
Small Molecules as Toll-like Receptor 4 Modulators Drug and In-House Computational Repurposing

 , ,
, ,  , and
, and
Abstract
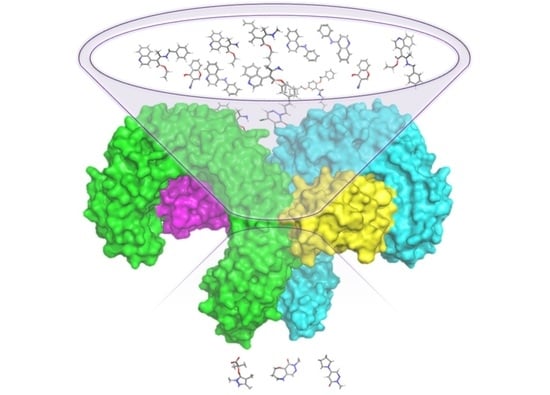
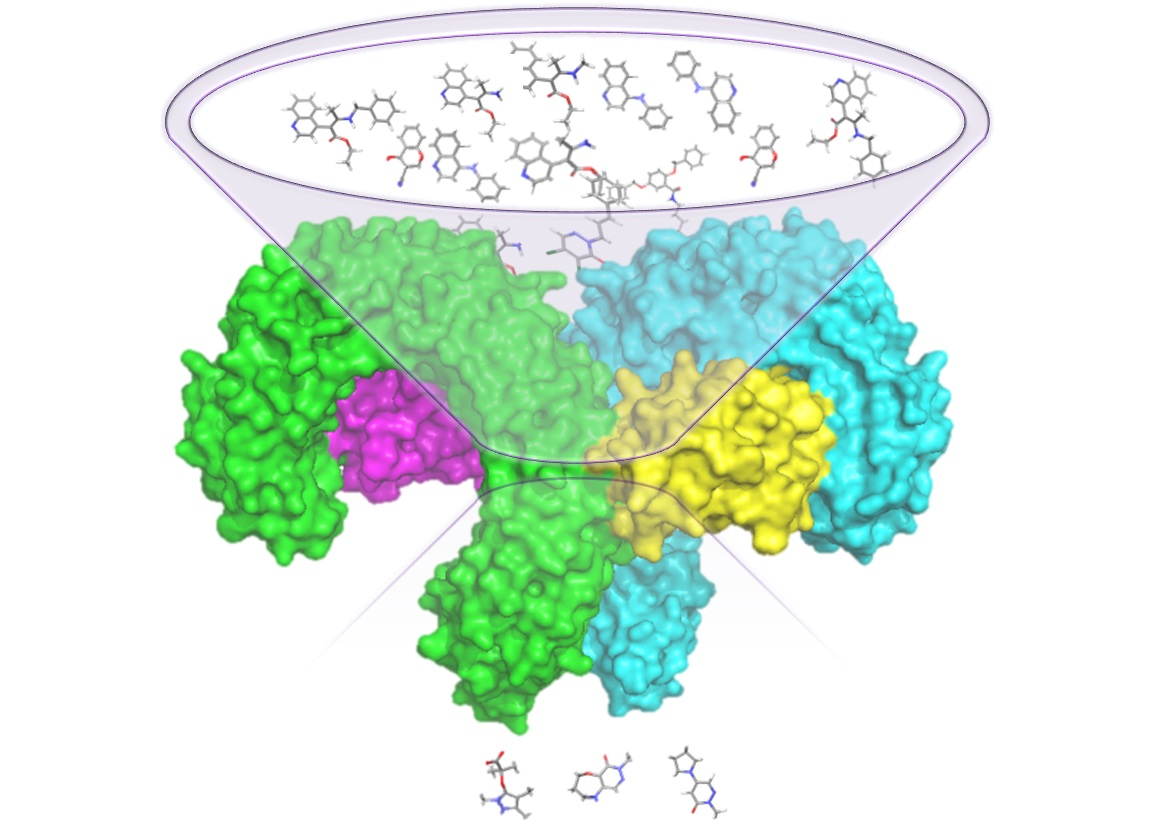
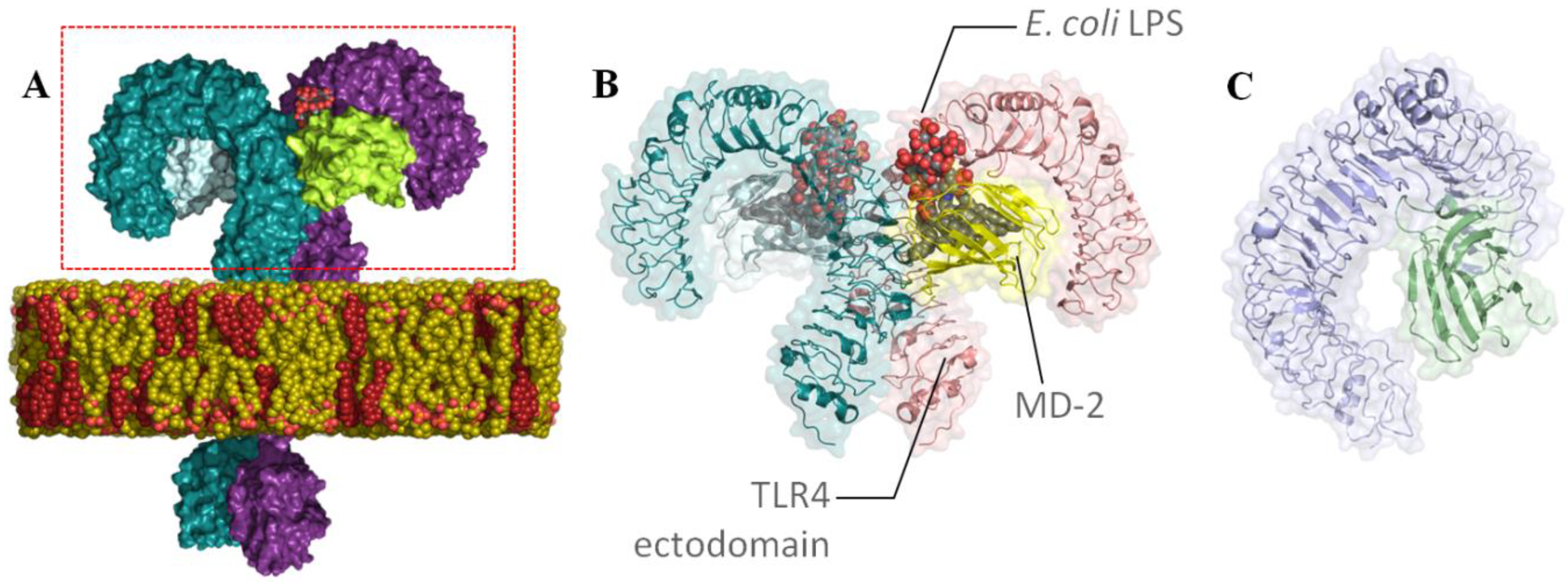
1. Introduction
2. Experimental Section
2.1. Computational Methods
2.1.1. Receptors
2.1.2. Databases
2.1.3. Library Preparation
- Lipophilicity of the molecules: a maximum logP of 6 was considered taking into account that the natural LPSs and the reported synthetic glycolipids have a very high logP: 29.14 ± 0.83, 14.35 ± 0.73, and 13.53 ± 0.47 for lipid IVa, P01, and ONO-4007, respectively. This limit is within a reasonable margin above the value of 5 according to Lipinski’s rule (oral bioavailability) [65].
