

Blood-Based Immune Protein Markers of Disease Progression in Murine Models of Acute and Chronic Inflammatory Bowel Disease


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. DSS-Induced Mouse Model
2.3. T Cell Transfer Mouse Model
2.4. Multiplexed Cytokine/Chemokine Analysis
2.5. Statistical Analysis
3. Results
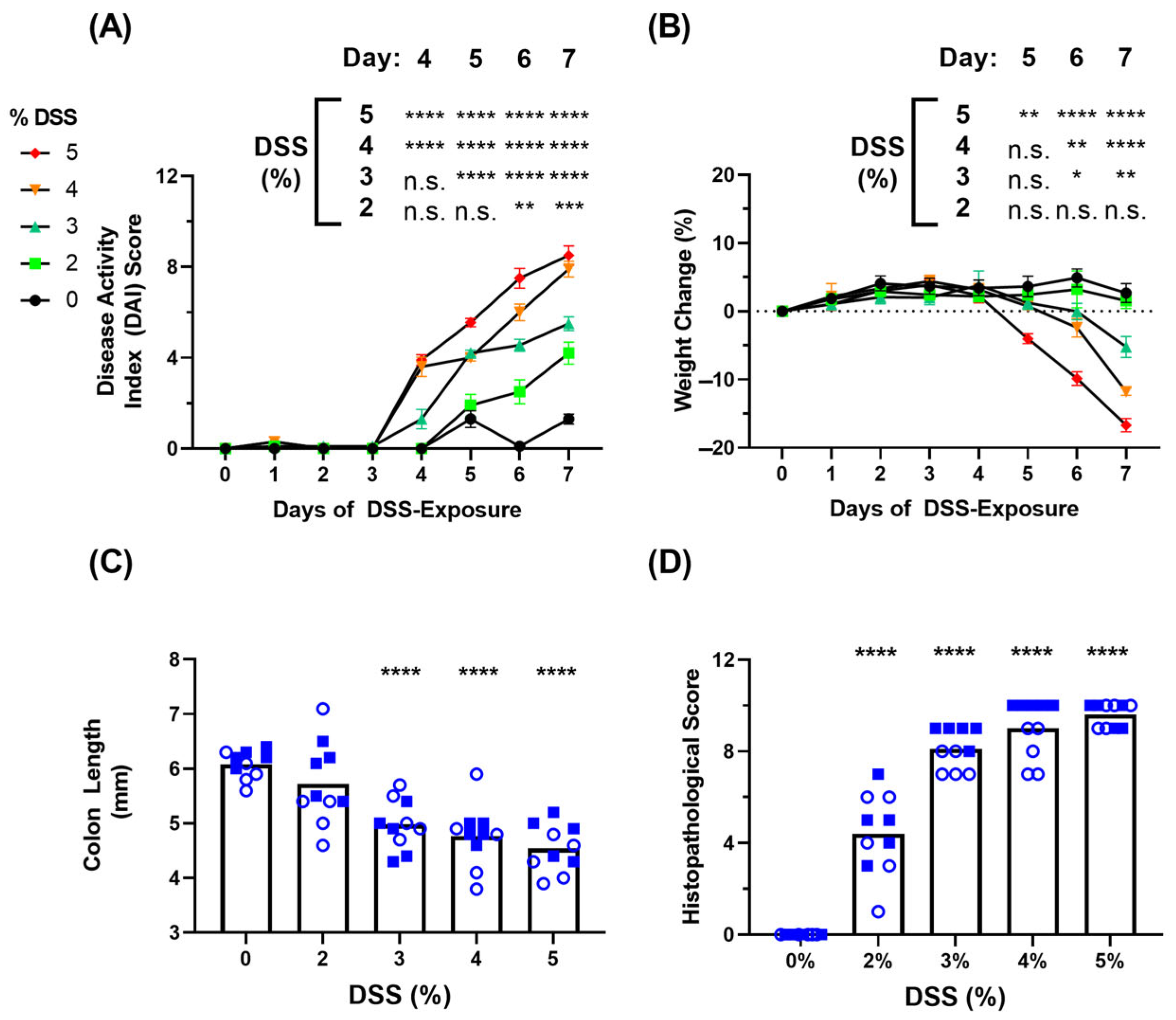
3.1. Clinical Symptoms in DSS-Induced Colitis Model
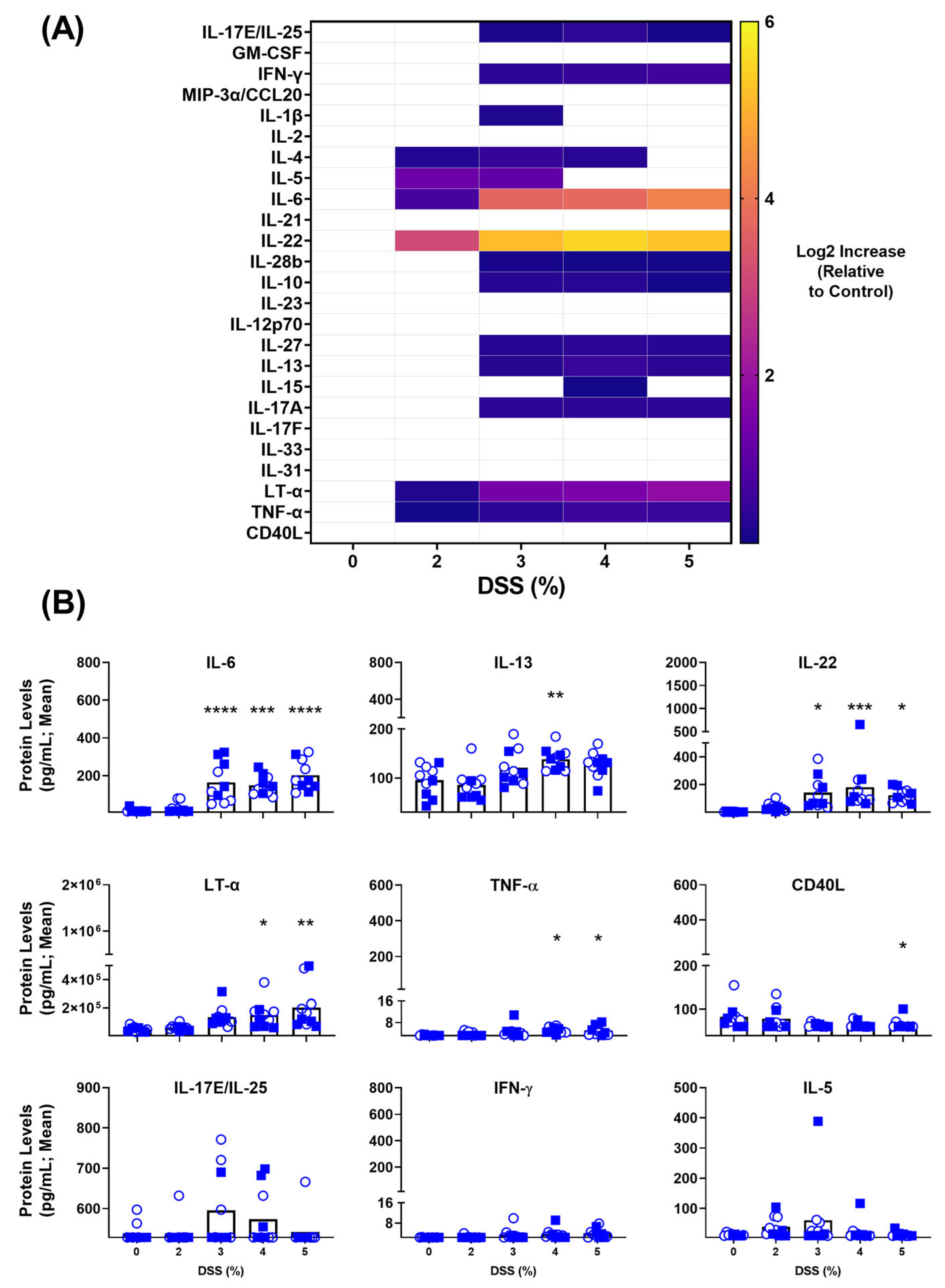
3.2. Blood-Based Immune Protein Markers in DSS-Induced Colitis Model
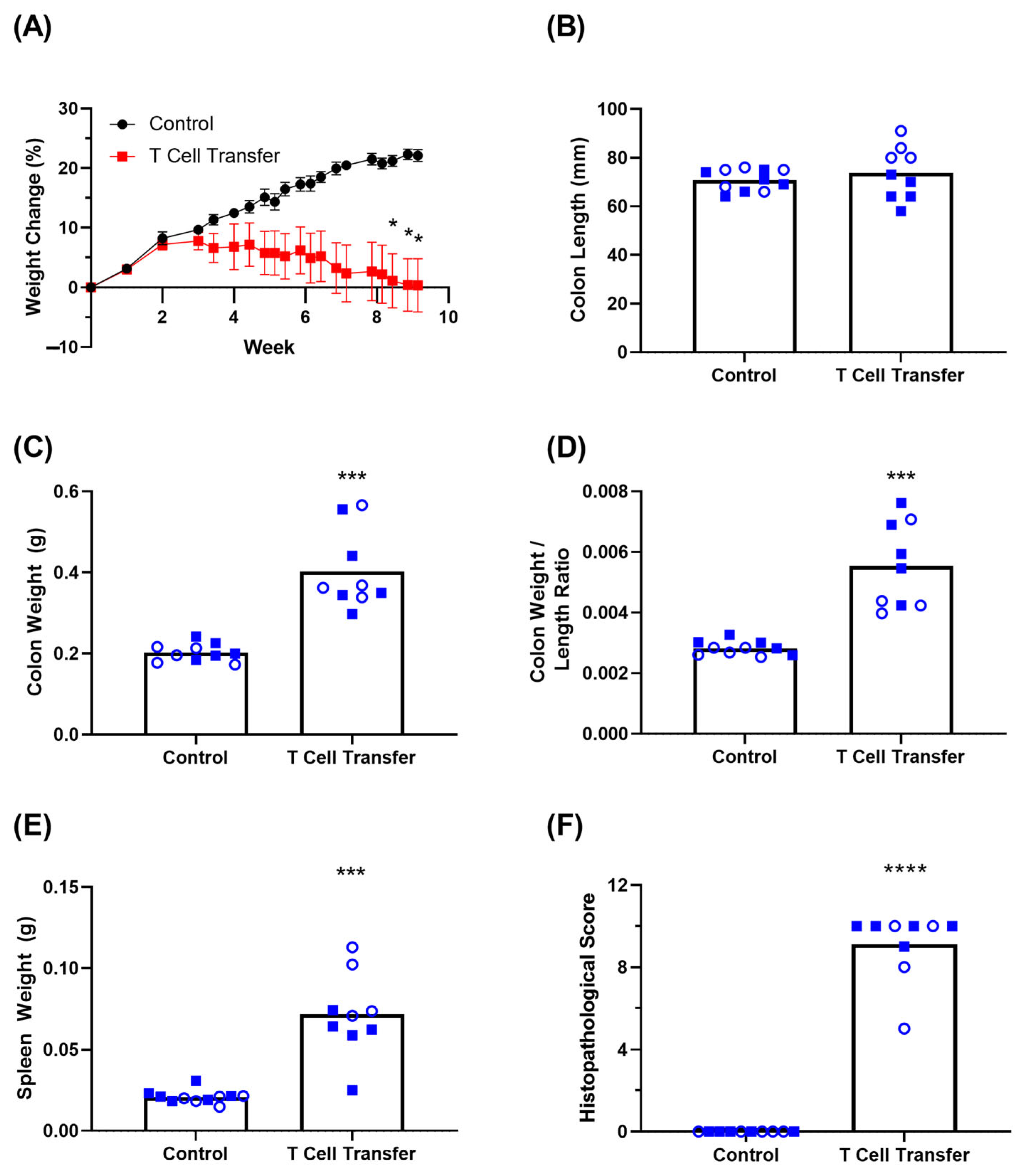
3.3. Clinical Symptoms in T Cell Transfer Colitis Model
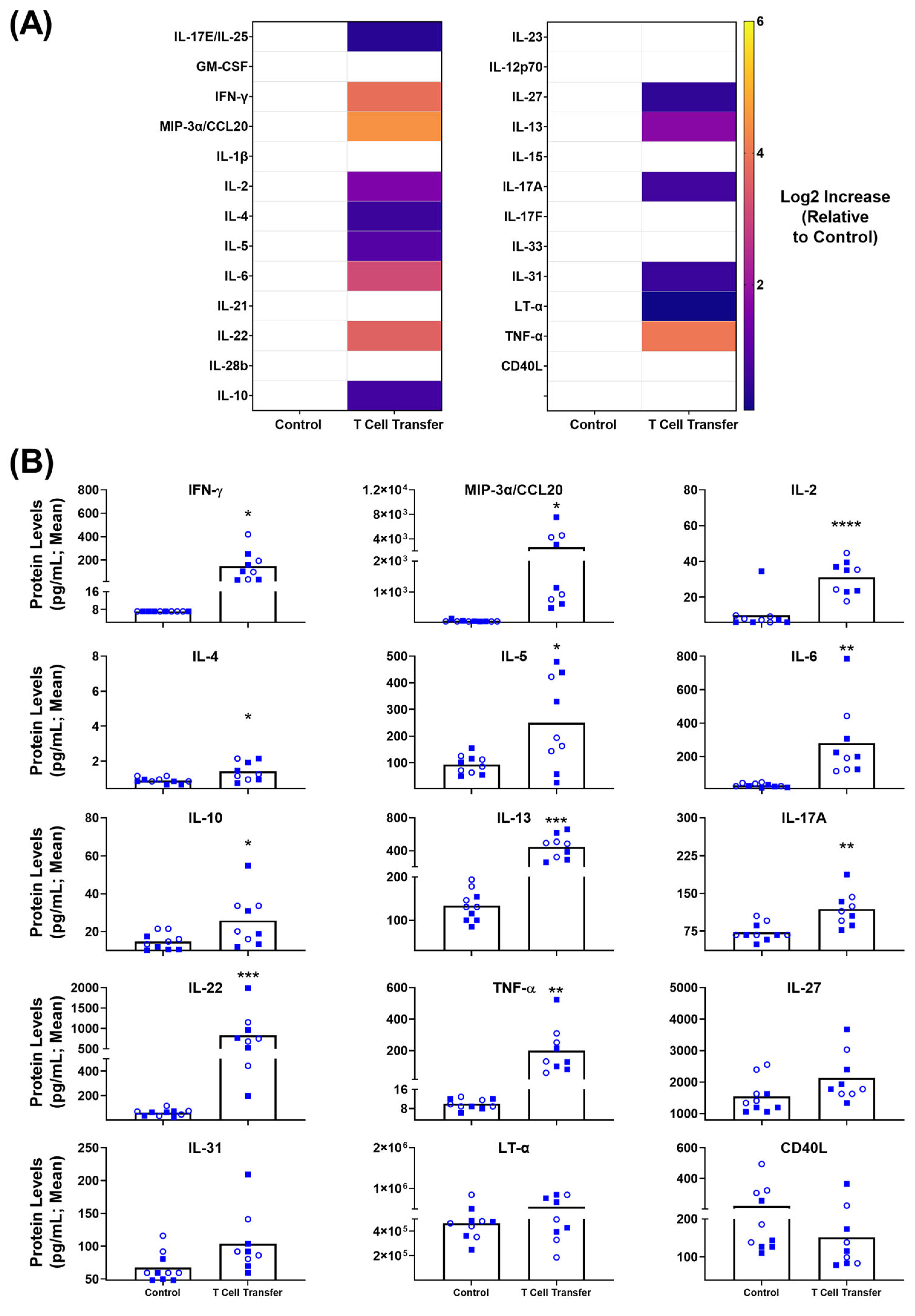
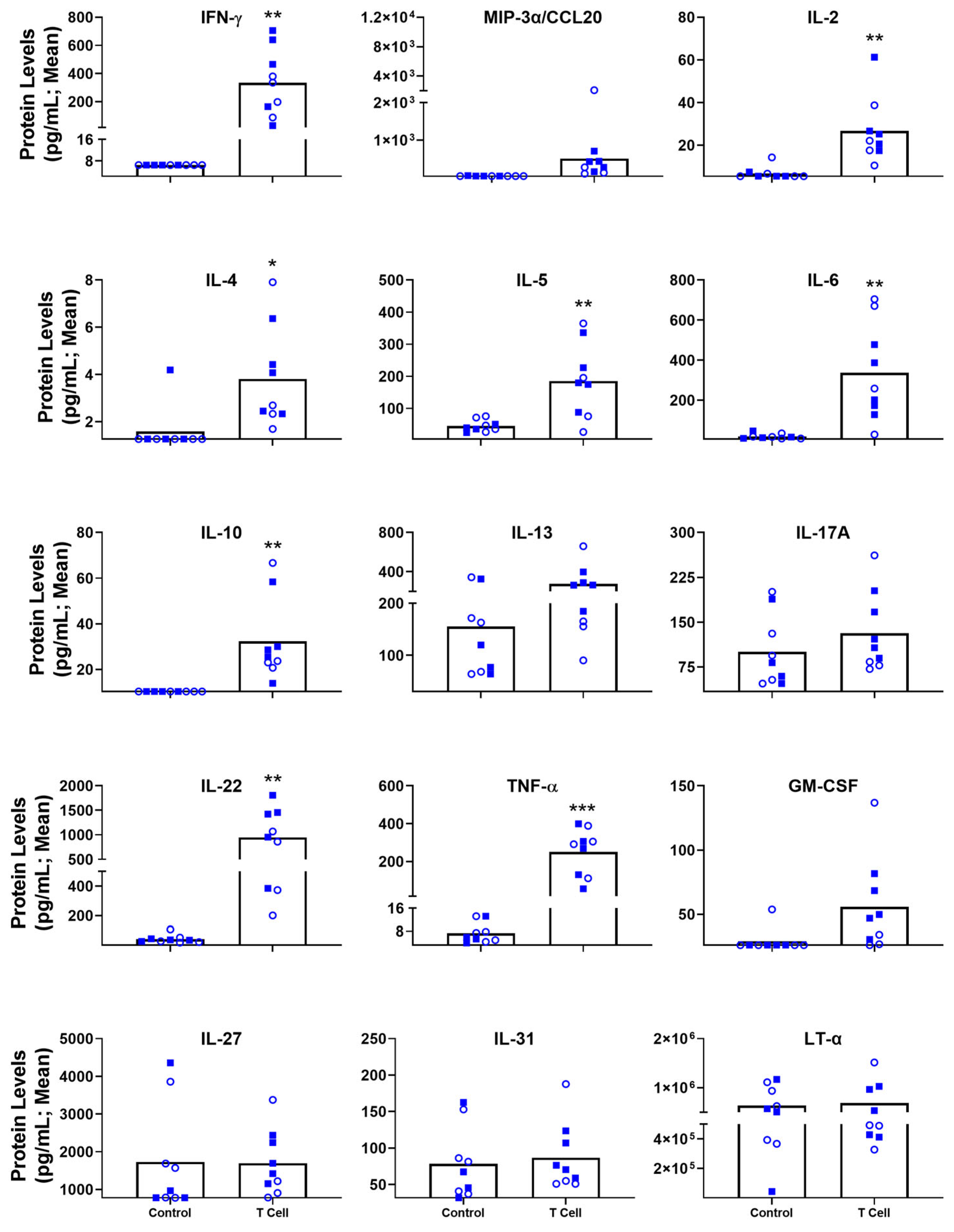
3.4. Blood-Based Immune Protein Markers in T Cell Transfer Colitis Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 Inflammatory Bowel Disease Collaborators The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [CrossRef] [PubMed]
- Manceur, A.M.; Ding, Z.; Muser, E.; Obando, C.; Voelker, J.; Pilon, D.; Kinkead, F.; Lafeuille, M.-H.; Lefebvre, P. Burden of Crohn’s disease in the United States: Long-term healthcare and work-loss related costs. J. Med. Econ. 2020, 23, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Falloon, K.A.; Fiocchi, C. Current Therapy in Inflammatory Bowel Disease: Why and How We Need to Change? Eur. Med. J. 2022, 6, 40–49. [Google Scholar] [CrossRef]
- Al-Bawardy, B.; Shivashankar, R.; Proctor, D.D. Novel and Emerging Therapies for Inflammatory Bowel Disease. Front. Pharmacol. 2021, 12, 651415. [Google Scholar] [CrossRef] [PubMed]
