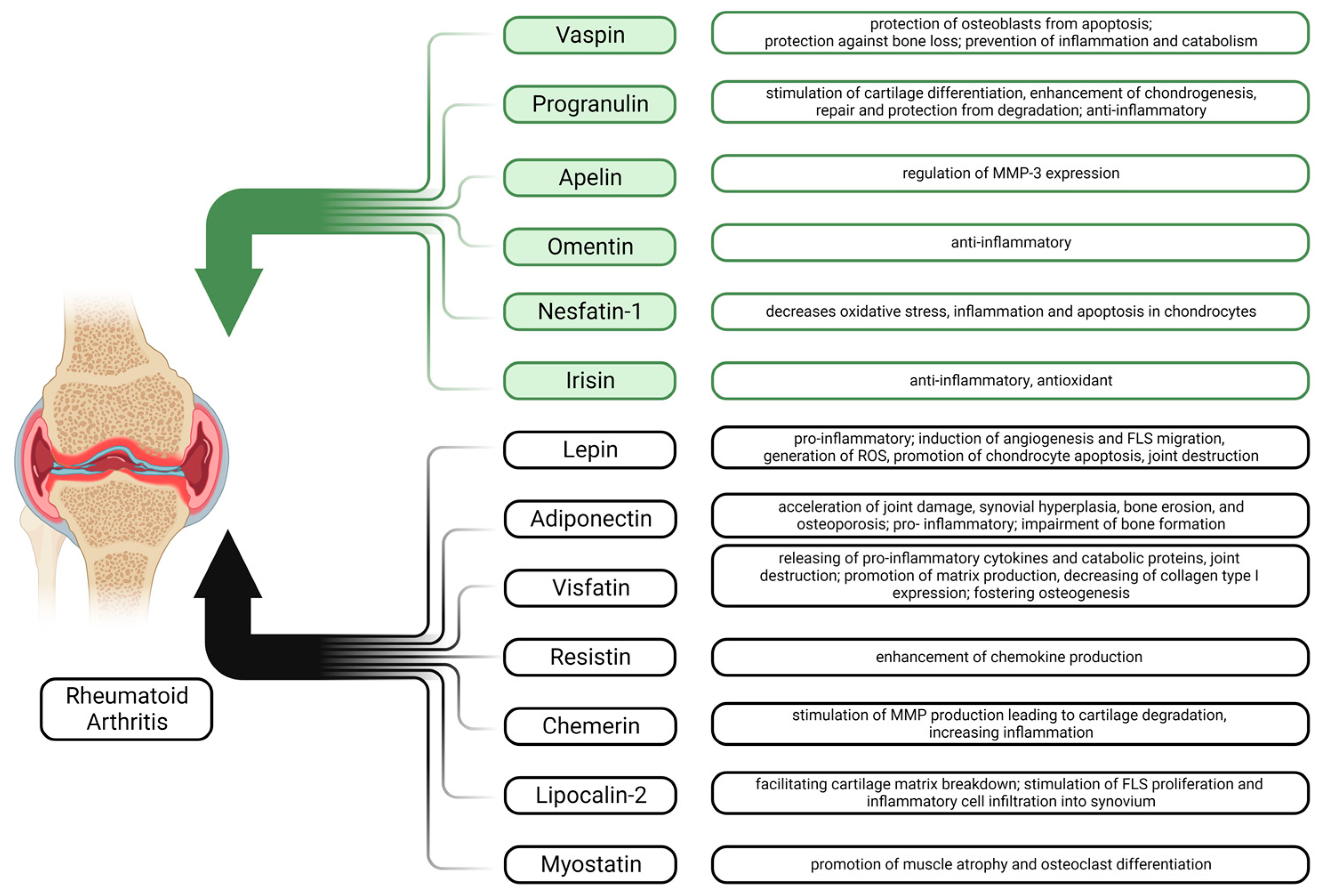

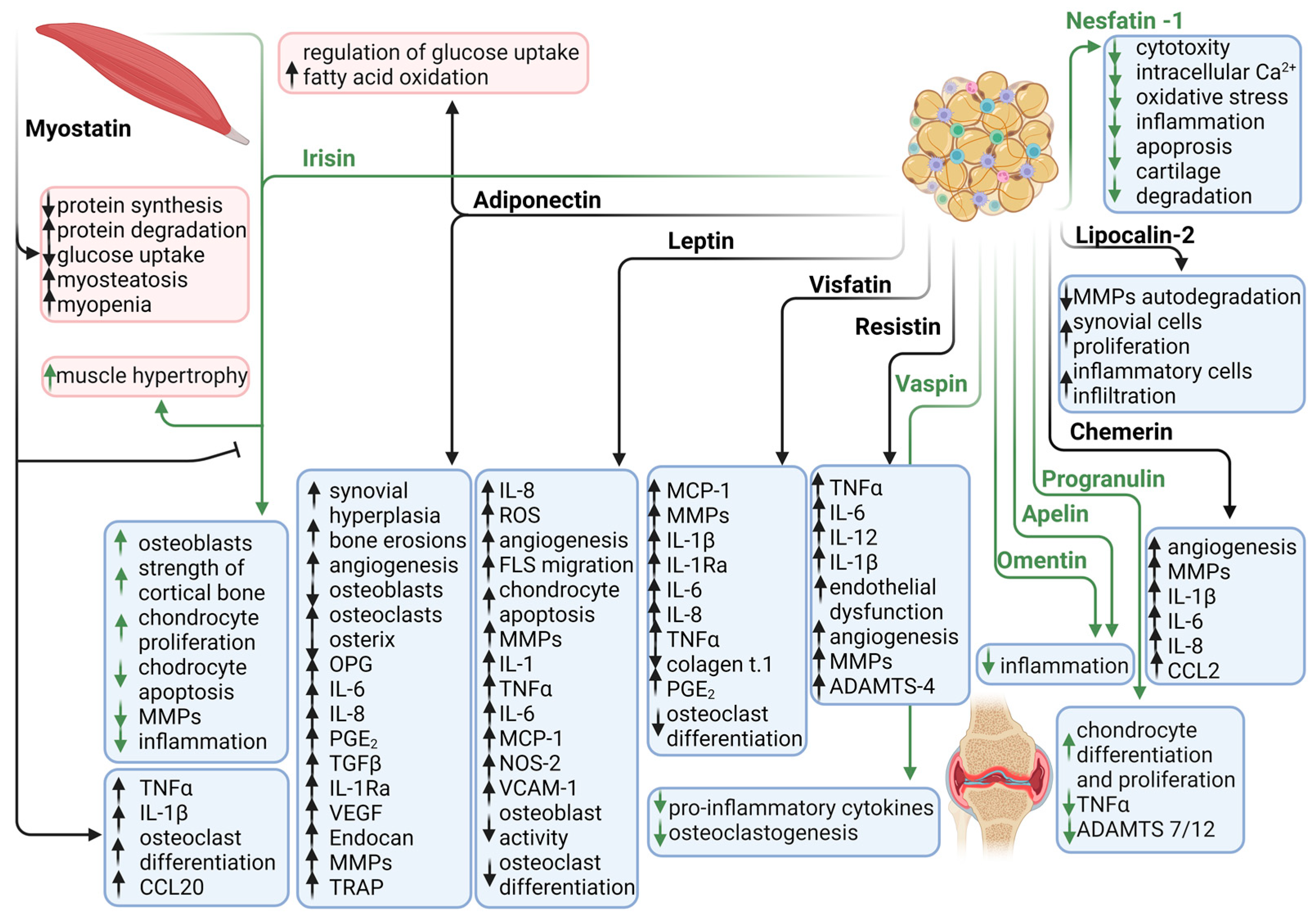

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Subjects | Results/Outcomes |
|---|---|---|
| Anders et al. (1999) [209] | 58 RA patients 16 controls without RA | No differences were observed in serum leptin levels between RA patients and healthy controls. No correlations existed between serum leptin levels and DAS. |
| Bokarewa et al. (2003) [198] | 76 RA patients 34 controls without RA | Plasma levels in RA patients were significantly greater than in controls. Leptin levels in RA patients were significantly greater in plasma than in SF samples, |
| Popa et al. (2005) [210] | 31 RA patients 19 controls without RA | No differences between RA patients and healthy controls in plasma leptin levels were observed. In RA patients, plasma leptin levels were inversely correlated with CRP and IL6 levels. |
| Hizmetli et al. (2005) [211] | 41 RA patients 25 controls without RA | No significant difference between RA patients and the control group was observed in plasma leptin levels. No correlation was observed between serum leptin level and disease duration, ESR, or CRP in RA patients. |
| Otero et al. (2006) [200] | 31 RA patients 18 controls without RA | Leptin serum levels in RA patients were significantly greater than in controls. A positive correlation was observed between serum leptin and CRP levels. |
| Gunaydin et al. (2006) [212] | 50 RA patients 34 controls without RA | Serum leptin levels were greater in RA patients than in controls. No correlations existed between serum leptin levels and disease duration, swollen and tender joint counts, DAS28, CRP, ESR, and serum TNFα levels. |
| Lee et al. (2007) [213] | 50 RA patients | Serum leptin levels were greater in RA patients with DAS28 > 3.2. There was a positive correlation between serum leptin levels and DAS28 and CRP. |
| Wislowska et al. (2007) [214] | 30 RA patients, 30 OA patients | There were no significant differences in serum leptin levels between RA and OA patients. No correlation existed between serum leptin level and disease duration, duration of morning stiffness, and DAS28 in RA patients. |
| Seven et al. (2009) [215] | 20 RA patients 25 controls without RA | Serum and SF leptin levels were significantly greater in RA patients than in the control group. RA patients with moderate disease activity (DAS > 2.7) had significantly greater leptin levels than those with low disease activity (DAS < 2.7) |
| Canoruç et al. (2009) [216] | 52 RA patients 52 controls without RA | Plasma leptin levels in RA patients were significantly greater than in controls. Plasma leptin level significantly correlated with ESR and swollen and tender joint count. |
| Targonska-Stepniak et al. (2010) [217] | 80 RA patients | A positive correlation existed between leptin levels and the DAS28. |
| Ismail et al. (2011) [218] | 40 RA patients 30 controls without RA | Serum leptin levels were greater in RA patients than in controls. A positive correlation was observed between serum leptin levels and the DAS28. |
| Yoshino et al. (2011) [219] | 141 RA patients 146 controls without RA | A positive correlation was observed between serum leptin and CRP levels. |
