

Characteristics of Sensory Neuron Dysfunction in Amyotrophic Lateral Sclerosis (ALS): Potential for ALS Therapy
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. An Overview of ALS
2.1. Clinical Characteristics of ALS
2.2. Motor Neuron Dysfunction in ALS
3. Clinical Research in Patients with ALS
3.1. Clinical Features of Sensory Neuron Dysfunction in ALS
3.2. Peripheral Nerve Abnormalities Observed in Patients with ALS
3.3. Pathological Alterations in Sensory Neurons in Patients with ALS
3.4. Abnormalities of the Somatosensory Cortex in Patients with ALS
4. Preclinical Research in ALS
4.1. Pathological Alterations in Sensory Neurons in Patients with ALS
4.2. Degeneration of DRG Neurons
4.3. Characteristic Abnormalities in Mesencephalic Trigeminal Neurons
4.4. Protein Aggregates and Mitochondrial Dysfunction
4.5. Glial Involvement
4.6. Excitotoxicity and Calcium Dysregulation
4.7. Oxidative Stress and Protein Homeostasis
4.8. Protein Changes in Sensory Neurons
4.9. Role of Cytoskeletal Dysregulation and Neuronal Transport
5. Targeting Proprioceptive Sensory Neurons as a Potential Therapy for ALS
5.1. Current ALS Drugs
5.2. Addressing DRG Neuron Abnormalities May Treat Motor Neuron Impairments in ALS
5.3. Potential for Gene Therapy Targeting TDP-43
5.4. Treatment Strategies for ALS Masticatory Abnormalities through the MesV
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Chalabi, A.; Hardiman, O. The Epidemiology of ALS: A Conspiracy of Genes, Environment and Time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Gonzalo, I.; Ruiz-Soto, M.; Berciano, J. Why Do Motor Neurons Degenerate? Actualization in the Pathogenesis of Amyotrophic Lateral Sclerosis. Neurología 2019, 34, 27–37. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Szabó, Á.; Spekker, E.; Polyák, H.; Tóth, F.; Vécsei, L. Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan–Kynurenine Metabolic System. Cells 2022, 11, 2607. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.; Saccon, R.A.; Fratta, P.; Fisher, E.M. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Hübers, A.; Ludolph, A.C.; Rosenbohm, A.; Pinkhardt, E.H.; Weishaupt, J.H.; Dorst, J. Amyotrophe Lateralsklerose. Eine Multisystemdegeneration. Nervenarzt 2016, 87, 179–188. [Google Scholar] [CrossRef]
- Müller, K.; Brenner, D.; Weydt, P.; Meyer, T.; Grehl, T.; Petri, S.; Grosskreutz, J.; Schuster, J.; Volk, A.E.; Borck, G.; et al. Comprehensive Analysis of the Mutation Spectrum in 301 German ALS Families. J. Neurol. Neurosurg. Psychiatry 2018, 89, 817–827. [Google Scholar] [CrossRef]
- Ruiz-Soto, M.; Riancho, J.; Tapia, O.; Lafarga, M.; Berciano, M.T. Satellite Glial Cells of the Dorsal Root Ganglion: A New “Guest/Physiopathological Target” in ALS. Front. Aging Neurosci. 2020, 12, 595751. [Google Scholar] [CrossRef]
- Yun, Y.; Ha, Y. CRISPR/Cas9-Mediated Gene Correction to Understand ALS. Int. J. Mol. Sci. 2020, 21, 3801. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Geser, F.; Martinez-Lage, M.; Robinson, J.; Uryu, K.; Neumann, M.; Brandmeir, N.J.; Xie, S.X.; Kwong, L.K.; Elman, L.; McCluskey, L.; et al. Clinical and Pathological Continuum of Multisystem TDP-43 Proteinopathies. Arch. Neurol. 2009, 66, 180–189. [Google Scholar] [CrossRef]
- McCombe, P.A.; Wray, N.R.; Henderson, R.D. Extra-motor Abnormalities in Amyotrophic Lateral Sclerosis: Another Layer of Heterogeneity. Expert Rev. Neurother. 2017, 17, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649. [Google Scholar] [CrossRef]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 Aggregation Induced by Oxidative Stress Causes Global Mitochondrial Imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef]
- De Marchi, F.; Franjkic, T.; Schito, P.; Russo, T.; Nimac, J.; Chami, A.A.; Mele, A.; Vidatic, L.; Kriz, J.; Julien, J.P.; et al. Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder. Biomedicines 2023, 11, 1599. [Google Scholar] [CrossRef]
- Tao, Q.Q.; Wei, Q.; Wu, Z.Y. Sensory Nerve Disturbance in Amyotrophic Lateral Sclerosis. Life Sci. 2018, 203, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Matamala, J.M.; Howells, J.; Dharmadasa, T.; Huynh, W.; Park, S.B.; Burke, D.; Kiernan, M.C. Excitability of Sensory Axons in Amyotrophic Lateral Sclerosis. Clin. Neurophysiol. 2018, 129, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, M.; Turkman, A.; Swash, M. Sensory Modulation of Fasciculation Discharge Frequency. Muscle Nerve 2019, 59, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Paz-Fajardo, L.; López de Munaín, A. Clinical and Preclinical Evidence of Somatosensory Involvement in Amyotrophic Lateral Sclerosis. Br. J. Pharmacol. 2021, 178, 1257–1268. [Google Scholar] [CrossRef]
