

Myosins and MyomiR Network in Patients with Obstructive Hypertrophic Cardiomyopathy
 , , , , ,
, , , , ,  , ,
, ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.1.1. Inclusion Criteria
2.1.2. Exclusion Criteria
2.2. Control Hearts
2.3. Sample Processing
2.4. Genetic Testing
2.5. Histology and Morphometry
2.6. ELISA
2.7. MS Analysis of Myosin Proteins
2.8. Laser-Capture Microdissection and RNA Extraction
2.9. Generation of Human Induced Pluripotent Stem Cells, Differentiation to Cardiomyocytes and Transfection
2.10. Extracellular Vesicles Isolation
2.11. RNA Extraction: Gene and miRNA Expression Analysis
2.12. RNA Sequencing
2.13. In Silico Analysis
2.14. Statistical Analysis
3. Results
3.1. Baseline Characteristics of Study Patients
3.2. Histology
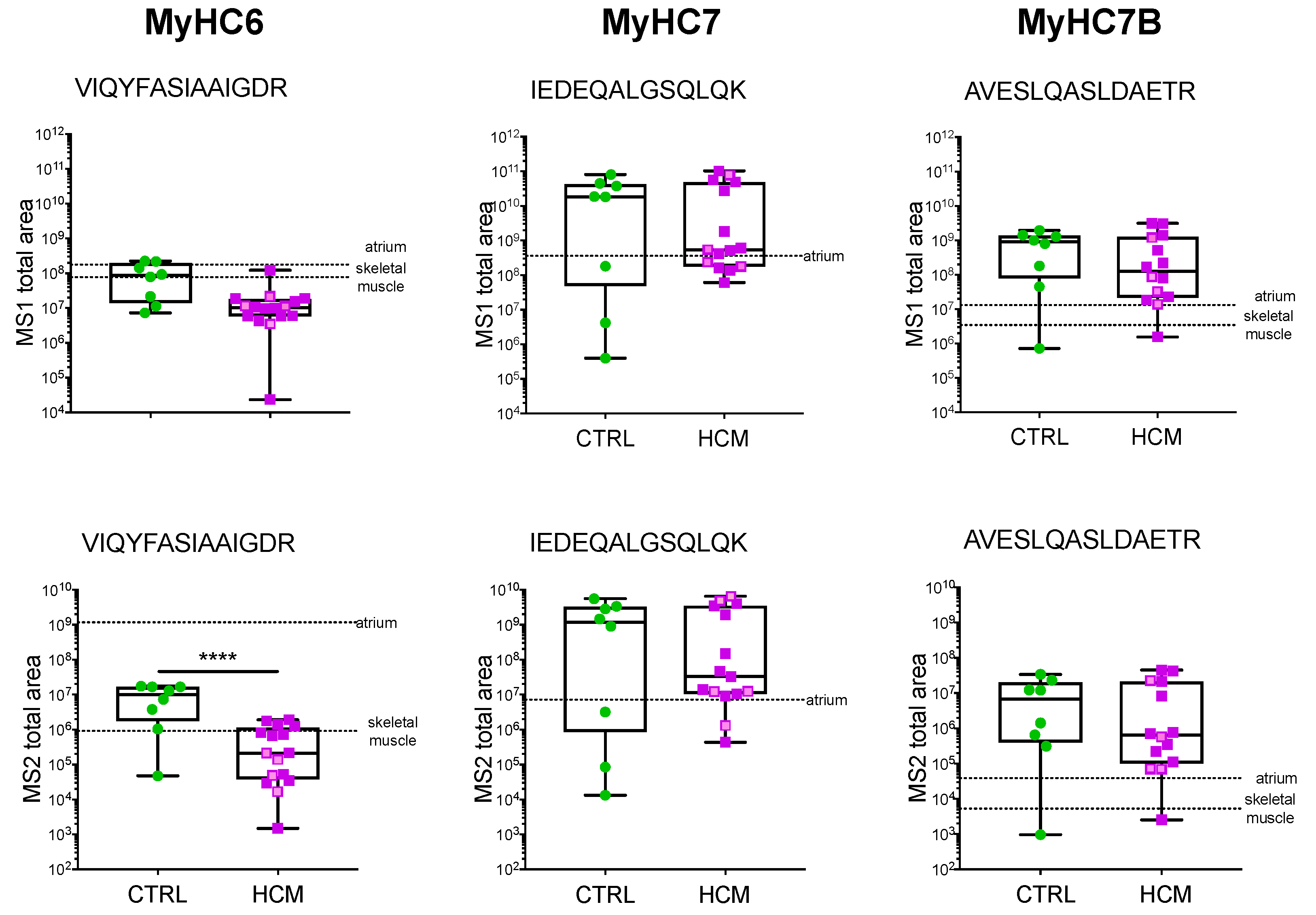
3.3. Myosin Isoforms in HCM Myocardium: Protein Analysis
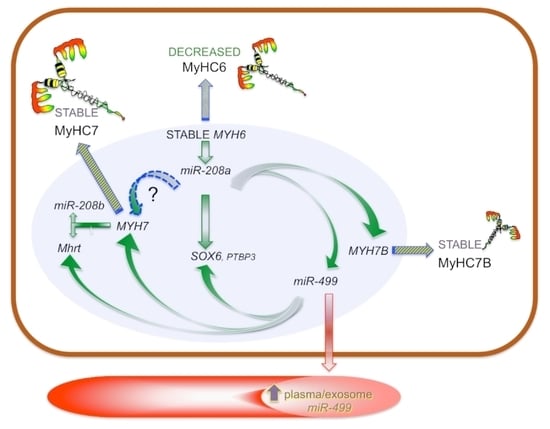
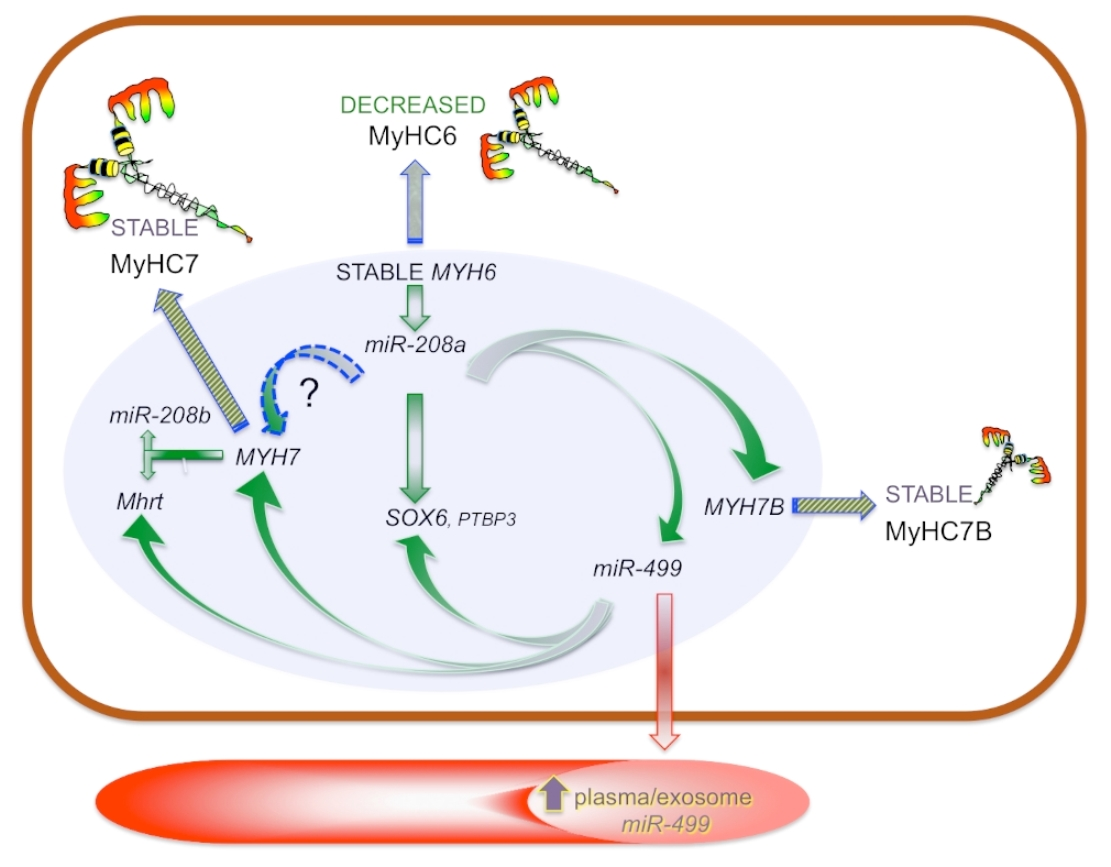
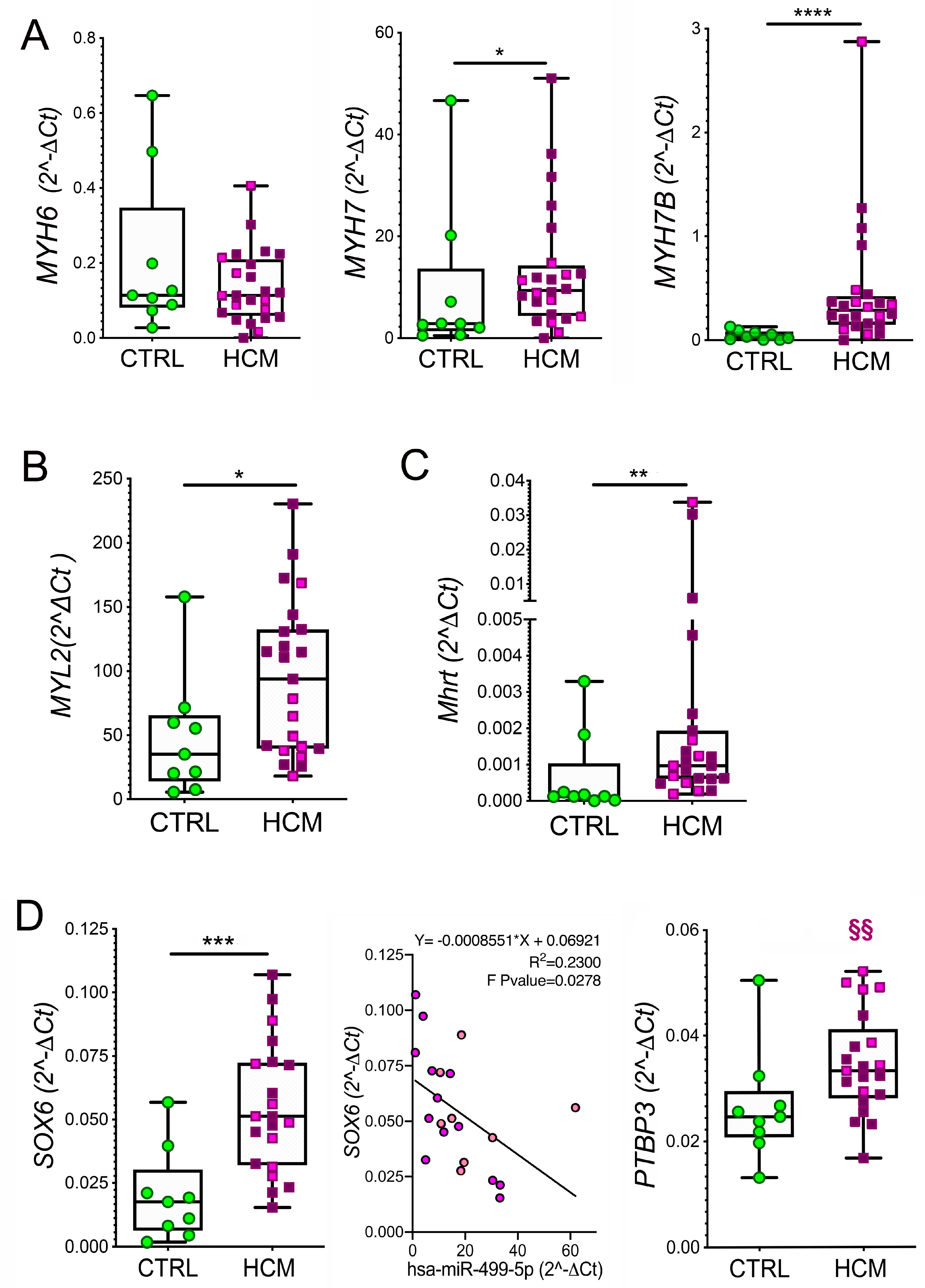
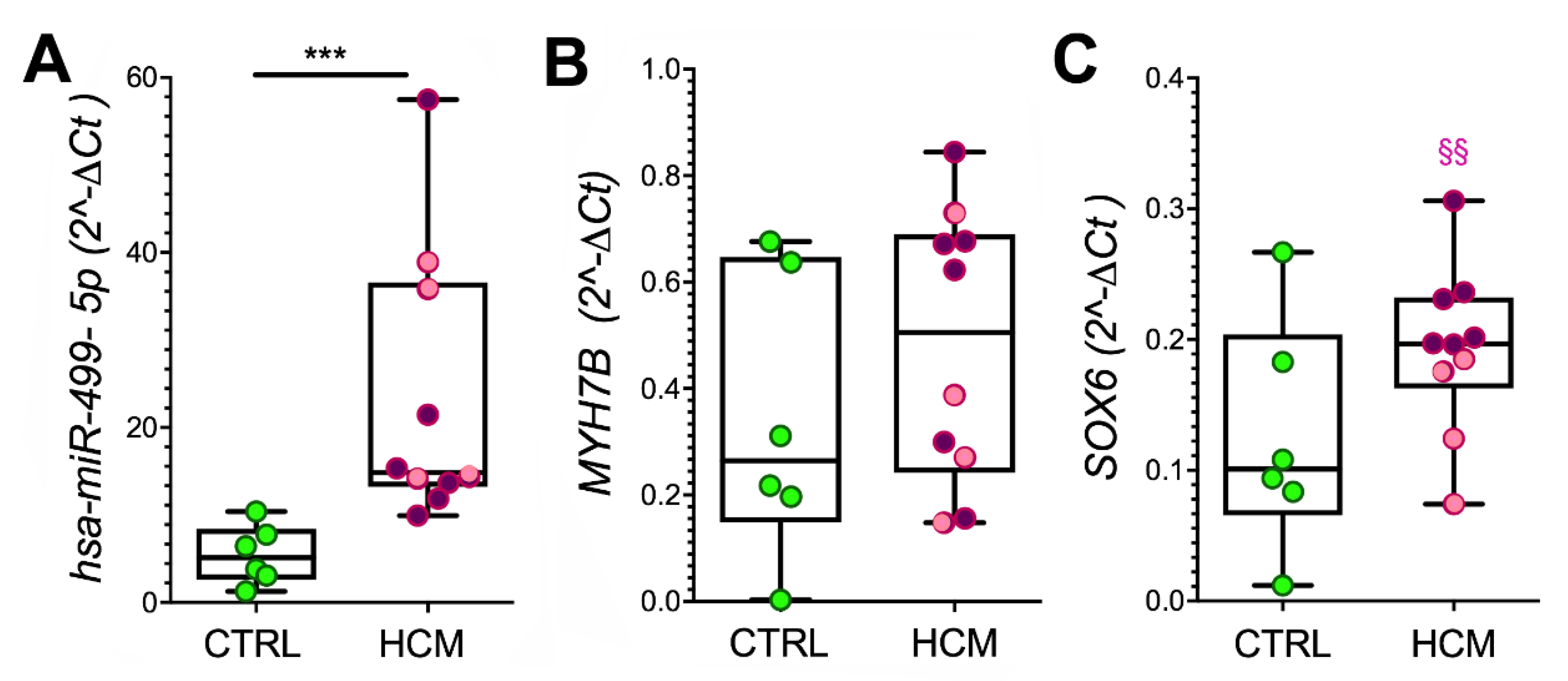
3.4. Myosin Isoforms in HCM and MyomiR Network in the Myocardium: Gene Expression Analysis
3.5. Expression of miR-499 Targets in the Myocardium: SOX6 and PTBP3
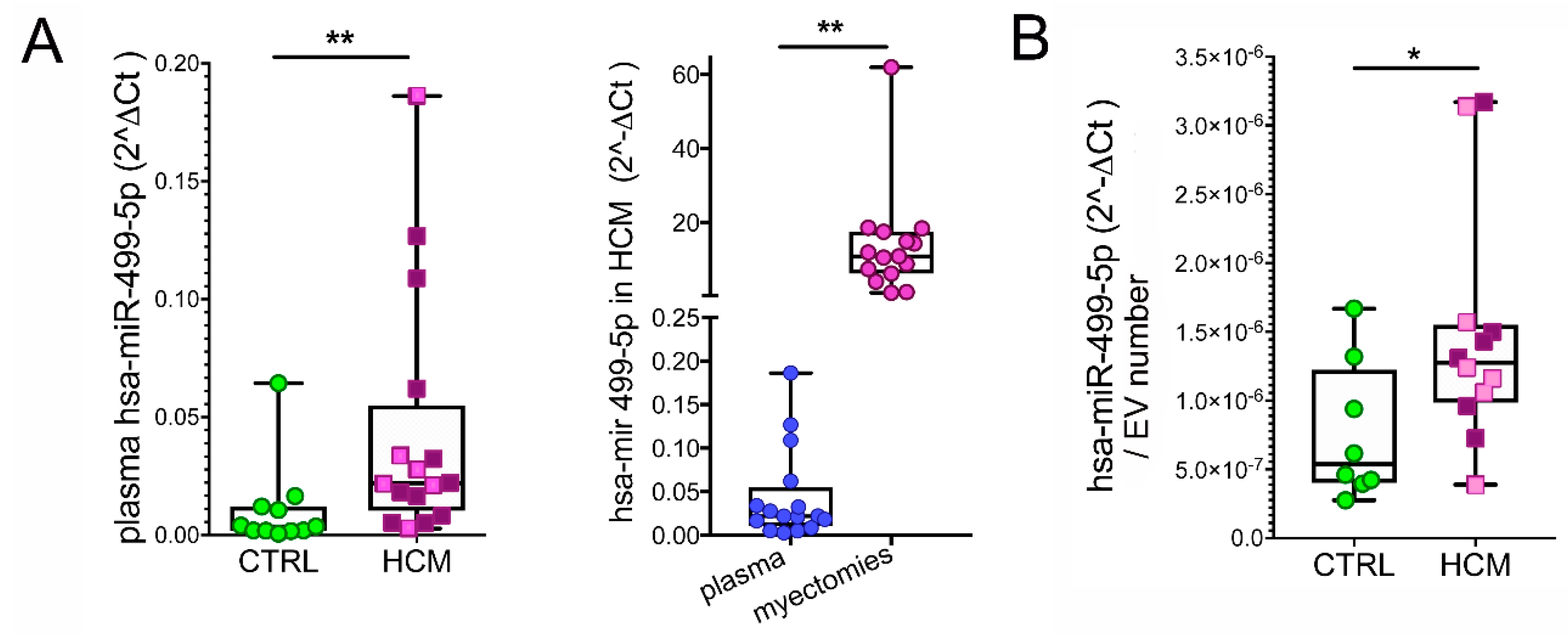
3.6. Myosins and MyomiR Network in Cardiomyocytes from Myectomies
3.7. MyomiRs in Plasma Samples
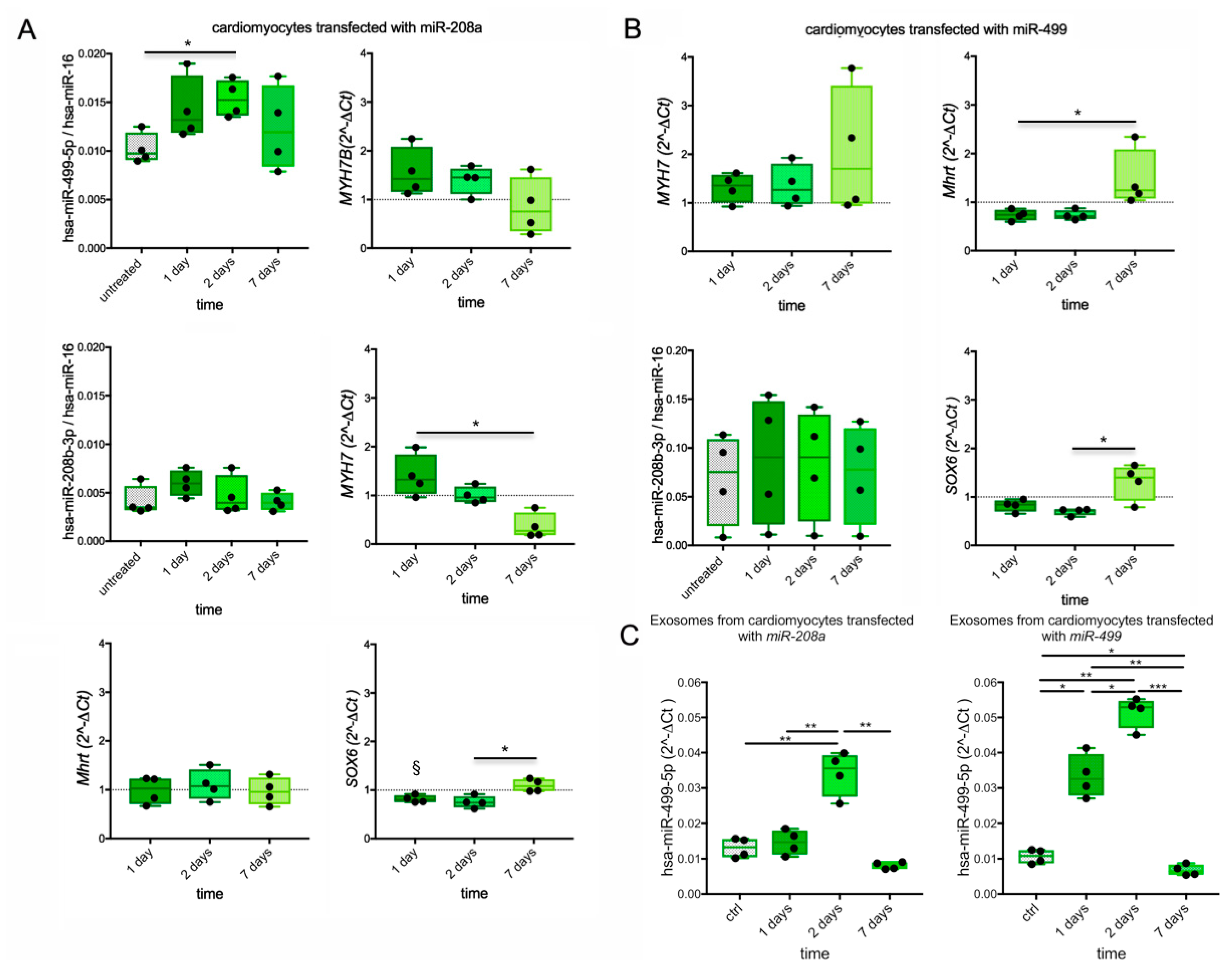
3.8. Myosin and MyomiR Network in Cardiomyocytes from Human iPSC
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olivotto, I.; Cecchi, F.; Poggesi, C.; Yacoub, M.H. Patterns of disease progression in hypertrophic cardiomyopathy: An individualized approach to clinical staging. Circ. Heart Fail. 2012, 5, 535–546. [Google Scholar] [CrossRef]
- Marian, A.J. Molecular Genetic Basis of Hypertrophic Cardiomyopathy. Circ. Res. 2021, 128, 1533–1553. [Google Scholar] [CrossRef]
- Lowes, B.D.; Minobe, W.; Abraham, W.T.; Rizeq, M.N.; Bohlmeyer, T.J.; Quaife, R.A.; Roden, R.L.; Dutcher, D.L.; Robertson, A.D.; Voelkel, N.F.; et al. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J. Clin. Investig. 1997, 100, 2315–2324. [Google Scholar] [CrossRef]
