

Novel Therapies for the Treatment of Cardiac Fibrosis Following Myocardial Infarction


Abstract
:
1. Introduction
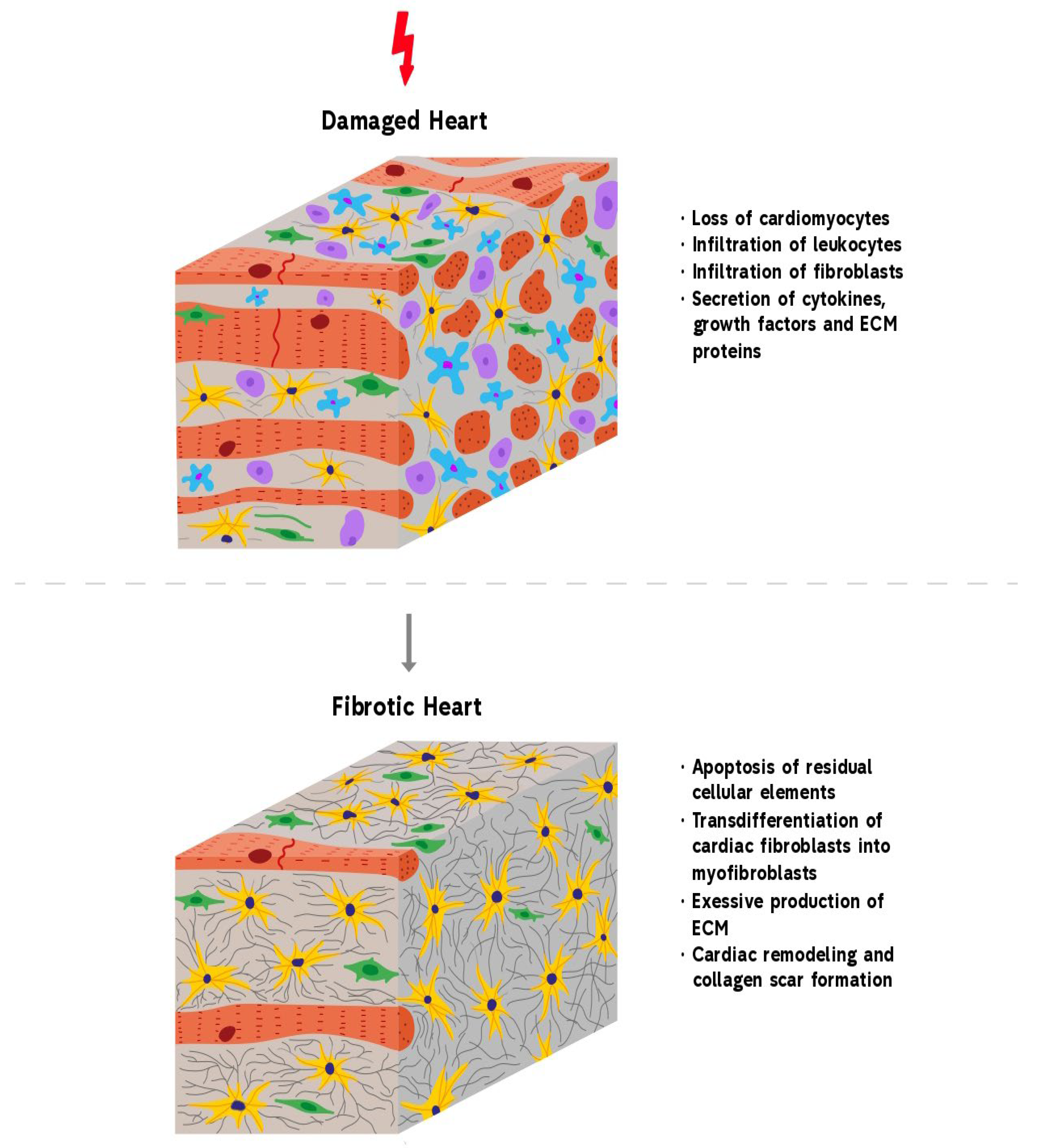
2. Cardiac Fibrosis
3. Molecular Mechanisms of Cardiac Fibrosis
3.1. The Role of TGF-β in Cardiac Fibrosis
3.2. The Role of RAAS in Cardiac Fibrosis
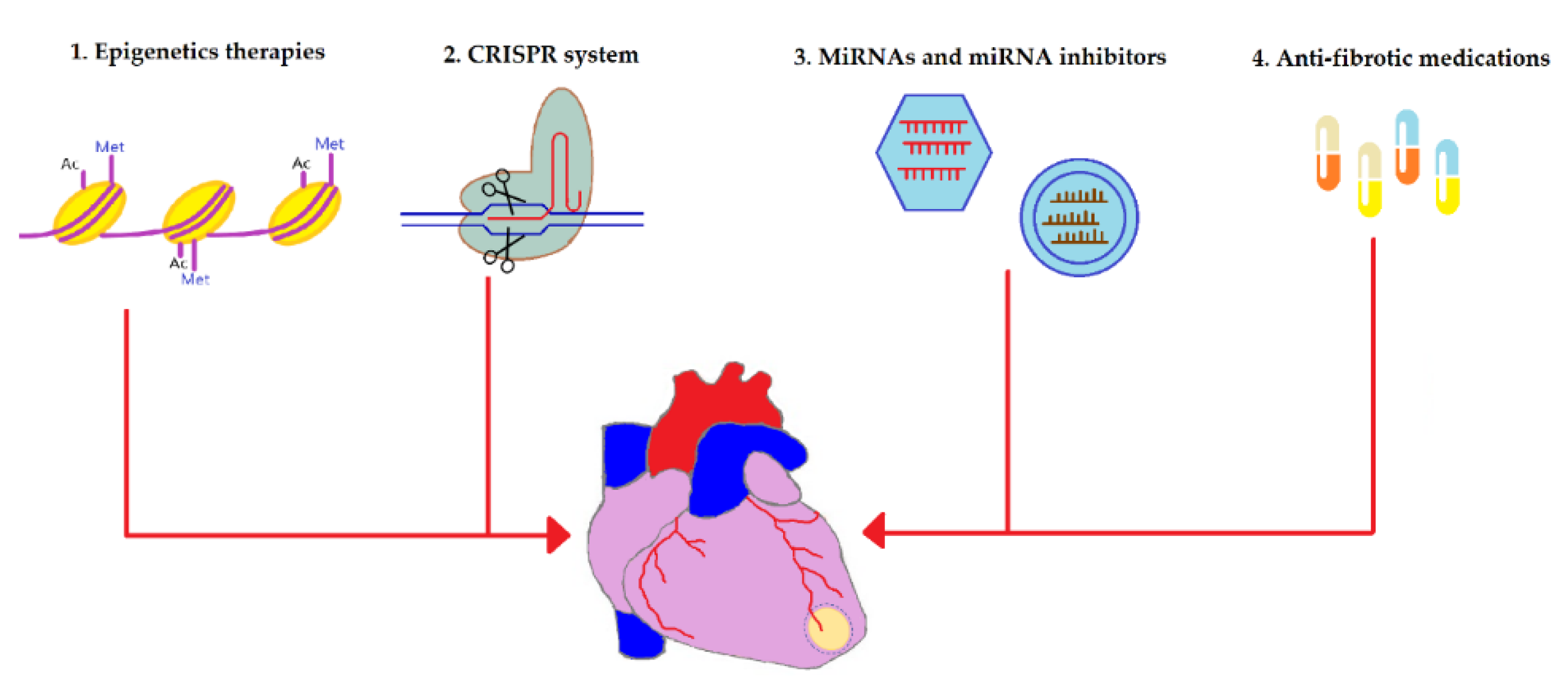
4. Anti-Fibrotic Therapies
4.1. Epigenetics
4.2. CRISPR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Treatment | Outcome | Reference |
| Epigenetics | Histone deacetylase inhibitor trichostatin A | Reduced fibrosis and improved systolic and diastolic functions a murine MI model | [59] |
| Class I and II Histone deacetylase inhibitor Rhein | Inhibited TGF-β1-induced fibroblast-to-myofibroblast transition and transcription of pro-fibrotic genes in primary human ventricular cardiac fibroblasts culture under sustained hypoxia | [61] | |
| Histone methyltransferase DOT1L inhibition | Alleviated fibrosis, reduced expression of collagen type I alpha1 and fibronectin 1 and improved cardiac function in mice with MI | [62] | |
| Histone methyltransferase G9a inhibition | Decreased expression of several fibrosis markers such as fibronectin, Smad3, and TGF-ß; reduced fibrotic area; increased angiogenesis; and preserved heart function in a rat model of MI | [63] | |
| CRISPR | CRISPR-Cas9-mediated inactivation of miR34a gene | Decreased fibrosis, enhanced proliferation cardiomyocytes, and improved heart function | [70] |
| CRISPR-mediated reprogramming of fibroblasts to cardiovascular progenitor cells | Differentiation of reprogrammed fibroblasts to endothelial cells, cardiomyocytes, and smooth muscle cells; reduced scar size; and restored cardiac function in a mouse model of MI | [71] | |
| CRISPR/Cas9-mediated integration of LEF1 gene into human umbilical cord blood-derived mesenchymal stem cells | Improved survival, enhanced cardiac function, increased vessel density, and decreased fibrosis after MI | [72] | |
| CRISPR/dCas9 activation system-induced overexpression of IL-10 in bone marrow-derived mesenchymal stem cells | Reduced scar tissue, improved heart function, suppresses cardiomyocyte apoptosis, and enhanced angiogenesis | [73] |
4.3. miRNAs
4.3.1. Pro-Fibrotic miRNAs
4.3.2. Anti-Fibrotic miRNAs
4.4. Anti-Fibrotic Medications
4.4.1. Renin–Angiotensin–Aldosterone System (RAAS) Inhibitors
4.4.2. TGF-β and CTGF Inhibitors
4.4.3. Gal-3 and NLRP3 Inflammasome Inhibitors
4.4.4. β-Adrenergic Receptor Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N. Heart disease and stroke statistics—2020 update: A report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Timmis, A.; Vardas, P.; Townsend, N.; Torbica, A.; Katus, H.; De Smedt, D.; Gale, C.P.; Maggioni, A.P.; Petersen, S.E.; Huculeci, R. European Society of Cardiology: Cardiovascular disease statistics 2021. Eur. Heart J. 2022, 43, 716–799. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, B.M.; Floege, J.; Boor, P. PDGF in organ fibrosis. Mol. Asp. Med. 2018, 62, 44–62. [Google Scholar] [CrossRef]
- Hashimoto, H.; Olson, E.N.; Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 2018, 15, 585–600. [Google Scholar] [CrossRef]
- Raziyeva, K.; Smagulova, A.; Kim, Y.; Smagul, S.; Nurkesh, A.; Saparov, A. Preconditioned and genetically modified stem cells for myocardial infarction treatment. Int. J. Mol. Sci. 2020, 21, 7301. [Google Scholar] [CrossRef]
- Kim, Y.; Zharkinbekov, Z.; Sarsenova, M.; Yeltay, G.; Saparov, A. Recent Advances in Gene Therapy for Cardiac Tissue Regeneration. Int. J. Mol. Sci. 2021, 22, 9206. [Google Scholar] [CrossRef]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- De Boer, R.A.; Heymans, S.; Backs, J.; Carrier, L.; Coats, A.J.; Dimmeler, S.; Eschenhagen, T.; Filippatos, G.; Gepstein, L.; Hulot, J.S. Targeted therapies in genetic dilated and hypertrophic cardiomyopathies: From molecular mechanisms to therapeutic targets. A position paper from the Heart Failure Association (HFA) and the Working Group on Myocardial Function of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2022, 24, 406–420. [Google Scholar]
- Raafs, A.G.; Verdonschot, J.A.; Henkens, M.T.; Adriaans, B.P.; Wang, P.; Derks, K.; Abdul Hamid, M.A.; Knackstedt, C.; van Empel, V.P.; Díez, J. The combination of carboxy-terminal propeptide of procollagen type I blood levels and late gadolinium enhancement at cardiac magnetic resonance provides additional prognostic information in idiopathic dilated cardiomyopathy–A multilevel assessment of myocardial fibrosis in dilated cardiomyopathy. Eur. J. Heart Fail. 2021, 23, 933–944. [Google Scholar]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.-F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2020, 19, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Nguyen, N.B.; Pezhouman, A.; Ardehali, R. Cardiac fibrosis: Potential therapeutic targets. Transl. Res. 2019, 209, 121–137. [Google Scholar] [CrossRef]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef]
