
Genetic Load of Alternations of Transcription Factor Genes in Non-Syndromic Deafness and the Associated Clinical Phenotypes: Experience from Two Tertiary Referral Centers

 , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Clinical Phenotyping
2.3. Molecular Genetic Testing
2.4. Statistical Analysis
3. Results
3.1. Distribution of TF Genes
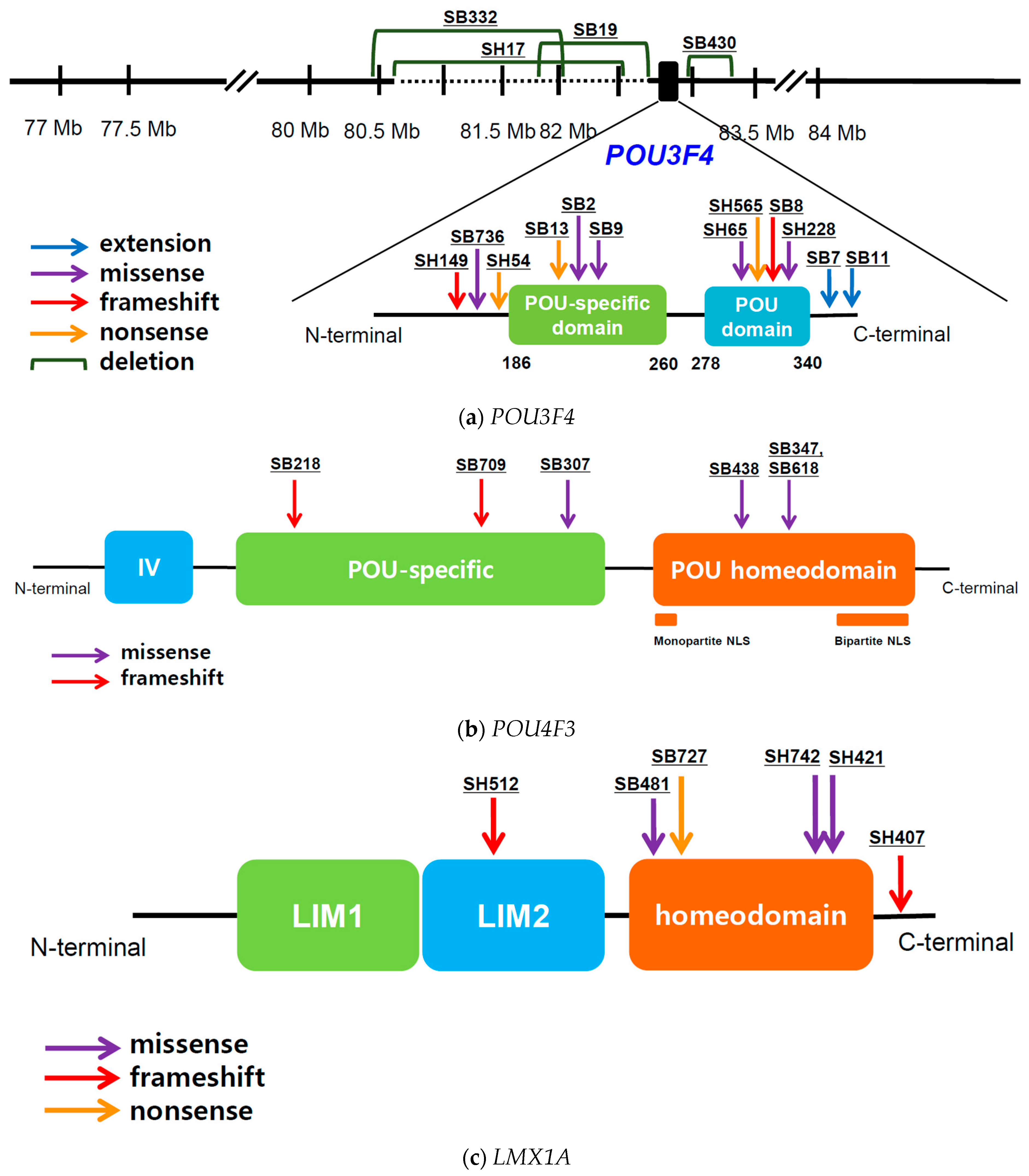
3.2. POU3F4
3.2.1. POU3F4: Genotype Profile
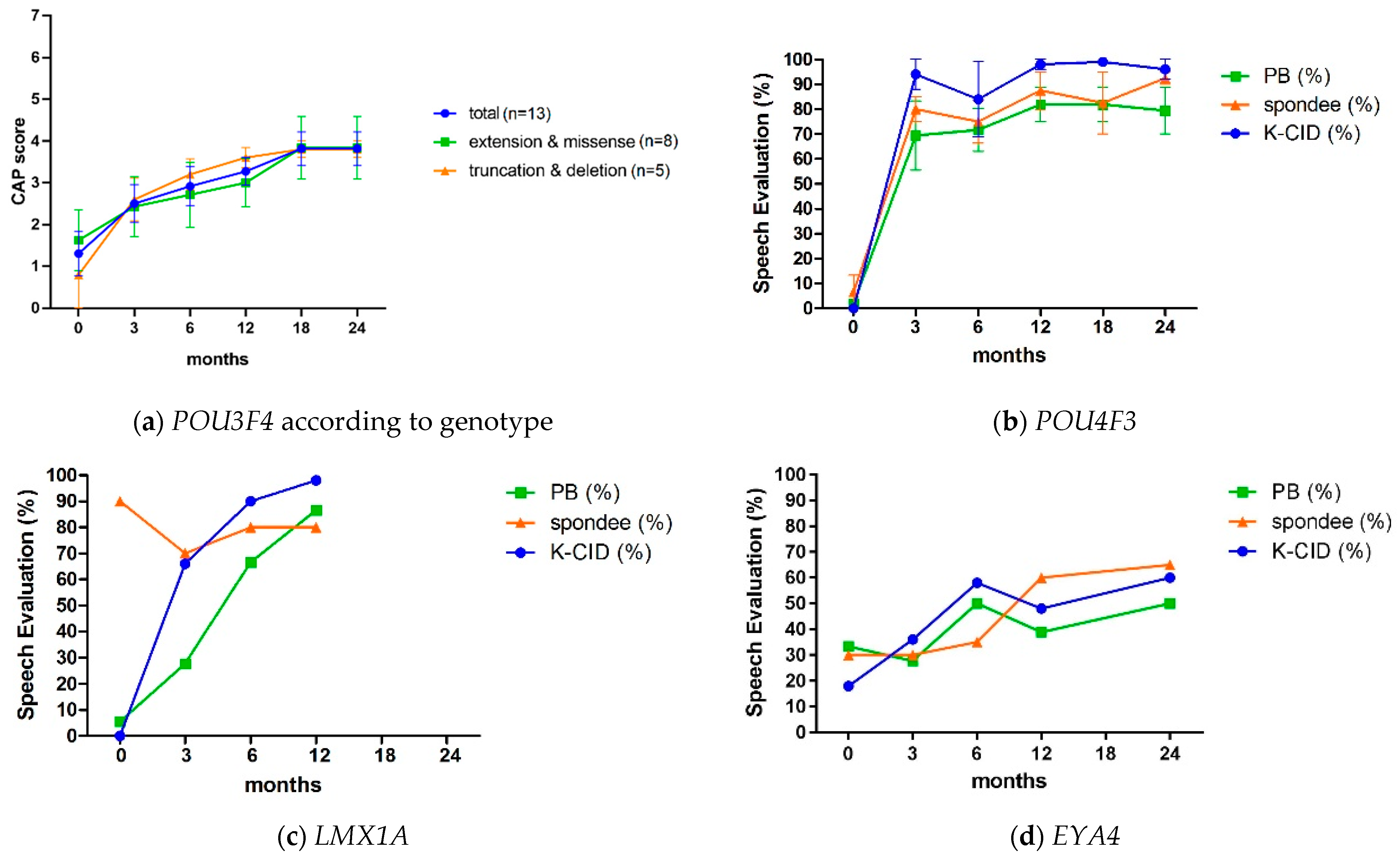
3.2.2. POU3F4: Audiological Profile and Cochlear Implantation Results
3.3. POU4F3
3.3.1. POU4F3: Genotype Profile
3.3.2. POU4F3: Audiological Profile and Cochlear Implantation Results
3.4. LMX1A
3.4.1. LMX1A: Genotype Profile
3.4.2. LMX1A: Audiological Profile and Cochlear Implantation Results
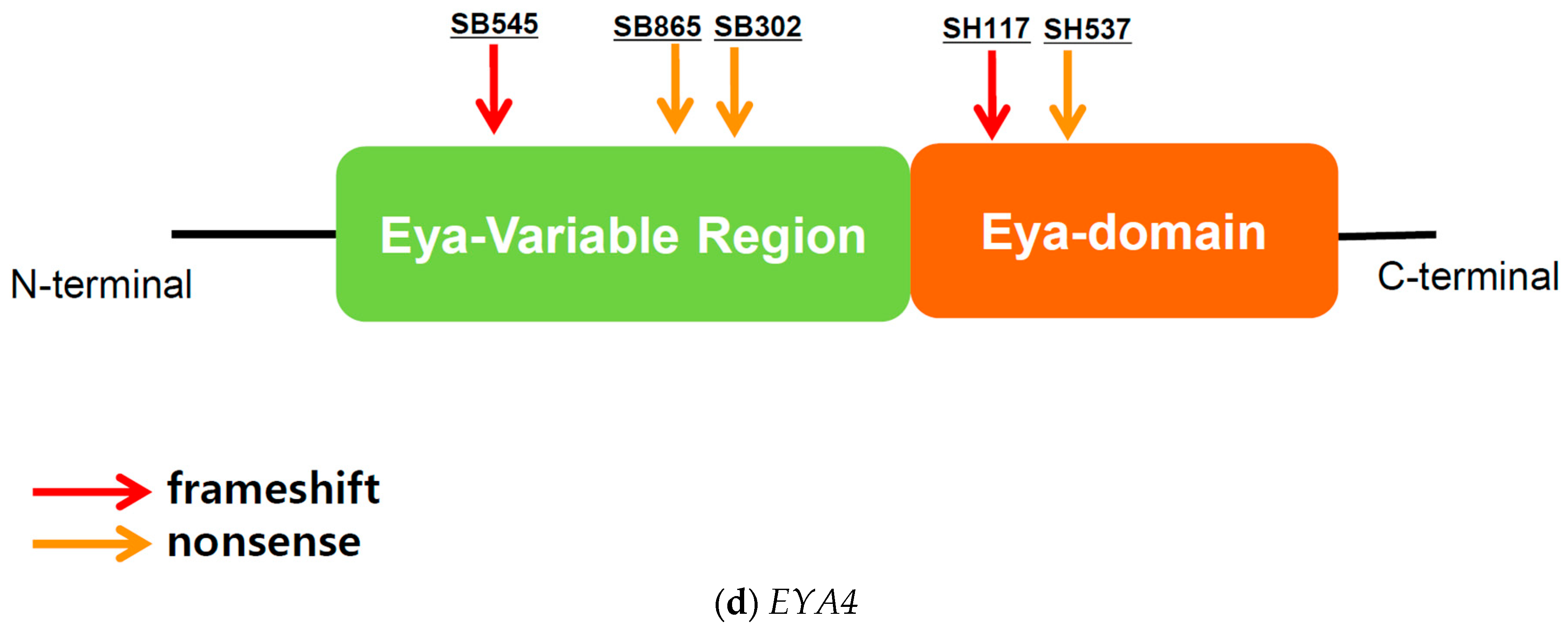
3.5. EYA4
3.5.1. EYA4: Genotype Profile
3.5.2. EYA4: Audiological Profile and Cochlear Implantation Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berninger, E.; Westling, B. Outcome of a universal newborn hearing-screening programme based on multiple transient-evoked otoacoustic emissions and clinical brainstem response audiometry. Acta Otolaryngol. 2011, 131, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, G.; Smith, R.J.H. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org (accessed on 1 February 2022).
- Delmaghani, S.; El-Amraoui, A. Inner Ear Gene Therapies Take Off: Current Promises and Future Challenges. J. Clin. Med. 2020, 9, 2309. [Google Scholar] [CrossRef]
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 12, 138–163. [Google Scholar] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef]