- Molecular weight (MW): we considered a wide range between 300 and 700 Da given the MW of the glycolipids targeting TLR4, with a reasonable margin above the value of 500 according to Lipinski’s rule.
- pH: only possible tautomers at the physiological pH were considered within a range of 7 ± 0.5.
- Prediction of favorable binding from at least two docking programs and in two different conformations of TLR4.
2.1.4. Protein Preparation
2.1.5. Receptor Grid Preparation
2.1.6. Docking
Structure-Based Virtual Screening (SBVS) with FLAP
SBVS with GLIDE
SBVS with AutoDock4 and AutoDock VINA
Ligand Redocking Using GLIDE
Molecular Redocking Using FLAP
2.2. Biological Characterization
3. Results and Discussion
3.1. Searching for TLR4/MD-2 Modulators: Virtual Screening
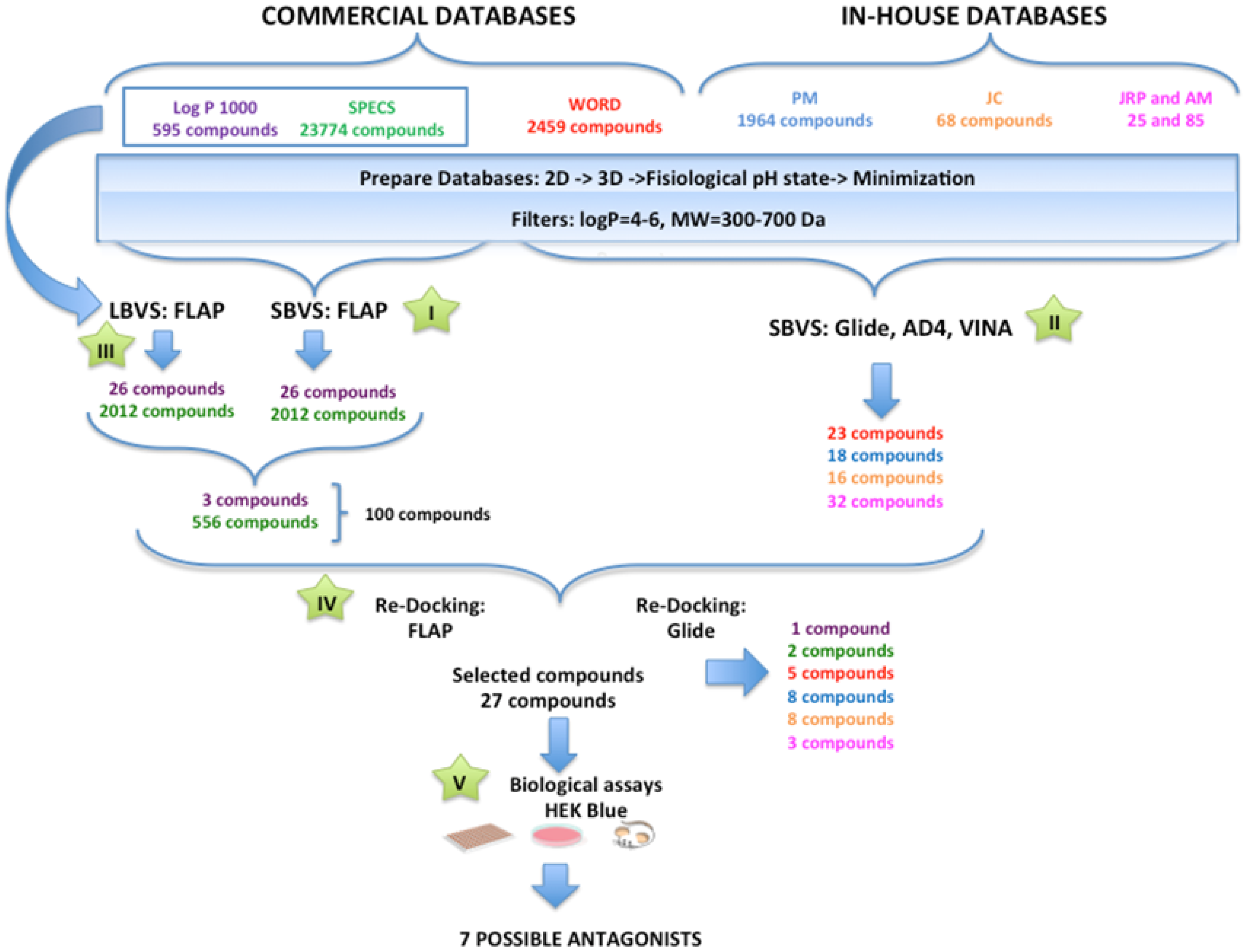
General Overview of the Virtual Screening Protocol
3.2. Performance of the Virtual Screening Study
3.2.1. Step I. Structure-Based Virtual Screening (SBVS) with FLAP
3.2.2. Step II. Structure-Based Virtual Screening (SBVS) with GLIDE, AutoDock, and VINA
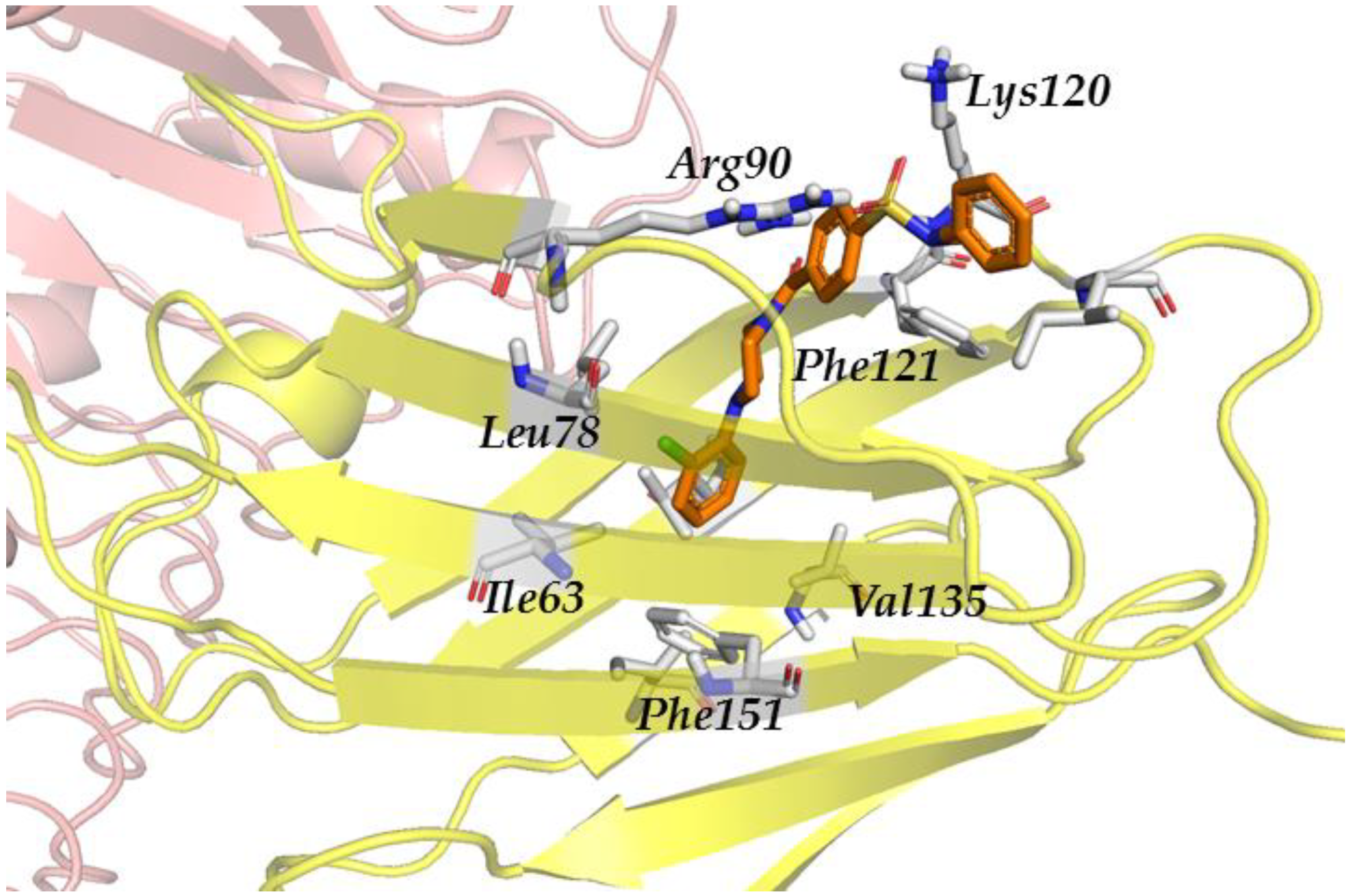
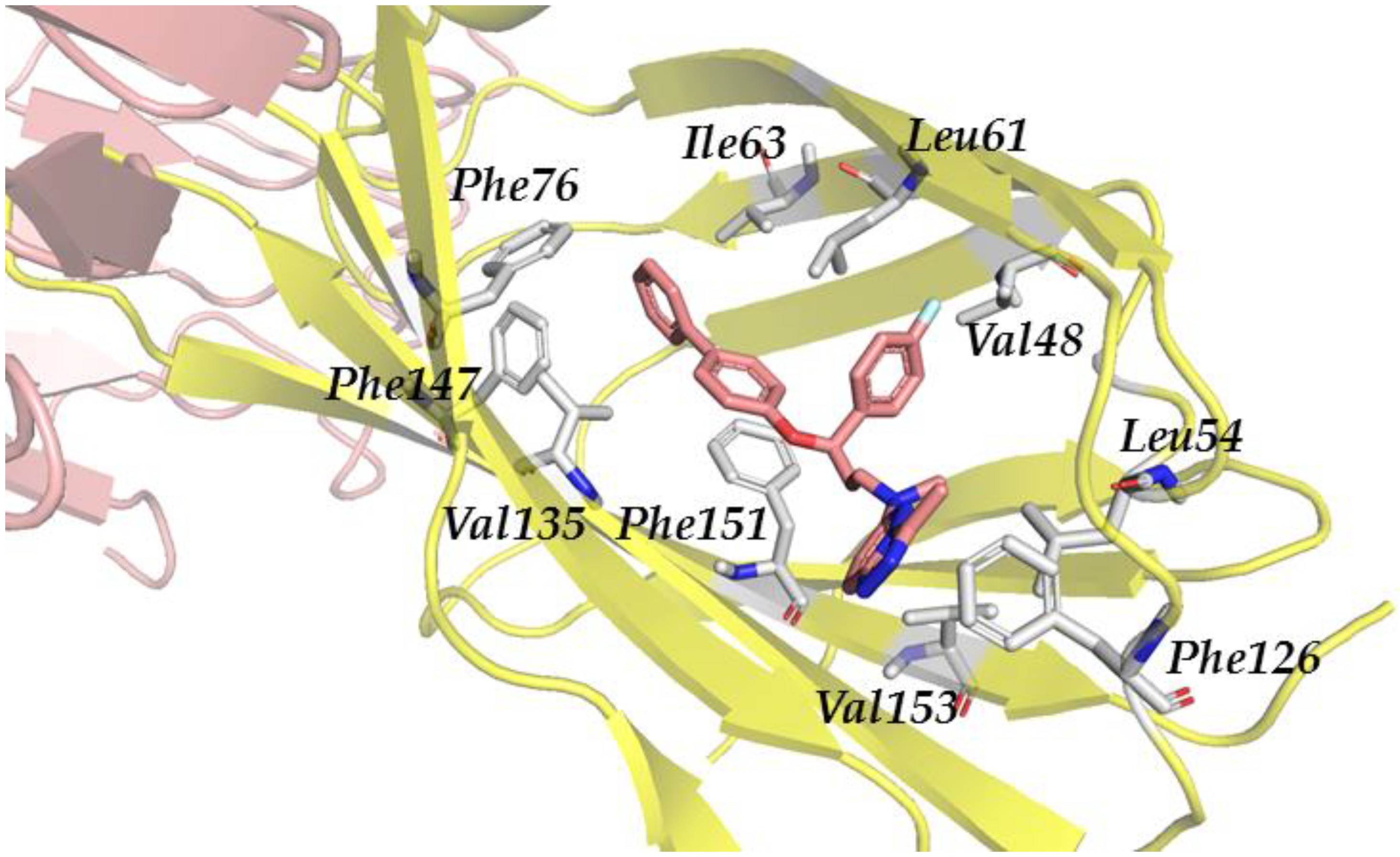
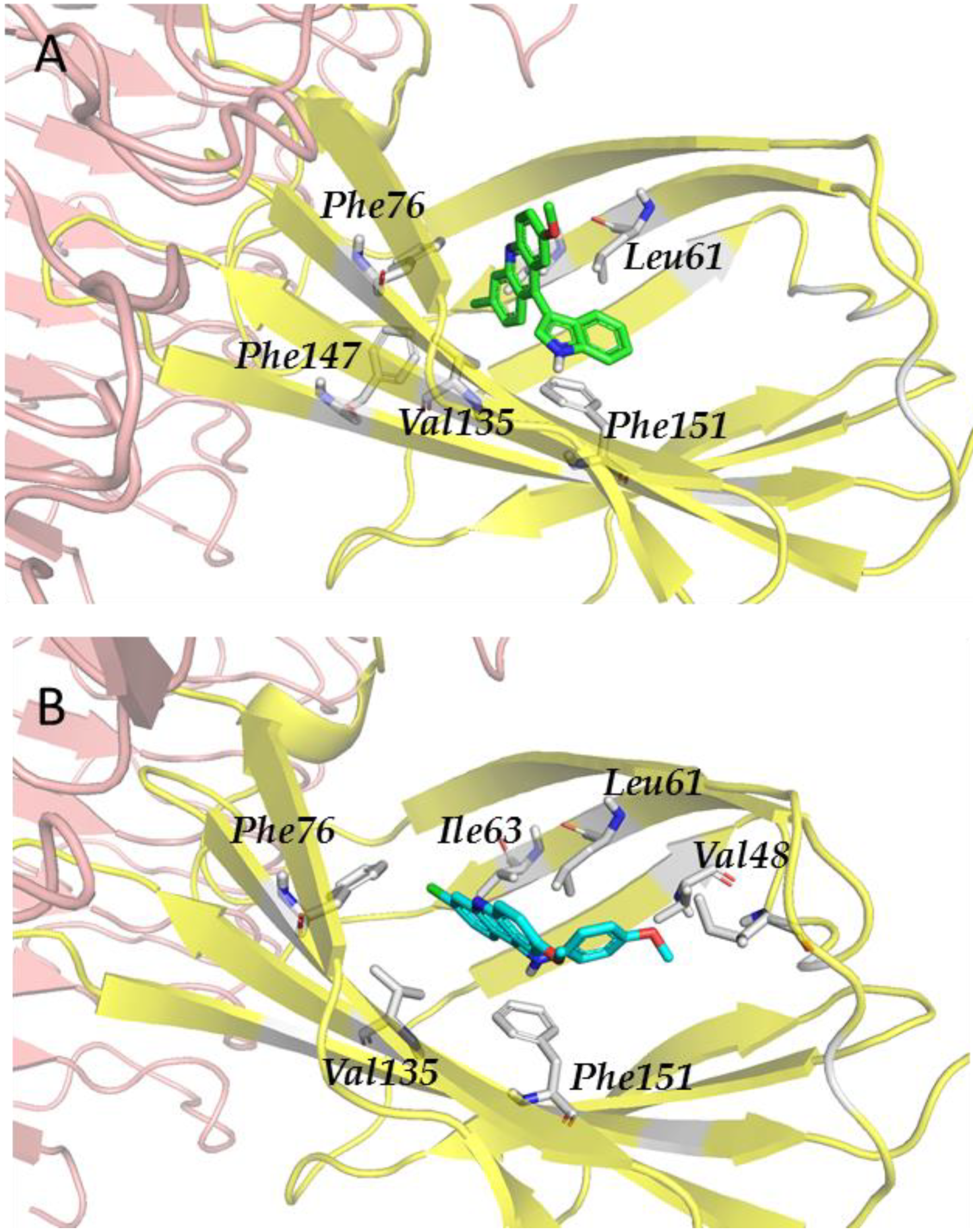
3.2.3. Step III. Ligand-Based Virtual Screening (LBVS) with FLAP
3.2.4. Step IV. Redocking with FLAP and GLIDE
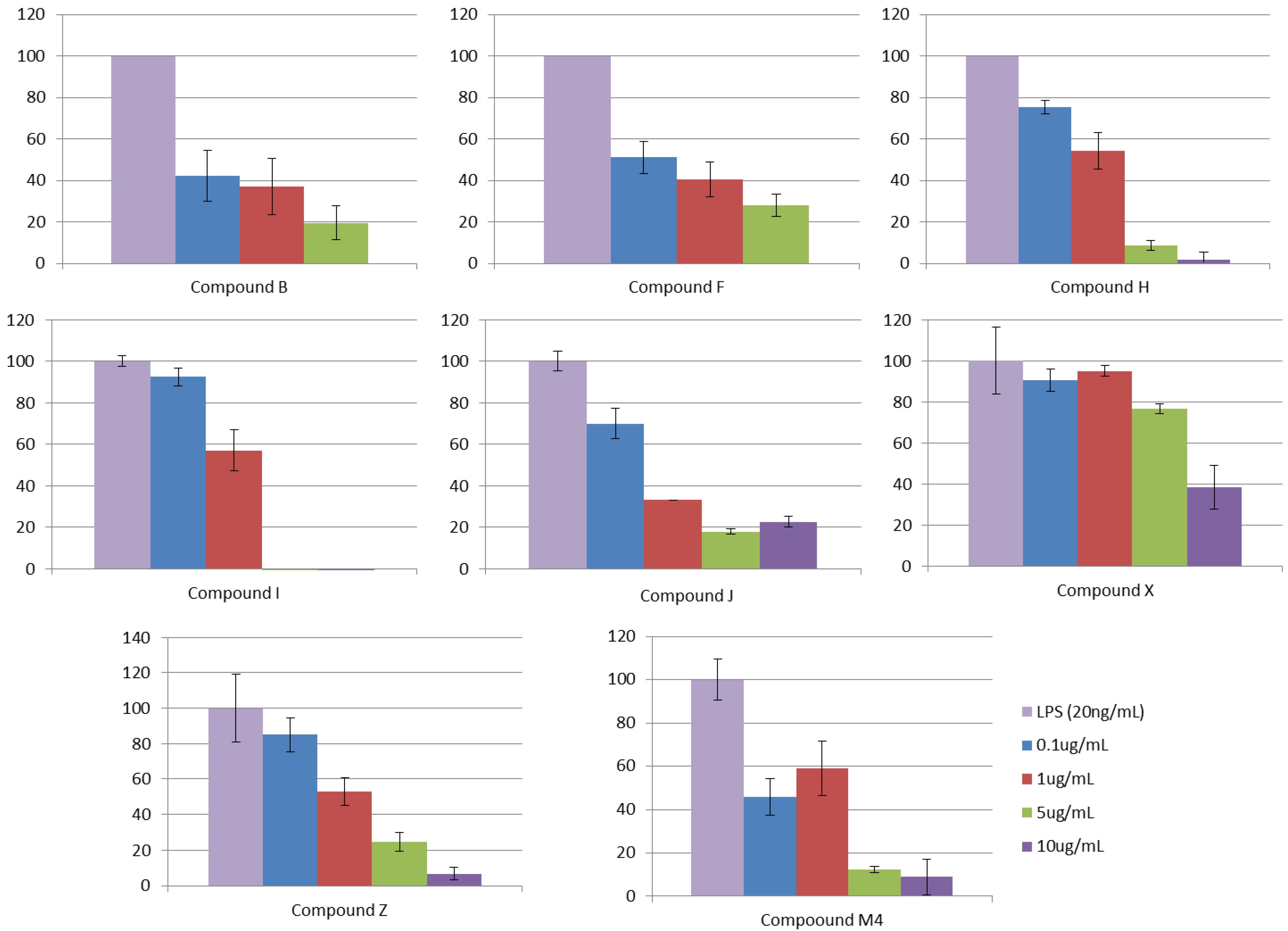
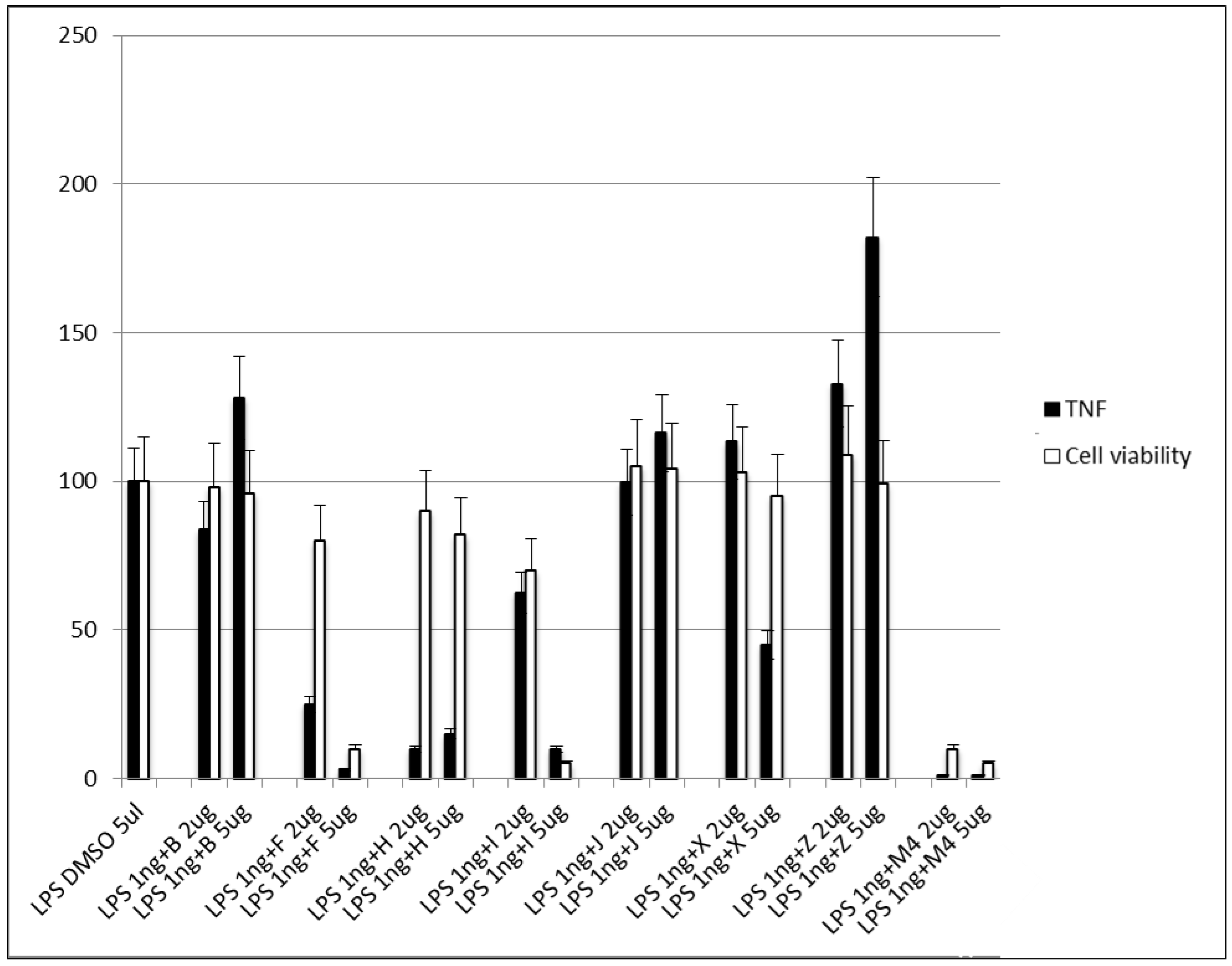
3.2.5. Step V. Biological Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mifsud, E.J.; Tan, A.C.; Jackson, D.C. TLR Agonists as Modulators of the Innate Immune Response and Their Potential as Agents Against Infectious Disease. Front. Immunol. 2014, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Lind, N.A.; Rael, V.E.; Pestal, K.; Liu, B.; Barton, G.M. Regulation of the nucleic acid-sensing Toll-like receptors. Nat. Rev. Immunol. 2022, 22, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Abdollahi-Roodsaz, S.; Dinarello, C.A.; O’Neill, L.; Netea, M.G. Toll-like receptors and chronic inflammation in rheumatic diseases: New developments. Nat. Rev. Rheumatol. 2016, 12, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Gooshe, M.; Aleyasin, A.R.; Abdolghaffari, A.H.; Rezaei, N. Toll like receptors: A new hope on the horizon to treat multiple sclerosis. Expert Rev. Clin. Immunol. 2014, 10, 1277–1279. [Google Scholar] [CrossRef]