- Kim, Y.; Nurakhayev, S.; Nurkesh, A.; Zharkinbekov, Z.; Saparov, A. Macrophage Polarization in Cardiac Tissue Repair following Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 2715. [Google Scholar] [CrossRef] [PubMed]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and fibrosis: Mitochondrial and metabolic control of cellular differentiation. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef] [PubMed]
- Disertori, M.; Masè, M.; Ravelli, F. Myocardial fibrosis predicts ventricular tachyarrhythmias. Trends Cardiovasc. Med. 2017, 27, 363–372. [Google Scholar] [CrossRef]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex relationship between cardiac fibroblasts and cardiomyocytes in health and disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef]
- Park, T.-J.; Park, J.H.; Lee, G.S.; Lee, J.-Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.-Y.; Oh, K.-J.; Han, B.-S. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Giacca, M. Cardiac regeneration after myocardial infarction: An approachable goal. Curr. Cardiol. Rep. 2020, 22, 1–8. [Google Scholar] [CrossRef]
- Yousefi, F.; Shabaninejad, Z.; Vakili, S.; Derakhshan, M.; Movahedpour, A.; Dabiri, H.; Ghasemi, Y.; Mahjoubin-Tehran, M.; Nikoozadeh, A.; Savardashtaki, A. TGF-β and WNT signaling pathways in cardiac fibrosis: Non-coding RNAs come into focus. Cell Commun. Signal. 2020, 18, 1–16. [Google Scholar] [CrossRef]
- Bacmeister, L.; Schwarzl, M.; Warnke, S.; Stoffers, B.; Blankenberg, S.; Westermann, D.; Lindner, D. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 2019, 114, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Kurose, H. Cardiac fibrosis and fibroblasts. Cells 2021, 10, 1716. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.; Jackson, S.P.; Moon, J.C.; Captur, G. Myocardial fibrosis in heart failure: Anti-fibrotic therapies and the role of cardiovascular magnetic resonance in drug trials. Cardiol. Ther. 2020, 9, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Karamitsos, T.D.; Arvanitaki, A.; Karvounis, H.; Neubauer, S.; Ferreira, V.M. Myocardial tissue characterization and fibrosis by imaging. Cardiovasc. Imaging 2020, 13, 1221–1234. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Umemura, M.; Narikawa, M.; Hikichi, M.; Osaw, K.; Fujita, T.; Yokoyama, U.; Ishigami, T.; Tamura, K.; Ishikawa, Y. Reactive fibrosis precedes doxorubicin-induced heart failure through sterile inflammation. ESC Heart Fail. 2020, 7, 588–603. [Google Scholar] [CrossRef]
- Hara, H.; Takeda, N.; Komuro, I. Pathophysiology and therapeutic potential of cardiac fibrosis. Inflamm. Regen. 2017, 37, 1–10. [Google Scholar] [CrossRef]
- Kyriakou, P.; Mouselimis, D.; Tsarouchas, A.; Rigopoulos, A.; Bakogiannis, C.; Noutsias, M.; Vassilikos, V. Diagnosis of cardiac amyloidosis: A systematic review on the role of imaging and biomarkers. BMC Cardiovasc. Disord. 2018, 18, 221. [Google Scholar] [CrossRef]
- Bonderman, D.; Pölzl, G.; Ablasser, K.; Agis, H.; Aschauer, S.; Auer-Grumbach, M.; Binder, C.; Dörler, J.; Duca, F.; Ebner, C. Diagnosis and treatment of cardiac amyloidosis: An interdisciplinary consensus statement. Wien. Klin. Wochenschr. 2020, 132, 742–761. [Google Scholar] [CrossRef]
- Heymans, S.; González, A.; Pizard, A.; Papageorgiou, A.P.; López-Andrés, N.; Jaisser, F.; Thum, T.; Zannad, F.; Díez, J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur. J. Heart Fail. 2015, 17, 764–771. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Scharf, G.M.; Kilian, K.; Cordero, J.; Wang, Y.; Grund, A.; Hofmann, M.; Froese, N.; Wang, X.; Kispert, A.; Kist, R.; et al. Inactivation of Sox9 in fibroblasts reduces cardiac fibrosis and inflammation. JCI Insight 2019, 5, e126721. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Peng, L.; Chen, Y. The effect of immune cell-derived exosomes in the cardiac tissue repair after myocardial infarction: Molecular mechanisms and pre-clinical evidence. J. Cell. Mol. Med. 2021, 25, 6500–6510. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Raziyeva, K.; Kim, Y.; Zharkinbekov, Z.; Kassymbek, K.; Jimi, S.; Saparov, A. Immunology of acute and chronic wound healing. Biomolecules 2021, 11, 700. [Google Scholar] [CrossRef] [PubMed]
- Ceauşu, Z.; Socea, B.; Costache, M.; Predescu, D.; Şerban, D.; Smarandache, C.G.; Pacu, I.; Alexandru, H.H.; Daviţoiu, A.M.; Jacotă-Alexe, F. Fibroblast involvement in cardiac remodeling and repair under ischemic conditions. Exp. Ther. Med. 2021, 21, 1. [Google Scholar] [CrossRef]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Li, W.; Zhuge, Y.; Xu, S.; Wang, Y.; Chen, Y.; Shen, G.; Wang, F. Inhibition of circHIPK3 prevents angiotensin II-induced cardiac fibrosis by sponging miR-29b-3p. Int. J. Cardiol. 2019, 292, 188–196. [Google Scholar] [CrossRef]