- Morano, I.; Hadicke, K.; Grom, S.; Koch, A.; Schwinger, R.H.; Bohm, M.; Bartel, S.; Erdmann, E.; Krause, E.G. Titin, myosin light chains and C-protein in the developing and failing human heart. J. Mol. Cell Cardiol. 1994, 26, 361–368. [Google Scholar] [CrossRef]
- Miyata, S.; Minobe, W.; Bristow, M.R.; Leinwand, L.A. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ. Res. 2000, 86, 386–390. [Google Scholar] [CrossRef]
- Nakao, K.; Minobe, W.; Roden, R.; Bristow, M.R.; Leinwand, L.A. Myosin heavy chain gene expression in human heart failure. J. Clin. Investig. 1997, 100, 2362–2370. [Google Scholar] [CrossRef]
- Peter, A.K.; Rossi, A.C.; Buvoli, M.; Ozeroff, C.D.; Crocini, C.; Perry, A.R.; Buvoli, A.E.; Lee, L.A.; Leinwand, L.A. Expression of Normally Repressed Myosin Heavy Chain 7b in the Mammalian Heart Induces Dilated Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e013318. [Google Scholar] [CrossRef]
- Reiser, P.J. Current understanding of conventional and novel co-expression patterns of mammalian sarcomeric myosin heavy chains and light chains. Arch. Biochem. Biophys. 2019, 662, 129–133. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef]
- Han, P.; Li, W.; Lin, C.H.; Yang, J.; Shang, C.; Nuernberg, S.T.; Jin, K.K.; Xu, W.; Lin, C.Y.; Lin, C.J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106. [Google Scholar] [CrossRef]
- Broadwell, L.J.; Smallegan, M.J.; Rigby, K.M.; Navarro-Arriola, J.S.; Montgomery, R.L.; Rinn, J.L.; Leinwand, L.A. Myosin 7b is a regulatory long noncoding RNA (lncMYH7b) in the human heart. J. Biol. Chem. 2021, 296. [Google Scholar] [CrossRef]
- van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J., Jr.; Olson, E.N. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 2009, 17, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.T.; Huang, Y.; Gilmore, J.; Srivastava, D. Elevated miR-499 levels blunt the cardiac stress response. PLoS ONE 2011, 6, e19481. [Google Scholar] [CrossRef]
- Sluijter, J.P.; van Mil, A.; van Vliet, P.; Metz, C.H.; Liu, J.; Doevendans, P.A.; Goumans, M.J. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arter. Thromb. Vasc. Biol. 2010, 30, 859–868. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef]