- Nelson, M.H.; Diven, M.A.; Huff, L.W.; Paulos, C.M. Harnessing the Microbiome to Enhance Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 368736. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar] [CrossRef]
- Kaur, A.; Baldwin, J.; Brar, D.; Salunke, D.B.; Petrovsky, N. Toll-like receptor (TLR) agonists as a driving force behind next-generation vaccine adjuvants and cancer therapeutics. Curr. Opin. Chem. Biol. 2022, 70, 102172. [Google Scholar] [CrossRef]
- Gambuzza, M.E.; Sofo, V.; Salmeri, F.M.; Soraci, L.; Marino, S.; Bramanti, P. Toll-like receptors in Alzheimer’s disease: A therapeutic perspective. CNS Neurol. Disor. Drug Targets 2014, 13, 1542–1558. [Google Scholar] [CrossRef]
- Garcísa, M.M.; Goicoechea, C.; Molina-Álvarez, M.; Pascual, D. Toll-like receptor 4: A promising crossroads in the diagnosis and treatment of several pathologies. Eur. J. Pharmacol. 2020, 874, 172975. [Google Scholar] [CrossRef]
- Ain, Q.U.; Batool, M.; Choi, S. TLR4-Targeting Therapeutics: Structural Basis and Computer-Aided Drug Discovery Approaches. Molecules 2020, 25, 627. [Google Scholar] [CrossRef]
- Smith, M.; García-Martínez, E.; Pitter, M.R.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 2018, 7, e1526250. [Google Scholar] [CrossRef] [PubMed]
- Marzabadi, C.H.; Franck, R.W. Small-Molecule Carbohydrate-Based Immunostimulants. Chem. Eur. J. 2017, 23, 1728–1742. [Google Scholar] [CrossRef] [PubMed]
- Alderson, M.R.; McGowan, P.; Baldridge, J.R.; Probst, P. TLR4 agonists as immunomodulatory agents. J. Endotoxin Res. 2006, 12, 313–319. [Google Scholar] [CrossRef]
