
Enrichment of Deleterious Mutated Genes Involved in Ciliary Function and Histone Modification in Brain Cancer Patient-Derived Xenograft Models


 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tumor Samples from Patients with Brain Tumors
2.2. Establishment of Brain Tumor–PDX Models
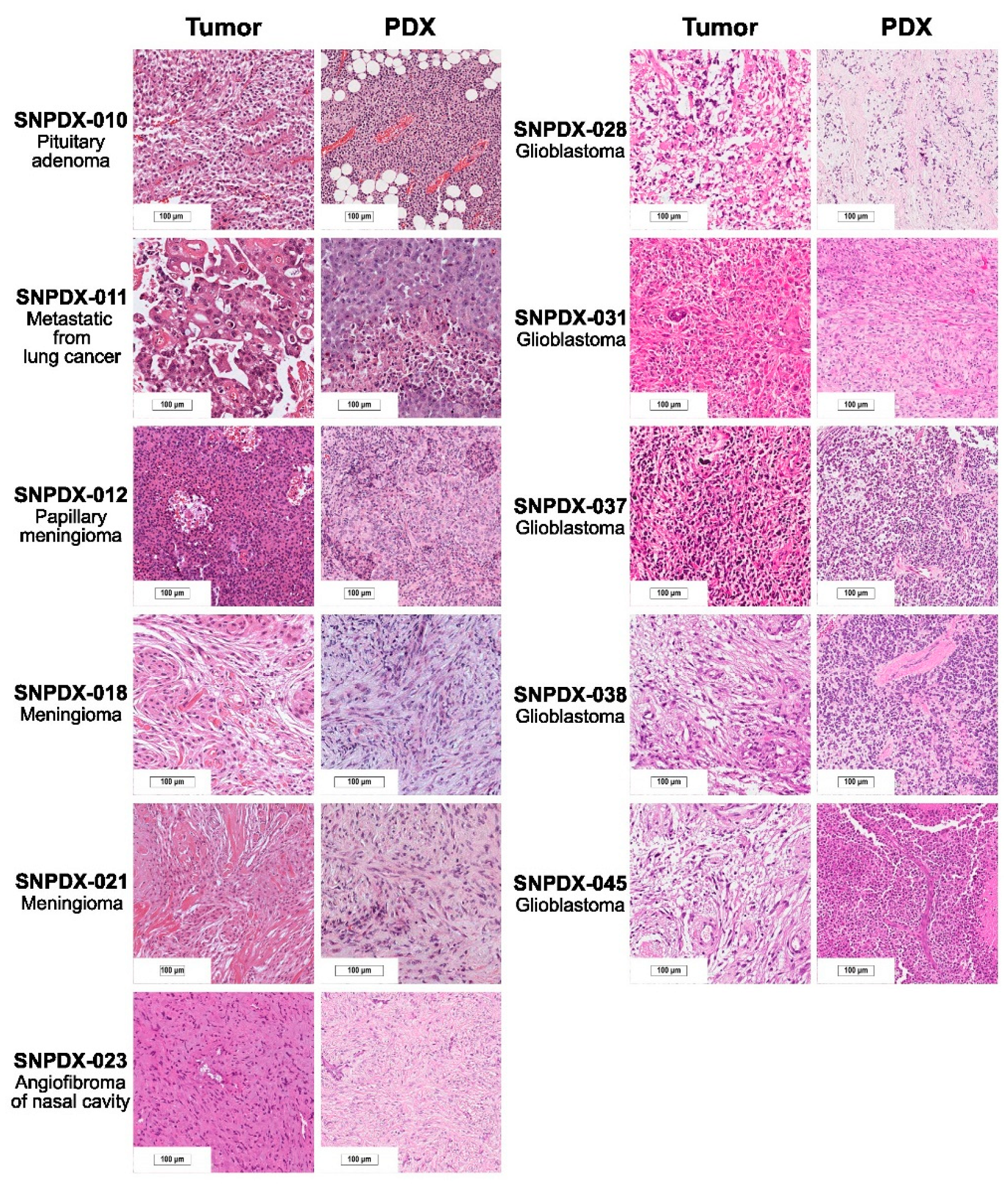
2.3. Histopathological Analysis
2.4. Mutational Analysis
2.5. Mutational Signature Analysis
2.6. Gene Ontology (GO) Term Enrichment Analysis
3. Results
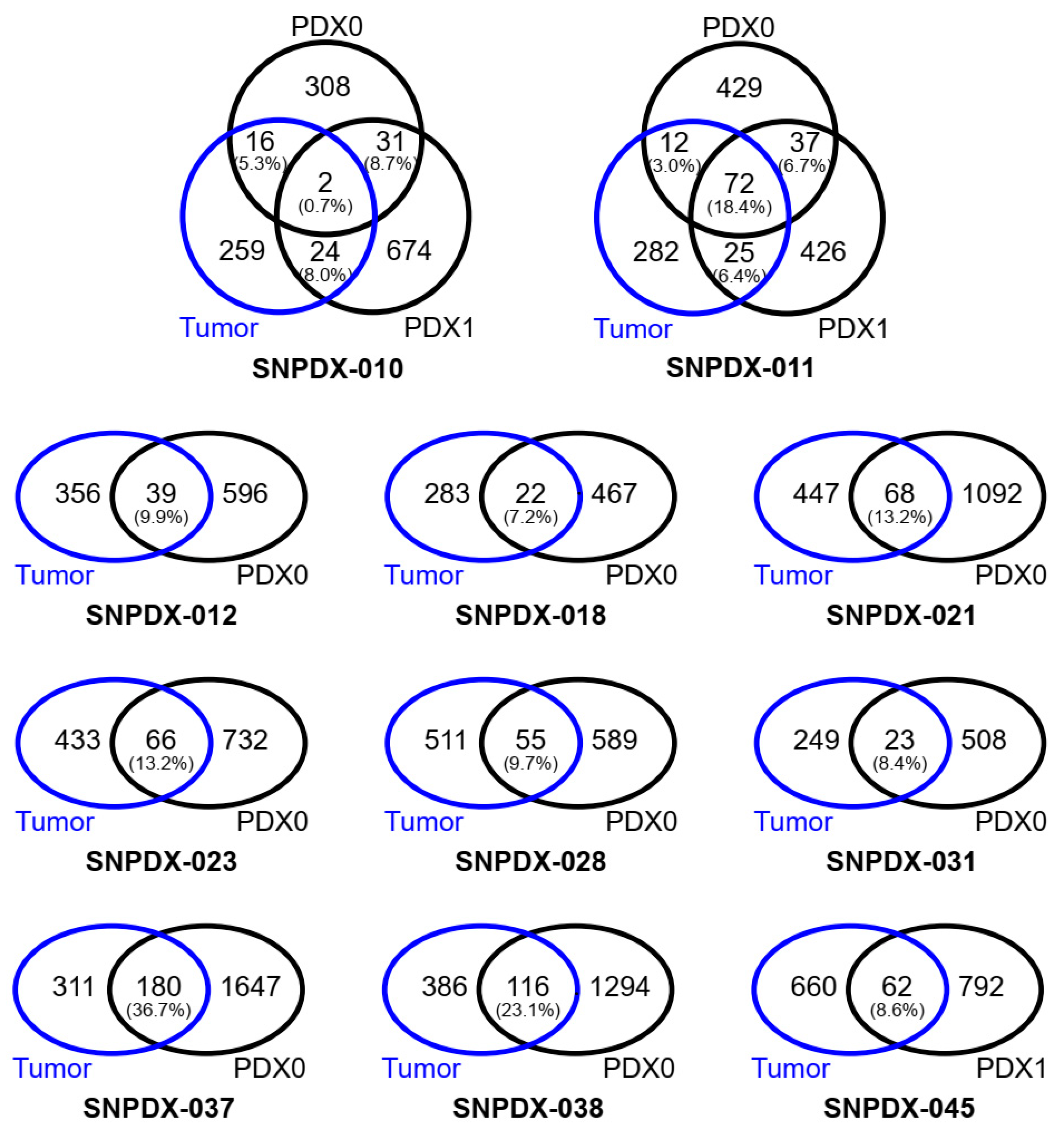
3.1. Deleterious Somatic Mutations Are Discordant between the Original Tumors and Xenograft Tissues
3.2. Germline Variations Are Concordant between Original Tumors and Xenograft Tissues
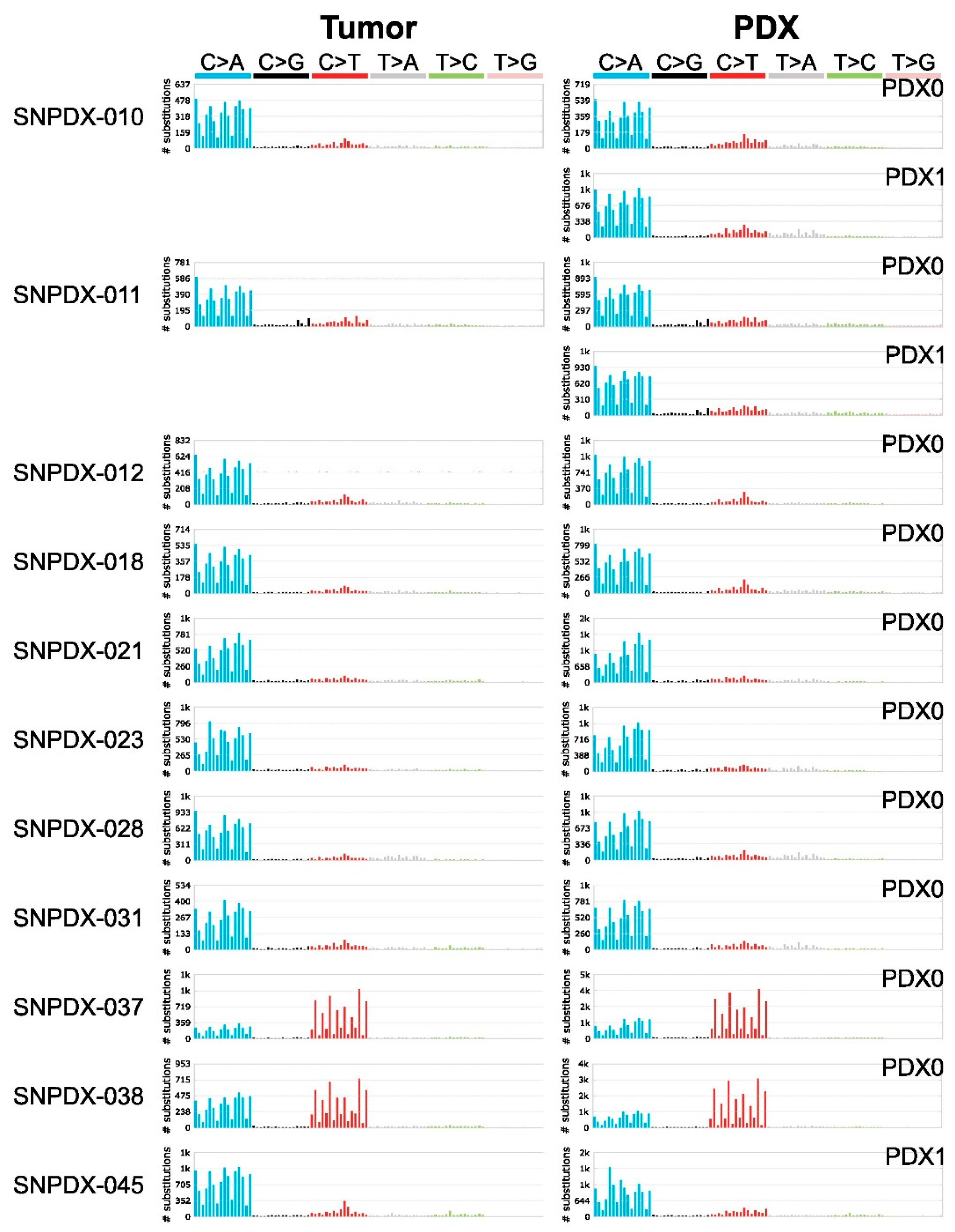
3.3. Mutational Signatures Are Well Conserved between Tumors and Xenografts, and Provide Information about the Source of Mutations
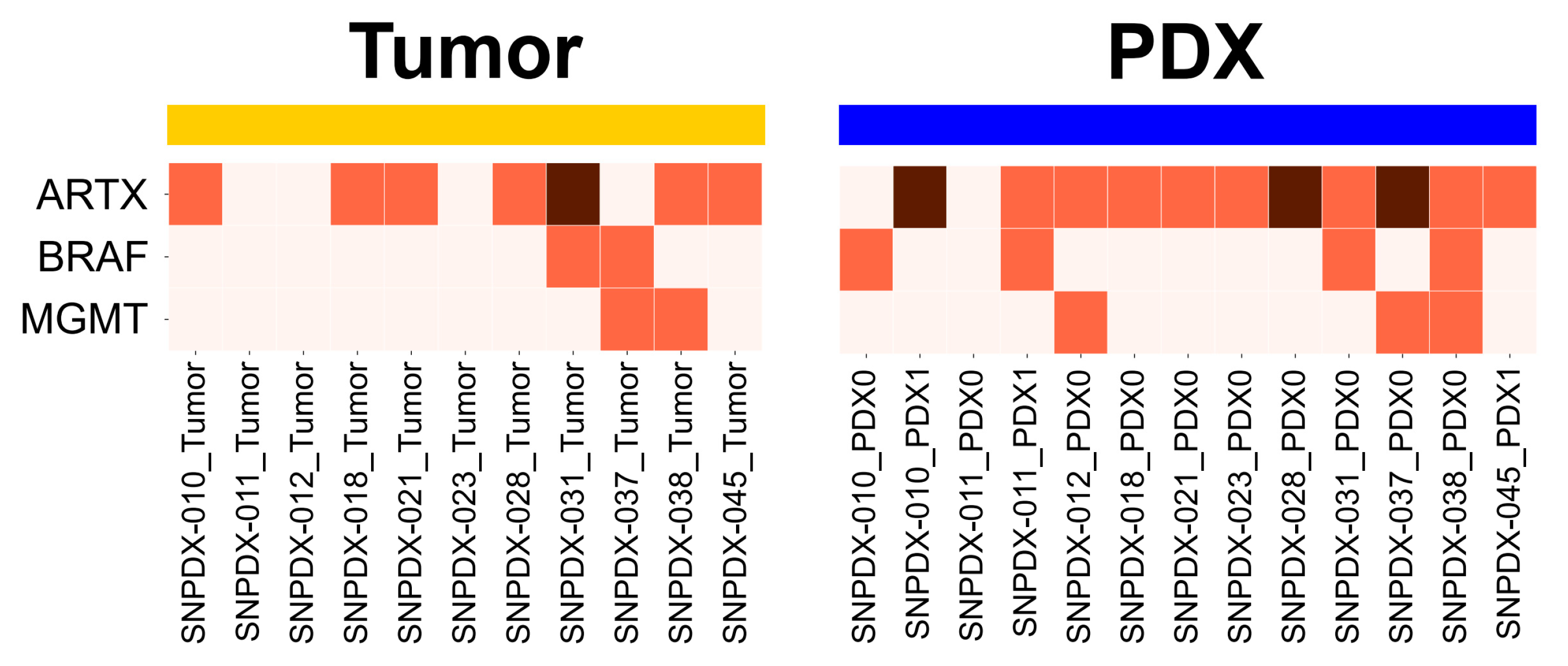
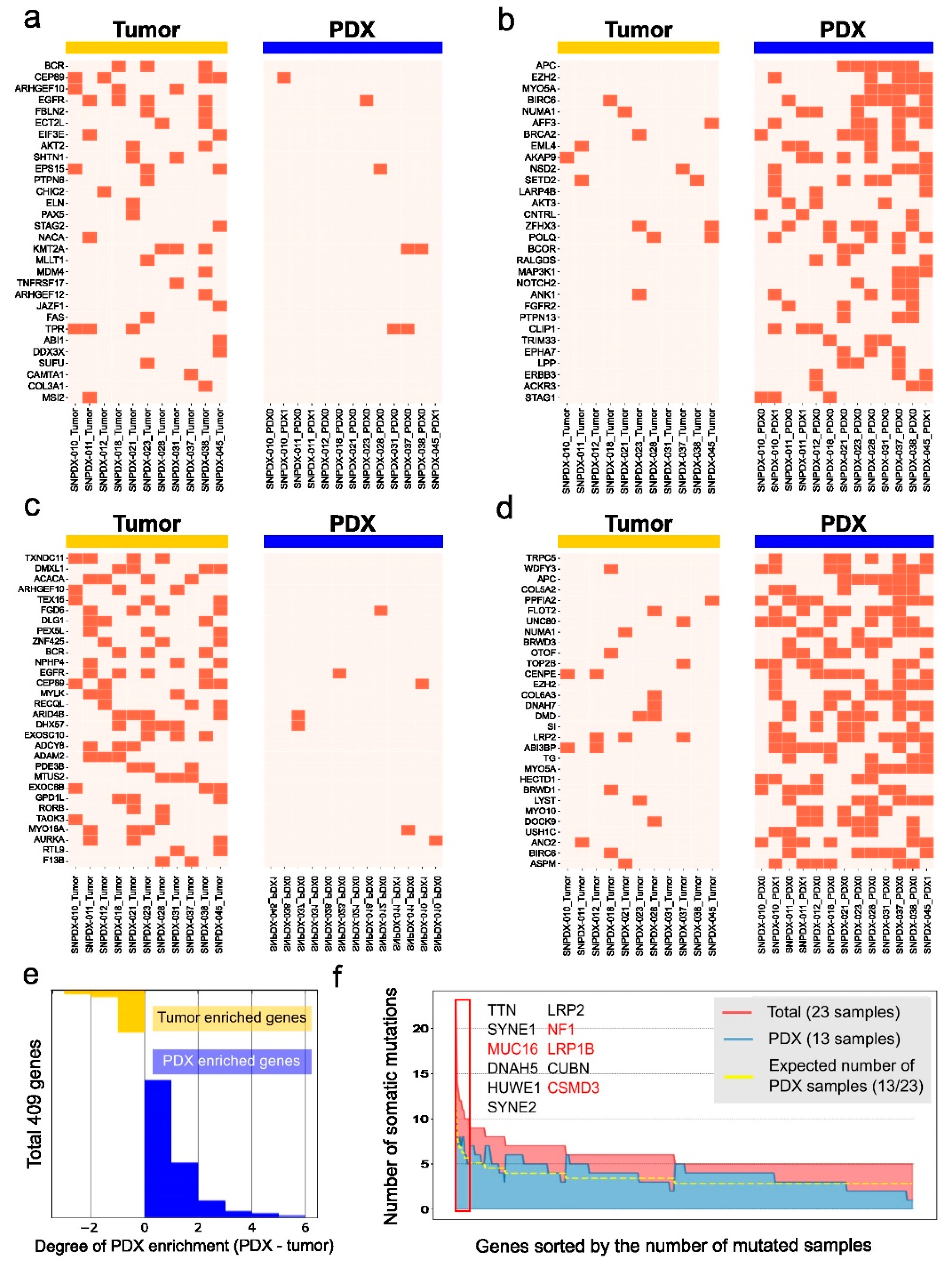
3.4. Brain Tumor-Associated Mutations Are Enriched in PDX Samples
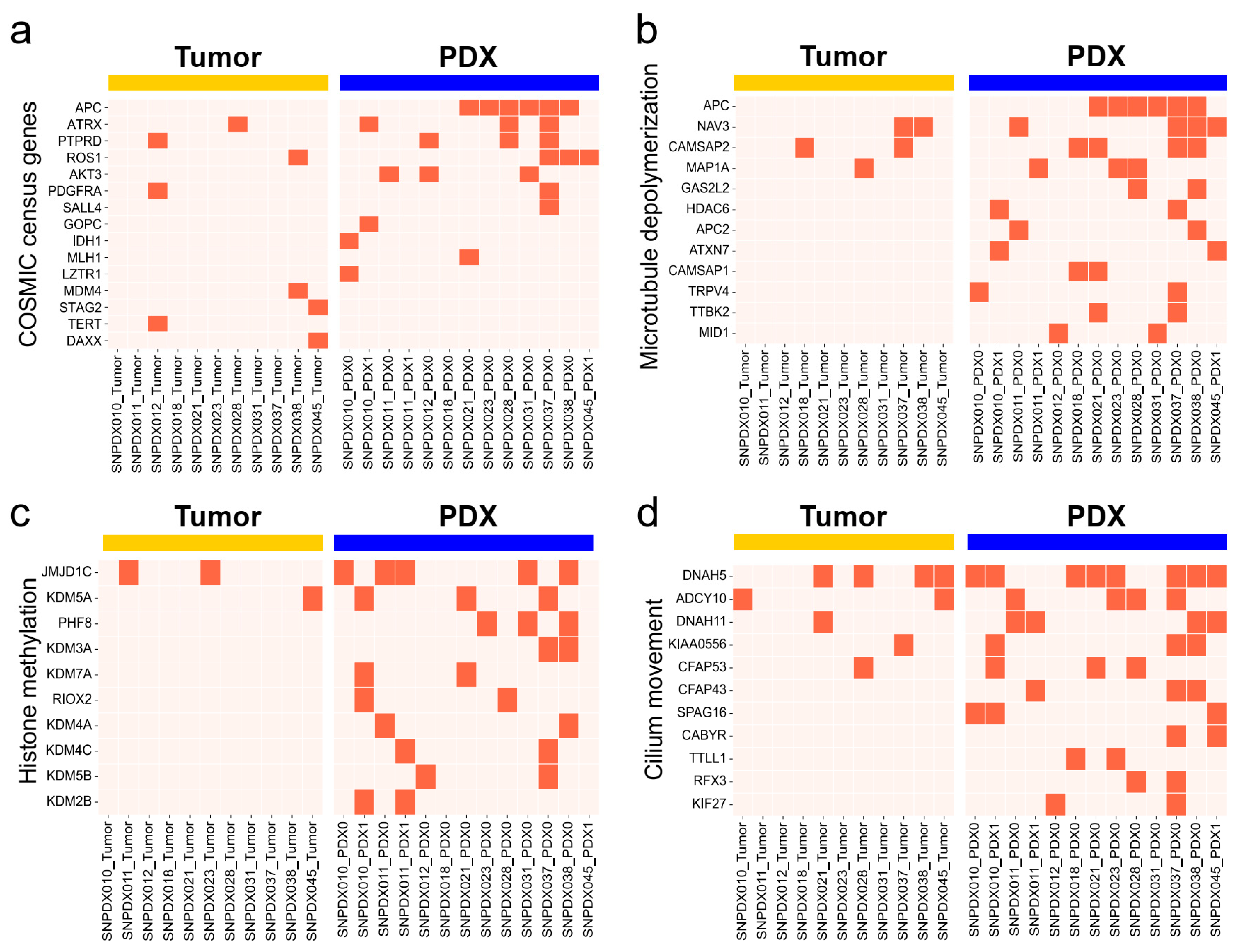
