

Exploiting TLK1 and Cisplatin Synergy for Synthetic Lethality in Androgen-Insensitive Prostate Cancer


Abstract
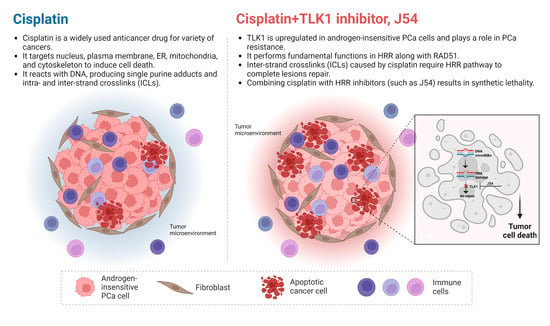
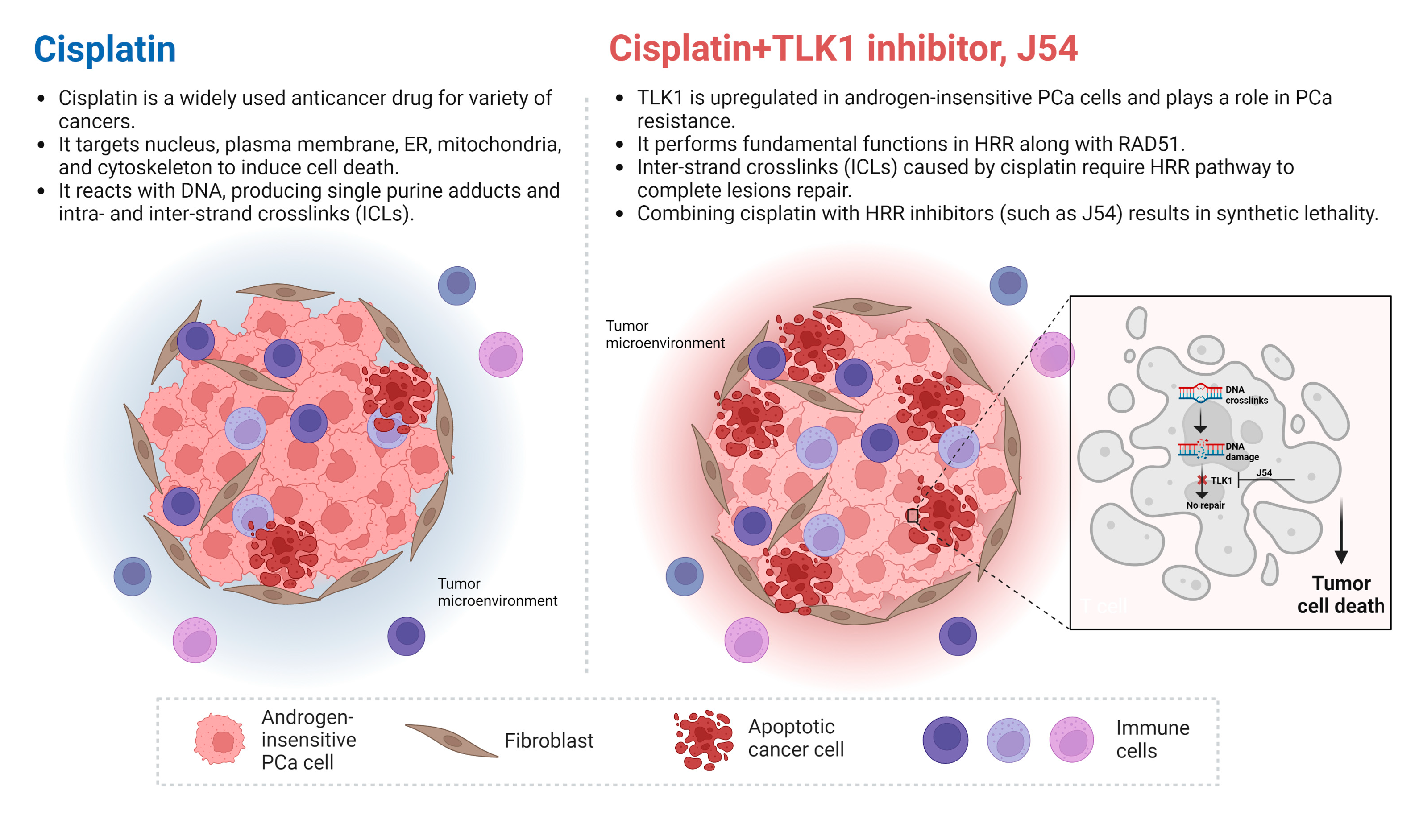
:
{kind=link}
{kind=link}
{kind=link}
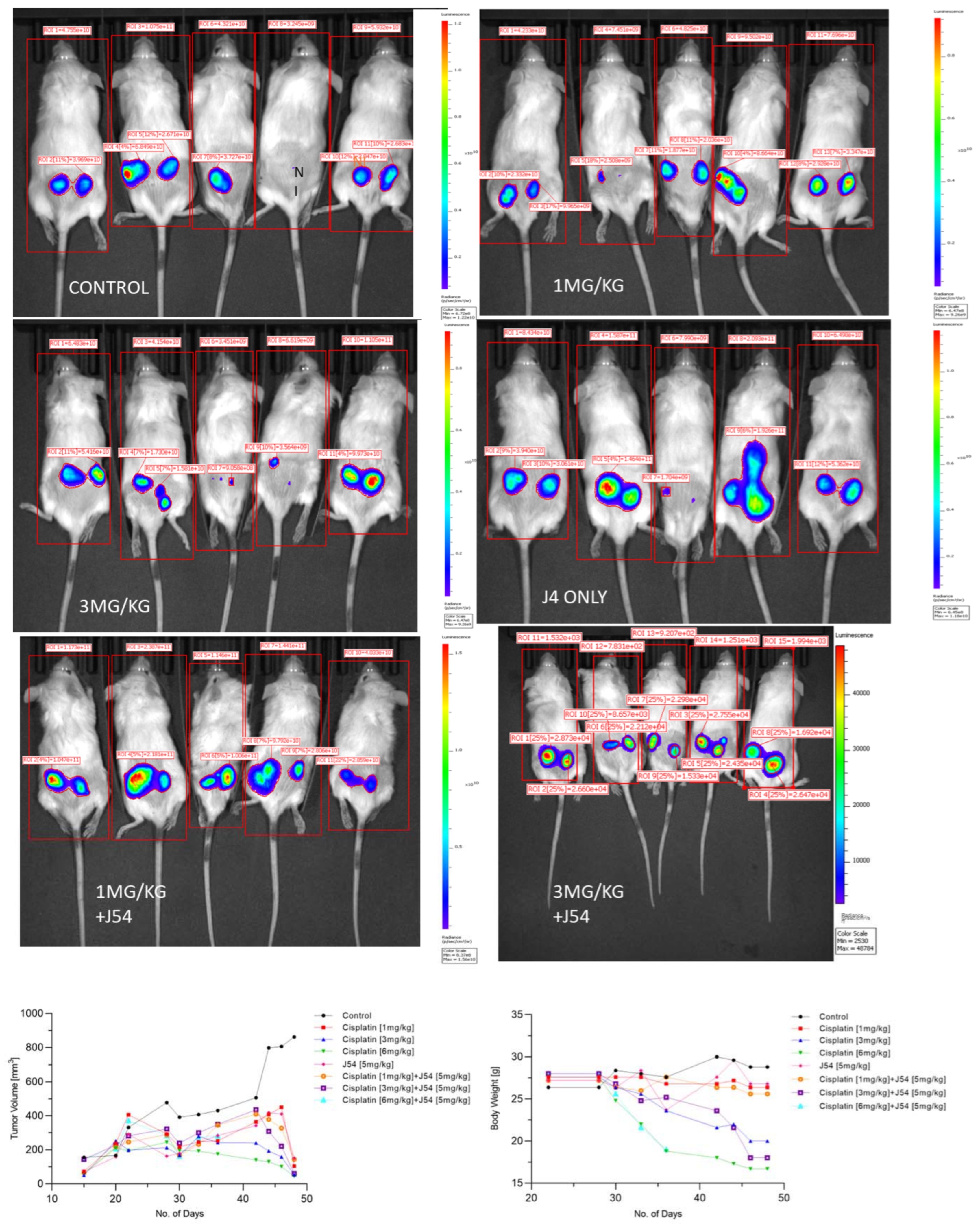{kind=link}
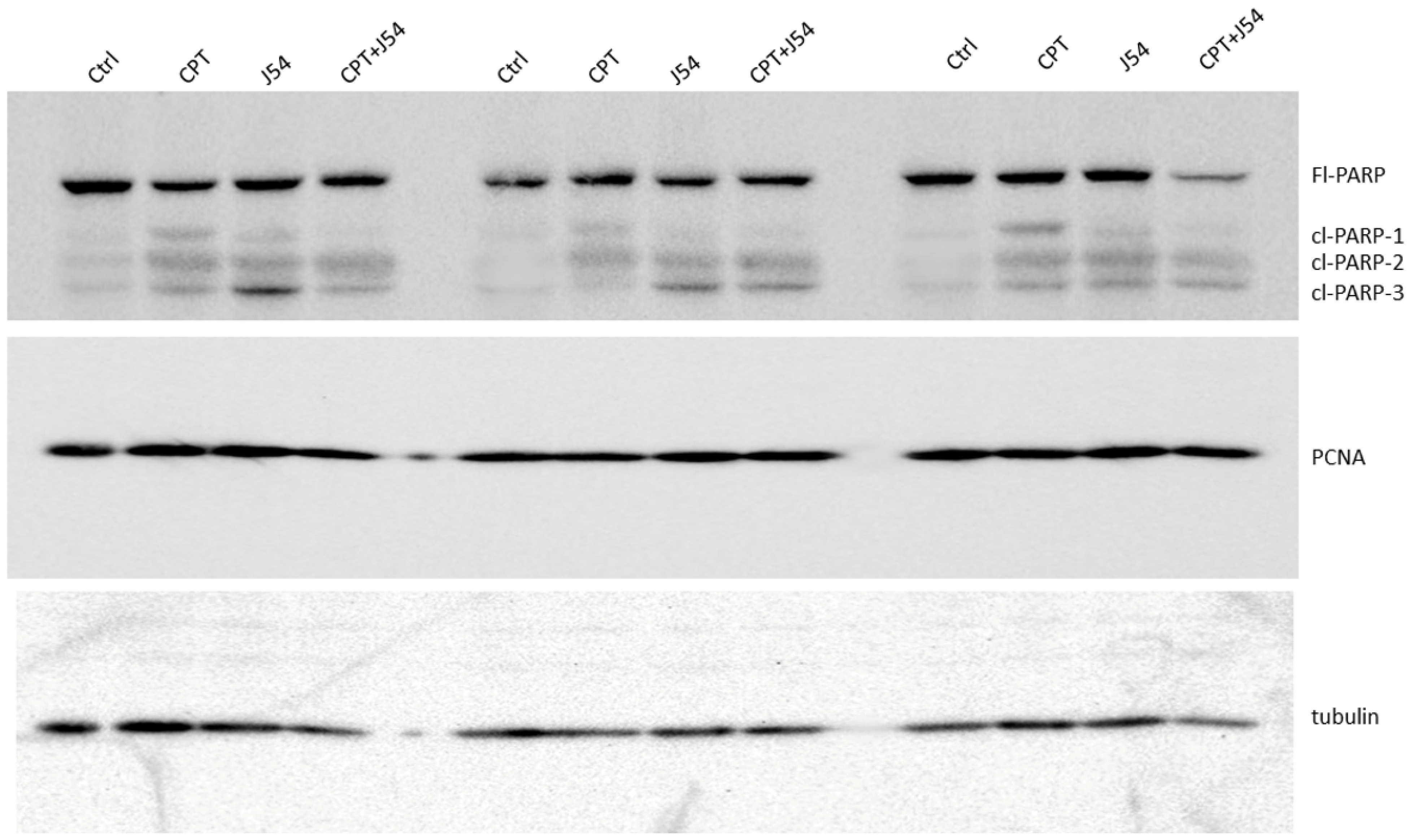{kind=link}
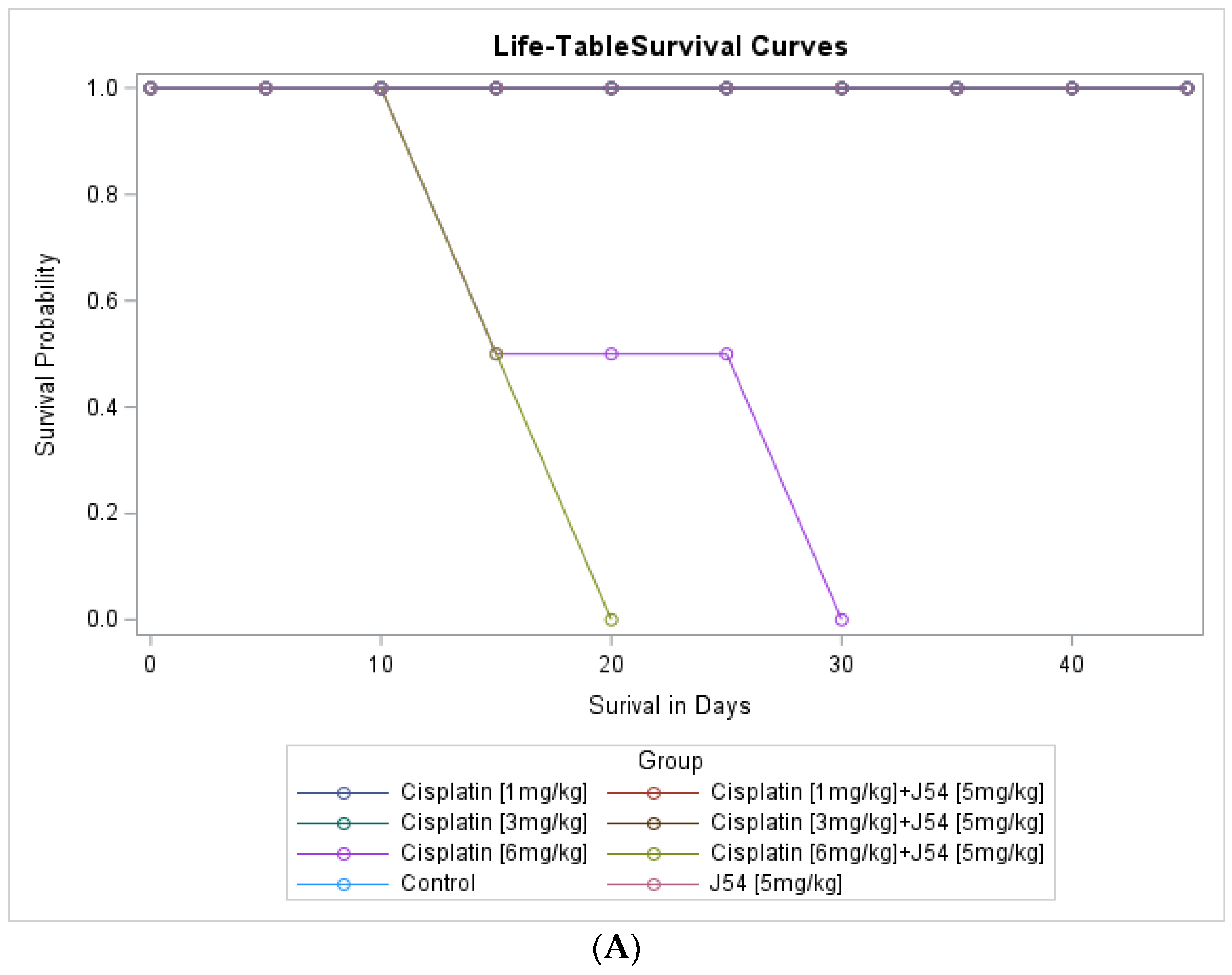{kind=link}
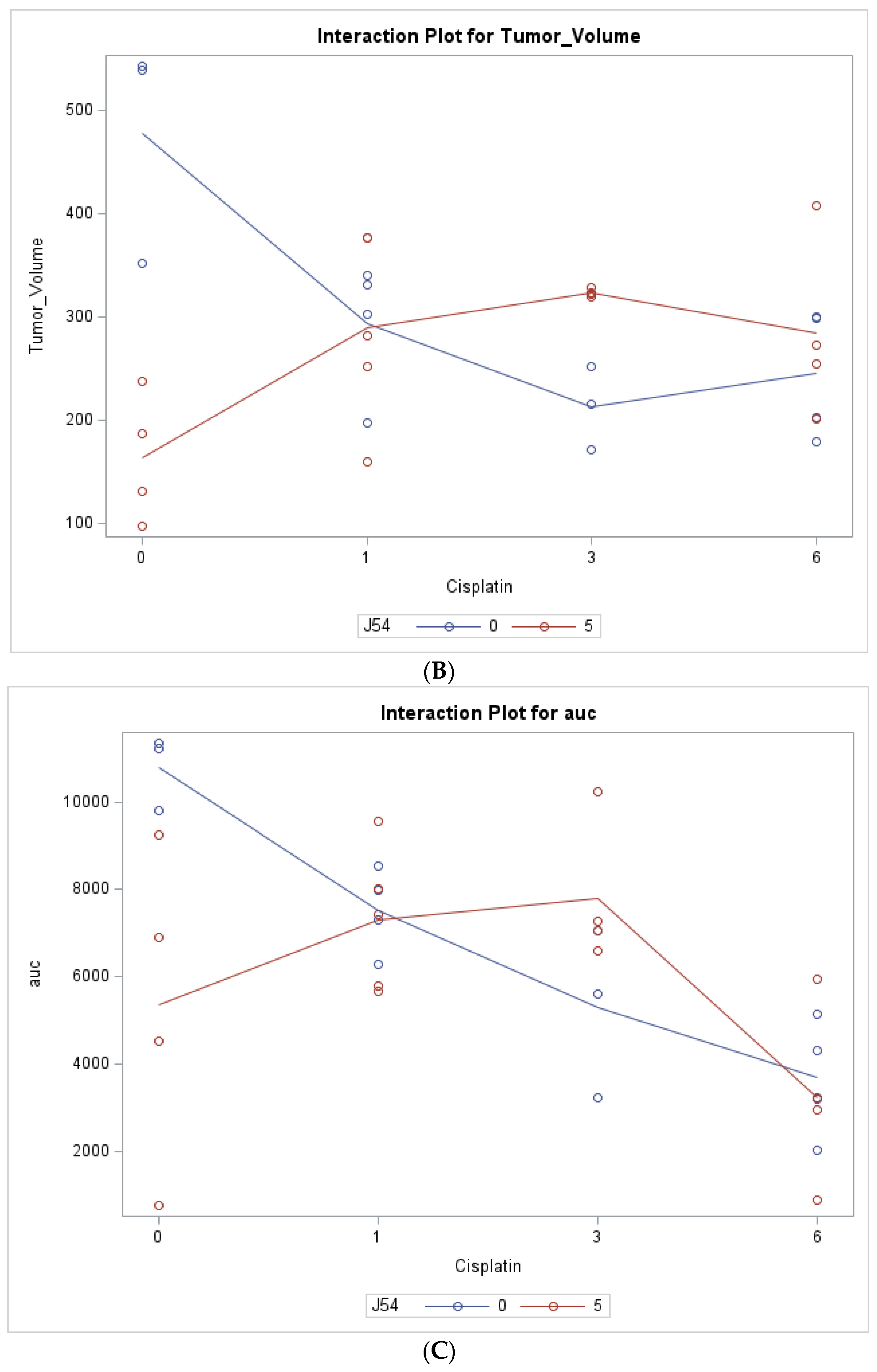{kind=link}
1. Introduction
2. Materials and Methods
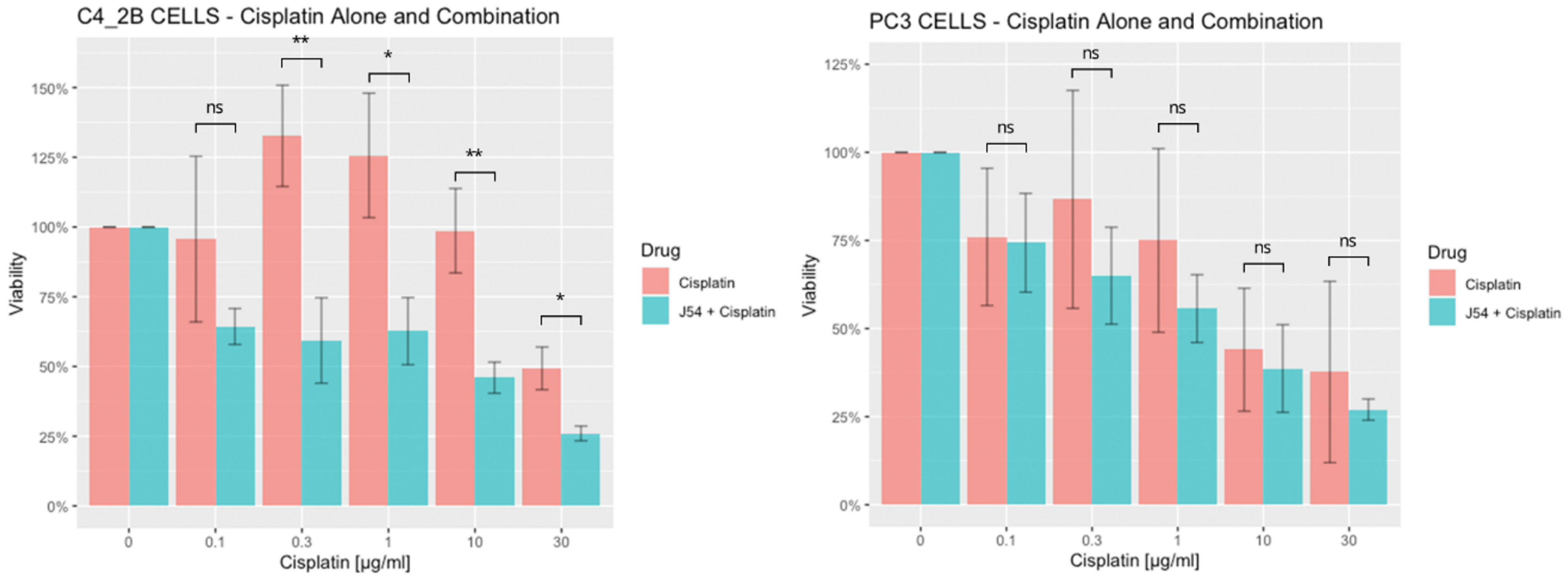
2.1. Cell Viability Assay
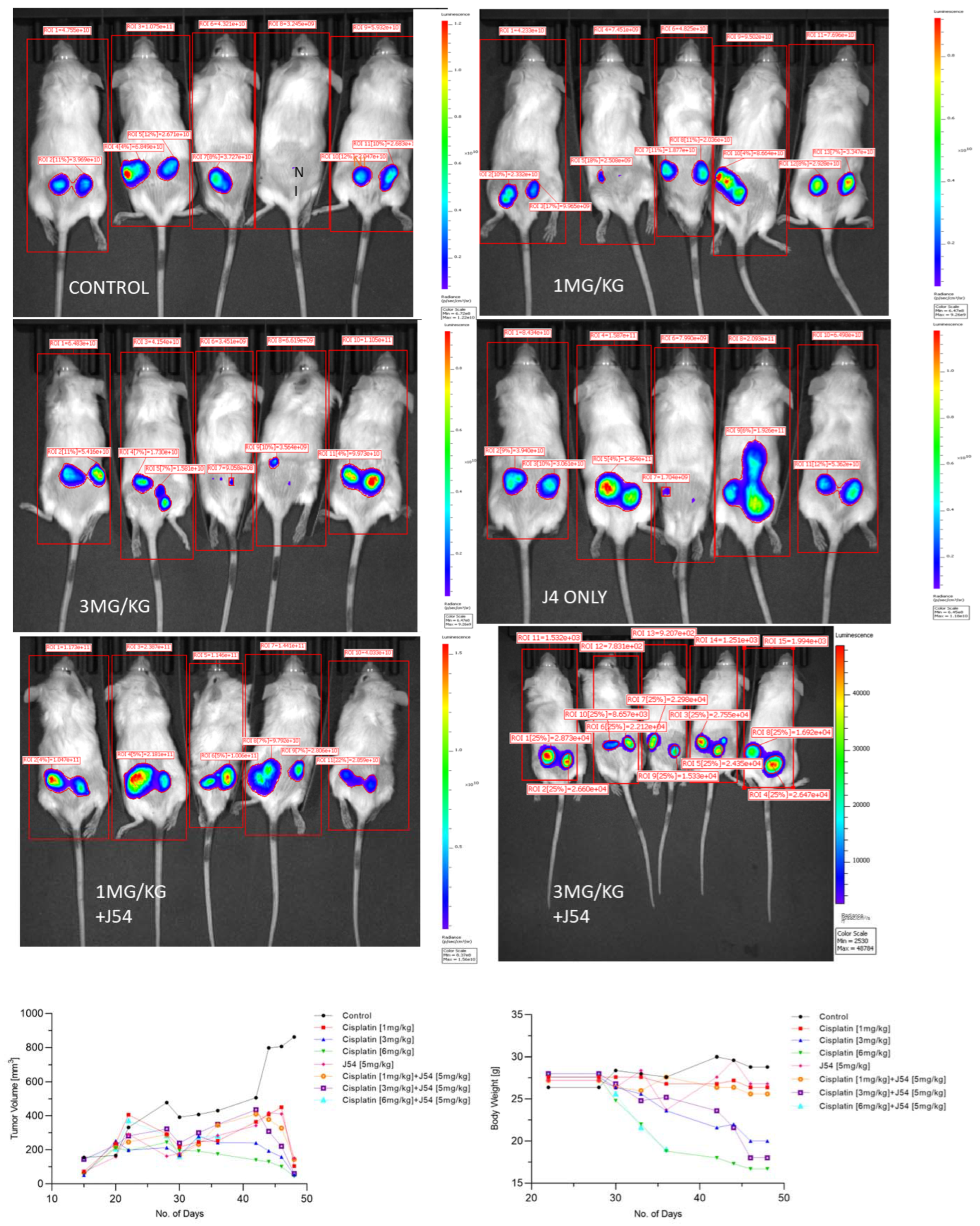
2.2. Animal Studies
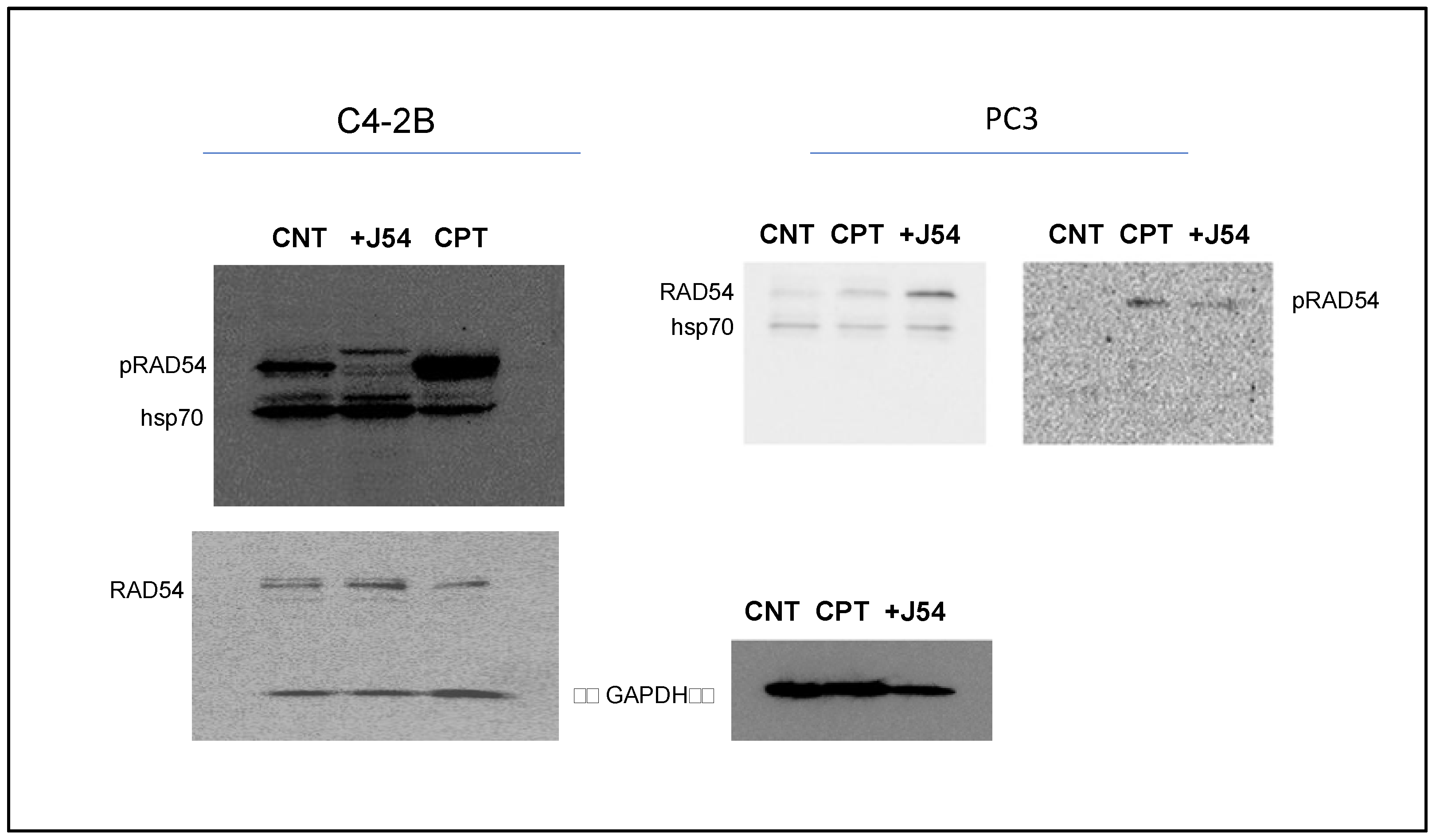
2.3. Western Blots
- a.
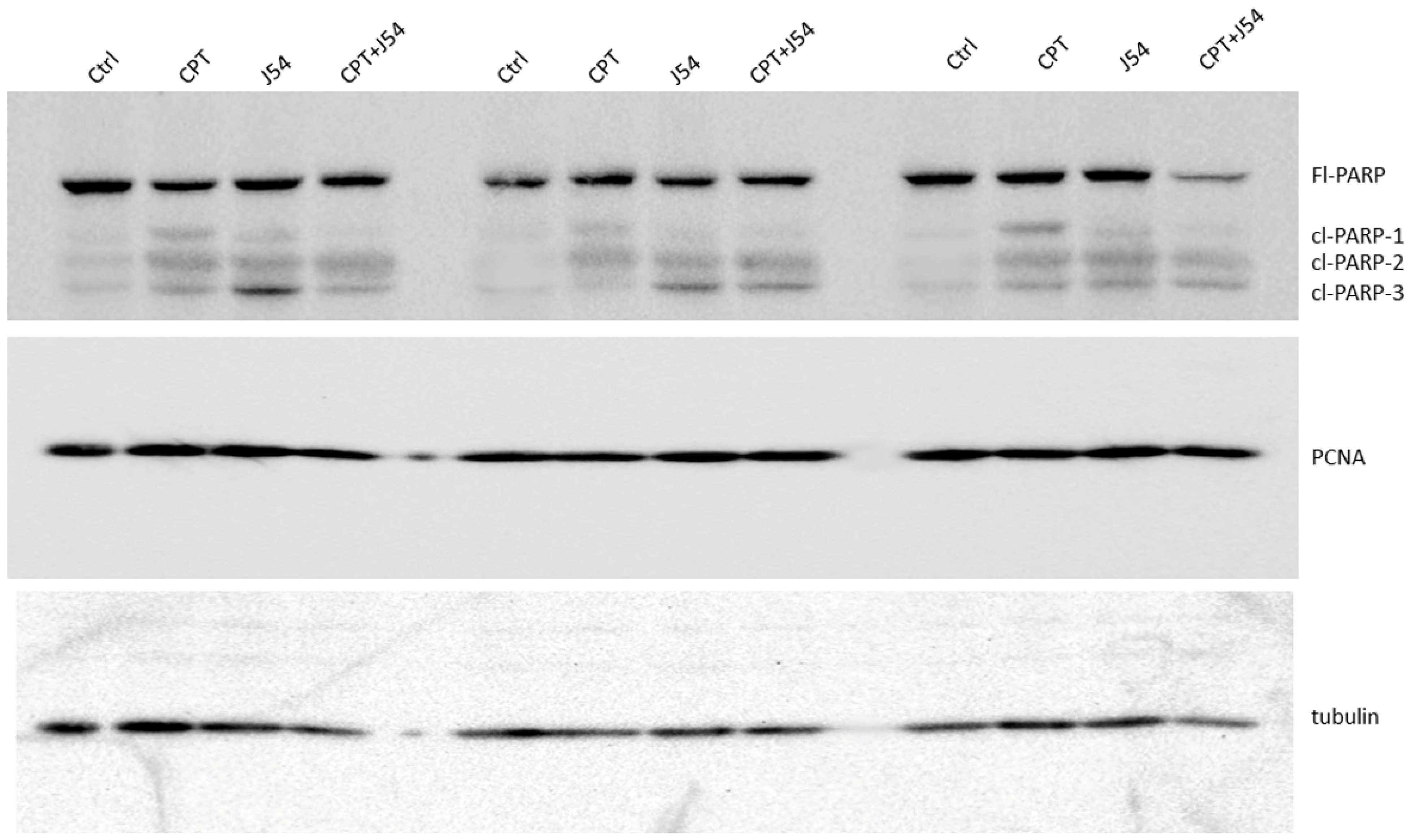