- Opal, S.M.; Laterre, P.F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The ACCESS randomized trial. Jama 2013, 309, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Zaffaroni, L.; Peri, F. Recent advances on Toll-like receptor 4 modulation: New therapeutic perspectives. Future Med. Chem. 2018, 10, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.; Hayashi, T.; Mathewson, R.D.; Nour, A.; Hayashi, Y.; Yao, S.; Tawatao, R.I.; Crain, B.; Tsigelny, I.F.; Kouznetsova, V.L.; et al. Identification of substituted pyrimido[5,4-b]indoles as selective Toll-like receptor 4 ligands. J. Med. Chem. 2013, 56, 4206–4223. [Google Scholar] [CrossRef] [PubMed]
- Neve, J.E.; Wijesekera, H.P.; Duffy, S.; Jenkins, I.D.; Ripper, J.A.; Teague, S.J.; Campitelli, M.; Garavelas, A.; Nikolakopoulos, G.; Le, P.V.; et al. Euodenine A: A small-molecule agonist of human TLR4. J. Med. Chem. 2014, 57, 1252–1275. [Google Scholar] [CrossRef]
- Shanmugam, A.; Rajoria, S.; George, A.L.; Mittelman, A.; Suriano, R.; Tiwari, R.K. Synthetic Toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PLoS ONE 2012, 7, e30839. [Google Scholar] [CrossRef]
- Park, S.J.; Kang, S.H.; Kang, Y.K.; Eom, Y.B.; Koh, K.O.; Kim, D.Y.; Youn, H.S. Inhibition of homodimerization of toll-like receptor 4 by 4-oxo-4-(2-oxo-oxazolidin-3-yl)-but-2-enoic acid ethyl ester. Int. Immunopharmacol. 2011, 11, 19–22. [Google Scholar] [CrossRef]
- Jin, G.H.; Li, H.; An, S.; Ryu, J.H.; Jeon, R. Design, synthesis and activity of benzothiazole-based inhibitors of NO production in LPS-activated macrophages. Bioorg. Med. Chem. Lett. 2010, 20, 6199–6202. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, T.; Ii, M.; Kitazaki, T.; Iizawa, Y.; Kimura, H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur. J. Pharmacol. 2008, 584, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Chavez, S.A.; Martinko, A.J.; Lau, C.; Pham, M.N.; Cheng, K.; Bevan, D.E.; Mollnes, T.E.; Yin, H. Development of beta-amino alcohol derivatives that inhibit Toll-like receptor 4 mediated inflammatory response as potential antiseptics. J. Med. Chem. 2011, 54, 4659–4669. [Google Scholar] [CrossRef] [PubMed]