3.5. Histone Methylation- and Cilium-Related Gene Mutations Are likely Enriched in PDX Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary Brain Tumours in Adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. CIMPACT-NOW Update 3: Recommended Diagnostic Criteria for “Diffuse Astrocytic Glioma, IDH-Wildtype, with Molecular Features of Glioblastoma, WHO Grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef]
- Ellison, D.W.; Hawkins, C.; Jones, D.T.W.; Onar-Thomas, A.; Pfister, S.M.; Reifenberger, G.; Louis, D.N. CIMPACT-NOW Update 4: Diffuse Gliomas Characterized by MYB, MYBL1, or FGFR1 Alterations or BRAFV600E Mutation. Acta Neuropathol. 2019, 137, 683–687. [Google Scholar] [CrossRef]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. CIMPACT-NOW Update 6: New Entity and Diagnostic Principle Recommendations of the CIMPACT-Utrecht Meeting on Future CNS Tumor Classification and Grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef]
- Louis, D.N.; Wesseling, P.; Paulus, W.; Giannini, C.; Batchelor, T.T.; Cairncross, J.G.; Capper, D.; Figarella-Branger, D.; Lopes, M.B.; Wick, W.; et al. CIMPACT-NOW Update 1: Not Otherwise Specified (NOS) and Not Elsewhere Classified (NEC). Acta Neuropathol. 2018, 135, 481–484. [Google Scholar] [CrossRef]
- Louis, D.N.; Aldape, K.; Brat, D.J.; Capper, D.; Ellison, D.W.; Hawkins, C.; Paulus, W.; Perry, A.; Reifenberger, G.; Figarella-Branger, D.; et al. CIMPACT-NOW (the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy): A New Initiative in Advancing Nervous System Tumor Classification. Brain Pathol. 2017, 27, 851–852. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D. Glioblastoma Heterogeneity and Cancer Cell Plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.R.; Khong, P.; Parkinson, J.F.; Howell, V.M.; Wheeler, H.R. Molecular Heterogeneity in Glioblastoma: Potential Clinical Implications. Front. Oncol. 2015, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Vaubel, R.A.; Tian, S.; Remonde, D.; Schroeder, M.A.; Mladek, A.C.; Kitange, G.J.; Caron, A.; Kollmeyer, T.M.; Grove, R.; Peng, S.; et