
Role of Ferroptosis in Regulating the Epithelial–Mesenchymal Transition in Pulmonary Fibrosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. EMT and Pulmonary Fibrosis
3. Ferroptosis and EMT
3.1. Discovery of Ferroptosis
3.2. Role of Ferroptosis in EMT
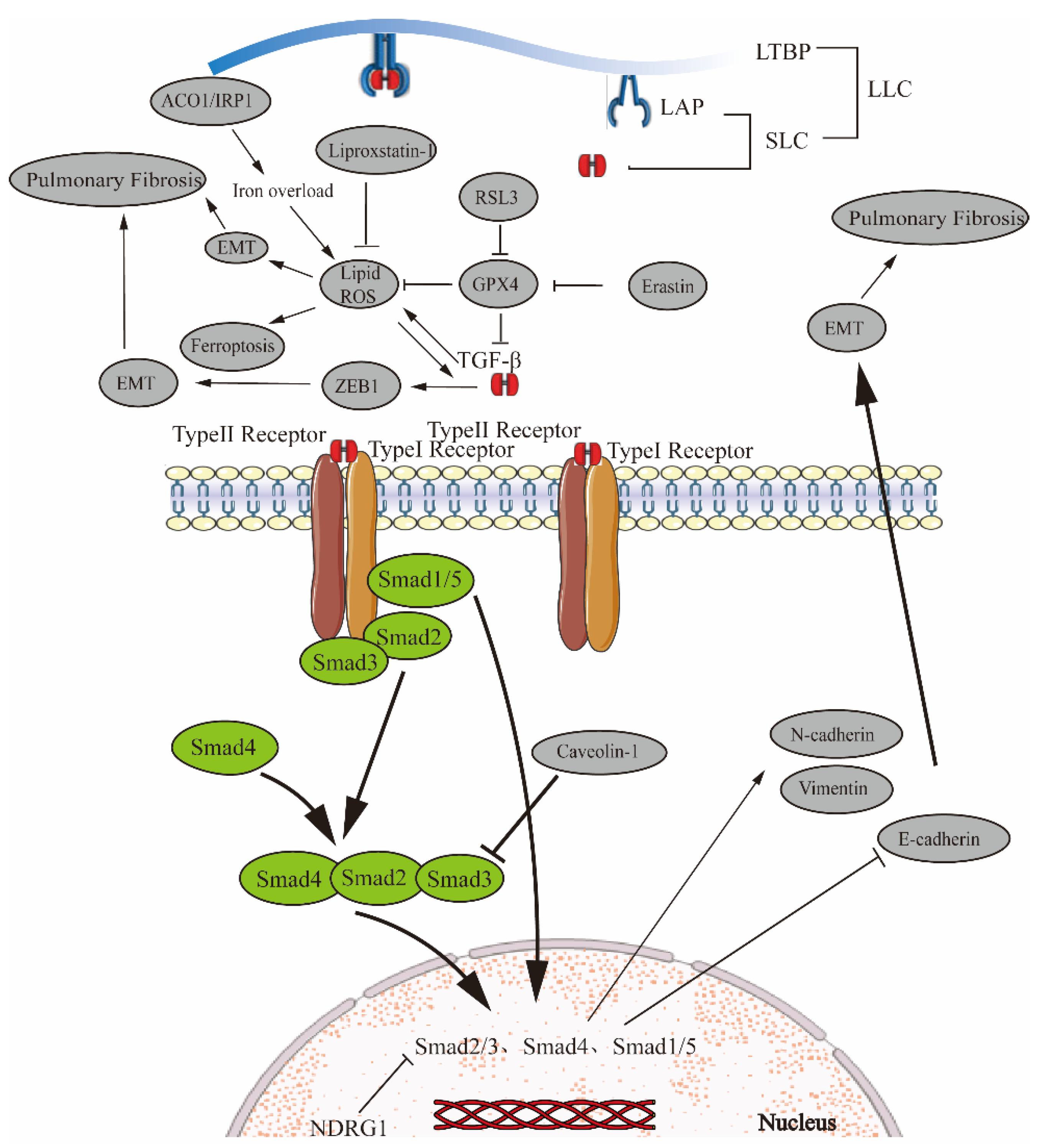
4. Multiple Signaling Pathways Associated with Ferroptosis Regulate EMT in Pulmonary Fibrosis
4.1. TGF-β/Smad Signaling Pathway
4.2. Nrf2 Signaling Pathway
4.3. Wnt-Related Signaling Pathway
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolters, P.J.; Blackwell, T.S.; Eickelberg, O.; Loyd, J.E.; Kaminski, N.; Jenkins, G.; Maher, T.M.; Molina-Molina, M.; Noble, P.W.; Raghu, G.; et al. Time for a change: Is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir. Med. 2018, 6, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Raghu, G. Idiopathic pulmonary fibrosis: Lessons from clinical trials over the past 25 years. Eur. Respir. J. 2017, 50, 1701209. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Bonella, F.; Wollin, L. Therapeutic targets in idiopathic pulmonary fibrosis. Respir. Med. 2017, 131, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Kishore, A.; Petrek, M. Roles of Macrophage Polarization and Macrophage-Derived miRNAs in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 678457. [Google Scholar] [CrossRef] [PubMed]
- Behr, J. The diagnosis and treatment of idiopathic pulmonary fibrosis. Dtsch. Arztebl. Int. 2013, 110, 875–881. [Google Scholar] [CrossRef]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial–Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef]
- Mylvaganam, R.J.; Bailey, J.I.; Sznajder, J.I.; Sala, M.A. Recovering from a pandemic: Pulmonary fibrosis after SARS-CoV-2 infection. Eur. Respir. Rev. 2021, 30, 210194. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, J.; Xie, W. The role of ferroptosis in acute lung injury. Mol. Cell Biochem. 2022, 477, 1453–1461. [Google Scholar] [CrossRef]
- Wang, F.; Kream, R.M.; Stefano, G.B. Long-Term Respiratory and Neurological Sequelae of COVID-19. Med. Sci. Monit. 2020, 26, e928996. [Google Scholar] [CrossRef] [PubMed]
- Čepelak, I.; Dodig, S.; Dodig, D. Ferroptosis: Regulated cell death. Arh. Hig. Rada. Toksikol. 2020, 71, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tai, W.; Lu, N.; Li, T.; Liu, Y.; Wu, W.; Li, Z.; Pu, L.; Zhao, X.; Zhang, T.; et al. lncRNA ZFAS1 promotes lung fibroblast-to-myofibroblast transition and ferroptosis via functioning as a ceRNA through miR-150-5p/SLC38A1 axis. Aging 2020, 12, 9085–9102. [Google Scholar] [CrossRef] [PubMed]
- Tao, N.; Li, K.; Liu, J. Molecular Mechanisms of Ferroptosis and Its Role in Pulmonary Disease. Oxid. Med. Cell Longev. 2020, 2020, 9547127. [Google Scholar] [CrossRef]
- Nguyen, N.; Xu, S.; Lam, T.Y.W.; Liao, W.; Wong, W.S.F.; Ge, R. ISM1 suppresses LPS-induced acute lung injury and post-injury lung fibrosis in mice. Mol. Med. 2022, 28, 72. [Google Scholar] [CrossRef]
- Yin, X.; Zhu, G.; Wang, Q.; Fu, Y.D.; Wang, J.; Xu, B. Ferroptosis, a New Insight Into Acute Lung Injury. Front. Pharmacol. 2021, 12, 709538. [Google Scholar] [CrossRef]