- Saleem, M.; Rahman, S.; Elijovich, F.; Laffer, C.L.; Ertuglu, L.A.; Masenga, S.K.; Kirabo, A. Sox6, A Potential Target for MicroRNAs in Cardiometabolic Disease. Curr. Hypertens. Rep. 2022, 24, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Nachtigall, P.G.; Dias, M.C.; Carvalho, R.F.; Martins, C.; Pinhal, D. MicroRNA-499 expression distinctively correlates to target genes sox6 and rod1 profiles to resolve the skeletal muscle phenotype in Nile tilapia. PLoS ONE 2015, 10, e0119804. [Google Scholar] [CrossRef]
- de Oliveira Silva, T.; Lino, C.A.; Miranda, J.B.; Balbino-Silva, C.S.; Lunardon, G.; Lima, V.M.; Jensen, L.; Donato, J., Jr.; Irigoyen, M.C.; Barreto-Chaves, M.L.M.; et al. The miRNA-143–3p-Sox6-Myh7 pathway is altered in obesogenic diet-induced cardiac hypertrophy. Exp. Physiol. 2022, 107, 892–905. [Google Scholar] [CrossRef]
- Bell, M.L.; Buvoli, M.; Leinwand, L.A. Uncoupling of expression of an intronic microRNA and its myosin host gene by exon skipping. Mol. Cell Biol. 2010, 30, 1937–1945. [Google Scholar] [CrossRef]
- McCarthy, J.J.; Esser, K.A.; Peterson, C.A.; Dupont-Versteegden, E.E. Evidence of MyomiR network regulation of beta-myosin heavy chain gene expression during skeletal muscle atrophy. Physiol. Genom. 2009, 39, 219–226. [Google Scholar] [CrossRef]
- Satoh, M.; Minami, Y.; Takahashi, Y.; Tabuchi, T.; Nakamura, M. Expression of microRNA-208 is Associated with Adverse Clinical Outcomes in Human Dilated Cardiomyopathy. J. Card. Fail. 2010, 16, 404–410. [Google Scholar] [CrossRef]
- Prodanovic, M.; Geeves, M.A.; Poggesi, C.; Regnier, M.; Mijailovich, S.M. Effect of Myosin Isoforms on Cardiac Muscle Twitch of Mice, Rats and Humans. Int. J. Mol. Sci. 2022, 23, 1135. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Whiffin, N.; Walsh, R.; Govind, R.; Edwards, M.; Ahmad, M.; Zhang, X.; Tayal, U.; Buchan, R.; Midwinter, W.; Wilk, A.E.; et al. CardioClassifier: Disease- and gene-specific computational decision support for clinical genome interpretation. Genet. Med. 2018, 20, 1246–1254. [Google Scholar] [CrossRef]