- Alulis, S.; Vadstrup, K.; Olsen, J.; Jørgensen, T.R.; Qvist, N.; Munkholm, P.; Borsi, A. The cost burden of Crohn’s disease and ulcerative colitis depending on biologic treatment status—A Danish register-based study. BMC Health Serv. Res. 2021, 21, 836. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, S.; Li, J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front. Med. 2021, 8, 765474. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Gerardi, V.; Papa, V.; Scaldaferri, F.; Rapaccini, G.L.; Gasbarrini, A.; Papa, A. Can We Predict the Efficacy of Anti-TNF-α Agents? Int. J. Mol. Sci. 2017, 18, 1973. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Hanauer, S.; Colombel, J.-F.; Sands, B.E.; Lukas, M.; Fedorak, R.N.; Lee, S.; Bressler, B.; et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 2013, 369, 711–721. [Google Scholar] [CrossRef]
- Vermeire, S.; O’Byrne, S.; Keir, M.; Williams, M.; Lu, T.T.; Mansfield, J.C.; Lamb, C.A.; Feagan, B.G.; Panes, J.; Salas, A.; et al. Etrolizumab as induction therapy for ulcerative colitis: A randomised, controlled, phase 2 trial. Lancet Lond. Engl. 2014, 384, 309–318. [Google Scholar] [CrossRef]
- Katsandegwaza, B.; Horsnell, W.; Smith, K. Inflammatory Bowel Disease: A Review of Pre-Clinical Murine Models of Human Disease. Int. J. Mol. Sci. 2022, 23, 9344. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 2014, 104, 1–15. [Google Scholar] [CrossRef]
- Ostanin, D.V.; Bao, J.; Koboziev, I.; Gray, L.; Robinson-Jackson, S.A.; Kosloski-Davidson, M.; Price, V.H.; Grisham, M.B. T cell transfer model of chronic colitis: Concepts, considerations, and tricks of the trade. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 296, G135–G146. [Google Scholar] [CrossRef]