- Riku, Y.; Seilhean, D.; Duyckaerts, C.; Boluda, S.; Iguchi, Y.; Ishigaki, S.; Iwasaki, Y.; Yoshida, M.; Sobue, G.; Katsuno, M. Pathway from TDP-43-Related Pathology to Neuronal Dysfunction in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. Int. J. Mol. Sci. 2021, 22, 3843. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD Pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef]
- Bowser, R.; Turner, M.R.; Shefner, J. Biomarkers in Amyotrophic Lateral Sclerosis: Opportunities and Limitations. Nat. Rev. Neurol. 2011, 7, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Krüger, T.; Lautenschläger, J.; Grosskreutz, J.; Rhode, H. Proteome Analysis of Body Fluids for Amyotrophic Lateral Sclerosis Biomarker Discovery. Proteom. Clin. Appl. 2013, 7, 123–135. [Google Scholar] [CrossRef]
- Collins, M.A.; An, J.; Hood, B.L.; Conrads, T.P.; Bowser, R.P. Label-Free LC-MS/MS Proteomic analysis of Cerebrospinal Fluid Identifies Protein/Pathway Alterations and Candidate Biomarkers for Amyotrophic Lateral Sclerosis. J. Proteome Res. 2015, 14, 4486–4501. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.H.; Wu, J.J.; Ren, H.M.; Wang, J.; Ding, Z.T.; Jiang, Y.P. Proteomic Analysis of Cerebrospinal Fluid in Amyotrophic Lateral Sclerosis. Exp. Ther. Med. 2016, 11, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.T.; Bowser, R. Fluid-Based Biomarkers for Amyotrophic Lateral Sclerosis. Neurotherapeutics 2017, 14, 119–134. [Google Scholar] [CrossRef]
- Barschke, P.; Oeckl, P.; Steinacker, P.; Ludolph, A.; Otto, M. Proteomic Studies in the Discovery of Cerebrospinal Fluid Biomarkers for Amyotrophic Lateral Sclerosis. Expert Rev. Proteom. 2017, 14, 769–777. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Thézénas, M.L.; Charles, P.D.; Evetts, S.; Hu, M.T.; Talbot, K.; Fischer, R.; Kessler, B.M.; Turner, M.R. Cerebrospinal Fluid Macrophage Biomarkers in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2018, 83, 258–268. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Recent Advances in the Diagnosis and Prognosis of Amyotrophic Lateral Sclerosis. Lancet Neurol. 2022, 21, 480–493. [Google Scholar] [CrossRef]
- Oh, S.; Jang, Y.; Na, C.H. Discovery of Biomarkers for Amyotrophic Lateral Sclerosis from Human Cerebrospinal Fluid Using Mass-Spectrometry-Based Proteomics. Biomedicines 2023, 11, 1250. [Google Scholar] [CrossRef] [PubMed]
- Jerusalem, F.; Pohl, C.; Karitzky, J.; Ries, F. ALS. Neurology 1996, 47 (Suppl. 4), S218–S220. [Google Scholar] [CrossRef]
- Rubio, M.A.; Herrando-Grabulosa, M.; Navarro, X. Sensory Involvement in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 15521. [Google Scholar] [CrossRef] [PubMed]
- Sassone, J.; Taiana, M.; Lombardi, R.; Porretta-Serapiglia, C.; Freschi, M.; Bonanno, S.; Marcuzzo, S.; Caravello, F.; Bendotti, C.; Lauria, G. ALS Mouse Model SOD1G93A Displays Early Pathology of Sensory Small Fibers Associated to Accumulation of a Neurotoxic Splice Variant of Peripherin. Hum. Mol. Genet. 2016, 25, 1588–1599. [Google Scholar] [CrossRef]
- Seki, S.; Yamamoto, T.; Quinn, K.; Spigelman, I.; Pantazis, A.; Olcese, R.; Wiedau-Pazos, M.; Chandler, S.H.; Venugopal, S. Circuit-Specific Early Impairment of Proprioceptive Sensory Neurons in the SOD1G93A Mouse Model for ALS. J. Neurosci. 2019, 39, 8798–8815. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, Y.; Seki, S.; Kawata, S.; Nishiura, A.; Kawamura, K.; Hiraoka, S.I.; Kogo, M.; Tanaka, S. Analysis of Feeding Behavior Characteristics in the Cu/Zn Superoxide Dismutase 1 (SOD1) SOD1G93A Mice Model for Amyotrophic Lateral Sclerosis (ALS). Nutrients 2023, 15, 1651. [Google Scholar] [CrossRef]
- Ferrea, S.; Junker, F.; Korth, M.; Gruhn, K.; Grehl, T.; Schmidt-Wilcke, T. Cortical Thinning of Motor and Non-Motor Brain Regions Enables Diagnosis of Amyotrophic Lateral Sclerosis and Supports Distinction Between Upper- and Lower-Motoneuron Phenotypes. Biomedicines 2021, 9, 1195. [Google Scholar] [CrossRef]
- Raghunathan, R.; Turajane, K.; Wong, L.C. Biomarkers in Neurodegenerative Diseases: Proteomics Spotlight on ALS and Parkinson’s disease. Int. J. Mol. Sci. 2022, 23, 9299. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Mitchell, J.D.; Borasio, G.D. Amyotrophic Lateral Sclerosis. Lancet 2007, 369, 2031–2041. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic Lateral Sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic Lateral Sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Akçimen, F.; Lopez, E.R.; Landers, J.E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B.J. Amyotrophic Lateral Sclerosis: Translating Genetic Discoveries into Therapies. Nat. Rev. Genet. 2023, 24, 642–658. [Google Scholar] [CrossRef] [PubMed]
- Ravnik Glavač, M.; Mezzavilla, M.; Dolinar, A.; Koritnik, B.; Glavač, D. Aberrantly Expressed Hsa_circ_0060762 and CSE1L as Potential Peripheral Blood Biomarkers for ALS. Biomedicines 2023, 11, 1316. [Google Scholar] [CrossRef]