- He, Y.; Shang, Y.; Li, Y.; Wang, M.; Yu, D.; Yang, Y.; Ning, S.; Chen, H. An 8-ferroptosis-related genes signature from Bronchoalveolar Lavage Fluid for prognosis in patients with idiopathic pulmonary fibrosis. BMC Pulm. Med. 2022, 22, 15. [Google Scholar] [CrossRef]
- He, J.; Li, X.; Yu, M. Bioinformatics Analysis Identifies Potential Ferroptosis Key Genes in the Pathogenesis of Pulmonary Fibrosis. Front. Genet. 2021, 12, 788417. [Google Scholar] [CrossRef]
- Ali, M.K.; Kim, R.Y.; Brown, A.C.; Donovan, C.; Vanka, K.S.; Mayall, J.R.; Liu, G.; Pillar, A.L.; Jones-Freeman, B.; Xenaki, D.; et al. Critical role for iron accumulation in the pathogenesis of fibrotic lung disease. J. Pathol. 2020, 251, 49–62. [Google Scholar] [CrossRef]
- Yuan, L.; Sun, Y.; Zhou, N.; Wu, W.; Zheng, W.; Wang, Y. Dihydroquercetin Attenuates Silica-Induced Pulmonary Fibrosis by Inhibiting Ferroptosis Signaling Pathway. Front. Pharmacol. 2022, 13, 845600. [Google Scholar] [CrossRef]
- Pei, Z.; Qin, Y.; Fu, X.; Yang, F.; Huo, F.; Liang, X.; Wang, S.; Cui, H.; Lin, P.; Zhou, G.; et al. Inhibition of ferroptosis and iron accumulation alleviates pulmonary fibrosis in a bleomycin model. Redox Biol. 2022, 57, 102509. [Google Scholar] [CrossRef]
- Han, Y.; Ye, L.; Du, F.; Ye, M.; Li, C.; Zhu, X.; Wang, Q.; Jiang, H.; Liu, Z.; Ma, J.; et al. Iron metabolism regulation of epithelial-mesenchymal transition in idiopathic pulmonary fibrosis. Ann. Transl. Med. 2021, 9, 1755. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Jia, J.; Zheng, J.; Zhou, Y.; Jia, D.; Wang, J. Recent Progress of Ferroptosis in Lung Diseases. Front. Cell Dev. Biol. 2021, 9, 789517. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Mizumura, K.; Gon, Y.; Shimizu, T.; Kozu, Y.; Shikano, S.; Iida, Y.; Hikichi, M.; Okamoto, S.; Tsuya, K.; et al. Iron-Dependent Mitochondrial Dysfunction Contributes to the Pathogenesis of Pulmonary Fibrosis. Front. Pharmacol. 2021, 12, 643980. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xu, P.; Ke, S.; Dong, H.; Zhan, M.; Hu, Q.; Li, J. Histone methyltransferase SETDB1 inhibits TGF-β-induced epithelial-mesenchymal transition in pulmonary fibrosis by regulating SNAI1 expression and the ferroptosis signaling pathway. Arch. Biochem. Biophys. 2022, 715, 109087. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef]
- Ruaro, B.; Salton, F.; Braga, L.; Wade, B.; Confalonieri, P.; Volpe, M.C.; Baratella, E.; Maiocchi, S.; Confalonieri, M. The History and Mystery of Alveolar Epithelial Type II Cells: Focus on Their Physiologic and Pathologic Role in Lung. Int. J. Mol. Sci. 2021, 22, 2566. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, X.; Hua, Y.; Zhao, Q.; Wang, K.; Zhen, L.; Wang, G.; Lü, J.; Luo, A.; Cho, W.C.; et al. YY1 mediates TGF-β1-induced EMT and pro-fibrogenesis in alveolar epithelial cells. Respir. Res. 2019, 20, 249. [Google Scholar] [CrossRef]
- Sha, Y.; Haensel, D.; Gutierrez, G.; Du, H.; Dai, X.; Nie, Q. Intermediate cell states in epithelial-to-mesenchymal transition. Phys. Biol. 2019, 16, 021001. [Google Scholar] [CrossRef]