- Kim, J.J.; Shajib, M.S.; Manocha, M.M.; Khan, W.I. Investigating Intestinal Inflammation in DSS-induced Model of IBD. J. Vis. Exp. JoVE 2012, 60, 3678. [Google Scholar] [CrossRef]
- Wang, N.; Kong, R.; Han, W.; Bao, W.; Shi, Y.; Ye, L.; Lu, J. Honokiol alleviates ulcerative colitis by targeting PPAR-γ–TLR4–NF-κB signaling and suppressing gasdermin-D-mediated pyroptosis in vivo and in vitro. Int. Immunopharmacol. 2022, 111, 109058. [Google Scholar] [CrossRef]
- Tian, W.; Wang, Q.; Wang, X.; Dong, D. Clophosome alleviate dextran sulphate sodium-induced colitis by regulating gut immune responses and maintaining intestinal integrity in mice. Clin. Exp. Pharmacol. Physiol. 2021, 48, 902–910. [Google Scholar] [CrossRef]
- Yan, Y.; Shao, M.; Qi, Q.; Xu, Y.; Yang, X.; Zhu, F.; He, S.; He, P.; Feng, C.; Wu, Y.; et al. Artemisinin analogue SM934 ameliorates DSS-induced mouse ulcerative colitis via suppressing neutrophils and macrophages. Acta Pharmacol. Sin. 2018, 39, 1633–1644. [Google Scholar] [CrossRef]
- Zhang, W.; Zou, G.; Li, B.; Du, X.; Sun, Z.; Yu, S.; Jiang, X. Fecal Microbiota Transplantation (FMT) Alleviates Experimental Colitis in Mice by Gut Microbiota Regulation. J. Microbiol. Biotechnol. 2020, 30, 1132–1141. [Google Scholar] [CrossRef]
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.E.; Conklin, L.S.; Centola, M.; Li, X. Distinct Cytokine Patterns Identified from Multiplex Profiles of Murine DSS and TNBS-Induced Colitis. Inflamm. Bowel Dis. 2009, 15, 341–352. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Chen, Y.-T.; Lo, C.-F.; Hsieh, C.-I.; Chiu, S.-Y.; Wu, C.-Y.; Yeh, Y.-S.; Hung, S.-H.; Cheng, P.-H.; Su, Y.-H.; et al. Early Detection of T cell Transfer-induced Autoimmune Colitis by In Vivo Imaging System. Sci. Rep. 2016, 6, 35635. [Google Scholar] [CrossRef]