- Tissue Western Blot: Western blots were performed in three biological replicates for the tumors excised from the different treatment groups, including the control (PBS), cisplatin (3 mg/kg), J54 (5 mg/kg) and the combination of the PC-3 grafted NOD SCID mice. The frozen tumor tissues were disrupted with the Bioruptor® Plus sonication device (Diagenode; Cat. No. B01020001), and homogenized and lyzed in the ice-cold RIPA lysis buffer system (Santa Cruz Biotechnology, Dallas, TX, USA; Cat. No. SC-24948). The samples were clarified via centrifugation at 13,000 rpm for 20 min in the refrigerated setting. The supernatant was collected, transferred into fresh 1.5 mL microfuge tubes, flash-frozen and stored at −80 °C until further use. The total protein concentration was measured using a Pierce™ BCA protein assay kit (Thermo Scientific, Waltham, MA, USA; Cat. No. 23225) with bovine serum albumin (BSA) as a standard control. An equal loading amount of 15 µg was calculated for each protein sample. The sample supernatant was denatured with 1X Laemmli Buffer for 10 min at 950C and separated using 12% Mini PROTEAN TGX protein gel (BioRad, Hercules, CA, USA; Cat. No. 4568084) at 100 volts for 120 min. The proteins were transferred to the Immun-Blot PVDF membrane (BioRad; Cat. No. 1620177) using a Mini Trans-Blot Cell (BioRad; Cat. No. 1703930) at 100 volts for 150–180 min on ice. The membrane was blocked with 5% non-fat dry milk (Cell Signaling Technology, Danvers, MA, USA; Cat. No. 9999S) in 1X Tris-buffered saline with Tween-20 (TBST) for 1 h at room temperature. Following blocking, the membrane was