al. Genomic and Phenotypic Characterization of a Broad Panel of Patient-Derived Xenografts Reflects the Diversity of Glioblastoma. Clin. Cancer Res. 2020, 26, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Grisdale, C.J.; Islam, S.A.; Bose, P.; Lever, J.; Zhao, E.Y.; Grinshtein, N.; Ma, Y.; Mungall, A.J.; Moore, R.A.; et al. Comprehensive Genomic Profiling of Glioblastoma Tumors, BTICs, and Xenografts Reveals Stability and Adaptation to Growth Environments. Proc. Natl. Acad. Sci. USA 2019, 116, 19098–19108. [Google Scholar] [CrossRef]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-Derived Xenografts Undergo Mouse-Specific Tumor Evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and Accurate Calling of Germline and Somatic Variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- PCAWG Mutational Signatures Working Group; PCAWG Consortium; Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Bergstrom, E.N.; Huang, M.N.; Mahto, U.; Barnes, M.; Stratton, M.R.; Rozen, S.G.; Alexandrov, L.B. SigProfilerMatrixGenerator: A Tool for Visualizing and Exploring Patterns of Small Mutational Events. BMC Genom. 2019, 20, 685. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for Large-Scale Genome and Gene Function Analysis with the PANTHER Classification System (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Moore, L.; Cagan, A.; Coorens, T.H.H.; Neville, M.D.C.; Sanghvi, R.; Sanders, M.A.; Oliver, T.R.W.; Leongamornlert, D.; Ellis, P.; Noorani, A.; et al. The Mutational Landscape of Human Somatic and Germline Cells. Nature 2021, 597, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, F.J.; Nik-Zainal, S.; Wuster, A.; Li, Y.; Conte, N.; Koike-Yusa, H.; Kumasaka, N.; Vallier, L.; Yusa, K.; Bradley, A. Mutational History of a Human Cell Lineage from Somatic to Induced Pluripotent Stem Cells. PLoS Genet. 2016, 12, e1005932. [Google Scholar] [CrossRef]
- Pich, O.; Muiños, F.; Lolkema, M.P.; Steeghs, N.; Gonzalez-Perez, A.; Lopez-Bigas, N. The Mutational Footprints of Cancer Therapies. Nat. Genet. 2019, 51, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal Evolution of Glioblastoma under Therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef]