- Yadav, V.; Mai, Y.; McCoubrey, L.E.; Wada, Y.; Tomioka, M.; Kawata, S.; Charde, S.; Basit, A.W. 5-Aminolevulinic Acid as a Novel Therapeutic for Inflammatory Bowel Disease. Biomedicines 2021, 9, 578. [Google Scholar] [CrossRef]
- Reinoso Webb, C.; den Bakker, H.; Koboziev, I.; Jones-Hall, Y.; Rao Kottapalli, K.; Ostanin, D.; Furr, K.L.; Mu, Q.; Luo, X.M.; Grisham, M.B. Differential Susceptibility to T Cell-Induced Colitis in Mice: Role of the Intestinal Microbiota. Inflamm. Bowel Dis. 2018, 24, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Valatas, V.; Vakas, M.; Kolios, G. The value of experimental models of colitis in predicting efficacy of biological therapies for inflammatory bowel diseases. Am. J. Physiol.-Gastrointest. Liver Physiol. 2013, 305, G763–G785. [Google Scholar] [CrossRef] [PubMed]
- Filippopoulou, F.; Habeos, G.I.; Rinotas, V.; Sophocleous, A.; Sykiotis, G.P.; Douni, E.; Chartoumpekis, D.V. Dexamethasone Administration in Mice Leads to Less Body Weight Gain over Time, Lower Serum Glucose, and Higher Insulin Levels Independently of NRF2. Antioxidants 2021, 11, 4. [Google Scholar] [CrossRef]
- Bábíčková, J.; Tóthová, Ľ.; Lengyelová, E.; Bartoňová, A.; Hodosy, J.; Gardlík, R.; Celec, P. Sex Differences in Experimentally Induced Colitis in Mice: A Role for Estrogens. Inflammation 2015, 38, 1996–2006. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.R.; Viney, J.L. Overview of Mouse Models of Inflammatory Bowel Disease and Their Use in Drug Discovery. Curr. Protoc. Pharmacol. 2009, 47, 5.57.1–5.57.19. [Google Scholar] [CrossRef]