- Gratal, P.; Mediero, A.; Lamuedra, A.; Matamoros-Recio, A.; Herencia, C.; Herrero-Beaumont, G.; Martín-Santamaría, S.; Largo, R. 6-shogaol treatment improves experimental knee OA exerting a pleiotropic effect over immune innate signaling response in chondrocytes. Br. J. Pharmacol. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- da Silva Rocha, S.F.L.; Olanda, C.G.; Fokoue, H.H.; Sant’Anna, C.M.R. Virtual Screening Techniques in Drug Discovery: Review and Recent Applications. Curr. Top. Med. Chem. 2019, 19, 1751–1767. [Google Scholar] [CrossRef]
- Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef]
- Schneider, G. Virtual screening: An endless staircase? Nat. Rev. Drug Discov. 2010, 9, 273–276. [Google Scholar] [CrossRef]
- Haga, J.H.; Ichikawa, K.; Date, S. Virtual Screening Techniques and Current Computational Infrastructures. Curr. Pharm. Des. 2016, 22, 3576–3584. [Google Scholar] [CrossRef]
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Yan, X.; Liao, C.; Liu, Z.; Hagler, A.T.; Gu, Q.; Xu, J. Chemical Structure Similarity Search for Ligand-based Virtual Screening: Methods and Computational Resources. Curr. Drug Targets 2016, 17, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Regidor, L.; Zarioh, M.; Ortega, L.; Martín-Santamaría, S. Virtual Screening Approaches towards the Discovery of Toll-Like Receptor Modulators. Int. J. Mol. Sci. 2016, 17, 1508. [Google Scholar] [CrossRef] [PubMed]
- Billod, J.M.; Lacetera, A.; Guzmán-Caldentey, J.; Martín-Santamaría, S. Computational Approaches to Toll-Like Receptor 4 Modulation. Molecules 2016, 21, 994. [Google Scholar] [CrossRef]
- Chong, C.R.; Sullivan, D.J., Jr. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2021. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2022, 27, 1075. [Google Scholar] [CrossRef]
- Shibayama, S.; Tanikawa, K.; Fujimoto, R.; Kimura, H. Effect of mergers and acquisitions on drug discovery: Perspective from a case study of a Japanese pharmaceutical company. Drug Discov. Today 2008, 13, 86–93. [Google Scholar] [CrossRef]
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.H. Drug repositioning and repurposing: Terminology and definitions in literature. Drug Discov. Today 2015, 20, 1027–1034. [Google Scholar] [CrossRef]