- Clark, J.G. Uses and abuses of hearing loss classification. ASHA 1981, 23, 493–500. [Google Scholar]
- Bahmad, F. Update on Hearing Loss; Bahmad, F., Ed.; IntechOpen Book Series; IntechOpen Limited: London, UK, 2015; p. 206. [Google Scholar]
- Liu, H.; Zhang, H.; Bentler, R.A.; Mo, L.; Han, D.; Zhang, L. Audiometric records analysis in a clinical population in China. ORL J. Otorhinolaryngol. Relat. Spec. 2011, 73, 237–245. [Google Scholar] [CrossRef]
- Pittman, A.L.; Stelmachowicz, P.G. Hearing loss in children and adults: Audiometric configuration, asymmetry, and progression. Ear Hear. 2003, 24, 198–205. [Google Scholar] [CrossRef]
- Lee, S.Y.; Han, S.C.; Han, J.H.; Kim, M.Y.; Oh, D.Y.; Kim, N.J.; Song, J.J.; Koo, J.W.; Lee, J.H.; Oh, S.H.; et al. Natural Course of Residual Hearing with Reference to GJB2 and SLC26A4 Genotypes: Clinical Implications for Hearing Rehabilitation. Ear Hear. 2021, 42, 644–653. [Google Scholar] [CrossRef]
- Sennaroglu, L.; Saatci, I. A new classification for cochleovestibular malformations. Laryngoscope 2002, 112, 2230–2241. [Google Scholar] [CrossRef]
- Lee, S.Y.; Choi, B.Y. Potential Implications of Slim Modiolar Electrodes for Severely Malformed Cochleae: A Comparison With the Straight Array With Circumferential Electrodes. Clin. Exp. Otorhinolaryngol. 2021, 14, 287–294. [Google Scholar] [CrossRef]
- Han, K.H.; Kim, A.R.; Kim, M.Y.; Ahn, S.; Oh, S.H.; Song, J.H.; Choi, B.Y. Establishment of a Flexible Real-Time Polymerase Chain Reaction-Based Platform for Detecting Prevalent Deafness Mutations Associated with Variable Degree of Sensorineural Hearing Loss in Koreans. PLoS ONE 2016, 11, e0161756. [Google Scholar] [CrossRef]