- Canosa, A.; Calvo, A.; Mora, G.; Moglia, C.; Brunetti, M.; Barberis, M.; Borghero, G.; Caponnetto, C.; Trojsi, F.; Spataro, R.; et al. The HFE p.H63D (p.His63Asp) Polymorphism Is a Modifier of ALS Outcome in Italian and French Patients with SOD1 Mutations. Biomedicines 2023, 11, 704. [Google Scholar] [CrossRef]
- Zufiría, M.; Gil-Bea, F.J.; Fernández-Torrón, R.; Poza, J.J.; Muñoz-Blanco, J.L.; Rojas-García, R.; Riancho, J.; López de Munain, A. ALS: A Bucket of Genes, Environment, Metabolism and Unknown Ingredients. Prog. Neurobiol. 2016, 142, 104–129. [Google Scholar] [CrossRef]
- Zhang, R.; Bracci, P.M.; Azhir, A.; Forrest, B.D.; McGrath, M.S. Macrophage-Targeted Sodium Chlorite (NP001) Slows Progression of Amyotrophic Lateral Sclerosis (ALS) through Regulation of Microbial Translocation. Biomedicines 2022, 10, 2907. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic Lateral Sclerosis: A Clinical Review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Wintz, K.; Post, J.; Langen, K.J.; Willbold, D.; Willuweit, A.; Kutzsche, J. Oral Treatment with d-RD2RD2 Impedes Early Disease Mechanisms in SOD1*G93A Transgenic Mice but Does Not Prolong Survival. Biomedicines 2023, 11, 995. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic Lateral Sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- Ma, X.R.; Prudencio, M.; Koike, Y.; Vatsavayai, S.C.; Kim, G.; Harbinski, F.; Briner, A.; Rodriguez, C.M.; Guo, C.; Akiyama, T.; et al. TDP-43 Represses Cryptic Exon Inclusion in the FTD-ALS Gene UNC13A. Nature 2022, 603, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Hu, J.; Arogundade, O.A.; Goginashvili, A.; Vazquez-Sanchez, S.; Diedrich, J.K.; Gu, J.; Blum, J.; Oung, S.; Ye, Q.; et al. Heat-Shock Chaperone HSPB1 Regulates Cytoplasmic TDP-43 Phase Separation and Liquid-to-Gel Transition. Nat. Cell. Biol. 2022, 24, 1378–1393. [Google Scholar] [CrossRef]
- Linseman, D.A.; Winter, A.N.; Wilkins, H.M. The 2-Oxoglutarate Carrier Is S-Nitrosylated in the Spinal Cord of G93A Mutant hSOD1 Mice Resulting in Disruption of Mitochondrial Glutathione Transport. Biomedicines 2022, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Hammad, M.; Silva, A.; Glass, J.; Sladky, J.T.; Benatar, M. Clinical, Electrophysiologic, and Pathologic Evidence for Sensory Abnormalities in ALS. Neurology 2007, 69, 2236–2242. [Google Scholar] [CrossRef]
- Cohen-Adad, J.; El Mendili, M.M.; Morizot-Koutlidis, R.; Lehéricy, S.; Meininger, V.; Blancho, S.; Rossignol, S.; Benali, H.; Pradat, P.F. Involvement of Spinal Sensory Pathway in ALS and Specificity of Cord Atrophy to Lower Motor Neuron Degeneration. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2013, 14, 30–38. [Google Scholar] [CrossRef]
- Isak, B.; Pugdahl, K.; Karlsson, P.; Tankisi, H.; Finnerup, N.B.; Furtula, J.; Johnsen, B.; Sunde, N.; Jakobsen, J.; Fuglsang-Frederiksen, A. Quantitative Sensory Testing and Structural Assessment of Sensory Nerve Fibres in Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2017, 373, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Urso, D.; Zoccolella, S.; Gnoni, V.; Logroscino, G. Amyotrophic Lateral Sclerosis—The Complex Phenotype—From an Epidemiological Perspective: A Focus on Extrapyramidal and Non-Motor Features. Biomedicines 2022, 10, 2537. [Google Scholar] [CrossRef]
- Nolano, M.; Provitera, V.; Manganelli, F.; Iodice, R.; Caporaso, G.; Stancanelli, A.; Marinou, K.; Lanzillo, B.; Santoro, L.; Mora, G. Non-Motor Involvement in Amyotrophic Lateral Sclerosis: New Insight From Nerve and Vessel Analysis in Skin Biopsy. Neuropathol. Appl. Neurobiol. 2017, 43, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Venugopal, S.; Majid, S.; Ahn, I.S.; Diamante, G.; Hong, J.; Yang, X.; Chandler, S.H. Single-cell RNA-seq Analysis of the Brainstem of Mutant SOD1 Mice Reveals Perturbed Cell Types and Pathways of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2020, 141, 104877. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, X.; Ding, X.; Song, M.; Sui, K. Analysis of Clinical and Electrophysiological Characteristics of 150 Patients With Amyotrophic Lateral Sclerosis in China. Neurol. Sci. 2019, 40, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Amoiridis, G.; Tsimoulis, D.; Ameridou, I. Clinical, Electrophysiologic, and Pathologic Evidence for Sensory Abnormalities in ALS. Neurology 2008, 2, 779. [Google Scholar] [CrossRef] [PubMed]
- Mulder, D.W.; Bushek, W.; Spring, E.; Karnes, J.; Dyck, P.J. Motor Neuron Disease (ALS): Evaluation of Detection Thresholds of Cutaneous Sensation. Neurology 1983, 33, 1625–1627. [Google Scholar] [CrossRef]
- Sonkodi, B.; Hortobágyi, T. Amyotrophic Lateral Sclerosis and Delayed Onset Muscle Soreness in Light of the Impaired Blink and Stretch Reflexes—Watch Out for Piezo2. Open Med. 2022, 17, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Mora, G.; Lauria, G. Pain in Amyotrophic Lateral Sclerosis. Lancet Neurol. 2017, 16, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.; Mills, K.; Donaghy, M. Progressive Sensory Nerve Dysfunction in Amyotrophic Lateral Sclerosis: A Prospective Clinical and Neurophysiological Study. J. Neurol. 1993, 240, 309–314. [Google Scholar] [CrossRef]