| Olama et al. (2012) [203] | 40 RA patients 30 controls without RA | Leptin serum levels in RA patients were significantly greater than in controls. Serum leptin level and synovial/serum leptin ratio significantly correlated with the RA duration, DAS28, ESR, CRP, TNFα, and IL-6. The serum leptin and the synovial/serum leptin ratio were significantly greater in women than in men and in erosive RA than non-erosive RA. The synovial/serum leptin ratio was positively associated with erosion in patients with RA. |
| Allam et al. (2012) [220] | 37 RA patients 34 controls without RA | Serum leptin levels were greater in RA patients than in controls. There was no correlation between serum leptin level and disease duration, duration of morning stiffness, VAS, number of swollen and tender joints, DAS28, ESR or CRP in patients with RA. |
| Mirfeizi et al. (2014) [221] | 54 RA patients (29 with erosion, 25 without erosion) | The two groups had no significant differences in mean serum leptin levels. |
| Abdalla et al. (2014) [222] | 60 RA patients 30 controls without RA | Serum leptin levels were greater in RA patients than in controls. No correlation between leptin levels and the DAS28. |
| Bustos Rivera-Bahena et al. (2015) [223] | 121 RA patients | A positive correlation existed between leptin serum levels and disease activity. |
| Oner et al. (2015) [224] | 106 RA patients 52 healthy controls 37 OA patients | There were no differences in serum leptin levels between groups. No correlation was observed between serum leptin levels and DAS28. |
| Dervisevic et al. (2018) [205] | 55 RA patients, 25 controls without RA | Serum leptin levels were greater in RA patients than in controls. A positive correlation existed between leptin serum levels and DAS28. |
| Wang et al. (2018) [204] | 54 RA patients, 46 controls without RA | Serum leptin levels were greater in RA patients than in controls. |
| Chihara et al. (2020) [167] | 136 RA patients 78 controls without RA | Serum leptin levels were greater in RA patients than in controls. |
| Rodriguez et al. (2021) [225] | 51 eRA patients 51 controls without RA | Serum leptin levels were significantly greater in patients with eRA than in healthy controls. |
| Reference | Subjects | Results/Outcomes |
|---|---|---|
| Senolt et al. (2006) [258] | 20 RA patients, 21 OA patients 23 healthy controls | Serum adiponectin levels were significantly greater in RA patients than in healthy controls and comparable with OA patients. SF adiponectin was significantly greater in RA than in OA patients and healthy controls. |
| Otero et al. (2006) [200] | 31 RA patients 18 controls without RA | Serum adiponectin levels in RA patients were significantly greater than in controls. A positive correlation was observed between serum adiponectin and CRP levels. |
| Laurberg et al. (2009) [263] | 114 RA patients, 35 OA patients 45 healthy controls | RA patients had greater plasma adiponectin concentration than healthy controls but lower plasma adiponectin than OA patients. |
| Ebina et al. (2009) [265] | 90 RA patients 42 controls without RA | RA severity was assessed by examining joint destruction on radiographs. Serum adiponectin levels were significantly greater in severe RA patients when compared to mild and control groups, suggesting increased joint destruction is linked to hyperadiponectinaemia in RA patients. |