- Shang, B.Q.; Li, M.L.; Quan, H.Y.; Hou, P.F.; Li, Z.W.; Chu, S.F.; Zheng, J.N.; Bai, J. Functional roles of circular RNAs during epithelial-to-mesenchymal transition. Mol. Cancer 2019, 18, 138. [Google Scholar] [CrossRef]
- Rubio, K.; Castillo-Negrete, R.; Barreto, G. Non-coding RNAs and nuclear architecture during epithelial-mesenchymal transition in lung cancer and idiopathic pulmonary fibrosis. Cell. Signal. 2020, 70, 109593. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Yang, Z.Z. Research progress of epithelial-interstitial transition involved in pulmonary fibrosis process. J. Med. Grad. Stud. 2021, 34, 519–523. [Google Scholar] [CrossRef]
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. EMT: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.L.; Chen, D.Q.; Vaziri, N.D.; Guo, Y.; Zhao, Y.Y. Small molecule inhibitors of epithelial-mesenchymal transition for the treatment of cancer and fibrosis. Med. Res. Rev. 2020, 40, 54–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell. Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Chen, H.X.; Wu, Y.P.; Li, W.; Shen, H.H.; Chen, Z.H. Ferroptosis in respiratory diseases. Sheng Li Xue Bao 2020, 72, 575–585. [Google Scholar] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lin, W.; Rao, T.; Zheng, J.; Zhang, T.; Zhang, M.; Lin, Z. Ferroptosis and Its Potential Role in the Nervous System Diseases. J. Inflamm. Res. 2022, 15, 1555–1574. [Google Scholar] [CrossRef]
- Leng, Y.; Luo, X.; Yu, J.; Jia, H.; Yu, B. Ferroptosis: A Potential Target in Cardiovascular Disease. Front. Cell Dev. Biol. 2021, 9, 813668. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Cao, Y.; Cao, W.; Jia, Y.; Lu, N. The Application of Ferroptosis in Diseases. Pharmacol. Res. 2020, 159, 104919. [Google Scholar] [CrossRef]
- Proneth, B.; Conrad, M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019, 26, 14–24. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Park, T.J.; Park, J.H.; Lee, G.S.; Lee, J.Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.J.; Han, B.S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.H.; Song, C.C.; Pantopoulos, K.; Wei, X.L.; Zheng, H.; Luo, Z. Mitochondrial oxidative stress mediated Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic. Biol. Med. 2022, 180, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Araya, J.; Yoshida, M.; Sakamoto, T.; Koumura, T.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; et al. Involvement of GPx4-Regulated Lipid Peroxidation in Idiopathic Pulmonary Fibrosis Pathogenesis. J. Immunol. 2019, 203, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wang, W.W.; Zhang, M.Z.; Ma, Z.X.; Qiu, X.R.; Shen, M.; Yin, X.X. ROS induces epithelial-mesenchymal transition via the TGF-β1/PI3K/Akt/mTOR pathway in diabetic nephropathy. Exp. Ther. Med. 2019, 17, 835–846. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, F.; Lv, X.; Wang, S.; Liao, X.; Yu, Y.; Dai, Q.; Zhang, Y.; Meng, J.; Hu, G.; et al. Mefunidone ameliorates diabetic kidney disease in STZ and db/db mice. FASEB J. 2021, 35, e21198. [Google Scholar] [CrossRef]