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170. [Google Scholar] [CrossRef]
- Kim, T.W.; Seo, J.N.; Suh, Y.H.; Park, H.J.; Kim, J.H.; Kim, J.Y.; Oh, K.I. Involvement of lymphocytes in dextran sulfate sodium-induced experimental colitis. World J. Gastroenterol. 2006, 12, 302–305. [Google Scholar] [CrossRef]
- Xue, X.; Falcon, D.M. The Role of Immune Cells and Cytokines in Intestinal Wound Healing. Int. J. Mol. Sci. 2019, 20, 6097. [Google Scholar] [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef]
- Imam, T.; Park, S.; Kaplan, M.H.; Olson, M.R. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front. Immunol. 2018, 9, 1212. [Google Scholar] [CrossRef]
- Weinhage, T.; Däbritz, J.; Brockhausen, A.; Wirth, T.; Brückner, M.; Belz, M.; Foell, D.; Varga, G. Granulocyte Macrophage Colony-Stimulating Factor–Activated CD39+/CD73+ Murine Monocytes Modulate Intestinal Inflammation via Induction of Regulatory T Cells. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 433–449.e1. [Google Scholar] [CrossRef]
- Andrews, C.; McLean, M.H.; Durum, S.K. IL-27 as a novel therapy for inflammatory bowel disease: A critical review of the literature. Inflamm. Bowel Dis. 2016, 22, 2255–2264. [Google Scholar] [CrossRef]
- Zhang, Q.; Putheti, P.; Zhou, Q.; Liu, Q.; Gao, W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. 2008, 19, 347–356. [Google Scholar] [CrossRef]