- Lee, S.Y.; Oh, D.Y.; Han, J.H.; Kim, M.Y.; Kim, B.; Kim, B.J.; Song, J.J.; Koo, J.W.; Lee, J.H.; Oh, S.H.; et al. Flexible Real-Time Polymerase Chain Reaction-Based Platforms for Detecting Deafness Mutations in Koreans: A Proposed Guideline for the Etiologic Diagnosis of Auditory Neuropathy Spectrum Disorder. Diagnostics 2020, 10, 672. [Google Scholar] [CrossRef]
- Lee, S.Y.; Joo, K.; Oh, J.; Han, J.H.; Park, H.R.; Lee, S.; Oh, D.Y.; Woo, S.J.; Choi, B.Y. Severe or Profound Sensorineural Hearing Loss Caused by Novel USH2A Variants in Korea: Potential Genotype-Phenotype Correlation. Clin. Exp. Otorhinolaryngol. 2020, 13, 113–122. [Google Scholar] [CrossRef]
- Lee, S.Y.; Choi, H.B.; Park, M.; Choi, I.S.; An, J.; Kim, A.; Kim, E.; Kim, N.; Han, J.H.; Kim, M.Y.; et al. Novel KCNQ4 variants in different functional domains confer genotype- and mechanism-based therapeutics in patients with nonsyndromic hearing loss. Exp. Mol. Med. 2021, 53, 1192–1204. [Google Scholar] [CrossRef]
- Lee, S.Y.; Han, J.H.; Carandang, M.; Kim, M.Y.; Kim, B.; Yi, N.; Kim, J.; Kim, B.J.; Oh, D.Y.; Koo, J.W.; et al. Novel genotype-phenotype correlation of functionally characterized LMX1A variants linked to sensorineural hearing loss. Hum. Mutat. 2020, 41, 1877–1883. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Korean Reference Genome Database. Available online: http://152.99.75.168:9090/KRGDB (accessed on 17 August 2022).