| Popa et al. (2009) [264] | 58 RA patients 58 controls without RA | Serum adiponectin levels were significantly greater in all RA patients than in controls. |
| Rho et al. (2010) [202] | 167 RA patients 91 controls without RA | Serum adiponectin levels were significantly greater in all RA patients than in controls. |
| Ozgen et al. (2010) [262] | 56RA patients 29 controls without RA | Serum adiponectin levels in RA patients were significantly grater than in controls. A positive correlation was observed between serum adiponectin and DAS28. |
| Alkady et al. (2011) [260] | 70 RA patients 30 controls without RA | The serum levels of adiponectin were significantly greater in RA patients compared to controls, and RA patients with active disease had significantly greater adiponectin levels than those in remission. Serum and SF adiponectin levels positively correlated with the duration of disease, ESR, CRP, and DAS28 in RA patients with active disease. |
| Klein-Wieringa et al. (2011) [271] | 253 RA patients | A positive correlation existed between serum levels of adiponectin and radiographic progression of RA over 4 years. |
| Giles et al. (2011) [272] | 152 RA patients | A positive correlation existed between serum adiponectin levels and erosive joint destruction. |
| Ozgen et al. (2010) [262] | 56 RA patients 29 controls without RA | Serum adiponectin was significantly greater in RA patients than in healthy controls. |
| Yoshino et al. (2011) [219] | 141 RA patients 146 controls without RA | Female, but not male, RA patients had significantly greater serum adiponectin concentrations than controls. Serum adiponectin levels were negatively associated with CRP levels. |
| Kang et al. (2013) [273] | 192 RA patients | There was a significant correlation between serum adiponectin levels and ESR. |
| Meyer et al. (2013) [274] | 632 RA patients | A positive association was observed between serum adiponectin levels and early radiographic RA progression. |
| Toussirot et al. (2013) [275] | 70 RA patients 51 controls without RA | Serum adiponectin levels were significantly greater in RA patients than in controls. In RA patients, HMW/total adiponectin correlated with the DAS28. |
| Bustos Rivera-Bahena et al. (2015) [223] | 121 RA patients | No correlation existed between serum adiponectin and DAS28. A negative correlation existed between serum adiponectin and TNFα. A positive correlation existed between serum adiponectin and IL-11β. |
| Chennareddy et al. (2016) [268] | 43 RA patients 25 controls without RA | Serum levels of adiponectin were significantly greater in RA patients when compared to the control group. There was no correlation with an erosive and non-erosive disease, disease duration, BMI, waist–hip ratio, and DAS28. |
| Khajoei et al. (2019) [261] | 90 RA patients 30 controls without RA | Serum adiponectin levels were greater in all RA patients than in controls. There was a positive correlation between serum adiponectin levels and DAS28 and ESR. |
| Zhang et al. (2020) [269] | 3693 obese subjects followed for up to 29 years. | High serum adiponectin levels at baseline were a significant risk factor for RA in the cohort of subjects with obesity. |