- Pugdahl, K.; Fuglsang-Frederiksen, A.; Johnsen, B.; de Carvalho, M.; Fawcett, P.R.; Labarre-Vila, A.; Liguori, R.; Nix, W.A.; Schofield, I.S. A Prospective Multicentre Study on Sural Nerve Action Potentials in ALS. Clin. Neurophysiol. 2008, 119, 1106–1110. [Google Scholar] [CrossRef]
- Isak, B.; Tankisi, H.; Johnsen, B.; Pugdahl, K.; Torvin MØLler, A.; Finnerup, N.B.; Christensen, P.B.; Fuglsang-Frederiksen, A. Involvement of Distal Sensory Nerves in Amyotrophic Lateral Sclerosis. Muscle Nerve 2016, 54, 1086–1092. [Google Scholar] [CrossRef]
- Hama, T.; Hirayama, M.; Hara, T.; Nakamura, T.; Atsuta, N.; Banno, H.; Suzuki, K.; Katsuno, M.; Tanaka, F.; Sobue, G. Discrimination of Spinal and Bulbar Muscular Atrophy from Amyotrophic Lateral Sclerosis Using Sensory Nerve Action Potentials. Muscle Nerve 2012, 45, 169–174. [Google Scholar] [CrossRef]
- Shimizu, T.; Bokuda, K.; Kimura, H.; Kamiyama, T.; Nakayama, Y.; Kawata, A.; Isozaki, E.; Ugawa, Y. Sensory Cortex Hyperexcitability Predicts Short Survival in Amyotrophic Lateral Sclerosis. Neurology 2018, 90, e1578–e1587. [Google Scholar] [CrossRef] [PubMed]
- Held, A.; Major, P.; Sahin, A.; Reenan, R.A.; Lipscombe, D.; Wharton, K.A. Circuit Dysfunction in SOD1-ALS Model First Detected in Sensory Feedback Prior to Motor Neuron Degeneration Is Alleviated by BMP Signaling. J. Neurosci. 2019, 39, 2347–2364. [Google Scholar] [CrossRef]
- Nardone, R.; Golaszewski, S.; Thomschewski, A.; Sebastianelli, L.; Versace, V.; Brigo, F.; Orioli, A.; Saltuari, L.; Höller, Y.; Trinka, E. Disinhibition of Sensory Cortex in Patients with Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2020, 23, 722. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Liu, W.; Li, Y.; Sun, B.; Li, Y.; Yang, F.; Wang, H.; Li, M.; Cui, F.; Huang, X. Cutaneous Somatic and Autonomic Nerve TDP-43 Deposition in Amyotrophic Lateral Sclerosis. J. Neurol. 2018, 265, 1753–1763. [Google Scholar] [CrossRef]
- Truini, A.; Biasiotta, A.; Onesti, E.; Di Stefano, G.; Ceccanti, M.; La Cesa, S.; Pepe, A.; Giordano, C.; Cruccu, G.; Inghilleri, M. Small-fibre Neuropathy Related to Bulbar and Spinal-onset in Patients with ALS. J. Neurol. 2015, 262, 1014–1018. [Google Scholar] [CrossRef]
- Iglesias, C.; Sangari, S.; El Mendili, M.M.; Benali, H.; Marchand-Pauvert, V.; Pradat, P.F. Electrophysiological and Spinal Imaging Evidences for Sensory Dysfunction in Amyotrophic Lateral Sclerosis. BMJ Open 2015, 24, e007659. [Google Scholar] [CrossRef]
- Isaacs, J.D.; Dean, A.F.; Shaw, C.E.; Al-Chalabi, A.; Mills, K.R.; Leigh, P.N. Amyotrophic Lateral Sclerosis with Sensory Neuropathy: Part of a Multisystem Disorder? J. Neurol. Neurosurg. Psychiatry 2007, 78, 750–753. [Google Scholar] [CrossRef]
- Heads, T.; Pollock, M.; Robertson, A.; Sutherland, W.H.; Allpress, S. Sensory Nerve Pathology in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 1991, 82, 316–320. [Google Scholar] [CrossRef]
- Riancho, J.; Castanedo-Vázquez, D.; Gil-Bea, F.; Tapia, O.; Arozamena, J.; Durán-Vían, C.; Sedano, M.J.; Berciano, M.T.; Lopez de Munain, A.; Lafarga, M. ALS-Derived Fibroblasts Exhibit Reduced Proliferation Rate, Cytoplasmic TDP-43 Aggregation and a Higher Susceptibility to DNA Damage. J. Neurol. 2020, 267, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Conte, A.; Del Grande, A.; Bisogni, G.; Romano, A.; Sabatelli, M. Sural Nerve Pathology in ALS Patients: A Single-Centre Experience. Neurol. Sci. 2012, 33, 1095–1099. [Google Scholar] [CrossRef]
- Hamada, M.; Hanajima, R.; Terao, Y.; Sato, F.; Okano, T.; Yuasa, K.; Furubayashi, T.; Okabe, S.; Arai, N.; Ugawa, Y. Median Nerve Somatosensory Evoked Potentials and Their High-Frequency Oscillations in Amyotrophic Lateral Sclerosis. Clin. Neurophysiol. 2007, 118, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Theys, P.A.; Peeters, E.; Robberecht, W. Evolution of Motor and Sensory Deficits in Amyotrophic Lateral Sclerosis Estimated by Neurophysiological Techniques. J. Neurol. 1999, 246, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Simone, I.L.; Tortelli, R.; Samarelli, V.; D’Errico, E.; Sardaro, M.; Difruscolo, O.; Calabrese, R.; Francesco, V.D.V.; Livrea, P.; de Tommaso, M. Laser Evoked Potentials in Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2010, 15, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.K.; Sutherland, N.M.; Zhang, S.; Hatzipetros, T.; Vieira, F.; Valdez, G. The ALS-inducing Factors, TDP43A315T and SOD1G93A, Directly Affect and Sensitize Sensory Neurons to Stress. Sci. Rep. 2018, 8, 16582. [Google Scholar] [CrossRef]
- Schäfer, M.K.; Bellouze, S.; Jacquier, A.; Schaller, S.; Richard, L.; Mathis, S.; Vallat, J.M.; Haase, G. Sensory Neuropathy in Progressive Motor Neuronopathy (pmn) Mice is Associated with Defects in Microtubule Polymerization and Axonal Transport. Brain Pathol. 2017, 27, 459–471. [Google Scholar] [CrossRef]