- Guan, R.; Wang, J.; Cai, Z.; Li, Z.; Wang, L.; Li, Y.; Xu, J.; Li, D.; Yao, H.; Liu, W.; et al. Hydrogen sulfide attenuates cigarette smoke-induced airway remodeling by upregulating SIRT1 signaling pathway. Redox Biol. 2020, 28, 101356. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar] [CrossRef]
- Li, J.; Yuan, J.; Li, Y.; Wang, J.; Xie, Q.; Ma, R.; Wang, J.; Ren, M.; Lu, D.; Xu, Z. d-Borneol enhances cisplatin sensitivity via autophagy dependent EMT signaling and NCOA4-mediated ferritinophagy. Phytomedicine 2022, 106, 154411. [Google Scholar] [CrossRef]
- Li, H.; Zhou, W.; Wei, H.; Li, L.; Wang, X.; Li, Y.; Li, S.; Li, C. Ferritinophagic Flux Was a Driving Force in Determination of Status of EMT, Ferroptosis, and NDRG1 Activation in Action of Mechanism of 2-Pyridylhydrazone Dithiocarbamate S-Acetic Acid. J. Oncol. 2021, 2021, 3015710. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Libby, P.; Tuomilehto, J.; Lip, G.Y.H.; Penninger, J.M.; Richardson, D.R.; Tang, D.; Zhou, H.; Wang, S.; et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol. Metab. 2021, 32, 444–462. [Google Scholar] [CrossRef]
- Guan, D.; Zhou, W.; Wei, H.; Wang, T.; Zheng, K.; Yang, C.; Feng, R.; Xu, R.; Fu, Y.; Li, C.; et al. Ferritinophagy-Mediated Ferroptosis and Activation of Keap1/Nrf2/HO-1 Pathway Were Conducive to EMT Inhibition of Gastric Cancer Cells in Action of 2,2′-Di-pyridineketone Hydrazone Dithiocarbamate Butyric Acid Ester. Oxid. Med. Cell. Longev. 2022, 2022, 3920664. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Li, C.; Li, Y.; Li, Y.; Wang, G.; Gao, F.; Li, C. The DpdtbA induced EMT inhibition in gastric cancer cell lines was through ferritinophagy-mediated activation of p53 and PHD2/hif-1α pathway. J. Inorg. Biochem. 2021, 218, 111413. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhang, Y.; Li, M.; Sun, Z.; Liu, T.; Zhao, M.; Li, Z. Single-Cell RNA-Seq Reveals the Promoting Role of Ferroptosis Tendency During Lung Adenocarcinoma EMT Progression. Front. Cell Dev. Biol. 2021, 9, 822315. [Google Scholar] [CrossRef]
- Lu, T.; Zhang, Z.; Pan, X.; Zhang, J.; Wang, X.; Wang, M.; Li, H.; Yan, M.; Chen, W. Caveolin-1 promotes cancer progression via inhibiting ferroptosis in head and neck squamous cell carcinoma. J. Oral Pathol. Med. 2022, 51, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, Y.; Xie, S.; Lai, Y.; Mo, C.; Zeng, T.; Kuang, S.; Zhou, C.; Zeng, Z.; Chen, Y.; et al. Isoliquiritigenin alleviates liver fibrosis through caveolin-1-mediated hepatic stellate cells ferroptosis in zebrafish and mice. Phytomedicine 2022, 101, 154117. [Google Scholar] [CrossRef] [PubMed]
- Strippoli, R.; Loureiro, J.; Moreno, V.; Benedicto, I.; Pérez Lozano, M.L.; Barreiro, O.; Pellinen, T.; Minguet, S.; Foronda, M.; Osteso, M.T.; et al. Caveolin-1 deficiency induces a MEK-ERK1/2-Snail-1-dependent epithelial-mesenchymal transition and fibrosis during peritoneal dialysis. EMBO Mol. Med. 2015, 7, 102–123. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, Y.; Shi, S.; Katoh, M.; Kitada, M.; Kanasaki, K.; Koya, D. Dipeptidyl peptidase-4 plays a pathogenic role in BSA-induced kidney injury in diabetic mice. Sci. Rep. 2019, 9, 7519. [Google Scholar] [CrossRef]