washed once with 1X TBST and incubated with mouse anti-PCNA (PC10) monoclonal antibodies (Santa Cruz Biotechnology; Cat. No. SC-56; 1:1000 dilution) and mouse anti-PARP-1 (F-2) monoclonal antibodies (Santa Cruz Biotechnology; Cat. No. SC-8007; 1:1000 dilution) or anti-Cl-CAS3 (Asp175) rabbit antibodies (Santa Cruz Biotechnology; Cat. No. SC-9661) in 5% BSA in 1X TBST overnight at 4 °C with gentle rocking. The next day, after washing four times with 1X TBST, the membrane was incubated with horse anti-mouse antibodies (Cell Signaling Technology; Cat. No. 7076S: 1:2000 dilution) labeled with horseradish peroxidase in 5% BSA in 1X TBST for 1–1.5 h at room temperature. After incubation, the membrane was washed four times with 1X TBST, and the reactive bands were detected using a Pierce™ ECL Western Blotting Substrate (Thermo Scientific; Cat. No. 32106) on the ChemiDoc MP Imaging System (BioRad; Cat. No. 12003154).
- b.
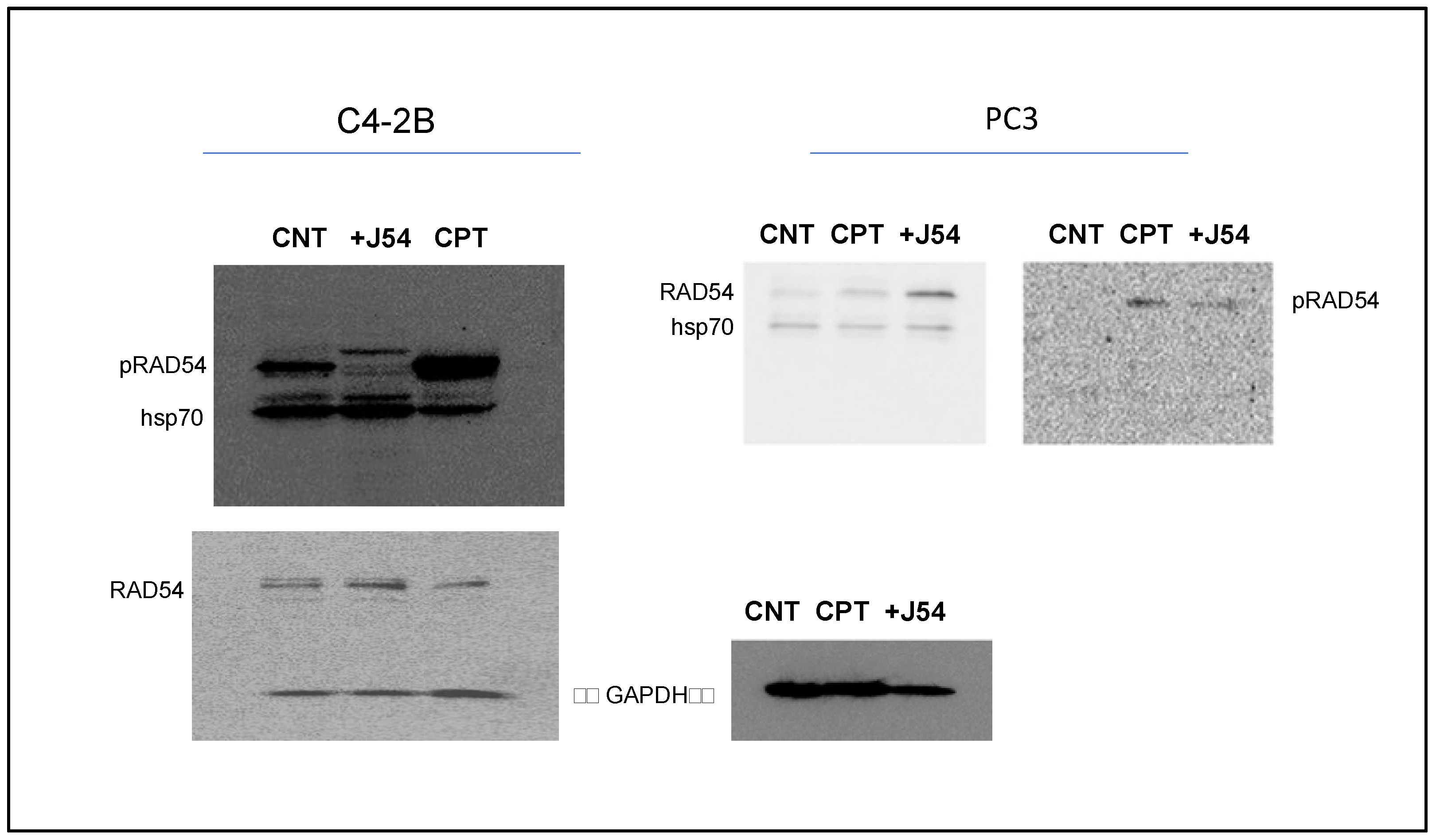
- Cell Western Blot: The Western blot for the PC-3 cells was performed as described above but with minor modifications. Briefly, 3 × 106 PC-3 cells (control and drug-treated) were collected, washed twice with ice-cold PBS and lyzed with the RIPA lysis buffer system. The lysate was vortexed and centrifuged at 13,000 rpm for 10 min to remove cell debris. The total protein was estimated, and 30 μg of the cell lysate was loaded onto an SDS-PAGE gel. The separated proteins were transferred to the membrane using a wet transfer apparatus. The complete transfer was ensured by checking the membrane for uniform background staining. The membrane was then incubated in a blocking solution (e.g., 5% non-fat milk in TBST) for 1 h at room temperature to block non-specific binding sites, followed by primary antibody (custom-made anti-pRAD54 rabbit polyclonal; Thermo Scientific; Cat. No. AB1991; 1:1000) incubation in a blocking solution overnight at 4 °C. The next day, the membrane was washed 3× with TBST for 10 min each to remove excess primary antibodies. Further, it was incubated with goat anti-rabbit HRP-conjugated secondary antibodies diluted in a blocking solution for 1 h at room temperature. Next, the membrane was washed 3× with TBST for 10 min each to remove excess secondary antibodies, and the bands were detected using the ECL chemiluminescent substrate.