- Krenning, G.; Zeisberg, E.M.; Kalluri, R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 2010, 225, 631–637. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Aujla, P.K.; Kassiri, Z. Diverse origins and activation of fibroblasts in cardiac fibrosis. Cell. Signal. 2021, 78, 109869. [Google Scholar] [CrossRef]
- Dobaczewski, M.; de Haan, J.J.; Frangogiannis, N.G. The extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J. Cardiovasc. Transl. Res. 2012, 5, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Cakir, S.N.; Whitehead, K.M.; Hendricks, H.K.; de Castro Brás, L.E. Novel Techniques Targeting Fibroblasts after Ischemic Heart Injury. Cells 2022, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Sygitowicz, G.; Maciejak-Jastrzębska, A.; Sitkiewicz, D. A review of the molecular mechanisms underlying cardiac fibrosis and atrial fibrillation. J. Clin. Med. 2021, 10, 4430. [Google Scholar] [CrossRef] [PubMed]
- Hamid, T.; Xu, Y.; Ismahil, M.A.; Rokosh, G.; Jinno, M.; Zhou, G.; Wang, Q.; Prabhu, S.D. Cardiac mesenchymal stem cells promote fibrosis and remodeling in heart failure: Role of PDGF signaling. JACC Basic Transl. Sci. 2022, 7, 465–483. [Google Scholar] [CrossRef]
- Liu, Y.; Yin, Z.; Xu, X.; Liu, C.; Duan, X.; Song, Q.; Tuo, Y.; Wang, C.; Yang, J.; Yin, S. Crosstalk between the activated Slit2–Robo1 pathway and TGF-β1 signalling promotes cardiac fibrosis. ESC Heart Fail. 2021, 8, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Noskovicova, N.; Schuster, R.; van Putten, S.; Ezzo, M.; Koehler, A.; Boo, S.; Coelho, N.M.; Griggs, D.; Ruminski, P.; McCulloch, C.A. Suppression of the fibrotic encapsulation of silicone implants by inhibiting the mechanical activation of pro-fibrotic TGF-β. Nat. Biomed. Eng. 2021, 5, 1437–1456. [Google Scholar] [CrossRef]
- Działo, E.; Czepiel, M.; Tkacz, K.; Siedlar, M.; Kania, G.; Błyszczuk, P. WNT/β-catenin signaling promotes TGF-β-mediated activation of human cardiac fibroblasts by enhancing IL-11 production. Int. J. Mol. Sci. 2021, 22, 10072. [Google Scholar] [CrossRef]
- Hanna, A.; Humeres, C.; Frangogiannis, N.G. The role of Smad signaling cascades in cardiac fibrosis. Cell. Signal. 2021, 77, 109826. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. TGF-β in fibrosis by acting as a conductor for contractile properties of myofibroblasts. Cell Biosci. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Parichatikanond, W.; Luangmonkong, T.; Mangmool, S.; Kurose, H. Therapeutic targets for the treatment of cardiac fibrosis and cancer: Focusing on TGF-β signaling. Front. Cardiovasc. Med. 2020, 7, 34. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Hill, M.A.; Sowers, J.R. Role of renin-angiotensin-aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension 2018, 72, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Hu, M.; Peng, J.; Zhang, X.; Sanders, Y.Y. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 2040622319862697. [Google Scholar] [CrossRef]
- Yoon, S.; Kang, G.; Eom, G.H. HDAC inhibitors: Therapeutic potential in fibrosis-associated human diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef]
- Fernández-Barrena, M.G.; Arechederra, M.; Colyn, L.; Berasain, C.; Avila, M.A. Epigenetics in hepatocellular carcinoma development and therapy: The tip of the iceberg. JHEP Rep. 2020, 2, 100167. [Google Scholar] [CrossRef] [PubMed]
- Prasher, D.; Greenway, S.C.; Singh, R.B. The impact of epigenetics on cardiovascular disease. Biochem. Cell Biol. 2020, 98, 12–22. [Google Scholar] [CrossRef]
- Soler-Botija, C.; Forcales, S.V.; Genís, A.B. Spotlight on epigenetic reprogramming in cardiac regeneration. Semin. Cell Dev. Biol. 2020, 97, 26–37. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Chen, S.; Zhou, J.; Li, J.; Cheng, Y. Epigenetics-based therapeutics for myocardial fibrosis. Life Sci. 2021, 271, 119186. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, P.; Wang, L.; Zhao, J.; Zhong, Z.; Wang, Y.; Xu, J. Inhibition of histone deacetylases prevents cardiac remodeling after myocardial infarction by restoring autophagosome processing in cardiac fibroblasts. Cell. Physiol. Biochem. 2018, 49, 1999–2011. [Google Scholar] [CrossRef]
- Zhang, L.X.; Du, J.; Zhao, Y.T.; Wang, J.; Zhang, S.; Dubielecka, P.M.; Wei, L.; Zhuang, S.; Qin, G.; Chin, Y.E. Transgenic overexpression of active HDAC4 in the heart attenuates cardiac function and exacerbates remodeling in infarcted myocardium. J. Appl. Physiol. 2018, 125, 1968–1978. [Google Scholar] [CrossRef]