- Kim, S.; Kang, S.W.; Joo, J.; Han, S.H.; Shin, H.; Nam, B.Y.; Park, J.; Yoo, T.H.; Kim, G.; Lee, P.; et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Feng, Z.; Chen, L.; Li, Y.; Bian, H.; Geng, J.; Zheng, Z.H.; Fu, X.; Pei, Z.; Qin, Y.; et al. TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models. Nat. Commun. 2022, 13, 676. [Google Scholar] [CrossRef]
- Yoshimatsu, Y.; Wakabayashi, I.; Kimuro, S.; Takahashi, N.; Takahashi, K.; Kobayashi, M.; Maishi, N.; Podyma-Inoue, K.A.; Hida, K.; Miyazono, K.; et al. TNF-α enhances TGF-β-induced endothelial-to-mesenchymal transition via TGF-β signal augmentation. Cancer Sci. 2020, 111, 2385–2399. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Qu, Z.; Liang, Y.; Xu, Y.; Zhou, D.; Li, D.; Zhang, Y.; Yin, S. Iguratimod decreases bleomycin-induced pulmonary fibrosis in association with inhibition of TNF-α in mice. Int. Immunopharmacol. 2021, 99, 107936. [Google Scholar] [CrossRef] [PubMed]
- Becerril, C.; Montaño, M.; Cisneros, J.; Mendoza-Milla, C.; Pardo, A.; Ortiz-Quintero, B.; Selman, M.; Ramos, C. Mesenchymal-Epithelial Transition in Fibroblasts of Human Normal Lungs and Interstitial Lung Diseases. Biomolecules 2021, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Diepenbruck, M.; Christofori, G. Epithelial-mesenchymal transition (EMT) and metastasis: Yes, no, maybe? Curr. Opin. Cell. Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial–Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef]
- He, R.; Liu, B.; Xiong, R.; Geng, B.; Meng, H.; Lin, W.; Hao, B.; Zhang, L.; Wang, W.; Jiang, W.; et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. 2022, 8, 43. [Google Scholar] [CrossRef]
- Ma, J.; Wang, J. Wnt5a regulates SiO(2)-induced ferroptosis in mouse alveolar macrophages by positive feedback. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2022, 38, 699–706. [Google Scholar] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef]
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018, 71–72, 112–127. [Google Scholar] [CrossRef]
- Katsuno, Y.; Derynck, R. Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-β family. Dev. Cell 2021, 56, 726–746. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Mo, N.; Li, Z. Ginsenosides: Potential therapeutic source for fibrosis-associated human diseases. J. Ginseng Res. 2020, 44, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Ligier, M.; Puisieux, A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 2018, 78, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; You, J.H.; Kim, M.S.; Roh, J.L. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol. 2020, 37, 101697. [Google Scholar] [CrossRef]