- Schoeberl, F.; Abicht, A.; Kuepper, C.; Voelk, S.; Sonnenfeld, S.; Tonon, M.; Schaub, A.; Scholz, V.; Kleinle, S.; Erdmann, H.; et al. Sensory Neuropathy Due to RFC1 in a Patient with ALS: More Than a Coincidence? J. Neurol. 2022, 269, 2774–2777. [Google Scholar] [CrossRef]
- Ringer, C.; Weihe, E.; Schütz, B. SOD1G93A Mutant Mice Develop a Neuroinflammation-Independent Dendropathy in Excitatory Neuronal Subsets of the Olfactory Bulb and Retina. J. Neuropathol. Exp. Neurol. 2017, 76, 769–778. [Google Scholar] [CrossRef]
- Park, J.H.; Chung, C.G.; Park, S.S.; Lee, D.; Kim, K.M.; Jeong, Y.; Kim, E.S.; Cho, J.H.; Jeon, Y.M.; Shen, C.J.; et al. Cytosolic Calcium Regulates Cytoplasmic Accumulation of TDP-43 Through Calpain-A and Importin α3. eLife 2020, 9, e60132. [Google Scholar] [CrossRef]
- Rumpf, S.; Bagley, J.A.; Thompson-Peer, K.L.; Zhu, S.; Gorczyca, D.; Beckstead, R.B.; Jan, L.Y.; Jan, Y.N. Drosophila Valosin-Containing Protein is Required for Dendrite Pruning Through a Regulatory Role in mRNA Metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 7331–7336. [Google Scholar] [CrossRef]
- Guo, Y.S.; Wu, D.X.; Wu, H.R.; Wu, S.Y.; Yang, C.; Li, B.; Bu, H.; Zhang, Y.S.; Li, C.Y. Sensory Involvement in the SOD1-G93A Mouse Model of Amyotrophic Lateral Sclerosis. Exp. Mol. Med. 2009, 41, 140–150. [Google Scholar] [CrossRef]
- Yan, H.D.; Lim, W.; Lee, K.W.; Kim, J. Sera from Amyotrophic Lateral Sclerosis Patients Reduce High-Voltage Activated Ca2+ Currents in Mice Dorsal Root Ganglion Neurons. Neurosci. Lett. 1997, 235, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.A.; Herrando-Grabulosa, M.; Vilches, J.J.; Navarro, X. Involvement of Sensory Innervation in the Skin of SOD1(G93A) ALS Mice. J. Peripher. Nerv. Syst. 2016, 21, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Chrast, R.; Schneider, B.L. Endoplasmic Reticulum and Mitochondria in Diseases of Motor and Sensory Neurons: A Broken Relationship? Cell Death Dis. 2018, 9, 333. [Google Scholar] [CrossRef]
- Ayers, J.I.; Fromholt, S.E.; O’Neal, V.M.; Diamond, J.H.; Borchelt, D.R. Prion-Like Propagation of Mutant SOD1 Misfolding and Motor Neuron Disease Spread Along Neuroanatomical Pathways. Acta Neuropathol. 2016, 131, 103–114. [Google Scholar] [CrossRef]
- Vaughan, S.K.; Kemp, Z.; Hatzipetros, T.; Vieira, F.; Valdez, G. Degeneration of Proprioceptive Sensory Nerve Endings in Mice Harboring Amyotrophic Lateral Sclerosis-Causing Mutations. J. Comp. Neurol. 2015, 523, 2477–2494. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Seki, S.; Ono, Y.; Enomoto, A.; Kogo, M. Persistent Sodium Conductance Contributes to Orexin-A-mediated Modulation of Membrane Excitability in Neonatal Rat Mesencephalic V Neurons. Neurosci. Lett. 2021, 753, 135846. [Google Scholar] [CrossRef]
- Chandler, S.H. Evidence for Excitatory Amino Acid Transmission Between Mesencephalic Nucleus of V Afferents and Jaw-Closer Motoneurons in the Guinea Pig. Brain Res. 1989, 477, 252–264. [Google Scholar] [CrossRef]
- Trueblood, P.R.; Levine, M.S.; Chandler, S.H. Dual-Component Excitatory Amino Acid-mediated Responses in Trigeminal Motoneurons and their Modulation by Serotonin In Vitro. J. Neurophysiol. 1996, 76, 2461–2473. [Google Scholar] [CrossRef]
- Venugopal, S.; Hsiao, C.F.; Sonoda, T.; Wiedau-Pazos, M.; Chandler, S.H. Homeostatic Dysregulation in Membrane Properties of Masticatory Motoneurons Compared with Oculomotor Neurons in a Mouse Model for Amyotrophic Lateral Sclerosis. J. Neurosci. 2015, 35, 707–720. [Google Scholar] [CrossRef]
- Venugopal, S.; Ghulam-Jhelani, Z.; Ahn, I.S.; Yang, X.; Wiedau, M.; Simmons, D.; Chandler, S.H. Early Deficits in GABA Inhibition Parallels an Increase in L-type Ca2+ Currents in the Jaw Motor Neurons of SOD1G93A Mouse Model for ALS. Neurobiol. Dis. 2023, 177, 105992. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Yamada, S. A Novel Hypothesis on Metal Dyshomeostasis and Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis: Potential Pathogenetic Mechanism and Therapeutic Implications. Eur. J. Pharmacol. 2021, 892, 173737. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial Bioenergetic Deficits in C9orf72 Amyotrophic Lateral Sclerosis Motor Neurons Cause Dysfunctional Axonal Homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Inoue, H.; Tsukita, K.; Iwasato, T.; Suzuki, Y.; Tomioka, M.; Tateno, M.; Nagao, M.; Kawata, A.; Saido, T.C.; Miura, M.; et al. The Crucial Role of Caspase-9 in the Disease Progression of a Transgenic ALS Mouse Model. EMBO J. 2003, 15, 6665–6674. [Google Scholar] [CrossRef]
- Nemtsova, Y.; Steinert, B.L.; Wharton, K.A. Compartment Specific Mitochondrial Dysfunction in Drosophila Knock-In Model of ALS Reversed by Altered Gene Expression of OXPHOS Subunits and Pro-Fission Factor Drp1. Mol. Cell. Neurosci. 2023, 125, 103834. [Google Scholar] [CrossRef]