| Vasileiadis et al. (2021) [232] | 70 RA patients | Both total and HMW adiponectin were positively associated with DAS28 and CRP in RA patients. |
| Baker et al. (2022) [276] | 2583 RA patients | Serum adiponectin levels were strongly associated with radiographic damage, seropositivity, longer disease duration, and circulating inflammatory cytokines. |
| Reference | Subjects | Results/Outcomes |
|---|---|---|
| Otero et al. (2006) [200] | 31 RA patients 18 controls without RA | Serum visfatin levels in RA patients were significantly greater than in controls. There was a positive correlation between serum visfatin and CRP levels. |
| Matsui et al. (2008) [286] | 22 RA patients 17 controls without RA | Serum visfatin levels in RA patients were significantly greater than in controls. |
| Rho et al. (2010) [202] | 167 RA patients 91 controls without RA | A positive correlation existed between visfatin serum levels and TNFα, IL-6, CRP, neutrophil count, MHAQ score, and Larsen score. |
| Alkady et al. (2011) [260] | 70 RA patients 30 controls without RA | Serum levels of visfatin were significantly greater in RA patients than in controls, and RA patients with active disease had significantly greater visfatin levels than those in remission. Serum and SF visfatin levels positively correlated with the duration of the disease, ESR, CRP, and DAS28 in RA patients with active disease. |
| Klein-Wieringa et al. (2011) [271] | 253 RA patients | A positive correlation existed between visfatin serum levels and the radiographic progression of RA for 4 years. |
| Ozgen et al. (2011) [288] | 26 RA patients 29 controls without RA | Serum visfatin levels in RA patients were significantly greater than in controls. |
| Meyer et al. (2013) [274] | 632 RA patients | No association between serum visfatin level and radiographic disease progression was observed. |
| Khalifa et al. (2013) [301] | 60 RA patients 20 controls without RA | There was a significant positive correlation between serum visfatin and ESR, CRP, IL-6, TNFα, DAS-28, and VAS pain score in RA patients. No correlation existed between serum visfatin and age of RA patients, disease duration, or BMI. |
| El-Hini et al. (2013) [287] | 40 RA patients 40 controls without RA | Serum visfatin levels in RA patients were significantly greater than in controls. A positive correlation was observed between serum visfatin and DAS28. |
| Sglunda et al. (2014) [166] | 40 eRA patients, 30 controls without RA | eRA patients had elevated serum visfatin levels compared to healthy controls, which decreased after three months of treatment. Circulating visfatin and visfatin level changes correlated with disease activity and improved over time, with a decrease in visfatin predicting improvement in DAS28. |
| Mirfeizi et al. (2014) [221] | 54 RA patients (29 with and 25 without erosion) | Serum visfatin levels were greater in patients with radiographic joint damage and dependent on the duration of the disease. |
| Mohammed Ali et al. (2020) [302] | 60 RA patients 30 controls without RA | Serum visfatin levels in RA patients were significantly greater than in controls. There was no correlation between serum visfatin levels and the DAS28. |
| Reference | Subjects | Results/Outcomes |