- Han, X.; Duan, X.; Liu, Z.; Long, Y.; Liu, C.; Zhou, J.; Li, N.; Qin, J.; Wang, Y. ZEB1 directly inhibits GPX4 transcription contributing to ROS accumulation in breast cancer cells. Breast Cancer Res. Treat. 2021, 188, 329–342. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Sun, L.; Dong, H.; Zhang, W.; Wang, N.; Ni, N.; Bai, X.; Liu, N. Lipid Peroxidation, GSH Depletion, and SLC7A11 Inhibition Are Common Causes of EMT and Ferroptosis in A549 Cells, but Different in Specific Mechanisms. DNA Cell Biol. 2021, 40, 172–183. [Google Scholar] [CrossRef]
- Liu, P.; Luo, G.; Dodson, M.; Schmidlin, C.J.; Wei, Y.; Kerimoglu, B.; Ooi, A.; Chapman, E.; Garcia, J.G.; Zhang, D.D. The NRF2-LOC344887 signaling axis suppresses pulmonary fibrosis. Redox Biol. 2021, 38, 101766. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, D.; Yue, F.; Zheng, M.; Kovacevic, Z.; Richardson, D.R. The iron chelators Dp44mT and DFO inhibit TGF-β-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J. Biol. Chem. 2012, 287, 17016–17028. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, K.; Zhang, Y.; Fan, M.; Li, A.; Zhou, J.; Yang, T.; Shi, P.; Li, D.; Zhang, G.; et al. Ferroptosis-Related Genes in Bronchoalveolar Lavage Fluid Serves as Prognostic Biomarkers for Idiopathic Pulmonary Fibrosis. Front. Med. 2021, 8, 693959. [Google Scholar] [CrossRef]
- Berry, T.M.; Moustafa, A.A. A novel treatment strategy to prevent Parkinson’s disease: Focus on iron regulatory protein 1 (IRP1). Int. J. Neurosci. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, J.; Lin, M.; Banday, M.M.; Patil, S.S.; Krishnamurthy, S.; Breitzig, M.; Soundararajan, R.; Galam, L.; Narala, V.R.; Johns, C.; et al. Aberrant Expression of ACO1 in Vasculatures Parallels Progression of Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2022, 13, 890380. [Google Scholar] [CrossRef]
- Wang, X.M.; Zhang, Y.; Kim, H.P.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F.; Ifedigbo, E.; Xu, X.; Oury, T.D.; Kaminski, N.; et al. Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 2006, 203, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Duan, L.; Yuan, S.; Zhuang, X.; Qiao, T.; He, J. Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-β1. J. Inflamm. 2019, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, Y.; Xiao, J.; Shang, J.; Tan, Q.; Ping, F.; Huang, W.; Wu, F.; Zhang, H.; Zhang, X. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020, 27, 2635–2650. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jang, J.; Park, S.M.; Yang, S.R. An Update on the Role of Nrf2 in Respiratory Disease: Molecular Mechanisms and Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 8406. [Google Scholar] [CrossRef]
- Audousset, C.; McGovern, T.; Martin, J.G. Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches —Pulmonary Disease/Asthma. Front. Physiol. 2021, 12, 727806. [Google Scholar] [CrossRef]
- Cameron, B.D.; Sekhar, K.R.; Ofori, M.; Freeman, M.L. The Role of Nrf2 in the Response to Normal Tissue Radiation Injury. Radiat. Res. 2018, 190, 99–106. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 7090534. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Deng, S.; Wu, W.; Li, Z.; Lei, W.; Wang, Y.; Vongphouttha, C.; Zhang, T.; Dong, Z. Rapamycin attenuates the paraquat-induced pulmonary fibrosis through activating Nrf2 pathway. J. Cell. Physiol. 2020, 235, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Qiang, Z.; Chai, D.; Peng, J.; Xia, Y.; Hu, R.; Jiang, H. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging 2020, 12, 12943–12959. [Google Scholar] [CrossRef]