- LeBlanc, V.G.; Trinh, D.L.; Aslanpour, S.; Hughes, M.; Livingstone, D.; Jin, D.; Ahn, B.Y.; Blough, M.D.; Cairncross, J.G.; Chan, J.A.; et al. Single-Cell Landscapes of Primary Glioblastomas and Matched Explants and Cell Lines Show Variable Retention of Inter- and Intratumor Heterogeneity. Cancer Cell 2022, 40, 379–392.e9. [Google Scholar] [CrossRef]
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hostetter, R.; Cleary, K.; Bigner, S.H.; Davidson, N.; Baylin, S.; Devilee, P. Mutations in the P53 Gene Occur in Diverse Human Tumour Types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased Expression of the Epidermal Growth Factor Receptor Gene in Malignant Gliomas Is Invariably Associated with Gene Amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef] [PubMed]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor Suppressor P53: Biology, Signaling Pathways, and Therapeutic Targeting. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment. Genes. Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic Pathways to Primary and Secondary Glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Goranci-Buzhala, G.; Mariappan, A.; Ricci-Vitiani, L.; Josipovic, N.; Pacioni, S.; Gottardo, M.; Ptok, J.; Schaal, H.; Callaini, G.; Rajalingam, K.; et al. Cilium Induction Triggers Differentiation of Glioma Stem Cells. Cell Rep. 2021, 36, 109656. [Google Scholar] [CrossRef]
- Mallm, J.-P.; Windisch, P.; Biran, A.; Gal, Z.; Schumacher, S.; Glass, R.; Herold-Mende, C.; Meshorer, E.; Barbus, M.; Rippe, K. Glioblastoma Initiating Cells Are Sensitive to Histone Demethylase Inhibition Due to Epigenetic Deregulation. Int. J. Cancer 2020, 146, 1281–1292. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, H.; Moon, H.E.; Yun, S.; Cho, S.W.; Park, H.R.; Park, S.-H.; Myung, K.; Kwon, T.; Paek, S.H. Enrichment of Deleterious Mutated Genes Involved in Ciliary Function and Histone Modification in Brain Cancer Patient-Derived Xenograft Models. Biomedicines 2023, 11, 2934. https://doi.org/10.3390/biomedicines11112934