- Pereira, J.D.; DuBreuil, D.M.; Devlin, A.C.; Held, A.; Sapir, Y.; Berezovski, E.; Hawrot, J.; Dorfman, K.; Chander, V.; Wainger, B.J. Human Sensorimotor Organoids Derived from Healthy and Amyotrophic Lateral Sclerosis Stem Cells form Neuromuscular Junctions. Nat. Commun. 2021, 12, 4744. [Google Scholar] [CrossRef] [PubMed]
- Pennetta, G.; Welte, M.A. Emerging Links between Lipid Droplets and Motor Neuron Diseases. Dev. Cell. 2018, 21, 427–432. [Google Scholar] [CrossRef]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in Lipid Metabolism of Spinal Cord Linked to Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 12, 11642. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct Roles for Motor Neuron Autophagy Early and Late in the SOD1G93A Mouse Model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef]
- Wiksten, M.; Väänänen, A.; Liesi, P. Selective Overexpression of Gamma1 Laminin in Astrocytes in Amyotrophic Lateral Sclerosis Indicates an Involvement in ALS Pathology. J. Neurosci. Res. 2007, 85, 2045–2058. [Google Scholar] [CrossRef]
- Peng, A.Y.T.; Agrawal, I.; Ho, W.Y.; Yen, Y.C.; Pinter, A.J.; Liu, J.; Phua, Q.X.C.; Koh, K.B.; Chang, J.C.; Sanford, E.; et al. Loss of TDP-43 in Astrocytes Leads to Motor Deficits by Triggering A1-like Reactive Phenotype and Triglial Dysfunction. Proc. Natl. Acad. Sci. USA 2020, 117, 29101–29112. [Google Scholar] [CrossRef]
- Stracher, A. Calpain Inhibitors as Therapeutic Agents in Nerve and Muscle Degeneration. Ann. N. Y. Acad. Sci. 1999, 884, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Dilliott, A.A.; Andary, C.M.; Stoltz, M.; Petropavlovskiy, A.A.; Farhan, S.M.K.; Duennwald, M.L. DnaJC7 in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 4076. [Google Scholar] [CrossRef] [PubMed]
- Lambert-Smith, I.A.; Saunders, D.N.; Yerbury, J.J. Proteostasis Impairment and ALS. Prog. Biophys. Mol. Biol. 2022, 174, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Krus, K.L.; Strickland, A.; Yamada, Y.; Devault, L.; Schmidt, R.E.; Bloom, A.J.; Milbrandt, J.; DiAntonio, A. Loss of Stathmin-2, a Hallmark of TDP-43-Associated ALS, Causes Motor Neuropathy. Cell Rep. 2022, 39, 111001. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Davis, A.A.; Tennant, P.; Wang, M.; Coleman, M.; Asress, S.; Adalbert, R.; Alexander, G.M.; Glass, J.D. The WldS Gene Modestly Prolongs Survival in the SOD1G93A fALS Mouse. Neurobiol. Dis. 2005, 19, 293–300. [Google Scholar] [CrossRef]
- Sábado, J.; Casanovas, A.; Tarabal, O.; Hereu, M.; Piedrafita, L.; Calderó, J.; Esquerda, J.E. Accumulation of Misfolded SOD1 in Dorsal Root Ganglion Degenerating Proprioceptive Sensory Neurons of Transgenic Mice with Amyotrophic Lateral Sclerosis. Biomed. Res. Int. 2014, 2014, 852163. [Google Scholar] [CrossRef]
- Taiana, M.; Sassone, J.; Lauria, G. Mutant SOD1 Accumulation in Sensory Neurons Does Not Associate with Endoplasmic Reticulum Stress Features: Implications for Differential Vulnerability of Sensory and Motor Neurons to SOD1 Toxicity. Neurosci. Lett. 2016, 627, 107–114. [Google Scholar] [CrossRef]
- Rojas, P.; de Hoz, R.; Ramírez, A.I.; Ferreras, A.; Salobrar-Garcia, E.; Muñoz-Blanco, J.L.; Urcelay-Segura, J.L.; Salazar, J.J.; Ramírez, J.M. Changes in Retinal OCT and Their Correlations with Neurological Disability in Early ALS Patients, a Follow-Up Study. Brain Sci. 2019, 9, 337. [Google Scholar] [CrossRef]
- Cho, K.I.; Yoon, D.; Yu, M.; Peachey, N.S.; Ferreira, P.A. Microglial Activation in an Amyotrophic Lateral Sclerosis-Like Model Caused by Ranbp2 Loss and Nucleocytoplasmic Transport Impairment in Retinal Ganglion Neurons. Cell. Mol. Life. Sci. 2019, 76, 3407–3432. [Google Scholar] [CrossRef]
- Ferreira, P.A. The Coming-of-Age of Nucleocytoplasmic Transport in Motor Neuron Disease and Neurodegeneration. Cell. Mol. Life. Sci. 2019, 76, 2247–2273. [Google Scholar] [CrossRef]
- Nagano, S.; Jinno, J.; Abdelhamid, R.F.; Jin, Y.; Shibata, M.; Watanabe, S.; Hirokawa, S.; Nishizawa, M.; Sakimura, K.; Onodera, O.; et al. TDP-43 Transports Ribosomal Protein mRNA to Regulate Axonal Local Translation in Neuronal Axons. Acta Neuropathol. 2020, 140, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Camdessanché, J.P.; Belzil, V.V.; Jousserand, G.; Rouleau, G.A.; Créac’h, C.; Convers, P.; Antoine, J.C. Sensory and Motor Neuronopathy in a Patient with the A382P TDP-43 Mutation. Orphanet. J. Rare Dis. 2011, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Yuan, Y.; Liu, Z.; Hou, X.; Li, W.; Wen, J.; Zhang, K.; Jiao, B.; Shen, L.; Jiang, H.; et al. Association of Variants in the KIF1A Gene with Amyotrophic Lateral Sclerosis. Transl. Neurodegener. 2022, 11, 46. [Google Scholar] [CrossRef]
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neuroscientist 2018, 24, 54–72. [Google Scholar] [CrossRef]