- Fan, Z.; Wirth, A.K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef]
- Zhang, Q.; Qu, H.; Chen, Y.; Luo, X.; Chen, C.; Xiao, B.; Ding, X.; Zhao, P.; Lu, Y.; Chen, A.F.; et al. Atorvastatin Induces Mitochondria-Dependent Ferroptosis via the Modulation of Nrf2-xCT/GPx4 Axis. Front. Cell Dev. Biol. 2022, 10, 806081. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Matsumoto, M.; Shindo, T.; Saigusa, D.; Kato, H.; Suzuki, K.; Sato, M.; Ishii, Y.; Shimokawa, H.; Igarashi, K. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J. Biol. Chem. 2020, 295, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Nishizawa, H.; Saiki, Y.; Matsumoto, M. The transcription factor BACH1 at the crossroads of cancer biology: From epithelial-mesenchymal transition to ferroptosis. J. Biol. Chem. 2021, 297, 101032. [Google Scholar] [CrossRef]
- Sato, M.; Matsumoto, M.; Saiki, Y.; Alam, M.; Nishizawa, H.; Rokugo, M.; Brydun, A.; Yamada, S.; Kaneko, M.K.; Funayama, R.; et al. BACH1 Promotes Pancreatic Cancer Metastasis by Repressing Epithelial Genes and Enhancing Epithelial-Mesenchymal Transition. Cancer Res. 2020, 80, 1279–1292. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, J.; Xie, X.; Nan, P.; Liu, F.; Sun, Y.; Zhao, X. BACH1 promotes the progression of esophageal squamous cell carcinoma by inducing the epithelial-mesenchymal transition and angiogenesis. Cancer Med. 2021, 10, 3413–3426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, L.; Davies, K.J.A.; Forman, H.J. Silencing Bach1 alters aging-related changes in the expression of Nrf2-regulated genes in primary human bronchial epithelial cells. Arch. Biochem. Biophys. 2019, 672, 108074. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Yamanaka, M.; Igarashi, K. Ferroptosis: Regulation by competition between NRF2 and BACH1 and propagation of the death signal. FEBS J. 2022. [Google Scholar] [CrossRef] [PubMed]
- Boyapally, R.; Pulivendala, G.; Bale, S.; Godugu, C. Niclosamide alleviates pulmonary fibrosis in vitro and in vivo by attenuation of epithelial-to-mesenchymal transition, matrix proteins & Wnt/β-catenin signaling: A drug repurposing study. Life Sci. 2019, 220, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Kishida, S.; Yamamoto, H.; Hino, S.; Ikeda, S.; Kishida, M.; Kikuchi, A. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability. Mol. Cell. Biol. 1999, 19, 4414–4422. [Google Scholar] [CrossRef] [PubMed]
- Burgy, O.; Königshoff, M. The WNT signaling pathways in wound healing and fibrosis. Matrix Biol. 2018, 68–69, 67–80. [Google Scholar] [CrossRef]
- Yang, F.; Hou, Z.F.; Zhu, H.Y.; Chen, X.X.; Li, W.Y.; Cao, R.S.; Li, Y.X.; Chen, R.; Zhang, W. Catalpol Protects Against Pulmonary Fibrosis Through Inhibiting TGF-β1/Smad3 and Wnt/β-Catenin Signaling Pathways. Front. Pharmacol. 2020, 11, 594139. [Google Scholar] [CrossRef]