- Agbulut, O.; Li, Z.; Mouly, V.; Butler-Browne, G.S. Analysis of skeletal and cardiac muscle from desmin knock-out and normal mice by high resolution separation of myosin heavy-chain isoforms. Biol. Cell 1996, 88, 131–135. [Google Scholar] [CrossRef]
- Arnostova, P.; Jedelsky, P.L.; Soukup, T.; Zurmanova, J. Electrophoretic mobility of cardiac myosin heavy chain isoforms revisited: Application of MALDI TOF/TOF analysis. J. Biomed. Biotechnol. 2011, 2011, 634253. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef]
- Schwenk, J.M.; Omenn, G.S.; Sun, Z.; Campbell, D.S.; Baker, M.S.; Overall, C.M.; Aebersold, R.; Moritz, R.L.; Deutsch, E.W. The Human Plasma Proteome Draft of 2017: Building on the Human Plasma PeptideAtlas from Mass Spectrometry and Complementary Assays. J. Proteome Res. 2017, 16, 4299–4310. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Stuart, C.A.; Stone, W.L.; Howell, M.E.; Brannon, M.F.; Hall, H.K.; Gibson, A.L.; Stone, M.H. Myosin content of individual human muscle fibers isolated by laser capture microdissection. Am. J. Physiol. Cell Physiol. 2016, 310, C381–C389. [Google Scholar] [CrossRef] [PubMed]
- Pistilli, D.; di Gioia, C.R.; D’Amati, G.; Sciacchitano, S.; Quaglione, R.; Quitadamo, R.; Casali, C.; Gallo, P.; Santorelli, F.M. Detection of deleted mitochondrial DNA in Kearns-Sayre syndrome using laser capture microdissection. Hum. Pathol. 2003, 34, 1058–1061. [Google Scholar] [CrossRef]
- Pianezzi, E.; Altomare, C.; Bolis, S.; Balbi, C.; Torre, T.; Rinaldi, A.; Camici, G.G.; Barile, L.; Vassalli, G. Role of somatic cell sources in the maturation degree of human induced pluripotent stem cell-derived cardiomyocytes. BBA-Mol. Cell Res. 2020, 1867, 118538. [Google Scholar] [CrossRef]
- Balbi, C.; Milano, G.; Fertig, T.E.; Lazzarini, E.; Bolis, S.; Taniyama, Y.; Sanada, F.; Di Silvestre, D.; Mauri, P.; Gherghiceanu, M.; et al. An exosomal-carried short periostin isoform induces cardiomyocyte proliferation. Theranostics 2021, 11, 5634–5649. [Google Scholar] [CrossRef]