- Stankovic, K.M.; Hennessey, A.M.; Herrmann, B.; Mankarious, L.A. Cochlear implantation in children with congenital X-linked deafness due to novel mutations in POU3F4 gene. Ann. Otol. Rhinol. Laryngol. 2010, 119, 815–822. [Google Scholar] [CrossRef]
- Smeds, H.; Wales, J.; Karltorp, E.; Anderlid, B.M.; Henricson, C.; Asp, F.; Anmyr, L.; Lagerstedt-Robinson, K.; Lofkvist, U. X-linked Malformation Deafness: Neurodevelopmental Symptoms Are Common in Children With IP3 Malformation and Mutation in POU3F4. Ear Hear. 2021, 43, 53–69. [Google Scholar] [CrossRef]
- Minowa, O.; Ikeda, K.; Sugitani, Y.; Oshima, T.; Nakai, S.; Katori, Y.; Suzuki, M.; Furukawa, M.; Kawase, T.; Zheng, Y.; et al. Altered cochlear fibrocytes in a mouse model of DFN3 nonsyndromic deafness. Science 1999, 285, 1408–1411. [Google Scholar] [CrossRef]
- de Kok, Y.J.; van der Maarel, S.M.; Bitner-Glindzicz, M.; Huber, I.; Monaco, A.P.; Malcolm, S.; Pembrey, M.E.; Ropers, H.H.; Cremers, F.P. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 1995, 267, 685–688. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.Y.; An, Y.H.; Song, J.J.; Koo, J.W.; Lee, J.H.; Oh, S.H.; Chang, S.O.; Kim, C.S.; Park, J.H. Clinical observations and molecular variables of patients with hearing loss and incomplete partition type III. Laryngoscope 2016, 126, E123–E128. [Google Scholar] [CrossRef]
- Jang, J.H.; Oh, J.; Han, J.H.; Park, H.R.; Kim, B.J.; Lee, S.; Kim, M.Y.; Lee, S.; Oh, D.Y.; Choung, Y.H.; et al. Identification of a Novel Frameshift Variant of POU3F4 and Genetic Counseling of Korean Incomplete Partition Type III Subjects Based on Detailed Genotypes. Genet. Test. Mol. Biomark. 2019, 23, 423–427. [Google Scholar] [CrossRef]
- Tian, H.; Wang, L.; Gao, F.; Liang, W.; Peng, K.A. Cochlear implantation using a custom guide catheter in 14 patients with incomplete partition type III. Clin. Otolaryngol. 2018, 43, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.S.; Shim, B.S.; Lee, K.S. Audiologic performance after cochlear implantation in children with X-linked deafness: Comparison with deaf children with a normal inner ear structure. Otol. Neurotol. 2013, 34, 544–548. [Google Scholar] [CrossRef]
- Chao, X.; Xiao, Y.; Zhang, F.; Luo, J.; Wang, R.; Liu, W.; Wang, H.; Xu, L. Cochlear Implantation in a Patient with a Novel POU3F4 Mutation and Incomplete Partition Type-III Malformation. Neural. Plast. 2020, 2020, 8829587. [Google Scholar] [CrossRef]
- Vahava, O.; Morell, R.; Lynch, E.D.; Weiss, S.; Kagan, M.E.; Ahituv, N.; Morrow, J.E.; Lee, M.K.; Skvorak, A.B.; Morton, C.C.; et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 1998, 279, 1950–1954. [Google Scholar] [CrossRef]
- Weiss, S.; Gottfried, I.; Mayrose, I.; Khare, S.L.; Xiang, M.; Dawson, S.J.; Avraham, K.B. The DFNA15 deafness mutation affects POU4F3 protein stability, localization, and transcriptional activity. Mol. Cell. Biol. 2003, 23, 7957–7964. [Google Scholar] [CrossRef]
- Cui, T.Y.; Gao, X.; Huang, S.S.; Sun, Y.Y.; Zhang, S.Q.; Jiang, X.X.; Yang, Y.Z.; Kang, D.Y.; Zhu, Q.W.; Yuan, Y.Y. Four Novel Variants in POU4F3 Cause Autosomal Dominant Nonsyndromic Hearing Loss. Neural. Plast. 2020, 2020, 6137083. [Google Scholar] [CrossRef]
- Freitas, E.L.; Oiticica, J.; Silva, A.G.; Bittar, R.S.; Rosenberg, C.; Mingroni-Netto, R.C. Deletion of the entire POU4F3 gene in a familial case of autosomal dominant non-syndromic hearing loss. Eur. J. Med. Genet. 2014, 57, 125–128. [Google Scholar] [CrossRef]