- Barbosa, D.M.; Fahlbusch, P.; Herzfeld de Wiza, D.; Jacob, S.; Kettel, U.; Al-Hasani, H.; Krüger, M.; Ouwens, D.M.; Hartwig, S.; Lehr, S. Rhein, a novel Histone Deacetylase (HDAC) inhibitor with antifibrotic potency in human myocardial fibrosis. Sci. Rep. 2020, 10, 4888. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, L.; Zhang, J.; Yang, X.; Liu, Y. Histone methyltransferase DOT1L mediates the TGF-β1/Smad3 signaling pathway through epigenetic modification of SYK in myocardial infarction. Hum. Cell 2022, 35, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.-H.; Luo, C.-W.; Chiang, J.Y.; Yip, H.-K. The combination of G9a histone methyltransferase inhibitors with erythropoietin protects heart against damage from acute myocardial infarction. Am. J. Transl. Res. 2020, 12, 3255. [Google Scholar]
- Liu, F.; Wen, Y.; Kang, J.; Wei, C.; Wang, M.; Zheng, Z.; Peng, J. Regulation of TLR4 expression mediates the attenuating effect of erythropoietin on inflammation and myocardial fibrosis in rat heart. Int. J. Mol. Med. 2018, 42, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, L.; Yi, H.; Wu, X. LRP6-CRISPR prevents activation of hepatic stellate cells and liver fibrogenesis in rats. Am. J. Transl. Res. 2020, 12, 397. [Google Scholar]
- Yu, L.; Wang, L.; Wu, X.; Yi, H. RSPO4-CRISPR alleviates liver injury and restores gut microbiota in a rat model of liver fibrosis. Commun. Biol. 2021, 4, 230. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M. High-fidelity CRISPR/Cas9-based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 3509. [Google Scholar] [CrossRef]
- Tan, Q.; Link, P.A.; Meridew, J.A.; Pham, T.X.; Caporarello, N.; Ligresti, G.; Tschumperlin, D.J. Spontaneous lung fibrosis resolution reveals novel antifibrotic regulators. Am. J. Respir. Cell Mol. Biol. 2021, 64, 453–464. [Google Scholar] [CrossRef]
- Nishiga, M.; Liu, C.; Qi, L.S.; Wu, J.C. The use of new CRISPR tools in cardiovascular research and medicine. Nat. Rev. Cardiol. 2022, 19, 505–521. [Google Scholar] [CrossRef]
- Park, H.; Kim, D.; Cho, B.; Byun, J.; Kim, Y.S.; Ahn, Y.; Hur, J.; Oh, Y.-K.; Kim, J. In vivo therapeutic genome editing via CRISPR/Cas9 magnetoplexes for myocardial infarction. Biomaterials 2022, 281, 121327. [Google Scholar] [CrossRef]
- Jiang, L.; Liang, J.; Huang, W.; Ma, J.; Park, K.H.; Wu, Z.; Chen, P.; Zhu, H.; Ma, J.-J.; Cai, W. CRISPR activation of endogenous genes reprograms fibroblasts into cardiovascular progenitor cells for myocardial infarction therapy. Mol. Ther. 2022, 30, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-M.; Lee, K.-H.; Shen, Y.-M.; Shin, T.-J.; Ryu, P.-D.; Choi, M.-C.; Kang, K.-S.; Cho, J.-Y. Transplantation of hMSCs genome edited with LEF1 improves cardio-protective effects in myocardial infarction. Mol. Ther. Nucleic Acids 2020, 19, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zheng, M.; Yu, M.; Bai, W.; Zuo, L.; Bu, X.; Liu, Y.; Xia, L.; Hu, J.; Liu, L. Transplantation of CRISPRa system engineered IL10-overexpressing bone marrow-derived mesenchymal stem cells for the treatment of myocardial infarction in diabetic mice. J. Biol. Eng. 2019, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, J.; Wu, M.; Kang, L.; Xu, B. The effector cells and cellular mediators of immune system involved in cardiac inflammation and fibrosis after myocardial infarction. J. Cell. Physiol. 2020, 235, 8996–9004. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, H.; Farahani, N.; Hosseingholi, E.Z.; Sathyapalan, T.; hossein Sahebkar, A. Harnessing CRISPR/Cas9 technology in cardiovascular disease. Trends Cardiovasc. Med. 2020, 30, 93–101. [Google Scholar] [CrossRef]
- Dexheimer, P.J.; Cochella, L. MicroRNAs: From Mechanism to Organism. Front. Cell Dev. Biol. 2020, 8, 409. [Google Scholar] [CrossRef]
- Ferrari, S.; Pesce, M. Cell-Based Mechanosensation, Epigenetics, and Non-Coding RNAs in Progression of Cardiac Fibrosis. Int. J. Mol. Sci. 2019, 21, 28. [Google Scholar] [CrossRef]
- Varzideh, F.; Kansakar, U.; Donkor, K.; Wilson, S.; Jankauskas, S.S.; Mone, P.; Wang, X.; Lombardi, A.; Santulli, G. Cardiac Remodeling After Myocardial Infarction: Functional Contribution of microRNAs to Inflammation and Fibrosis. Front. Cardiovasc. Med. 2022, 9, 863238. [Google Scholar] [CrossRef]