- Gubernatorova, E.O.; Tumanov, A.V. Tumor Necrosis Factor and Lymphotoxin in Regulation of Intestinal Inflammation. Biochem. Biokhimiia 2016, 81, 1309–1325. [Google Scholar] [CrossRef]
- Senhaji, N.; Kojok, K.; Darif, Y.; Fadainia, C.; Zaid, Y. The Contribution of CD40/CD40L Axis in Inflammatory Bowel Disease: An Update. Front. Immunol. 2015, 6, 529. [Google Scholar] [CrossRef]
- Meitei, H.T.; Jadhav, N.; Lal, G. CCR6-CCL20 axis as a therapeutic target for autoimmune diseases. Autoimmun. Rev. 2021, 20, 102846. [Google Scholar] [CrossRef]
- Datsi, A.; Steinhoff, M.; Ahmad, F.; Alam, M.; Buddenkotte, J. Interleukin-31: The “itchy” cytokine in inflammation and therapy. Allergy 2021, 76, 2982–2997. [Google Scholar] [CrossRef]
- Do, J.; Visperas, A.; Sanogo, Y.O.; Bechtel, J.J.; Dvorina, N.; Kim, S.; Jang, E.; Stohlman, S.A.; Shen, B.; Fairchild, R.L.; et al. An IL-27/Lag3 axis enhances Foxp3+ regulatory T cell suppressive function and therapeutic efficacy. Mucosal Immunol. 2016, 9, 137–145. [Google Scholar] [CrossRef]
- Porter, R.J.; Andrews, C.; Brice, D.P.; Durum, S.K.; McLean, M.H. Can We Target Endogenous Anti-inflammatory Responses as a Therapeutic Strategy for Inflammatory Bowel Disease? Inflamm. Bowel Dis. 2018, 24, 2123–2134. [Google Scholar] [CrossRef]
- Rusu, I.; Mennillo, E.; Bain, J.L.; Li, Z.; Sun, X.; Ly, K.M.; Rosli, Y.Y.; Naser, M.; Wang, Z.; Advincula, R.; et al. Microbial signals, MyD88, and lymphotoxin drive TNF-independent intestinal epithelial tissue damage. J. Clin. Investig. 2022, 132, e154993. [Google Scholar] [CrossRef]