- He, L.; Pang, X.; Chen, P.; Wu, H.; Yang, T. Mutation in the Hair Cell Specific Gene POU4F3 Is a Common Cause for Autosomal Dominant Nonsyndromic Hearing Loss in Chinese Hans. Neural. Plast. 2016, 2016, 9890827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.Z.; Li, Y.; Xia, L.; Peng, Y.; He, C.F.; Jiang, L.; Feng, Y.; Xia, K.; Liu, X.Z.; Mei, L.Y.; et al. Exome sequencing identifies POU4F3 as the causative gene for a large Chinese family with non-syndromic hearing loss. J. Hum. Genet 2017, 62, 317–320. [Google Scholar] [CrossRef]
- Gao, X.; Xu, J.C.; Wang, W.Q.; Yuan, Y.Y.; Bai, D.; Huang, S.S.; Wang, G.J.; Su, Y.; Li, J.; Kang, D.Y.; et al. A Missense Mutation in POU4F3 Causes Midfrequency Hearing Loss in a Chinese ADNSHL Family. Biomed. Res. Int. 2018, 2018, 5370802. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Lin, Y.H.; Lu, Y.C.; Liu, T.C.; Chen, C.Y.; Hsu, C.J.; Chen, P.L.; Wu, C.C. A novel missense variant in the nuclear localization signal of POU4F3 causes autosomal dominant non-syndromic hearing loss. Sci. Rep. 2017, 7, 7551. [Google Scholar] [CrossRef] [PubMed]
- Kitano, T.; Miyagawa, M.; Nishio, S.Y.; Moteki, H.; Oda, K.; Ohyama, K.; Miyazaki, H.; Hidaka, H.; Nakamura, K.I.; Murata, T.; et al. POU4F3 mutation screening in Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis identified novel variants associated with autosomal dominant hearing loss. PLoS ONE 2017, 12, e0177636. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, F.; Xiao, Y.; Jin, Y.; Zheng, Q.; Wang, H.; Xu, L. Identification of two novel mutations in POU4F3 gene associated with autosomal dominant hearing loss in Chinese families. J. Cell. Mol. Med. 2020, 24, 6978–6987. [Google Scholar] [CrossRef]
- Miyake, K.; Shirai, K.; Nishiyama, N.; Kawaguchi, S.; Ohta, Y.; Kawano, A.; Usami, S.I.; Kitano, T.; Tsukahara, K. Cochlear implantation in a patient with a POU4F3 mutation. Clin. Case Rep. 2021, 9, 298–303. [Google Scholar] [CrossRef]
- Eppsteiner, R.W.; Shearer, A.E.; Hildebrand, M.S.; Deluca, A.P.; Ji, H.; Dunn, C.C.; Black-Ziegelbein, E.A.; Casavant, T.L.; Braun, T.A.; Scheetz, T.E.; et al. Prediction of cochlear implant performance by genetic mutation: The spiral ganglion hypothesis. Hear. Res. 2012, 292, 51–58. [Google Scholar] [CrossRef]
- Wesdorp, M.; de Koning Gans, P.A.M.; Schraders, M.; Oostrik, J.; Huynen, M.A.; Venselaar, H.; Beynon, A.J.; van Gaalen, J.; Piai, V.; Voermans, N.; et al. Heterozygous missense variants of LMX1A lead to nonsyndromic hearing impairment and vestibular dysfunction. Hum. Genet 2018, 137, 389–400. [Google Scholar] [CrossRef]
- Schrauwen, I.; Chakchouk, I.; Liaqat, K.; Jan, A.; Nasir, A.; Hussain, S.; Nickerson, D.A.; Bamshad, M.J.; Ullah, A.; Ahmad, W.; et al. A variant in LMX1A causes autosomal recessive severe-to-profound hearing impairment. Hum. Genet 2018, 137, 471–478. [Google Scholar] [CrossRef]
- Ozieblo, D.; Lee, S.Y.; Leja, M.L.; Sarosiak, A.; Baldyga, N.; Skarzynski, H.; Kim, Y.; Han, J.H.; Yoo, H.S.; Park, M.H.; et al. Update on CD164 and LMX1A genes to strengthen their causative role in autosomal dominant hearing loss. Hum. Genet 2022, 141, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Yuan, Y.; Liu, Y.; Zhu, Q.; Dai, P. A novel EYA4 mutation causing hearing loss in a Chinese DFNA family and genotype-phenotype review of EYA4 in deafness. J. Transl. Med. 2015, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Makishima, T.; Madeo, A.C.; Brewer, C.C.; Zalewski, C.K.; Butman, J.A.; Sachdev, V.; Arai, A.E.; Holbrook, B.M.; Rosing, D.R.; Griffith, A.J. Nonsyndromic hearing loss DFNA10 and a novel mutation of EYA4: Evidence for correlation of normal cardiac phenotype with truncating mutations of the Eya domain. Am. J. Med. Genet. A 2007, 143, 1592–1598. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, A.R.; Kim, S.H.; Choi, B.Y. Identification of a novel truncation mutation of EYA4 in moderate degree hearing loss by targeted exome sequencing. Eur. Arch. Otorhinolaryngol. 2016, 273, 1123–1129. [Google Scholar] [CrossRef]