|---|---|---|
| Otero et al. (2006) [200] | 31 RA patients 18 controls without RA | Serum resistin levels in RA patients were significantly greater than in controls. There was no correlation between serum resistin and CRP level. |
| Migita et al. (2006) [316] | 42 RA patients 38 controls without RA | Serum resistin levels in RA patients were significantly greater than in controls. Serum resistin levels were correlated with RA disease activity markers, CRP, ESR, and TNFα levels. |
| Forsblad d’Elia et al. (2008) [321] | 90 RA patients 30 controls without RA | Serum resistin levels were not significantly different between patients and healthy controls. Serum resistin levels were correlated with IL-1Ra, CRP, TNFα, ICTP, glucocorticosteroids, Larsen score, and inversely with BMD, hip, and TLM. |
| Rho et al. (2009) [202] | 167 RA patients 91 controls without RA | Serum resistin levels were significantly greater in all RA patients than in the control group. |
| Canoruç et al. (2009) [216] | 52 RA patients 52 controls without RA | Plasma resistin levels in RA patients were significantly greater than in controls. |
| Kassem et al. (2010) [315] | 30 RA patients 15 controls without RA | Serum and SF resistin levels in RA patients were significantly greater than in controls and in active RA greater than in non-active patients. A significant correlation existed between serum resistin levels and CRP, ESR, RF, and disease activity. A significant correlation existed between SF resistin levels and CRP, ESR, RF, and synovial leukocytic count. |
| Yoshino et al. (2011) [219] | 141 RA patients 146 controls without RA | Serum resistin levels were not significantly different between patients and healthy controls. A positive correlation was observed between serum resistin and CRP levels. |
| Alkady et al. (2011) [260] | 70 RA patients 30 controls without RA | No significant difference in serum resistin levels was observed between RA patients and controls. |
| Yoshino et al. (2011) [219] | 141 RA patients 146 controls without RA | A positive correlation was observed between serum resistin and CRP levels. |
| Fadda et al. (2013) [317] | 25 RA patients 25 OA patients | Resistin levels were greater in serum and SF of RA patients when compared to OA patients. There was a correlation between serum resistin levels, Larsen score, and total leukocyte count. A correlation existed between SF resistin levels and RF, ACPA, and Larsen score. |
| Kang et al. (2013) [273] | 192 RA patients | A significant correlation existed between serum resistin levels, ESR, CRP, and disease duration. |
| Hammad et al. (2014) [319] | 30 RA patients 30 controls without RA | Serum resistin levels in RA patients were significantly greater than in controls. |
| Bustos Rivera-Bahena et al. (2015) [223] | 121 RA patients | A positive correlation was observed between resistin serum levels and the DAS28. |
| Vuolteenaho et al. (2022) [322] | 99 RA patients | High resistin levels were associated with active inflammatory disease and predicted rapid radiological progression during 5-year follow-up. |
| Arias-de la Rosa et al. (2022) [206] | 150 RA patients 50 controls without RA | Serum resistin levels in RA patients were significantly greater than in controls. A positive correlation was observed between resistin serum levels and the DAS28. |