- Burrello, J.; Biemmi, V.; Dei Cas, M.; Amongero, M.; Bolis, S.; Lazzarini, E.; Bollini, S.; Vassalli, G.; Paroni, R.; Barile, L. Sphingolipid composition of circulating extracellular vesicles after myocardial ischemia. Sci. Rep. UK 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Larios, J.; Mercier, V.; Roux, A.; Gruenberg, J. ALIX- and ESCRT-III-dependent sorting of tetraspanins to exosomes. J. Cell Biol. 2020, 219, e201904113. [Google Scholar] [CrossRef]
- Willms, E.; Johansson, H.J.; Mager, I.; Lee, Y.; Blomberg, K.E.M.; Sadik, M.; Alaarg, A.; Smith, C.I.E.; Lehtio, J.; Andaloussi, S.E.L.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. UK 2016, 6. [Google Scholar] [CrossRef]
- Lee, S.S.; Won, J.H.; Lim, G.J.; Han, J.; Lee, J.Y.; Cho, K.O.; Bae, Y.K. A novel population of extracellular vesicles smaller than exosomes promotes cell proliferation. Cell Commun. Signal. 2019, 17, 1–15. [Google Scholar] [CrossRef]
- Lombardi, M.; Lazzeroni, D.; Benedetti, G.; Bertoli, G.; Lazarevic, D.; Riba, M.; De Cobelli, F.; Rimoldi, O.; d’Amati, G.; Olivotto, I.; et al. Plasmatic and myocardial microRNA profiles in patients with Hypertrophic Cardiomyopathy. Clin. Transl. Med. 2021, 11, e435. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Coats, C.J.; Heywood, W.E.; Virasami, A.; Ashrafi, N.; Syrris, P.; Dos Remedios, C.; Treibel, T.A.; Moon, J.C.; Lopes, L.R.; McGregor, C.G.A.; et al. Proteomic Analysis of the Myocardium in Hypertrophic Obstructive Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e001974. [Google Scholar] [CrossRef] [PubMed]
- Helmke, S.M.; Yen, C.Y.; Cios, K.J.; Nunley, K.; Bristow, M.R.; Duncan, M.W.; Perryman, M.B. Simultaneous quantification of human cardiac alpha- and beta-myosin heavy chain proteins by MALDI-TOF mass spectrometry. Anal. Chem. 2004, 76, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Ritter, O.; Luther, H.P.; Haase, H.; Baltas, L.G.; Baumann, G.; Schulte, H.D.; Morano, I. Expression of atrial myosin light chains but not alpha-myosin heavy chains is correlated in vivo with increased ventricular function in patients with hypertrophic obstructive cardiomyopathy. J. Mol. Med. 1999, 77, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Reiser, P.J.; Moravec, C.S. Sex differences in myosin heavy chain isoforms of human failing and nonfailing atria. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H265–H272. [Google Scholar] [CrossRef][Green Version]