- Schonberger, J.; Wang, L.; Shin, J.T.; Kim, S.D.; Depreux, F.F.; Zhu, H.; Zon, L.; Pizard, A.; Kim, J.B.; Macrae, C.A.; et al. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat. Genet. 2005, 37, 418–422. [Google Scholar] [CrossRef]
- Shinagawa, J.; Moteki, H.; Nishio, S.Y.; Ohyama, K.; Otsuki, K.; Iwasaki, S.; Masuda, S.; Oshikawa, C.; Ohta, Y.; Arai, Y.; et al. Prevalence and clinical features of hearing loss caused by EYA4 variants. Sci. Rep. 2020, 10, 3662. [Google Scholar] [CrossRef]
- Morin, M.; Borreguero, L.; Booth, K.T.; Lachgar, M.; Huygen, P.; Villamar, M.; Mayo, F.; Barrio, L.C.; Santos Serrao de Castro, L.; Morales, C.; et al. Insights into the pathophysiology of DFNA10 hearing loss associated with novel EYA4 variants. Sci. Rep. 2020, 10, 6213. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Timing of HL | Genotype | Age at HL Detection | Type of HL | Audiogram Configuration | Degree of HL (Most Recent) | Asymmetry | HL Progression | Final Aural Rehabilitation | Age at CI | Allele Frequency e |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SB2-1 | M | prelingual | POU3F4[NM_000307.4]c.626A>G:p.Gln229Arg | 1 month | SNHL | Flat | profound | No | No | B) CI | R) 2 yr, L) 12 mo | absent |
| SB2-2 | M | prelingual | POU3F4[NM_000307.4]c.626A>G:p.Gln229Arg | 6 months | SNHL | Flat | profound | No | No | B) CI | R) 6 yr, L) 7 yr | absent |
| SB7 | M | prelingual | POU3F4[NM_000307.4]c.1060delA:p.Thr354Glnfs*115 | 12 months | SNHL | Flat | profound | No | No | CI | 2 yr | absent |
| SB8 | M | postlingual | POU3F4[NM_000307.4]c.950dupT:p.Leu317Phefs*12 | 35 months | MHL | Mixed HL | severe | No | Yesd (2.25 dB HL/yr) | HA | (-) | absent |
| SB9 | M | postlingual | POU3F4[NM_000307.4]c.632C>T:p.Thr211Met | 3 years | MHL | Mixed HL | severe | Yes (24 dB) | Yes (0.7 dB HL/yr) | HA | (-) | absent |
| SB11 | M | prelingual | POU3F4[NM_000307.4]c.1084T>C:p.X362Argext*113 | 3 years | SNHL | Flat | profound | No | No | CI | 12 yr | absent |
| SB13 | M | prelingual | POU3F4[NM_000307.4]c.623T>A:p.Leu208* | 15 months | SNHL | Flat | profound | No | Yes (0.8 dB HL/yr) | B) CI | R) 6 yr, L) 2yr | absent |
| SH17 | M | prelingual | Xq21.2, 80851535-82597832 bp | 1 month | SNHL | Flat | profound | No | Yes (4.7 dB HL/yr) | B) CI | R) 13 mo, L) 25 mo | absent |
| SB19 | M | prelingual | Xq21.2, 81810457-82810060 bp | 14 months | SNHL | Flat | profound | No | No | CI | 29 yr | absent |
| SH54 | M | postlingual | POU3F4[NM_000307.4]c.540C>A:p.Cys180* | 1 month | MHL | Mixed HL | severe | No | Yes (1.4 dB HL/yr) | HA | (-) | absent |
| SH65 | M | prelingual | POU3F4[NM_000307.4]c.910C>A:p.Pro303His | 1 month | SNHL | Downsloping | severe | Yes (15 dB) | Yes (0.5 dB HL/yr) | CI | 3 yr | absent |
| SH149 | M | prelingual | POU3F4[NM_000307.4]c.458delC:p.Pro153Leufs*88 | 3 months | MHL | Mixed HL | profound | Yes (18 dB) | Yes (2.5 dB HL/yr) | CI | 3 yr | absent |
| SH228 | M | prelingual | POU3F4[NM_000307.4]c.989G>A:p.Arg330Lys | unknown a | MHL | Mixed HL | severe | No | No | HA | (-) | absent |
| SB332 | M | prelingual | Xq21.2, deletion | 1 month | MHL | N/A b | severe | No | No | HA | (-) | absent |
| SB430 | M | prelingual | Xq21.2, deletion | 1 month | MHL | N/A b | severe | No | No | CI | 21 mo | absent |
| SH565-1 | M | prelingual | POU3F4[NM_000307.4]c.958G>T:p.Glu320* | 1 month | MHL | Mixed HL | moderate | No | No | HA | (-) | absent |
| SH565-2 | M | prelingual | POU3F4[NM_000307.4]c.958G>T:p.Glu320* | 2 month | MHL | Mixed HL | moderately severe | No | No | HA | (-) | absent |