| Reference | Subjects | Results/Outcomes |
|---|---|---|
| Ozgen et al. (2010) [262] | 56 RA patients 29 controls without RA | Serum vaspin levels were significantly greater in RA patients than in healthy controls. |
| Wahba et al. (2021) [335] | 150 RA patients 150 controls without RA | Serum levels of chemerin and vaspin were greater, while those of apelin and omentin were lower, in RA patients than in healthy controls. A significant positive correlation existed between vaspin serum levels and BMI, DAS28, ESR, CRP, RF, and anti-CCP antibodies. |
| Senolt et al. (2010) [334] | 33 RA patients, 33 OA patients | SF vaspin levels were significantly greater in RA patients than in OA patients. A positive correlation existed between vaspin SF levels and DAS28. SF omentin levels were significantly lower in RA patients than in OA patients. |
| Ha et al. (2013) [336] | 71 RA patients 42 controls without RA | Plasma chemerin levels in RA patients were significantly greater than in controls. There was a positive correlation between plasma chemerin levels and DAS28. |
| Yamamoto et al. (2014) [337] | 56 RA patients, 31 OA patients 417 controls without RA | Serum PGRN levels in RA patients were significantly greater than in controls. SF PGRN levels were significantly greater in RA patients than in OA patients. |
| Cerezo et al. (2015) [338] | 47 RA patients, 42 OA patients 41 controls without RA | Serum PGRN levels correlated with DAS28 and HAQ scores in RA patients. The SF PGRN levels were significantly greater in RA than in healthy controls and OA patients. |
| Chen et al. (2016) [339] | 80 RA patients, 60 controls without RA | Serum PGRN levels in RA patients were significantly greater than in controls. A significant positive correlation existed between serum PGRN and DAS28, ESR, and CRP in RA patients. |
| Fouad et al. (2019) [340] | 52 RA patients, 19 controls without RA | Serum PGRN levels in RA patients were significantly greater than in controls. A significant positive correlation existed between serum PGRN, DAS28, and ESR in RA patients. |
| Kvlividze et al. 2019) [341] | 110 RA patients, 60 controls without RA | RA patients with severe disease had greater serum nesfatin-1 levels, positively correlated with greater CRP and ESR concentrations. |
| Gamal et al. (2020) [342] | 58 RA patients, 30 controls without RA | Serum irisin levels were significantly lower in RA patients than in controls. Serum irisin levels in RA-poor sleepers were significantly lower than in RA-good sleepers. Serum irisin levels in RA patients were associated with total Pittsburgh Sleep Quality Index scores. Irisin serum levels of RA patients were negatively associated with disease duration, morning stiffness duration, and DAS28. |
| Gonzalez-Ponce et al. (2021) [343] | 210 RA patients | A positive correlation was found between chemerin and DAS 28, CRP, and swollen joints counts. |
| Vazquez-Villegas et al. (2021) [344] | 82 RA patients | Elevated chemerin levels were associated with functional disability in RA. |
| Zhang et al. (2021) [345] | 40 RA patients, 15 controls without RA | Nesfatin-1 levels in the synovium were significantly greater in RA patients than in controls. Nesfatin-1 levels positively correlated with IL-1β and TNFα levels in the synovium of patients with RA. |