- O’Reilly, S. MicroRNAs in fibrosis: Opportunities and challenges. Arthritis Res. Ther. 2016, 18, 11. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Abak, A.; Talebi, S.F.; Shoorei, H.; Branicki, W.; Taheri, M.; Dilmaghani, N.A. Role of miRNA and lncRNAs in organ fibrosis and aging. Biomed. Pharmacother. 2021, 143, 112132. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Deng, Y.; Li, H. MicroRNA-223 regulates cardiac fibrosis after myocardial infarction by targeting RASA1. Cell. Physiol. Biochem. 2018, 46, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, J.-W.; Lin, J.-Y.; Miao, R.; Zhong, J.-C. Roles of microRNA-122 in cardiovascular fibrosis and related diseases. Cardiovasc. Toxicol. 2020, 20, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.J.F.; Biessen, E.A.L.; Hohl, M.; Weber, C.; van der Vorst, E.P.C.; Santovito, D. Small Things Matter: Relevance of MicroRNAs in Cardiovascular Disease. Front. Physiol. 2020, 11, 793. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Shao, Y.; Guo, H.C.; Zhi, Y.; Qiao, B.; Ma, K.; Lai, Y.Q.; Du, J.; Li, Y. MicroRNA-27b-3p downregulates FGF1 and aggravates pathological cardiac remodelling. Cardiovasc. Res. 2021, 118, 2139–2151. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, E.; Das, S. Non-coding RNAs in cardiac remodeling: Diversity in composition and function. Curr. Opin. Physiol. 2022, 26, 100534. [Google Scholar] [CrossRef]
- Fu, Q.; Lu, Z.; Fu, X.; Ma, S.; Lu, X. MicroRNA 27b promotes cardiac fibrosis by targeting the FBW7/Snail pathway. Aging 2019, 11, 11865–11879. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Yao, Y.; Song, W. Downregulation of miR-96 suppresses the profibrogenic functions of cardiac fibroblasts induced by angiotensin II and attenuates atrial fibrosis by upregulating KLF13. Hum. Cell 2020, 33, 337–346. [Google Scholar] [CrossRef]
- Yu, Y.H.; Zhang, Y.H.; Ding, Y.Q.; Bi, X.Y.; Yuan, J.; Zhou, H.; Wang, P.X.; Zhang, L.L.; Ye, J.T. MicroRNA-99b-3p promotes angiotensin II-induced cardiac fibrosis in mice by targeting GSK-3β. Acta Pharmacol. Sin. 2021, 42, 715–725. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, Y.; Tang, Y.; Dai, H.; OuYang, Y.; Li, C.; Yu, M. MiRNA-1202 promotes the TGF-β1-induced proliferation, differentiation and collagen production of cardiac fibroblasts by targeting nNOS. PLoS ONE 2021, 16, e0256066. [Google Scholar] [CrossRef]
- Chen, C.; Ponnusamy, M.; Liu, C.; Gao, J.; Wang, K.; Li, P. MicroRNA as a Therapeutic Target in Cardiac Remodeling. BioMed Res. Int. 2017, 2017, 1278436. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, H.; Gao, W.; Zhang, L.; Ye, Y.; Yuan, L.; Ding, Z.; Wu, J.; Kang, L.; Zhang, X.; et al. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics 2018, 8, 2565–2582. [Google Scholar] [CrossRef]
- Tian, H.B.; Li, S.H.; Hu, K.Q.; Zan, Y.S.; Zhang, X.L.; Su, G.H. MicroRNA-150 alleviates acute myocardial infarction through regulating cardiac fibroblasts in ventricular remodeling. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7611–7618. [Google Scholar] [CrossRef]
- Shen, J.; Xing, W.; Gong, F.; Wang, W.; Yan, Y.; Zhang, Y.; Xie, C.; Fu, S. MiR-150-5p retards the progression of myocardial fibrosis by targeting EGR1. Cell Cycle 2019, 18, 1335–1348. [Google Scholar] [CrossRef]
- Chiasson, V.; Takano, A.P.C.; Guleria, R.S.; Gupta, S. Deficiency of MicroRNA miR-1954 Promotes Cardiac Remodeling and Fibrosis. J. Am. Heart Assoc. 2019, 8, e012880. [Google Scholar] [CrossRef]
- Yang, X.; Yu, T.; Zhang, S. MicroRNA-489 suppresses isoproterenol-induced cardiac fibrosis by downregulating histone deacetylase 2. Exp. Ther. Med. 2020, 19, 2229–2235. [Google Scholar] [CrossRef]
- Li, J.; Salvador, A.M.; Li, G.; Valkov, N.; Ziegler, O.; Yeri, A.; Yang Xiao, C.; Meechoovet, B.; Alsop, E.; Rodosthenous, R.S.; et al. Mir-30d Regulates Cardiac Remodeling by Intracellular and Paracrine Signaling. Circ. Res. 2021, 128, e1–e23. [Google Scholar] [CrossRef]
- Cui, S.; Liu, Z.; Tao, B.; Fan, S.; Pu, Y.; Meng, X.; Li, D.; Xia, H.; Xu, L. miR-145 attenuates cardiac fibrosis through the AKT/GSK-3β/β-catenin signaling pathway by directly targeting SOX9 in fibroblasts. J. Cell. Biochem. 2021, 122, 209–221. [Google Scholar] [CrossRef]
- Liu, M.; de Juan Abad, B.L.; Cheng, K. Cardiac fibrosis: Myofibroblast-mediated pathological regulation and drug delivery strategies. Adv. Drug Deliv. Rev. 2021, 173, 504–519. [Google Scholar] [CrossRef]
- Leader, C.J.; Moharram, M.; Coffey, S.; Sammut, I.A.; Wilkins, G.W.; Walker, R.J. Myocardial global longitudinal strain: An early indicator of cardiac interstitial fibrosis modified by spironolactone, in a unique hypertensive rat model. PLoS ONE 2019, 14, e0220837. [Google Scholar] [CrossRef]
- Kovács, Z.Z.; Szűcs, G.; Freiwan, M.; Kovács, M.G.; Márványkövi, F.M.; Dinh, H.; Siska, A.; Farkas, K.; Kovács, F.; Kriston, A. Comparison of the antiremodeling effects of losartan and mirabegron in a rat model of uremic cardiomyopathy. Sci. Rep. 2021, 11, 17495. [Google Scholar] [CrossRef]
- Khder, Y.; Shi, V.; McMurray, J.J.V.; Lefkowitz, M.P. Sacubitril/Valsartan (LCZ696) in Heart Failure. Handb. Exp. Pharmacol. 2017, 243, 133–165. [Google Scholar] [CrossRef]
- Burke, R.M.; Lighthouse, J.K.; Mickelsen, D.M.; Small, E.M. Sacubitril/Valsartan Decreases Cardiac Fibrosis in Left Ventricle Pressure Overload by Restoring PKG Signaling in Cardiac Fibroblasts. Circ. Heart Fail. 2019, 12, e005565. [Google Scholar] [CrossRef]