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of Amyotrophic Lateral Sclerosis: Seeking Therapeutic Targets in the Era of Gene Therapy. J. Hum. Genet. 2023, 68, 131–152. [Google Scholar] [CrossRef]
- Farah, C.A.; Nguyen, M.D.; Julien, J.P.; Leclerc, N. Altered Levels and Distribution of Microtubule-Associated Proteins Before Disease Onset in a Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurochem. 2003, 84, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for Amyotrophic Lateral Sclerosis (ALS)/Motor Neuron Disease (MND). Cochrane Database Syst. Rev. 2012, 2012, CD001447. [Google Scholar] [CrossRef]
- Rothstein, J.D. Edaravone: A New Drug Approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef]
- Yoshino, H.; Kimura, A. Investigation of the Therapeutic Effects of Edaravone, a Free Radical Scavenger, on Amyotrophic Lateral Sclerosis (Phase II Study). Amyotroph. Lateral. Scler. 2006, 7, 241–245. [Google Scholar] [CrossRef]
- Sever, B.; Ciftci, H.; DeMirci, H.; Sever, H.; Ocak, F.; Yulug, B.; Tateishi, H.; Tateishi, T.; Otsuka, M.; Fujita, M.; et al. Comprehensive Research on Past and Future Therapeutic Strategies Devoted to Treatment of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 2400. [Google Scholar] [CrossRef] [PubMed]
- Fels, J.A.; Dash, J.; Leslie, K.; Manfredi, G.; Kawamata, H. Effects of PB-TURSO on the Transcriptional and Metabolic Landscape of Sporadic ALS Fibroblasts. Ann. Clin. Transl. Neurol. 2022, 9, 1551–1564. [Google Scholar] [CrossRef]
- Oki, R.; Izumi, Y.; Fujita, K.; Miyamoto, R.; Nodera, H.; Sato, Y.; Sakaguchi, S.; Nokihara, H.; Kanai, K.; Tsunemi, T.; et al. Japan Early-Stage Trial of Ultrahigh-Dose Methylcobalamin for ALS (JETALS) Collaborators. Efficacy and Safety of Ultrahigh-Dose Methylcobalamin in Early-Stage Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 2022, 79, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods. 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Brownstone, R.M.; Lancelin, C. Escape from Homeostasis: Spinal Microcircuits and Progression of Amyotrophic Lateral Sclerosis. J. Neurophysiol. 2018, 119, 1782–1794. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 Regulates its mRNA Levels Through a Negative Feedback Loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef]
- Neumann, M.; Mackenzie, I.R.; Cairns, N.J.; Boyer, P.J.; Markesbery, W.R.; Smith, C.D.; Taylor, J.P.; Kretzshmar, H.A.; Kimonis, V.E.; Forman, M.S. TDP-43 in the Ubiquitin Pathology of Frontotemporal Dementia with VCP Gene Mutations. J. Neuropathol. Exp. Neurol. 2007, 66, 152–157. [Google Scholar] [CrossRef]
- Hasegawa, K.; Yasuda, T.; Shiraishi, C.; Fujiwara, K.; Przedborski, S.; Mochizuki, H.; Yoshikawa, K. Promotion of Mitochondrial Biogenesis by Necdin Protects Neurons Against Mitochondrial Insults. Nat. Commun. 2016, 7, 10943. [Google Scholar] [CrossRef]
- Seki, S.; Tanaka, S.; Yamada, S.; Tsuji, T.; Enomoto, A.; Ono, Y.; Chandler, S.H.; Kogo, M. Neuropeptide Y Modulates Membrane Excitability in Neonatal Rat Mesencephalic V Neurons. J. Neurosci. Res. 2020, 98, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Morimoto, T.; Iwata, K.; Inoue, T.; Masuda, Y.; Kato, T.; Hidaka, O. Putative Feed-Forward Control of Jaw-Closing Muscle Activity During Rhythmic Jaw Movements in the Anesthetized Rabbit. J. Neurophysiol. 2001, 86, 2834–2844. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.; Seki, S.; Terman, D.H.; Pantazis, A.; Olcese, R.; Wiedau-Pazos, M.; Chandler, S.H. Resurgent Na+ Current Offers Noise Modulation in Bursting Neurons. PLoS Comput. Biol. 2019, 15, e1007154. [Google Scholar] [CrossRef] [PubMed]
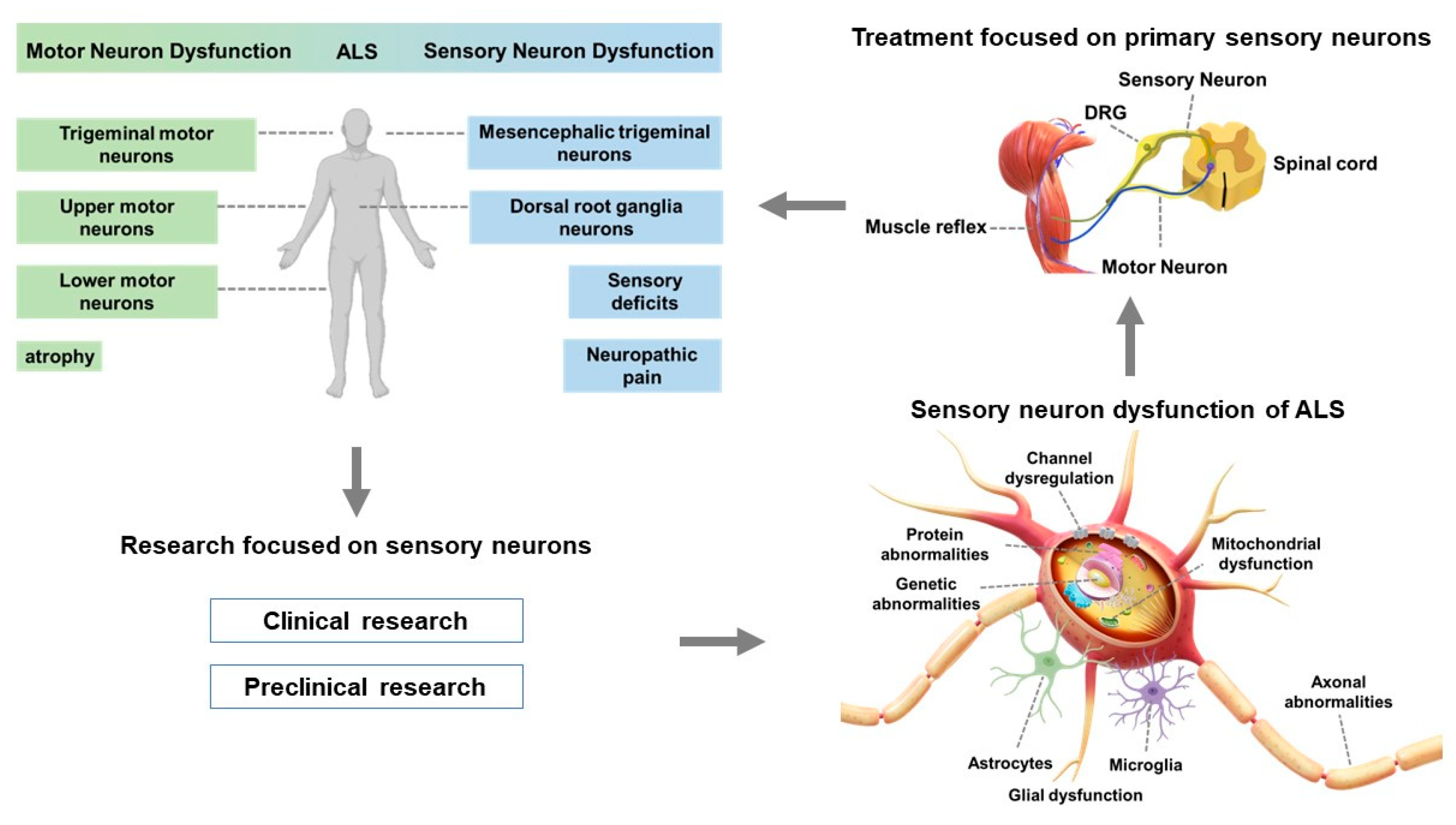
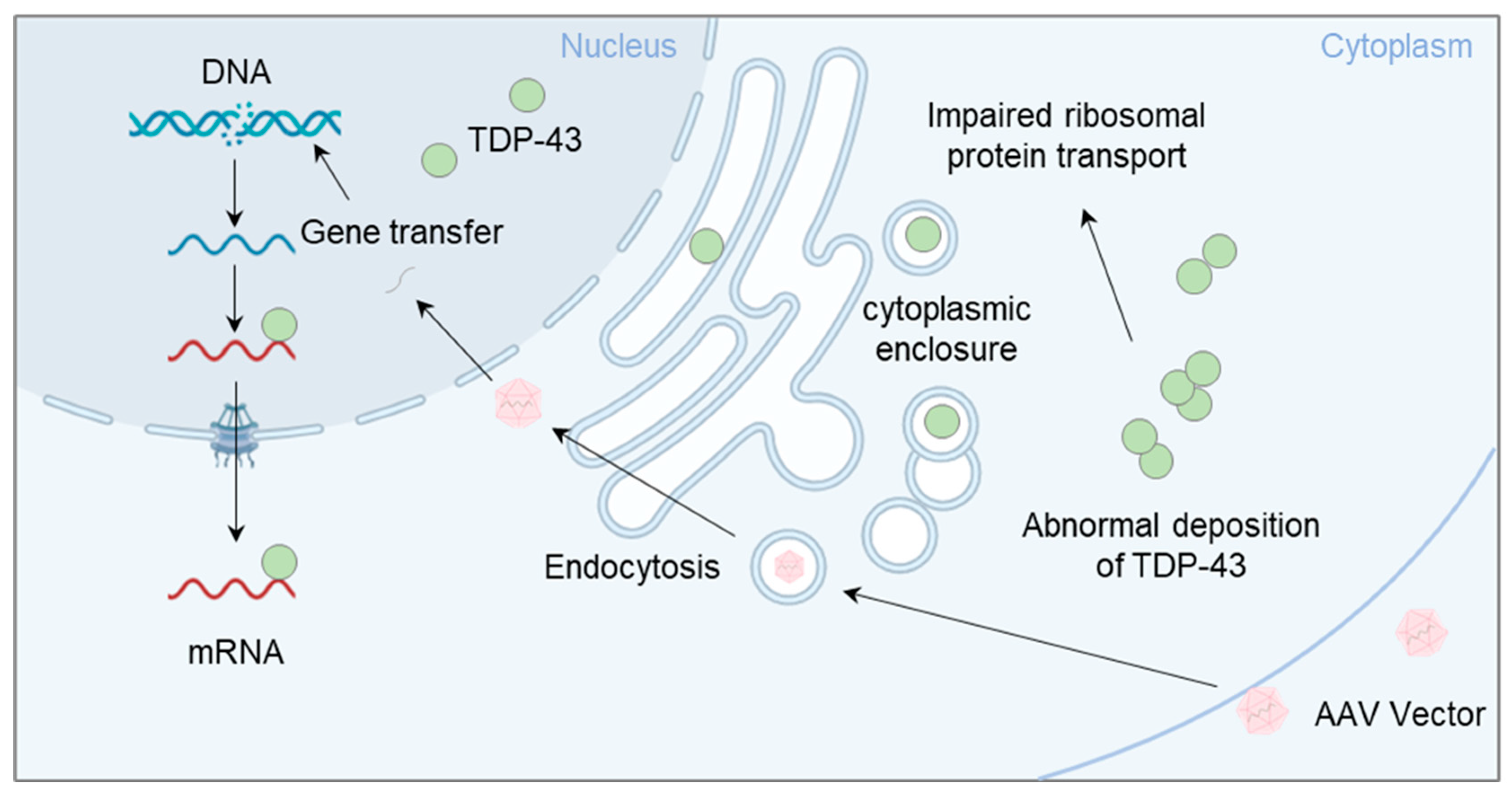


{kind=link}
{kind=link}
{kind=link}
| Type of Sensory Neuron | Dysfunction | Number of Patients with ALS | References |
|---|---|---|---|
| Median nerve | Attenuation of postsynaptic high-frequency somatosensory evoked potential bursts in patients with ALS with a long duration of the disease. | 20 | [73] |
| Sensory system | A total of 14.7% of patients with ALS have damaged sensory systems. | 150 | [62] |
| Sensory cortex | Sensory cortex overexcitation is associated with shorter survival in patients with ALS. | 145 | [71] |
| Intraepidermal nerve fibres | Decreased intraepidermal nerve fibres density with aggregation of TDP-43 in patients with ALS. | 18 | [74] |
| Epidermal nerve fibres | Loss of intraepidermal nerve fibres. | 41 | [60] |
| Intraepidermal nerve fibres | Increased axonal expansion rate and negative growth-related proteins in intraepidermal nerve fibres. | 32 | [58] |
| Intraepidermal nerve fibres | In 85% of patients with ALS, quantitative sensory testing showed abnormal thermal pain thresholds and skin biopsies showed decreased intraepidermal nerve fibre density. | 24 | [75] |
| Ascending sensory fibres | Anatomical damage to ascending sensory fibres in 60% of patients with solitary ALS. | 21 | [76] |