| Murillo-Saich et al. (2021) [346] | 84 RA patients, 127 controls without RA | MSTN serum levels were significantly greater in the RA group than in the controls. Greater MSTN levels correlated with low skeletal muscle mass index. A positive correlation existed between MSTN serum levels and DAS28, CRP and ESR. |
| Lin et al. (2022) [347] | 344 RA patients, 118 controls without RA | In a prospective cohort of consecutive RA patients with a one-year follow-up, it was found that RA patients had greater serum MSTN levels at baseline when compared to healthy controls. Elevated serum MSTN and myopenia are risk factors for radiographic progression in RA. RA patients with elevated MSTN overlapping myopenia had the highest levels of DAS28, SDAI, CDAI, and HAQ-DI at 3, 6, and 12 months, with the lowest proportion of CDAI remission at 3 months, 9 months, and 12 months. They also had the highest proportion of physical dysfunction at 3, 9, and 12 months. |
| Gonzalez-Ponce et al. (2022) [348] | 161 RA patients, 72 controls without RA | MSTN serum levels were significantly greater in the RA group than in the controls. Elevated MSTN levels in RA patients increased the risk of cachexia. |
| Soliman et al. (2022) [349] | 60 RA patients, 30 controls without RA | Serum irisin levels were significantly lower in RA patients than in controls. Serum irisin levels in RA patients negatively correlated with DAS28, CRP, ESR, and HAQ-DI. |
| Gamez-Nava et al. (2022) [350] | 148 RA patients, 97 controls without RA | Serum irisin levels were significantly lower in RA patients than in controls. Low serum irisin levels were associated with the presence of vertebral fractures in female patients with RA. |
| Arias-de la Rosa et al. (2022) [206] | 150 RA patients 50 controls without RA | Serum omentin and vaspin levels in RA patients were significantly higher than in controls. A positive correlation was observed between omentin serum levels and the DAS28. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilski, J.; Schramm-Luc, A.; Szczepanik, M.; Mazur-Biały, A.I.; Bonior, J.; Luc, K.; Zawojska, K.; Szklarczyk, J. Adipokines in Rheumatoid Arthritis: Emerging Biomarkers and Therapeutic Targets. Biomedicines 2023, 11, 2998. https://doi.org/10.3390/biomedicines11112998
Bilski J, Schramm-Luc A, Szczepanik M, Mazur-Biały AI, Bonior J, Luc K, Zawojska K, Szklarczyk J. Adipokines in Rheumatoid Arthritis: Emerging Biomarkers and Therapeutic Targets. Biomedicines. 2023; 11(11):2998. https://doi.org/10.3390/biomedicines11112998
Chicago/Turabian StyleBilski, Jan, Agata Schramm-Luc, Marian Szczepanik, Agnieszka Irena Mazur-Biały, Joanna Bonior, Kevin Luc, Klaudia Zawojska, and Joanna Szklarczyk. 2023. "Adipokines in Rheumatoid Arthritis: Emerging Biomarkers and Therapeutic Targets" Biomedicines 11, no. 11: 2998. https://doi.org/10.3390/biomedicines11112998
APA StyleBilski, J., Schramm-Luc, A., Szczepanik, M., Mazur-Biały, A. I., Bonior, J., Luc, K., Zawojska, K., & Szklarczyk, J. (2023). Adipokines in Rheumatoid Arthritis: Emerging Biomarkers and Therapeutic Targets. Biomedicines, 11(11), 2998. https://doi.org/10.3390/biomedicines11112998