- Vaskova, E.; Ikeda, G.; Tada, Y.; Wahlquist, C.; Mercola, M.; Yang, P.C. Sacubitril/Valsartan Improves Cardiac Function and Decreases Myocardial Fibrosis Via Downregulation of Exosomal miR-181a in a Rodent Chronic Myocardial Infarction Model. J. Am. Heart Assoc. 2020, 9, e015640. [Google Scholar] [CrossRef]
- Wang, L.; Liu, C.; Chen, X.; Li, P. Alamandine attenuates long-term hypertension-induced cardiac fibrosis independent of blood pressure. Mol. Med. Rep. 2019, 19, 4553–4560. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.M.; de Souza-Neto, F.P.; Jesus, I.C.G.; Gonçalves, G.K.; Santuchi, M.C.; Sanches, B.L.; de Alcântara-Leonídio, T.C.; Melo, M.B.; Vieira, M.A.R.; Guatimosim, S.; et al. Alamandine improves cardiac remodeling induced by transverse aortic constriction in mice. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H352–H363. [Google Scholar] [CrossRef]
- Fu, J.; Li, F.; Tang, Y.; Cai, L.; Zeng, C.; Yang, Y.; Yang, J. The Emerging Role of Irisin in Cardiovascular Diseases. J. Am. Heart Assoc. 2021, 10, e022453. [Google Scholar] [CrossRef]
- Chen, R.R.; Fan, X.H.; Chen, G.; Zeng, G.W.; Xue, Y.G.; Liu, X.T.; Wang, C.Y. Irisin attenuates angiotensin II-induced cardiac fibrosis via Nrf2 mediated inhibition of ROS/ TGFβ1/Smad2/3 signaling axis. Chem. Biol. Interact. 2019, 302, 11–21. [Google Scholar] [CrossRef]
- Pan, J.A.; Zhang, H.; Lin, H.; Gao, L.; Zhang, H.L.; Zhang, J.F.; Wang, C.Q.; Gu, J. Irisin ameliorates doxorubicin-induced cardiac perivascular fibrosis through inhibiting endothelial-to-mesenchymal transition by regulating ROS accumulation and autophagy disorder in endothelial cells. Redox Biol. 2021, 46, 102120. [Google Scholar] [CrossRef]
- Ambari, A.M.; Setianto, B.; Santoso, A.; Radi, B.; Dwiputra, B.; Susilowati, E.; Tulrahmi, F.; Doevendans, P.A.; Cramer, M.J. Angiotensin Converting Enzyme Inhibitors (ACEIs) Decrease the Progression of Cardiac Fibrosis in Rheumatic Heart Disease Through the Inhibition of IL-33/sST2. Front. Cardiovasc. Med. 2020, 7, 115. [Google Scholar] [CrossRef]
- Wang, L.; Tan, A.; An, X.; Xia, Y.; Xie, Y. Quercetin Dihydrate inhibition of cardiac fibrosis induced by angiotensin II In Vivo and In Vitro. Biomed. Pharm. 2020, 127, 110205. [Google Scholar] [CrossRef]
- Li, M.; Qi, C.; Song, R.; Xiong, C.; Zhong, X.; Song, Z.; Ning, Z.; Song, X. Inhibition of Long Noncoding RNA SNHG20 Improves Angiotensin II-Induced Cardiac Fibrosis and Hypertrophy by Regulating the MicroRNA 335/Galectin-3 Axis. Mol. Cell Biol. 2021, 41, e0058020. [Google Scholar] [CrossRef]
- Ock, S.; Ham, W.; Kang, C.W.; Kang, H.; Lee, W.S.; Kim, J. IGF-1 protects against angiotensin II-induced cardiac fibrosis by targeting αSMA. Cell Death Dis. 2021, 12, 688. [Google Scholar] [CrossRef] [PubMed]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 2012, 5, S24. [Google Scholar] [CrossRef] [PubMed]
- Meagher, P.B.; Lee, X.A.; Lee, J.; Visram, A.; Friedberg, M.K.; Connelly, K.A. Cardiac fibrosis: Key role of integrins in cardiac homeostasis and remodeling. Cells 2021, 10, 770. [Google Scholar] [CrossRef] [PubMed]
- Vainio, L.E.; Szabó, Z.; Lin, R.; Ulvila, J.; Yrjölä, R.; Alakoski, T.; Piuhola, J.; Koch, W.J.; Ruskoaho, H.; Fouse, S.D.; et al. Connective Tissue Growth Factor Inhibition Enhances Cardiac Repair and Limits Fibrosis After Myocardial Infarction. JACC Basic Transl. Sci. 2019, 4, 83–94. [Google Scholar] [CrossRef]
- Aimo, A.; Cerbai, E.; Bartolucci, G.; Adamo, L.; Barison, A.; Lo Surdo, G.; Biagini, S.; Passino, C.; Emdin, M. Pirfenidone is a cardioprotective drug: Mechanisms of action and preclinical evidence. Pharmacol. Res. 2020, 155, 104694. [Google Scholar] [CrossRef]
- Graziani, F.; Varone, F.; Crea, F.; Richeldi, L. Treating heart failure with preserved ejection fraction: Learning from pulmonary fibrosis. Eur. J. Heart Fail. 2018, 20, 1385–1391. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Ding, C.; Wilson, E.; Marcus, G.M.; Olgin, J.E. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm. 2010, 7, 1438–1445. [Google Scholar] [CrossRef]
- Li, C.; Han, R.; Kang, L.; Wang, J.; Gao, Y.; Li, Y.; He, J.; Tian, J. Pirfenidone controls the feedback loop of the AT1R/p38 MAPK/renin-angiotensin system axis by regulating liver X receptor-α in myocardial infarction-induced cardiac fibrosis. Sci. Rep. 2017, 7, 40523. [Google Scholar] [CrossRef]