Jeong H, Moon HE, Yun S, Cho SW, Park HR, Park S-H, Myung K, Kwon T, Paek SH. Enrichment of Deleterious Mutated Genes Involved in Ciliary Function and Histone Modification in Brain Cancer Patient-Derived Xenograft Models. Biomedicines. 2023; 11(11):2934. https://doi.org/10.3390/biomedicines11112934
Chicago/Turabian StyleJeong, Hyeongsun, Hyo Eun Moon, Seongmin Yun, Seung Woo Cho, Hye Ran Park, Sung-Hye Park, Kyungjae Myung, Taejoon Kwon, and Sun Ha Paek. 2023. "Enrichment of Deleterious Mutated Genes Involved in Ciliary Function and Histone Modification in Brain Cancer Patient-Derived Xenograft Models" Biomedicines 11, no. 11: 2934. https://doi.org/10.3390/biomedicines11112934
APA StyleJeong, H., Moon, H. E., Yun, S., Cho, S. W., Park, H. R., Park, S.-H., Myung, K., Kwon, T., & Paek, S. H. (2023). Enrichment of Deleterious Mutated Genes Involved in Ciliary Function and Histone Modification in Brain Cancer Patient-Derived Xenograft Models. Biomedicines, 11(11), 2934. https://doi.org/10.3390/biomedicines11112934