- Chuang, L.-S.; Villaverde, N.; Hui, K.Y.; Mortha, A.; Rahman, A.; Levine, A.P.; Haritunians, T.; Evelyn Ng, S.M.; Zhang, W.; Hsu, N.-Y.; et al. A Frameshift in CSF2RB Predominant Among Ashkenazi Jews Increases Risk for Crohn’s Disease and Reduces Monocyte Signaling via GM-CSF. Gastroenterology 2016, 151, 710–723.e2. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.P.; Pontikos, N.; Schiff, E.R.; Jostins, L.; Speed, D.; Lovat, L.B.; Barrett, J.C.; Grasberger, H.; Plagnol, V.; Segal, A.W. Genetic Complexity of Crohn’s Disease in Two Large Ashkenazi Jewish Families. Gastroenterology 2016, 151, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Castro-Dopico, T.; Fleming, A.; Dennison, T.W.; Ferdinand, J.R.; Harcourt, K.; Stewart, B.J.; Cader, Z.; Tuong, Z.K.; Jing, C.; Lok, L.S.C.; et al. GM-CSF Calibrates Macrophage Defense and Wound Healing Programs during Intestinal Infection and Inflammation. Cell Rep. 2020, 32, 107857. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weight Loss (%) | Stool Consistency | Fecal Blood Occurrence | |
|---|---|---|---|
| 0 | <0 | Normal | No blood on feces |
| 1 | 1–5 | Soft, but still formed | Slight traces of blood on feces |
| 2 | 5–10 | Very soft | Pronounced traces of blood on feces |
| 3 | 10–20 | N/A | N/A |
| 4 | >20 | Diarrhea | Gross rectal bleeding |
| Inflammatory Cell Infiltrate | Intestinal Architecture | Extent of Organ Inflammation (%) | |
|---|---|---|---|
| 0 | Normal | Normal | None |
| 1 | Mild/Mucosal | Focal erosion | 0–25 |
| 2 | Moderate/Mucosal and submucosal | Erosions/focal ulcerations | 25–50 |
| 3 | Marked/Transmural | Extended ulcerations/ granulation/ pseudopolyps | 50–75 |
| 4 | N/A | N/A | 75–100 |
| Acute (DSS) | Chronic (T Cell Transfer) | |
|---|---|---|
| Timeline | Days | Weeks |