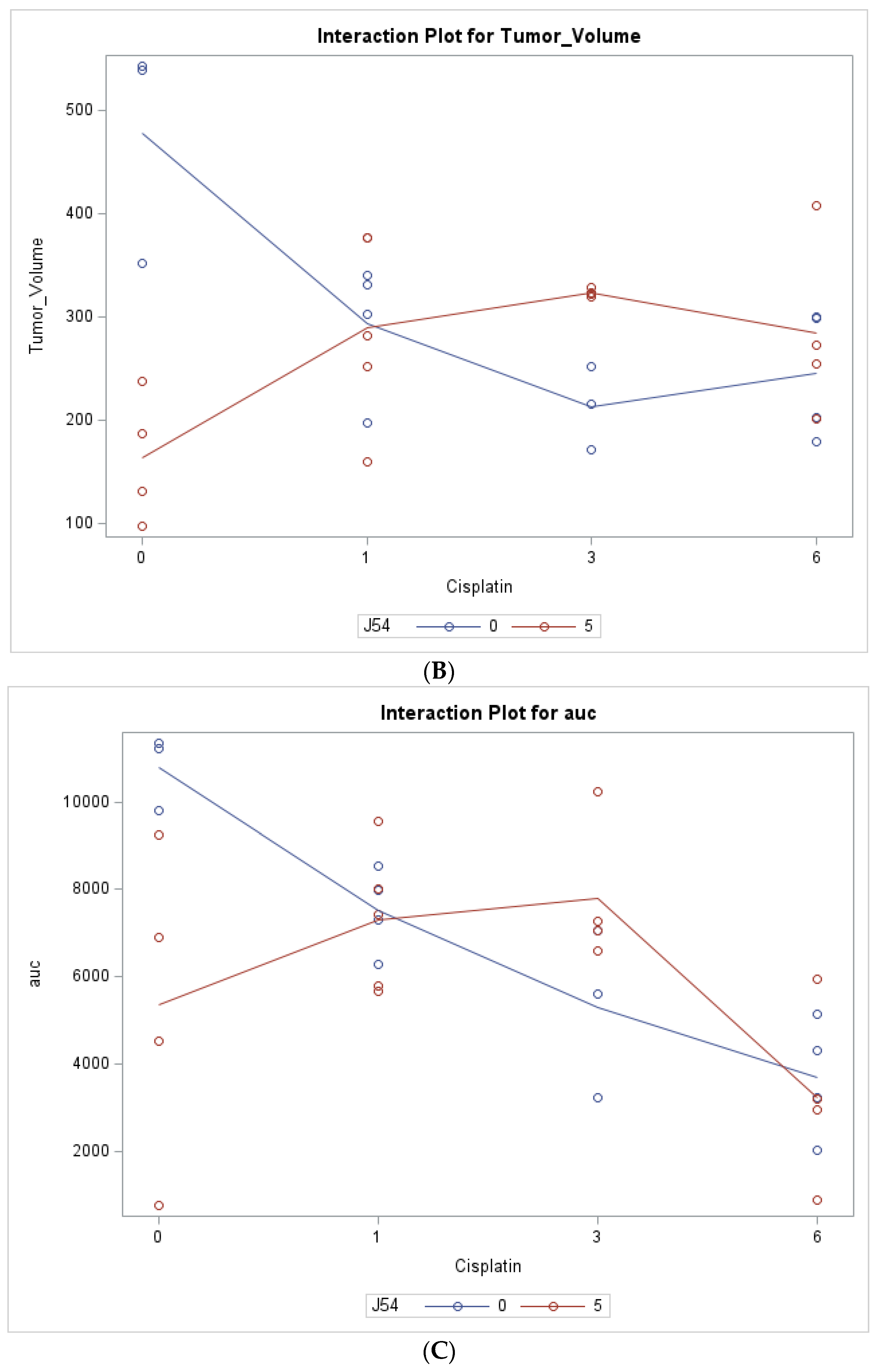
2.4. Statistical Analysis
2.5. Data Availability
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic lethality in cancer therapeutics: The next generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis from the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Tsujino, T.; Takai, T.; Hinohara, K.; Gui, F.; Tsutsumi, T.; Bai, X.; Miao, C.; Feng, C.; Gui, B.; Sztupinszki, Z.; et al. CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. bioRxiv 2022. preprint. [Google Scholar] [CrossRef]
- Adams, M.; Ashton, N.; Paquet, N.; O’Byrne, K.; Richard, D. Mechanisms of cisplatin resistance: DNA repair and cellular implications. Adv. Drug Resist. Res. 2014, 4, 1–37. [Google Scholar]
- Kiss, R.C.; Xia, F.; Acklin, S. Targeting DNA Damage Response and Repair to Enhance Therapeutic Index in Cisplatin-Based Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 8199. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; Jones, D.; Lee, S.H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. J. Natl. Cancer Inst. 2017, 109, djx059. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.; Omlin, A.; Higano, C.; Sweeney, C.; Martinez Chanza, N.; Mehra, N.; Kuppen, M.C.P.; Beltran, H.; Conteduca, V.; Vargas Pivato de Almeida, D.; et al. Activity of Platinum-Based Chemotherapy in Patients With Advanced Prostate Cancer with and without DNA Repair Gene Aberrations. JAMA Netw. Open 2020, 3, e2021692. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes. Environ. 2016, 38, 9. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef]
- Winter, C.; Albers, P. Testicular germ cell tumors: Pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2011, 7, 43–53. [Google Scholar] [CrossRef]
- Perše, M. Cisplatin Mouse Models: Treatment, Toxicity and Translatability. Biomedicines 2021, 9, 1406. [Google Scholar] [CrossRef]
- Ormerod, M.G.; Orr, R.M.; Peacock, J.H. The role of apoptosis in cell killing by cisplatin: A flow cytometric study. Br. J. Cancer 1994, 69, 93–100. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhzad, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 17356–17361. [Google Scholar] [CrossRef] [PubMed]
- Mota, J.M.; Barnett, E.; Nauseef, J.T.; Nguyen, B.; Stopsack, K.H.; Wibmer, A.; Flynn, J.R.; Heller, G.; Danila, D.C.; Rathkopf, D.; et al. Platinum-Based Chemotherapy in Metastatic Prostate Cancer with DNA Repair Gene Alterations. JCO Precis. Oncol. 2020, 4, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Selemenakis, P.; Sharma, N.; Uhrig, M.E.; Katz, J.; Kwon, Y.; Sung, P.; Wiese, C. RAD51AP1 and RAD54L Can Underpin Two Distinct RAD51-Dependent Routes of DNA Damage Repair via Homologous Recombination. Front. Cell Dev. Biol. 2022, 10, 866601. [Google Scholar] [CrossRef]
- Sillje, H.H.; Takahashi, K.; Tanaka, K.; Van Houwe, G.; Nigg, E.A. Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. Embo J. 1999, 18, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett. 2019, 453, 131–141. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G.; Fowler, M.; De Benedetti, A. Translation of the radioresistance kinase TLK1B is induced by gamma-irradiation through activation of mTOR and phosphorylation of 4E-BP1. BMC Mol. Biol. 2004, 5, 1. [Google Scholar] [CrossRef]
- Groth, A.; Lukas, J.; Nigg, E.A.; Sillje, H.H.; Wernstedt, C.; Bartek, J.; Hansen, K. Human Tousled like kinases are targeted by an ATM- and Chk1-dependent DNA damage checkpoint. Embo J. 2003, 22, 1676–1687. [Google Scholar] [CrossRef]
- Li, Y.; DeFatta, R.; Anthony, C.; Sunavala, G.; De Benedetti, A. A translationally regulated Tousled kinase phosphorylates histone H3 and confers radioresistance when overexpressed. Oncogene 2001, 20, 726–738. [Google Scholar] [CrossRef]
- Sen, S.P.; De Benedetti, A. TLK1B promotes repair of UV-damaged DNA through chromatin remodeling by Asf1. BMC Mol. Biol. 2006, 7, 37. [Google Scholar] [CrossRef]
- Takayama, Y.; Kokuryo, T.; Yokoyama, Y.; Ito, S.; Nagino, M.; Hamaguchi, M.; Senga, T. Silencing of Tousled-like kinase 1 sensitizes cholangiocarcinoma cells to cisplatin-induced apoptosis. Cancer Lett. 2010, 296, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Kwon, Y.; Shabestari, A.B.; Chikhale, R.; Chen, J.; Wiese, C.; Sung, P.; De Benedetti, A. TLK1-mediated RAD54 phosphorylation spatio-temporally regulates Homologous Recombination Repair. Nucleic Acids Res. 2023, 51, 8643–8662. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.-D.; Li, X.; Rolfsmeier, M.; Zhang, X.-P. Rad54: The Swiss Army knife of homologous recombination? Nucleic Acids Res. 2006, 34, 4115–4125. [Google Scholar] [CrossRef]