- Allarakhia, M. Open-source approaches for the repurposing of existing or failed candidate drugs: Learning from and applying the lessons across diseases. Drug Des. Dev. Ther. 2013, 7, 753–766. [Google Scholar] [CrossRef]
- Allison, M. NCATS launches drug repurposing program. Nat. Biotechnol. 2012, 30, 571–572. [Google Scholar] [CrossRef]
- Marusina, K.; Welsch, D.J.; Rose, L.; Brock, D.; Bahr, N. The CTSA Pharmaceutical Assets Portal–a public–private partnership model for drug repositioning. Drug Discov. Today 2012, 8, 77–83. [Google Scholar] [CrossRef][Green Version]
- Murteira, S.; Ghezaiel, Z.; Karray, S.; Lamure, M. Drug reformulations and repositioning in pharmaceutical industry and its impact on market access: Reassessment of nomenclature. J. Mark Access Health Policy 2013, 1, 21131. [Google Scholar] [CrossRef] [PubMed]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Fagan, S.C. Drug Repurposing for Drug Development in Stroke. Pharmacotherapy 2010, 30, 51S–54S. [Google Scholar] [CrossRef]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor Activity of Thalidomide in Refractory Multiple Myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef]
- Yang, T.-J.; Yang, T.-S.; Liang, H.-M. Thalidomide and congenital abnormalities. Lancet 1963, 281, 552–553. [Google Scholar] [CrossRef]
- Klett, J.; Reeves, J.; Oberhauser, N.; Perez-Regidor, L.; Martin-Santamaria, S. Modulation of toll-like receptor 4. Insights from x-ray crystallography and molecular modeling. Curr. Top. Med. Chem. 2014, 14, 2672–2683. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Ohto, U.; Fukase, K.; Miyake, K.; Satow, Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 2007, 316, 1632–1634. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: New York, NY, USA, 2015.
- Martel, S.; Gillerat, F.; Carosati, E.; Maiarelli, D.; Tetko, I.V.; Mannhold, R.; Carrupt, P.A. Large, chemically diverse dataset of logP measurements for benchmarking studies. Eur. J. Pharm. Sci. 2013, 48, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.specs.net/ (accessed on 6 September 2022).