| Leg sensory nerve | A total of 27% of patients with ALS had abnormal action potential. Amplitude, and 91% had pathological abnormalities of the leg sensory nerve. | 103 | [56] |
| Sensory neuron in sural | Sensory neuropathy and axonal degeneration. | 5 | [77] |
| Median, radial, and sural nerves | Asymptomatic decline in sensory nerve function. | 19 | [67] |
| Sensory nerves | Early axonal atrophy, increased remyelination, and predominance of smaller fibre diameters. | [78] |
| Animal Model | Animals | Characteristics of the Model | References |
|---|---|---|---|
| SOD1G93A | Mouse | Marked muscle weakness is observed at 15 weeks of age in SOD1G93A transgenic mice with familial ALS | [33] |
| dSOD1G85R | Drosophila | SOD1G85R knock-in model shows severe motor deficits with apparent degeneration of motor neurons, providing a better understanding of the contribution of multiple cell types in ALS | [72] |
| C9orf72 ALS | Drosophila | Transgenic Drosophila model overexpressing human TDP-43 shows reduced lifespan, reduced motor activity, increased morphological defects in motor neurons, a loss of neurons, and axonal damage | [88] |
| TDP-43A315T | Mouse | A315T mutant TDP-43 transgenic mouse model with marked early-onset progressive neurodegeneration resulting in reduced motor performance, spatial memory and deinhibition, and reduced grip strength due to muscle atrophy | [84] |
| TDP-43 (TBPH) | Drosophila | Transgenic fly that exhibits an adult locomotor defect and shares many features with human proteins | [89] |
| Type of Sensory Neuron | Dysfunction | Type of Animals | Genes | References |
|---|---|---|---|---|
| MesV | Firing abnormality | Mouse | SOD1 G93A | [36] |
| DRG | Decrease in SGNF density | Mouse | SOD1 G93A | [33] |
| C4da | TDP-43 accumulation in the cytoplasm via calcium-calpain-A-importin α3 pathway | Drosophila | C9orf72 ALS | [88] |
| DRG | SOD1 accumulation in SGCs | Mouse | SOD1 G93A | [8] |
| MesV | Firing abnormality with sodium channel dysfunction | Mouse | SOD1 G93A | [35] |
| Non-motor neuron | Neurotrophic BMP pathway | Drosophila | dSOD1G85R | [72] |
| DRG | Shorter and less complex neurites | Mouse | TDP43 A315TSOD1 G93A | [84] |
| Long-projection sensory neurons | Defects in MAM signalling | Mouse | TDP43 A315T | [93] |
| Olfactory bulb and retina | Neuronal vacuolisation in olfaction and vision pathways | Mouse | SOD1 G93A | [87] |
| DRG | Accumulation of a peripherin splice variant | Mouse | SOD1 G93A | [34] |
| DRG | Accumulation of misfolded protein | Mouse | dSOD1G85R | [94] |
| Ia and II proprioceptive nerve | Early disturbances in muscle spindles before motor neuron symptoms | Mouse | TDP43 A315T SOD1 G93A | [95] |
| C4da | Localised accumulation, predominantly of TDP-43 | Drosophila | TDP-43 (TBPH) | [89] |
| DRG | Mitochondrial abnormalities | Mouse | SOD1 G93A | [90] |
| DRG | Reduction of high-voltage-activated Ca2+ current | Mouse | ICR mice injected with sera from patients with ALS | [91] |
| Genes | Type of Sensory Neuron | Function | References |
|---|---|---|---|
| SOD1 | DRG | Encodes an itinerant enzyme that catalyses the conversion of superoxide to hydrogen peroxide and dioxygen. | [117] |
| KIF1A | DRG | Encodes the kinesin 3 motor that transports presynaptic vesicle precursors and dense core vesicles. | [123] |
| TARDBP | DRG | Mainly localised in the nucleus and regulates RNA processing. | [122] |
| FUS | VNC | RNA-binding protein. | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seki, S.; Kitaoka, Y.; Kawata, S.; Nishiura, A.; Uchihashi, T.; Hiraoka, S.-i.; Yokota, Y.; Isomura, E.T.; Kogo, M.; Tanaka, S. Characteristics of Sensory Neuron Dysfunction in Amyotrophic Lateral Sclerosis (ALS): Potential for ALS Therapy. Biomedicines 2023, 11, 2967. https://doi.org/10.3390/biomedicines11112967
Seki S, Kitaoka Y, Kawata S, Nishiura A, Uchihashi T, Hiraoka S-i, Yokota Y, Isomura ET, Kogo M, Tanaka S. Characteristics of Sensory Neuron Dysfunction in Amyotrophic Lateral Sclerosis (ALS): Potential for ALS Therapy. Biomedicines. 2023; 11(11):2967. https://doi.org/10.3390/biomedicines11112967
Chicago/Turabian StyleSeki, Soju, Yoshihiro Kitaoka, Sou Kawata, Akira Nishiura, Toshihiro Uchihashi, Shin-ichiro Hiraoka, Yusuke Yokota, Emiko Tanaka Isomura, Mikihiko Kogo, and Susumu Tanaka. 2023. "Characteristics of Sensory Neuron Dysfunction in Amyotrophic Lateral Sclerosis (ALS): Potential for ALS Therapy" Biomedicines 11, no. 11: 2967. https://doi.org/10.3390/biomedicines11112967
APA StyleSeki, S., Kitaoka, Y., Kawata, S., Nishiura, A., Uchihashi, T., Hiraoka, S.-i., Yokota, Y., Isomura, E. T., Kogo, M., & Tanaka, S. (2023). Characteristics of Sensory Neuron Dysfunction in Amyotrophic Lateral Sclerosis (ALS): Potential for ALS Therapy. Biomedicines, 11(11), 2967. https://doi.org/10.3390/biomedicines11112967