- Singh, V.; Bhoir, S.; Chikhale, R.V.; Hussain, J.; Dwyer, D.; Bryce, R.A.; Kirubakaran, S.; De Benedetti, A. Generation of phenothiazine with potent anti-TLK1 activity for prostate cancer therapy. Iscience 2020, 23, 101474. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Castri, P.; Lee, Y.J.; Ponzio, T.; Maric, D.; Spatz, M.; Bembry, J.; Hallenbeck, J. Poly(ADP-ribose) polymerase-1 and its cleavage products differentially modulate cellular protection through NF-kappaB-dependent signaling. Biochim. Biophys. Acta 2014, 1843, 640–651. [Google Scholar] [CrossRef]
- Los, M.; Mozoluk, M.; Ferrari, D.; Stepczynska, A.; Stroh, C.; Renz, A.; Herceg, Z.; Wang, Z.Q.; Schulze-Osthoff, K. Activation and caspase-mediated inhibition of PARP: A molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol. Biol. Cell 2002, 13, 978–988. [Google Scholar] [CrossRef]
- Ronald, S.; Awate, S.; Rath, A.; Carroll, J.; Galiano, F.; Dwyer, D.; Kleiner-Hancock, H.; Mathis, J.M.; Vigod, S.; De Benedetti, A. Phenothiazine Inhibitors of TLKs Affect Double-Strand Break Repair and DNA Damage Response Recovery and Potentiate Tumor Killing with Radiomimetic Therapy. Genes. Cancer 2013, 4, 39–53. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. Int. J. Cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef]
- Ramakrishnan Geethakumari, P.; Schiewer, M.J.; Knudsen, K.E.; Kelly, W.K. PARP Inhibitors in Prostate Cancer. Curr. Treat. Options Oncol. 2017, 18, 37. [Google Scholar] [CrossRef]
- Stopsack, K.H. Efficacy of PARP Inhibition in Metastatic Castration-Resistant Prostate Cancer Is very Different with Non-BRCA DNA Repair Alterations: Reconstructing Prespecified Endpoints for Cohort B from the Phase 3 PROfound Trial of Olaparib. Eur. Urol. 2021, 79, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Löbrich, M. Misrepair of radiation-induced DNA double-strand breaks and its relevance for tumorigenesis and cancer treatment (review). Int. J. Oncol. 2002, 21, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Gumulec, J.; Balvan, J.; Sztalmachova, M.; Raudenska, M.; Dvorakova, V.; Knopfova, L.; Polanska, H.; Hudcova, K.; Ruttkay-Nedecky, B.; Babula, P.; et al. Cisplatin-resistant prostate cancer model: Differences in antioxidant system, apoptosis and cell cycle. Int. J. Oncol. 2014, 44, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef]
- Carli, M.; Kolachalam, S.; Longoni, B.; Pintaudi, A.; Baldini, M.; Aringhieri, S.; Fasciani, I.; Annibale, P.; Maggio, R.; Scarselli, M. Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences. Pharmaceuticals 2021, 14, 238. [Google Scholar] [CrossRef]
- Dayabandara, M.; Hanwella, R.; Ratnatunga, S.; Seneviratne, S.; Suraweera, C.; de Silva, V.A. Antipsychotic-associated weight gain: Management strategies and impact on treatment adherence. Neuropsychiatr. Dis. Treat. 2017, 13, 2231–2241. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhoir, S.; Ogundepo, O.; Yu, X.; Shi, R.; De Benedetti, A. Exploiting TLK1 and Cisplatin Synergy for Synthetic Lethality in Androgen-Insensitive Prostate Cancer. Biomedicines 2023, 11, 2987. https://doi.org/10.3390/biomedicines11112987
Bhoir S, Ogundepo O, Yu X, Shi R, De Benedetti A. Exploiting TLK1 and Cisplatin Synergy for Synthetic Lethality in Androgen-Insensitive Prostate Cancer. Biomedicines. 2023; 11(11):2987. https://doi.org/10.3390/biomedicines11112987
Chicago/Turabian StyleBhoir, Siddhant, Oluwatobi Ogundepo, Xiuping Yu, Runhua Shi, and Arrigo De Benedetti. 2023. "Exploiting TLK1 and Cisplatin Synergy for Synthetic Lethality in Androgen-Insensitive Prostate Cancer" Biomedicines 11, no. 11: 2987. https://doi.org/10.3390/biomedicines11112987
APA StyleBhoir, S., Ogundepo, O., Yu, X., Shi, R., & De Benedetti, A. (2023). Exploiting TLK1 and Cisplatin Synergy for Synthetic Lethality in Androgen-Insensitive Prostate Cancer. Biomedicines, 11(11), 2987. https://doi.org/10.3390/biomedicines11112987