- Abraham, W.T.; Gilbert, E.M.; Lowes, B.D.; Minobe, W.A.; Larrabee, P.; Roden, R.L.; Dutcher, D.; Sederberg, J.; Lindenfeld, J.A.; Wolfel, E.E.; et al. Coordinate changes in Myosin heavy chain isoform gene expression are selectively associated with alterations in dilated cardiomyopathy phenotype. Mol. Med. 2002, 8, 750–760. [Google Scholar] [PubMed]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Kraft, T.; Montag, J.; Radocaj, A.; Brenner, B. Hypertrophic Cardiomyopathy: Cell-to-Cell Imbalance in Gene Expression and Contraction Force as Trigger for Disease Phenotype Development. Circ. Res. 2016, 119, 992–995. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.A.; Broadwell, L.J.; Buvoli, M.; Leinwand, L.A. Nonproductive Splicing Prevents Expression of MYH7b Protein in the Mammalian Heart. J. Am. Heart Assoc. 2021, 10, e020965. [Google Scholar] [CrossRef]
- Lee, L.A.; Karabina, A.; Broadwell, L.J.; Leinwand, L.A. The ancient sarcomeric myosins found in specialized muscles. Skelet. Muscle 2019, 9, 7. [Google Scholar] [CrossRef]
- Matkovich, S.J.; Hu, Y.; Eschenbacher, W.H.; Dorn, L.E.; Dorn, G.W., 2nd. Direct and indirect involvement of microRNA-499 in clinical and experimental cardiomyopathy. Circ. Res. 2012, 111, 521–531. [Google Scholar] [CrossRef]
- Liu, J.; Wang, D.Z. An epigenetic “LINK(RNA)” to pathological cardiac hypertrophy. Cell Metab. 2014, 20, 555–557. [Google Scholar] [CrossRef]
- Kingston, E.R.; Bartel, D.P. Global analyses of the dynamics of mammalian microRNA metabolism. Genome Res. 2019, 29, 1777–1790. [Google Scholar] [CrossRef]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef]
- Wang, J.; Xu, L.; Tian, L.; Sun, Q. Circulating microRNA-208 family as early diagnostic biomarkers for acute myocardial infarction: A meta-analysis. Medicine 2021, 100, e27779. [Google Scholar] [CrossRef]
- Baulina, N.; Pisklova, M.; Kiselev, I.; Chumakova, O.; Zateyshchikov, D.; Favorova, O. Circulating miR-499a-5p Is a Potential Biomarker of MYH7-Associated Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 3791. [Google Scholar] [CrossRef] [PubMed]