- Zhang, E.; Geng, X.; Shan, S.; Li, P.; Li, S.; Li, W.; Yu, M.; Peng, C.; Wang, S.; Shao, H.; et al. Exosomes derived from bone marrow mesenchymal stem cells reverse epithelial-mesenchymal transition potentially via attenuating Wnt/β-catenin signaling to alleviate silica-induced pulmonary fibrosis. Toxicol. Mech. Methods 2021, 31, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Zhang, Y.; Shi, W.; Ma, L.; Xu, T.; Chang, P.; Dong, L. Mesenchymal stromal cells can repair radiation-induced pulmonary fibrosis via a DKK-1-mediated Wnt/β-catenin pathway. Cell Tissue Res. 2021, 384, 87–97. [Google Scholar] [CrossRef]
- Menou, A.; Duitman, J.; Crestani, B. The impaired proteases and anti-proteases balance in Idiopathic Pulmonary Fibrosis. Matrix Biol. 2018, 68–69, 382–403. [Google Scholar] [CrossRef] [PubMed]
- Chekmarev, J.; Azad, M.G.; Richardson, D.R. The Oncogenic Signaling Disruptor, NDRG1: Molecular and Cellular Mechanisms of Activity. Cells 2021, 10, 2382. [Google Scholar] [CrossRef]
- Luo, C.; Xu, W.; Tang, X.; Liu, X.; Cheng, Y.; Wu, Y.; Xie, Z.; Wu, X.; He, X.; Wang, Q.; et al. Canonical Wnt signaling works downstream of iron overload to prevent ferroptosis from damaging osteoblast differentiation. Free Radic. Biol. Med. 2022, 188, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, L.; Shang, W.; Yang, Z.; Li, T.; Liu, F.; Shao, W.; Lv, L.; Chai, L.; Qu, L.; et al. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting GPX4 in gastric cancer. Cell Death Differ. 2022, 29, 2190–2202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, G.; Condello, S.; Huang, H.; Cardenas, H.; Tanner, E.J.; Wei, J.; Ji, Y.; Li, J.; Tan, Y.; et al. Frizzled-7 Identifies Platinum-Tolerant Ovarian Cancer Cells Susceptible to Ferroptosis. Cancer Res. 2021, 81, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Barriga, M.; Benitez, R.; Ferraz-de-Paula, V.; Garcia-Frutos, M.; Caro, M.; Robledo, G.; O’Valle, F.; Campos-Salinas, J.; Delgado, M. Protective role of cortistatin in pulmonary inflammation and fibrosis. Br. J. Pharmacol. 2021, 178, 4368–4388. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, H.; Xiao, H.; Luo, T.; Lin, H.; Deng, J. Role of Ferroptosis in Regulating the Epithelial–Mesenchymal Transition in Pulmonary Fibrosis. Biomedicines 2023, 11, 163. https://doi.org/10.3390/biomedicines11010163
Ling H, Xiao H, Luo T, Lin H, Deng J. Role of Ferroptosis in Regulating the Epithelial–Mesenchymal Transition in Pulmonary Fibrosis. Biomedicines. 2023; 11(1):163. https://doi.org/10.3390/biomedicines11010163
Chicago/Turabian StyleLing, Hong, Hong Xiao, Ting Luo, Huicai Lin, and Jiang Deng. 2023. "Role of Ferroptosis in Regulating the Epithelial–Mesenchymal Transition in Pulmonary Fibrosis" Biomedicines 11, no. 1: 163. https://doi.org/10.3390/biomedicines11010163
APA StyleLing, H., Xiao, H., Luo, T., Lin, H., & Deng, J. (2023). Role of Ferroptosis in Regulating the Epithelial–Mesenchymal Transition in Pulmonary Fibrosis. Biomedicines, 11(1), 163. https://doi.org/10.3390/biomedicines11010163