| Immunity | Innate i | Adaptive |
| Disease Activity Index (DAI) | ✓ | ✗ |
| Weight Loss * | ✓ | ✓ |
| Fecal Consistency | ✓ | ✗ |
| Fecal Blood | ✓ | ✗ |
| Colon size ** | ✓ | ✓ |
| Histopathological Scoring | ✓ | ✓ |
| Systemically Elevated Immune Markers | IL-6, IL-22, IL-13, LT-α, TNF-α (IFN-γ) | IFN-γ, MIP-3α/CCL20, IL-2, IL-4, IL-5, IL-6, IL-22, IL-10, IL-13, IL-17A, TNF-α (IL-31, IL-27, GM-CSF t) |
| Systemically Downregulated Immune Marker | CD40L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renner, T.M.; Agbayani, G.; Dudani, R.; McCluskie, M.J.; Akache, B. Blood-Based Immune Protein Markers of Disease Progression in Murine Models of Acute and Chronic Inflammatory Bowel Disease. Biomedicines 2023, 11, 140. https://doi.org/10.3390/biomedicines11010140
Renner TM, Agbayani G, Dudani R, McCluskie MJ, Akache B. Blood-Based Immune Protein Markers of Disease Progression in Murine Models of Acute and Chronic Inflammatory Bowel Disease. Biomedicines. 2023; 11(1):140. https://doi.org/10.3390/biomedicines11010140
Chicago/Turabian StyleRenner, Tyler Milston, Gerard Agbayani, Renu Dudani, Michael J. McCluskie, and Bassel Akache. 2023. "Blood-Based Immune Protein Markers of Disease Progression in Murine Models of Acute and Chronic Inflammatory Bowel Disease" Biomedicines 11, no. 1: 140. https://doi.org/10.3390/biomedicines11010140
APA StyleRenner, T. M., Agbayani, G., Dudani, R., McCluskie, M. J., & Akache, B. (2023). Blood-Based Immune Protein Markers of Disease Progression in Murine Models of Acute and Chronic Inflammatory Bowel Disease. Biomedicines, 11(1), 140. https://doi.org/10.3390/biomedicines11010140