| SB736 | M | prelingual | POU3F4[NM_000307.4]c.626A>G:p.Gln229Arg | 12 months | MHL | Mixed HL | profound | No | No | B) CI c | 10 yr | absent |
| SB218 | F | postlingual | POU4F3[NM_002700.2] c.564dupA:p.Ala189Serfs*26 | 30 years | SNHL | U-shaped | moderate | No | Yes (1.6 dB HL/yr) | B) MEI | (-) | absent |
| SB307 | F | postlingual | POU4F3[NM_002700.2] c.743T>C:p.Leu248Pro | 26 years | SNHL | U-shaped | moderately severe | No | Yes | HA | (-) | absent |
| SB347-1 | F | postlingual | POU4F3[NM_002700.2] c.952G>A:p.Val318Met | 16 years | MHL | Mixed HL | profound | No | Yes (16.7 dB HL/yr) | CI | 36 yr | absent |
| SB347-2 | F | postlingual | POU4F3[NM_002700.2] c.952G>A:p.Val318Met | 20 years | SNHL | Flat | profound | No | Yes | CI | 52 yr | absent |
| SB438-1 | F | postlingual | POU4F3[NM_002700.2] c.879C>A:p.Phe293Leu | unknown a | SNHL | Downsloping | mild | No | Yes | HA | (-) | absent |
| SB438-2 | F | postlingual | POU4F3[NM_002700.2] c.879C>A:p.Phe293Leu | 37 years | SNHL | Downsloping | moderately severe | No | Yes (2.3 dB HL/yr) | HA | (-) | absent |
| SB618-1 | M | prelingual | POU4F3[NM_002700.2] c.952G>A:p.Val318Met | unknown a | SNHL | U-shaped | moderate | No | Yes (5 dB HL/yr) | HA | (-) | absent |
| SB618-2 | M | unknown a | POU4F3[NM_002700.2] c.952G>A:p.Val318Met | unknown a | SNHL | U-shaped | severe | Yes (16 dB) | unknown a | HA | (-) | absent |
| SB618-3 | F | unknown a | POU4F3[NM_002700.2] c.952G>A:p.Val318Met | unknown a | SNHL | Downsloping | R) severe, L) profound | Yes (44 dB) | unknown a | HA | (-) | absent |
| SB709 | F | postlingual | POU4F3[NM_002700.2] c.662_675del:p.Gly221Glufs*77 | 39 years | SNHL | U-shaped | profound | No | Yes (10.6 dB HL/yr) | B) CI c | 36 yr | absent |
| SB481 | M | prelingual | LMX1A[NM_177398.4] c.595A>G:p.Arg199Gly | 1 months | SNHL | N/A b | R) profound, L) severe | Yes (15 dB) | unknown a | HA | (-) | absent |
| SB727 | F | postlingual | LMX1A[NM_177398.4] c.622C>T:p.Arg208* | 13 years | SNHL | Downsloping | R) moderate, L) profound | Yes (36 dB) | Yes | CI | 32 yr | absent |
| SH407 | F | postlingual | LMX1A[NM_177398.4] c.887dup:p.Gln297Thrfs*41 | 20 years | SNHL | Downsloping | R) moderately severe, L) moderate | Yes (18 dB) | fluctuation | HA | (-) | absent |
| SB742-1 | F | prelingual | LMX1A[NM_177398.4] c.719A>G:p.Gln240Arg | 2 months | SNHL | N/A b | R) moderate, L) profound | Yes (45 dB) | No | HA | (-) | absent |
| SB742-2 | F | postlingual | LMX1A[NM_177398.4] c.719A>G:p.Gln240Arg | 20 years | SNHL | Downsloping | moderately severe | No | Yes | (-) | (-) | absent |
| SH421-1 | F | prelingual | LMX1A[NM_177398.4] c.721G>A:p.Val241Met | 4 months | SNHL | N/A b | moderate | No | unknown a | HA | (-) | absent |
| SH421-2 | M | postlingual | LMX1A[NM_177398.4] c.721G>A:p.Val241Met | 17 years | SNHL | Downsloping | R) profound, L) moderate | Yes (61 dB) | No | HA | (-) | absent |
| SH512-1 | F | prelingual | LMX1A[NM_177398.4] c.331del:p.Gln111Argfs*7 | 3 months | SNHL | N/A b | moderate | No | unknown a | (-) | (-) | absent |
| SH512-2 | F | prelingual | LMX1A[NM_177398.4] c.331del:p.Gln111Argfs*7 | 1 year | SNHL | Downsloping | severe | No | unknown a | HA | (-) | absent |
| SB302-1 | F | postlingual | EYA4[NM_004100.5] c.697C>T:p.Gln233* | 35 years | SNHL | U-shaped | moderate | No | Yes | (-) | (-) | 1/3444 (KRGDB) |
| SB302-2 | F | postlingual | EYA4[NM_004100.5] c.697C>T:p.Gln233* | 40 years | SNHL | Flat | severe | No | Yes | HA | (-) | 1/3444 (KRGDB) |
| SB545 | F | postlingual | EYA4[NM_004100.5] c.208+1del | 50 years | MHL | Downsloping | severe | No | Yes | CI | 80 yr | absent |