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Staderini, M.; Cabezas, N.; Bolognesi, M.L.; Menendez, J.C.; Carlos, J. Solvent- and Chromatography-Free Amination of π-Deficient nitrogen Heterocycles under Microwave Irradiation. A Fast, Efficient and Green Route to 9-Aminoacridines, 4-Aminoquinolines and 4-Aminoquinazolines and Its Application to the Synthesis of the Drugs Amsacrine and Bistacrine; Elsevier: Kidlington, UK, 2013; Volume 69. [Google Scholar]
- Barroso, S.; Blay, G.; Munoz, M.C.; Pedro, J.R. Highly enantioselective nitrone cycloadditions with 2-alkenoyl pyridine N-oxides catalyzed by Cu(II)-BOX complexes. Org. Lett. 2011, 13, 402–405. [Google Scholar] [CrossRef]
- Blay, G.; Fernández, I.; Muñoz, M.C.; Pedro, J.R.; Vila, C. Synthesis of functionalized indoles with a trifluoromethyl-substituted stereogenic tertiary carbon atom through an enantioselective Friedel-Crafts alkylation with beta-trifluoromethyl-alpha,beta-enones. Chem. Eur. J. 2010, 16, 9117–9122. [Google Scholar] [CrossRef]
- Vilanova, C.; Torijano-Gutierrez, S.; Diaz-Oltra, S.; Murga, J.; Falomir, E.; Carda, M.; Alberto Marco, J. Design and synthesis of pironetin analogue/combretastatin A-4 hybrids containing a 1,2,3-triazole ring and evaluation of their cytotoxic activity. Eur. J. Med. Chem. 2014, 87, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2015-4: Maestro, v.; Schrödinger, LLC: New York, NY, USA, 2015.
- Schrödinger Release 2015-4: LigPrep, v.; Schrödinger, LLC: New York, NY, USA, 2015.
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R. Integrated modeling program, applied chemical theory (IMPACT). J. Med. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2015-4: Epik, v. Schrödinger, LLC: New York, NY, USA, 2015.
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Ciaramelli, C.; Calabrese, V.; Sestito, S.E.; Pérez-Regidor, L.; Klett, J.; Oblak, A.; Jerala, R.; Piazza, M.; Martín-Santamaría, S.; Peri, F. Glycolipid-based TLR4 Modulators and Fluorescent Probes: Rational Design, Synthesis, and Biological Properties. Chem. Biol. Drug Des. 2016, 88, 217–229. [Google Scholar] [CrossRef]
- Small-Molecule Drug Discovery Suite 2015-4: GLIDE, v. Schrödinger, LLC: New York, NY, USA, 2015.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. GLIDE: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef]
- Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. GRID-based three-dimensional pharmacophores I: FLAPpharm, a novel approach for pharmacophore elucidation. J. Chem. Inf. Model. 2012, 52, 2587–2598. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- MOE 2011.10, Chemical Computing Group Inc.: Montreal, QC, Canada, 2011.
- Allinger, N.L.; Yuh, Y.H.; Lii, J.H. Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 1989, 111, 8551–8566. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Berdini, V.; Hartshorn, M.J.; Mooij, W.T.; Murray, C.W.; Taylor, R.D.; Watson, P. Virtual screening using protein-ligand docking: Avoiding artificial enrichment. J. Chem. Inf. Comput. Sci. 2004, 44, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking Sets for Molecular Docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Ve, T.; Vajjhala, P.R.; Hedger, A.; Croll, T.; Dimaio, F.; Horsefield, S.; Yu, X.; Lavrencic, P.; Hassan, Z.; Morgan, G.P.; et al. Structural basis of TIR-domain-assembly formation in MAL- and MyD88-dependent TLR4 signaling. Nat. Struct. Mol. Biol. 2017, 24, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Matamoros-Recio, A.; Franco-González, J.F.; Pérez-Regidor, L.; Billod, J.M.; Guzmán-Caldentey, J.; Martín-Santamaría, S. Full-Atom Model of the Agonist LPS-Bound Toll-like Receptor 4 Dimer in a Membrane Environment. Chem. Eur. J. 2021, 27, 15406–15425. [Google Scholar] [CrossRef]
- Oberhauser, N. Lipophilicity in Computer-Aided Drug Design: New Tools and Applications; University of Geneve (Switzerland): Geneve, Switzerland, 2015. [Google Scholar]
- Koo, J.E.; Park, Z.Y.; Kim, N.D.; Lee, J.Y. Sulforaphane inhibits the engagement of LPS with TLR4/MD2 complex by preferential binding to Cys133 in MD2. Biochem. Biophys. Res. Commun. 2013, 434, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Panneerselvam, S.; Shah, M.; Choi, S. Insights into the species-specific TLR4 signaling mechanism in response to Rhodobacter sphaeroides lipid A detection. Sci. Rep. 2015, 5, 7657. [Google Scholar] [CrossRef]
- Available online: https://go.drugbank.com/drugs/DB01081 (accessed on 6 September 2022).