- Min, P.K.; Chan, S.Y. The biology of circulating microRNAs in cardiovascular disease. Eur. J. Clin. Investig. 2015, 45, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Azibani, F.; Devaux, Y.; Coutance, G.; Schlossarek, S.; Polidano, E.; Fazal, L.; Merval, R.; Carrier, L.; Solal, A.C.; Chatziantoniou, C.; et al. Aldosterone inhibits the fetal program and increases hypertrophy in the heart of hypertensive mice. PLoS ONE 2012, 7, e38197. [Google Scholar] [CrossRef]
- Lombardi, M.; Lazzeroni, D.; Pisano, A.; Girolami, F.; Alfieri, O.; La Canna, G.; d’Amati, G.; Olivotto, I.; Rimoldi, O.E.; Foglieni, C.; et al. Mitochondrial Energetics and Ca2+-Activated ATPase in Obstructive Hypertrophic Cardiomyopathy. J. Clin. Med. 2020, 9, 1799. [Google Scholar] [CrossRef]
- Claes, G.R.; van Tienen, F.H.; Lindsey, P.; Krapels, I.P.; Helderman-van den Enden, A.T.; Hoos, M.B.; Barrois, Y.E.; Janssen, J.W.; Paulussen, A.D.; Sels, J.W.; et al. Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. Eur. Heart J. 2016, 37, 1815–1822. [Google Scholar] [CrossRef]
- Trivedi, D.V.; Adhikari, A.S.; Sarkar, S.S.; Ruppel, K.M.; Spudich, J.A. Hypertrophic cardiomyopathy and the myosin mesa: Viewing an old disease in a new light. Biophys. Rev. 2018, 10, 27–48. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic Data | N = 23 |
|---|---|
| Age (years), M (SD) | 59 (9) |
| Gender (male), N (%) | 13 (56.5) |
| BMI (Kg/m2), M (SD) | 27.9 (5.1) |
| Creatinine (mg/dL), M (SD) | 0.84 (0.33) |
| GFR (mL/min) | 110.9 (44.8) |
| Troponin T (ng/L) M (SD) | 545.7 (461) |
| Clinical data | |
| Family history of HCM, N (%) | 6 (26) |
| Family history of SCD, N (%) | 4 (17) |
| NYHA ≥ III, N (%) | 9 (39) |
| Angina, N (%) | 6 (26) |
| Syncope, N (%) | 6 (26) |
| Atrial fibrillation (previous history), N (%) | 4 (17) |
| NSVT, N (%) | 1 (4) |
| ICD, N (%) | 0 |
| Medical Therapy | |
| Beta-blockers, N (%) | 21 (91) |
| Antiarrhythmic drugs, N (%) | 5 (21) |
| Diuretics, N (%) | 18 (74) |
| RAAS-i, N (%) | 3 (13) |
| Echocardiographic Structural Data. | |
|---|---|
| IVS thickness (mm), M (SD) | 21 (5.6) |
| PW thickness (mm), M (SD) | 12 (4) |