| SB865 | F | postlingual | EYA4[NM_004100.5] c.578dup:p.Tyr193* | 10 years | MHL | Downsloping | severe | Yes (54 dB) | No | HA | (-) | absent |
| SH537 | F | postlingual | EYA4[NM_004100.5] c.1468G>T:p.Glu490* | 45 years | SNHL | Flat | moderately severe | No | Yes | HA | (-) | 1/3444 (KRGDB) |
| SH117-1 | F | postlingual | EYA4[NM_004100.5] c.1194del:p.Met401Trpfs*3 | 15 years | SNHL | Downsloping | moderate | No | Yes | HA | (-) | absent |
| SH117-2 | M | postlingual | EYA4[NM_004100.5] c.1194del:p.Met401Trpfs*3 | unknown a | SNHL | Downsloping | severe | No | unknown a | (-) | (-) | absent |
| SH117-3 | F | postlingual | EYA4[NM_004100.5] c.1194del:p.Met401Trpfs*3 | unknown a | SNHL | Downsloping | moderate | No | unknown a | (-) | (-) | absent |
| Patient | Genotype | dbSNP ID (dbSNP v151) | Zygosity | Inheritance | ACMG Guideline | |
|---|---|---|---|---|---|---|
| Classification | Criteria | |||||
| SH149 | POU3F4[NM_000307.5]c.458delC:p.Pro153Leufs*88 | absent | hemizygote | XLR | Likely pathogenic | PVS1, PM2, PP4 |
| SH228 | POU3F4[NM_000307.5]c.989G>A:p.Arg330Lys | absent | hemizygote | XLR | Likely pathogenic | PS2_moderate, PM2, PP3, PP4 |
| SH565 | POU3F4[NM_000307.5]c.958G>T:p.Glu320* | absent | hemizygote | XLR | Likely pathogenic | PVS1, PM2, PP4 |
| SB736 | POU3F4[NM_000307.5]c.499C>T:p.Arg167* | rs111033345 | hemizygote | XLR | Pathogenic | PVS1, PM2, PP1_strong, PM3_strong, PP4 |
| SH512 | LMX1A[NM_177398.4] c.331del:p.Gln111Argfs*7 | absent | heterozygote | AD | Likely pathogenic | PVS1, PM2 |
| SB302 | EYA4[NM_004100.5] c.697C>T:p.Gln233* | rs1583346685 | heterozygote | AD | Likely pathogenic | PVS1, PM2 |
| SB545 | EYA4[NM_004100.5] c.208+1del | absent | heterozygote | AD | Likely pathogenic | PVS1, PM2, PP3 |
| SB865 | EYA4[NM_004100.5] c.578dup:p.Tyr193* | absent | heterozygote | AD | Likely pathogenic | PVS1, PM2, PP1_mod |
| SH537 | EYA4[NM_004100.5] c.1468G>T:p.Glu490* | rs1305000119 | heterozygote | AD | Likely pathogenic | PVS1, PM2 |
| Cochlear Implantees | Preoperative | Post-CI 3 Months | Post-CI 6 Months | Post-CI 12 Months | Post-CI 18 Months | Post-CI 24 Months |
|---|---|---|---|---|---|---|
| POU3F4 (n = 13) | 1.3 | 2.6 | 3.0 | 3.4 * | 3.7 * | 3.8 * |
| GJB2 control ** (n = 26) | 1.1 | 2.2 | 3.8 | 5.8 | 6.2 | 6.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, H.D.; Han, J.H.; Lee, S.M.; Choi, D.H.; Lee, S.-Y.; Choi, B.Y. Genetic Load of Alternations of Transcription Factor Genes in Non-Syndromic Deafness and the Associated Clinical Phenotypes: Experience from Two Tertiary Referral Centers. Biomedicines 2022, 10, 2125. https://doi.org/10.3390/biomedicines10092125
Jo HD, Han JH, Lee SM, Choi DH, Lee S-Y, Choi BY. Genetic Load of Alternations of Transcription Factor Genes in Non-Syndromic Deafness and the Associated Clinical Phenotypes: Experience from Two Tertiary Referral Centers. Biomedicines. 2022; 10(9):2125. https://doi.org/10.3390/biomedicines10092125
Chicago/Turabian StyleJo, Hyung Dong, Jin Hee Han, So Min Lee, Dong Hwa Choi, Sang-Yeon Lee, and Byung Yoon Choi. 2022. "Genetic Load of Alternations of Transcription Factor Genes in Non-Syndromic Deafness and the Associated Clinical Phenotypes: Experience from Two Tertiary Referral Centers" Biomedicines 10, no. 9: 2125. https://doi.org/10.3390/biomedicines10092125
APA StyleJo, H. D., Han, J. H., Lee, S. M., Choi, D. H., Lee, S.-Y., & Choi, B. Y. (2022). Genetic Load of Alternations of Transcription Factor Genes in Non-Syndromic Deafness and the Associated Clinical Phenotypes: Experience from Two Tertiary Referral Centers. Biomedicines, 10(9), 2125. https://doi.org/10.3390/biomedicines10092125