- Adamo, L.; Staloch, L.J.; Rocha-Resende, C.; Matkovich, S.J.; Jiang, W.; Bajpai, G.; Weinheimer, C.J.; Kovacs, A.; Schilling, J.D.; Barger, P.M.; et al. Modulation of subsets of cardiac B lymphocytes improves cardiac function after acute injury. JCI Insight 2018, 3, e120137. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, C.; Song, Q.; Li, Y.; Da, X.; Yu, Y.; Li, H.; Clark, I.M.; Chen, Q.; Wang, Q.K. ADAMTS16 activates latent TGF-β, accentuating fibrosis and dysfunction of the pressure-overloaded heart. Cardiovasc. Res. 2020, 116, 956–969. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Shinde, A.V.; Hanna, A.; Alex, L.; Hernández, S.C.; Li, R.; Chen, B.; Conway, S.J.; Frangogiannis, N.G. Smad7 effects on TGF-β and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J. Clin. Investig. 2022, 132, e146926. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, L.Y.; Liu, Z.Q.; Jiang, D.; Wu, S.Y.; Guo, Y.Q.; Tao, H.M.; Sun, M.; You, L.N.; Qin, S.; et al. TNAP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF-β1/Smads and activating P53 signaling pathways. Cell Death Dis. 2020, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ye, J.; Zhou, Y.; Huang, J.; Liu, X.; Huang, B.; Zhu, L.; Wu, B.; Zhang, G.; Cai, Y. Baicalin inhibits pressure overload-induced cardiac fibrosis through regulating AMPK/TGF-β/Smads signaling pathway. Arch. Biochem. Biophys. 2018, 640, 37–46. [Google Scholar] [CrossRef]
- Liu, M.; Li, W.; Wang, H.; Yin, L.; Ye, B.; Tang, Y.; Huang, C. CTRP9 Ameliorates Atrial Inflammation, Fibrosis, and Vulnerability to Atrial Fibrillation in Post-Myocardial Infarction Rats. J. Am. Heart Assoc. 2019, 8, e013133. [Google Scholar] [CrossRef]
- Cao, Z.Q.; Yu, X.; Leng, P. Research progress on the role of gal-3 in cardio/cerebrovascular diseases. Biomed. Pharm. 2021, 133, 111066. [Google Scholar] [CrossRef]
- Li, S.; Li, S.; Hao, X.; Zhang, Y.; Deng, W. Perindopril and a Galectin-3 Inhibitor Improve Ischemic Heart Failure in Rabbits by Reducing Gal-3 Expression and Myocardial Fibrosis. Front. Physiol. 2019, 10, 267. [Google Scholar] [CrossRef]
- Xu, G.R.; Zhang, C.; Yang, H.X.; Sun, J.H.; Zhang, Y.; Yao, T.T.; Li, Y.; Ruan, L.; An, R.; Li, A.Y. Modified citrus pectin ameliorates myocardial fibrosis and inflammation via suppressing galectin-3 and TLR4/MyD88/NF-κB signaling pathway. Biomed. Pharm. 2020, 126, 110071. [Google Scholar] [CrossRef]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, C.; Chen, Z.; Liu, L.; Jiang, J.; Wu, Z.; Zhao, M.; Chen, Y. NLRP3: A Novel Mediator in Cardiovascular Disease. J. Immunol. Res. 2018, 2018, 5702103. [Google Scholar] [CrossRef]
- Gao, R.; Shi, H.; Chang, S.; Gao, Y.; Li, X.; Lv, C.; Yang, H.; Xiang, H.; Yang, J.; Xu, L.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int. Immunopharmacol. 2019, 74, 105575. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.F.; Li, X.; Xiang, H.Y.; Yang, H.; Lv, C.Y.; Sun, X.L.; Chen, H.Z.; Gao, Y.; Yang, J.S.; Luo, W.; et al. The covalent NLRP3-inflammasome inhibitor Oridonin relieves myocardial infarction induced myocardial fibrosis and cardiac remodeling in mice. Int. Immunopharmacol. 2021, 90, 107133. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Koch, W.J. Targeting β3-Adrenergic Receptors in the Heart: Selective Agonism and β-Blockade. J. Cardiovasc. Pharmacol. 2017, 69, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Michel, L.Y.; Farah, C.; Balligand, J.-L. The beta3 adrenergic receptor in healthy and pathological cardiovascular tissues. Cells 2020, 9, 2584. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, L.; Li, X.; Xue, Y.; Wang, B.; Lv, Z.; Chen, J.; Sun, D.; Zheng, Q. β3-Adrenoreceptor stimulation protects against myocardial infarction injury via eNOS and nNOS activation. PLoS ONE 2014, 9, e98713. [Google Scholar] [CrossRef]
- Rossello, X.; Piñero, A.; Fernández-Jiménez, R.; Sánchez-González, J.; Pizarro, G.; Galán-Arriola, C.; Lobo-Gonzalez, M.; Vilchez, J.P.; García-Prieto, J.; García-Ruiz, J.M. Mirabegron, a Clinically Approved β3 adrenergic receptor agonist, does not reduce infarct size in a swine model of reperfused myocardial infarction. J. Cardiovasc. Transl. Res. 2018, 11, 310–318. [Google Scholar] [CrossRef]

| Class of Micro-RNA | Example | Treatment | Outcome | Reference |
|---|---|---|---|---|
| Pro-fibrotic | MiR-27b | Inhibition | Lowered expression of collagen I and III, and MMP9; significantly reduced fibrotic tissue formation in rat MI model | [86] |
| MiR-96 | Inhibition | Suppressed angiotensin II-induced proliferation of, migration of and collagen production by murine cardiac fibroblasts; decreased inflammatory cells infiltration; and reduced collagen I and III deposition in atrial tissue | [87] | |
| MiR-1202 | Inhibition | Decreased expression of collagen I and III, α-SMA, and Smad2/3 | [89] | |
| MiR-150 | Inhibition | Reduced expression of col1α1, col1α2, col3, and α-SMA; improved survival of cardiomyocytes in rat model of MI | [92] | |
| Anti-fibrotic | MiR-150-5p | Use of miRNA mimic | Suppressed the expression of MMP-13, collagen I and III; induced apoptosis of human myocardial fibrosis cells after Trypanosoma cruzi infection | [93] |