| IVS/PW ratio, M (SD) | 1.99 (0.88) |
| LV-EDD (mm), M (SD) | 43.6 (7.3) |
| BSA (m2), M (SD) | 1.92 (0.26) |
| LA volume indexed, M (SD) | 45.90 (18.0) |
| Systolic function | |
| LV-EF (%), M (SD) | 67 (9) |
| TDI s’ peak (cm/s), M (SD) | 9 (2) |
| Diastolic function | |
| E peak (cm/s), M (SD) | 64 (27) |
| A peak (cm/s), M (SD) | 78 (37) |
| E/A ratio, M (SD) | 1.0 (0.83) |
| DT (ms), M (SD) | 184 (84) |
| IVRT (ms), M (SD) | 78 (20) |
| TDI e’ peak (cm/s), M (SD) | 9 (2) |
| TDI, a’ peak (cm/s), M (SD) | 12 (4) |
| E/e’ ratio | 8 (5) |
| PAPs (mmHg), M (SD) | 31 (7) |
| Mitral regurgitation ≥ grade 3, N (%) | 8 (35) |
| SAM-related LVOT-max gradient at rest (mmHg), M (SD) | 80 (29) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foglieni, C.; Lombardi, M.; Lazzeroni, D.; Zerboni, R.; Lazzarini, E.; Bertoli, G.; Pisano, A.; Girolami, F.; Andolfo, A.; Magagnotti, C.; et al. Myosins and MyomiR Network in Patients with Obstructive Hypertrophic Cardiomyopathy. Biomedicines 2022, 10, 2180. https://doi.org/10.3390/biomedicines10092180
Foglieni C, Lombardi M, Lazzeroni D, Zerboni R, Lazzarini E, Bertoli G, Pisano A, Girolami F, Andolfo A, Magagnotti C, et al. Myosins and MyomiR Network in Patients with Obstructive Hypertrophic Cardiomyopathy. Biomedicines. 2022; 10(9):2180. https://doi.org/10.3390/biomedicines10092180
Chicago/Turabian StyleFoglieni, Chiara, Maria Lombardi, Davide Lazzeroni, Riccardo Zerboni, Edoardo Lazzarini, Gloria Bertoli, Annalinda Pisano, Francesca Girolami, Annapaola Andolfo, Cinzia Magagnotti, and et al. 2022. "Myosins and MyomiR Network in Patients with Obstructive Hypertrophic Cardiomyopathy" Biomedicines 10, no. 9: 2180. https://doi.org/10.3390/biomedicines10092180
APA StyleFoglieni, C., Lombardi, M., Lazzeroni, D., Zerboni, R., Lazzarini, E., Bertoli, G., Pisano, A., Girolami, F., Andolfo, A., Magagnotti, C., Peretto, G., Sartorio, C. L., Olivotto, I., La Canna, G., Alfieri, O., Rimoldi, O. E., Barile, L., d’Amati, G., & Camici, P. G. (2022). Myosins and MyomiR Network in Patients with Obstructive Hypertrophic Cardiomyopathy. Biomedicines, 10(9), 2180. https://doi.org/10.3390/biomedicines10092180