- Lewis, S.S.; Hutchinson, M.R.; Rezvani, N.; Loram, L.C.; Zhang, Y.; Maier, S.F.; Rice, K.C.; Watkins, L.R. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1β. Neuroscience 2010, 165, 569–583. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Zhang, Y.; Shridhar, M.; Evans, J.H.; Buchanan, M.M.; Zhao, T.X.; Slivka, P.F.; Coats, B.D.; Rezvani, N.; Wieseler, J.; et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav. Immun. 2010, 24, 83–95. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Northcutt, A.L.; Hiranita, T.; Wang, X.; Lewis, S.; Thomas, J.; van Steeg, K.; Kopajtic, T.A.; Loram, L.; Sfregola, C.; et al. Opioid activation of Toll-Like receptor 4 contributes to drug reinforcement. J. Neurosci. 2012, 32, 11187–11200. [Google Scholar] [CrossRef]
- Ishinaga, H.; Takeuchi, K.; Kishioka, C.; Suzuki, S.; Basbaum, C.; Majima, Y. Pranlukast inhibits NF-kappaB activation and MUC2 gene expression in cultured human epithelial cells. Pharmacology 2005, 73, 89–96. [Google Scholar] [CrossRef]
- Woszczek, G.; Chen, L.-Y.; Alsaaty, S.; Nagineni, S.; Shelhamer, J.H. Concentration dependent non-CysLT(1) receptor mediated inhibitory activity of leukotriene receptor antagonists. J. Immunol. 2010, 184, 2219–2225. [Google Scholar] [CrossRef]
- Thivierge, M.; Stankova, J.; Rola-Pleszczynski, M. Toll-like receptor agonists differentially regulate cysteinyl-leukotriene receptor 1 expression and function in human dendritic cells. J. Allergy Clin. Immunol. 2006, 117, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA. 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Goto, Y.; Arigami, T.; Kitago, M.; Nguyen, S.L.; Narita, N.; Ferrone, S.; Morton, D.L.; Irie, R.F.; Hoon, D.S. Activation of Toll-like receptors 2, 3, and 4 on human melanoma cells induces inflammatory factors. Mol. Cancer. Ther. 2008, 7, 3642–3653. [Google Scholar] [CrossRef]
- Rogava, M.; Braun, A.D.; van der Sluis, T.C.; Shridhar, N.; Tüting, T.; Gaffal, E. Tumor cell intrinsic Toll-like receptor 4 signaling promotes melanoma progression and metastatic dissemination. Int. J. Cancer 2022, 150, 142–151. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antagonist | Glob-Sum | Antagonist | Glob-Sum |
|---|---|---|---|
| Paclitaxel | 3.245 | 6-Shogaol | 2.498 |
| JSH | 2.714 | Isoxanthohumol | 2.465 |
| Curcumin | 2.695 | Isoquiritigenine | 2.239 |
| 1D10G | 2.669 | Cinnamaldehyde | 2.179 |
| CAPE | 2.621 | C34 | 2.136 |
| Xanthohumol | 2.611 | OSL7 | 1.799 |
| JTT705 | 2.513 | Sulforaphane | 1.707 |
| Template | Compound | Glob-Sum | H | N1 | DRY | O |
|---|---|---|---|---|---|---|
| 6-Shogaol | 152 | 1.326 | 0.663 | 0.508 | 0.224 | 0.239 |
| 481 | 1.742 | 0.598 | 0.271 | 0.254 | 0.702 | |
| Xanthohumol | 568 | 1.269 | 0.699 | 0.283 | 0.340 | 0.124 |
| 19,907 | 1.912 | 0.703 | 0.368 | 0.508 | 0.359 | |
| Paclitaxel | 383 | 0.847 | 0.505 | 0.175 | 0.134 | 0.337 |
| 20,513 | 1.022 | 0.565 | 0.171 | 0.105 | 0.321 | |
| 1D10G | 368 | 1.152 | 0.579 | 0.203 | 0.181 | 0.310 |
| 20,700 | 1.857 | 0.654 | 0.306 | 0.260 | 0.734 | |
| JSH | 492 | 1.165 | 0.598 | 0.359 | 0.229 | 0.144 |
| 21,315 | 1.421 | 0.515 | 0.371 | 0.304 | 0.329 | |
| Isoliquiritigenin | 42 | 1.181 | 0.637 | 0.364 | 0.195 | 0.010 |
| 120 | 1.706 | 0.750 | 0.431 | 0.343 | 0.294 | |
| Isoxanthohumol | 138 | 1.054 | 0.638 | 0.234 | 0.308 | 0.010 |
| 28 | 1.493 | 0.634 | 0.430 | 0.304 | 0.305 | |
| CAPE | 575 | 1.149 | 0.625 | 0.242 | 0.181 | 0.243 |
| 22,298 | 1.528 | 0.587 | 0.230 | 0.159 | 0.700 | |
| Curcumin | 548 | 1.041 | 0.631 | 0.242 | 0.204 | 0.010 |
| 23,010 | 1.562 | 0.519 | 0.264 | 0.173 | 0.623 | |
| Sulforaphane | 46 | 1.104 | 0.650 | 0.361 | 0.166 | 0.000 |
| 3203 | 1.184 | 0.684 | 0.296 | 0.000 | 0.000 | |
| Cinnamaldehyde | 40 | 1.489 | 0.684 | 0.581 | 0.383 | 0.000 |
| 23,599 | 1.500 | 0.580 | 0.673 | 0.273 | 0.000 | |
| OSL7 | 35 | 1.007 | 0.648 | 0.295 | 0.128 | 0.000 |
| 1171 | 1.285 | 0.702 | 0.445 | 0.191 | 0.000 | |
| C34 | 187 | 1.142 | 0.506 | 0.205 | 0.137 | 0.512 |
| 10,959 | 1.428 | 0.560 | 0.216 | 0.102 | 0.903 | |
| JTT705 | 439 | 1.033 | 0.539 | 0.391 | 0.188 | 0.010 |
| 14,650 | 1.127 | 0.592 | 0.347 | 0.188 | 0.000 |
| Compound | Structure | LogP a | LogP b | Drug-Likeness c | LogS d |
|---|---|---|---|---|---|
| ID-5382 (B) |  | 2.77 ± 0.49 | 4.13 | Yes; 0 violations | −5.49 |
| MS21 (F) |  | 5.89 ± 0.40 | 6.331 | Yes; 0 violations | −6.29 |
| MS32 (H) |  | 4.20 ± 0.83 | 6.434 | Yes; 0 violations | −5.96 |
| MS35 (I) |  | 4.68 ± 0.84 | 6.728 | Yes; 1 violation: MLOGP > 4.15 | −6.37 |
| MS45 (J) |  | 5.10 ± 0.45 | 5.712 | Yes; 1 violation: MLOGP > 4.15 | −5.54 |
| PM1090 (X) |  | 5.70 ± 0.89 | 6.063 | Yes; 1 violation: MLOGP > 4.15 | −6.48 |
| PM1200 (Z) |  | 6.29 ± 0.45 | 6.116 | Yes; 0 violations | −6.35 |
| Sorafenib (M4) |  | 6.13 ± 0.36 | 5.89 | Yes; 0 violations | −5.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Regidor, L.; Guzmán-Caldentey, J.; Oberhauser, N.; Punzón, C.; Balogh, B.; Pedro, J.R.; Falomir, E.; Nurisso, A.; Mátyus, P.; Menéndez, J.C.; et al. Small Molecules as Toll-like Receptor 4 Modulators Drug and In-House Computational Repurposing. Biomedicines 2022, 10, 2326. https://doi.org/10.3390/biomedicines10092326
Pérez-Regidor L, Guzmán-Caldentey J, Oberhauser N, Punzón C, Balogh B, Pedro JR, Falomir E, Nurisso A, Mátyus P, Menéndez JC, et al. Small Molecules as Toll-like Receptor 4 Modulators Drug and In-House Computational Repurposing. Biomedicines. 2022; 10(9):2326. https://doi.org/10.3390/biomedicines10092326
Chicago/Turabian StylePérez-Regidor, Lucía, Joan Guzmán-Caldentey, Nils Oberhauser, Carmen Punzón, Balázs Balogh, José R. Pedro, Eva Falomir, Alessandra Nurisso, Péter Mátyus, J. Carlos Menéndez, and et al. 2022. "Small Molecules as Toll-like Receptor 4 Modulators Drug and In-House Computational Repurposing" Biomedicines 10, no. 9: 2326. https://doi.org/10.3390/biomedicines10092326
APA StylePérez-Regidor, L., Guzmán-Caldentey, J., Oberhauser, N., Punzón, C., Balogh, B., Pedro, J. R., Falomir, E., Nurisso, A., Mátyus, P., Menéndez, J. C., de Andrés, B., Fresno, M., & Martín-Santamaría, S. (2022). Small Molecules as Toll-like Receptor 4 Modulators Drug and In-House Computational Repurposing. Biomedicines, 10(9), 2326. https://doi.org/10.3390/biomedicines10092326