| MiR-1954 | Overexpression | Alleviated fibrotic cardiac remodeling, reduced systolic blood pressure, and decreased the expression of cardiac hypertrophy (NppA, NppB, beta-MHC) and fibrotic marker genes (col1a1, col3a1 and col4) in angiotensin II-induced model of cardiac hypertrophy in mice | [94] | |
| MiR-489 | Use of miRNA mimic | Downregulated pro-fibrotic markers such as col1a1, α-SMA, and HDAC2 in an isoproterenol-induced rat model of cardiac fibrosis and inhibited the viability and differentiation of cardiac fibroblasts in vitro | [95] | |
| MiR-30d | Overexpression | Reduced cardiac fibrosis, downregulated fibrosis marker genes such as α-SMA, inhibited cardiomyocyte apoptosis, and improved left ventricular function in a murine model of MI | [96] | |
| MiR-145 | Overexpression | Reduced fibrous tissue formation and collagen synthesis; enhanced left ventricular function in rat model of MI | [97] |
| Group of Inhibitors | Drugs/Inhibitors | Outcome | Reference |
|---|---|---|---|
| RAAS inhibitors | Sacubitril/Valsartan | Blocked cardiac fibroblasts activation and proliferation via downregulation of exosomal miR-181a | [101,102,103] |
| Alamandine | Decreased the density of cardiac fibrosis and reduced expression of fibrotic proteins, increased of ERK1/2 phosphorylation and restored the level of AMPKα phosphorylation preventing cardiac hypertrophy and fibrosis | [104,105] | |
| Irisin | Ameliorated cardiac perivascular fibrosis via regulating ROS accumulation and activating Nrf2-antioxidant signaling pathway and inhibiting pro-fibrotic TGFβ1-Smad3 signaling | [107,108] | |
| Angiotensin converting enzyme inhibitors | Reduced the proliferation of fibroblasts and expression of a-SMA as well as improved diastolic functions, reduced the inflammatory response and the level of sST2 providing cardioprotection against fibrosis | [37,109] | |
| TGF-beta and CTGF inhibitors | Pamrevlumab | Reduced MI-induced fibrosis in in the remote, nonischemic myocardium, reduced basal and TGF-β1–induced αSMA and collagen-1 expression, and genes related to fibrosis | [115] |
| Pirfenidone | Reduced scar size and myocardial fibrosis in the border zone, with better preserved LV systolic function and slowed down the progression toward HF | [118,119] | |
| Tetramisole | Reduced the expression of collagen-related genes and MI-induced fibrosis | [123] | |
| Baicalin | Inhibited cell proliferation, collagen synthesis, fibronectin, and CTGF protein expression in cardiac fibroblasts through reduction of TGF-β/Smads signaling pathway | [124] | |
| C1q/tumor necrosis factor-related protein-9 | Reduced atrial inflammation and fibrosis via inhibition of TLR4/MyD88/NF-κB signaling pathway, TGF-β, collagen deposition, in early phase of MI, decreased the expression of IL-1β, IL-18 and TNF-α | [127,128] | |
| Galectin-3 and NLRP3 inflammasome Inhibitor | Modified citrus pectin | Decreased myocardial fibrosis, myocardial Gal-3, collagen type I, and collagen type III gene and protein expressions via inhibiting TLR4/MyD88/NF-κB signaling pathway, decreased expression of IL-1β, IL-18, and TNF-α | [127,128] |
| MCC950 | Attenuated myocardial fibrosis and improved cardiac remodeling as well as inhibited NLRP3 and reduced caspase-1 activity with further downregulation of IL-1β and IL-18 | [131] | |
| Oridonin | Reduced myocardial fibrosis, decreased expression of IL-1β and IL-18 as well as infiltration by myocardial macrophages and neutrophils | [133] | |
| β3AR inhibitors | BRL37344 Agonist | Attenuated fibrosis, decreased scar area, altered the phosphorylation of endothelial NOS, and increased neuronal NOS expression | [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raziyeva, K.; Kim, Y.; Zharkinbekov, Z.; Temirkhanova, K.; Saparov, A. Novel Therapies for the Treatment of Cardiac Fibrosis Following Myocardial Infarction. Biomedicines 2022, 10, 2178. https://doi.org/10.3390/biomedicines10092178
Raziyeva K, Kim Y, Zharkinbekov Z, Temirkhanova K, Saparov A. Novel Therapies for the Treatment of Cardiac Fibrosis Following Myocardial Infarction. Biomedicines. 2022; 10(9):2178. https://doi.org/10.3390/biomedicines10092178
Chicago/Turabian StyleRaziyeva, Kamila, Yevgeniy Kim, Zharylkasyn Zharkinbekov, Kamila Temirkhanova, and Arman Saparov. 2022. "Novel Therapies for the Treatment of Cardiac Fibrosis Following Myocardial Infarction" Biomedicines 10, no. 9: 2178. https://doi.org/10.3390/biomedicines10092178
APA StyleRaziyeva, K., Kim, Y., Zharkinbekov, Z., Temirkhanova, K., & Saparov, A. (2022). Novel Therapies for the Treatment of Cardiac Fibrosis Following Myocardial Infarction. Biomedicines, 10(9), 2178. https://doi.org/10.3390/biomedicines10092178