Abstract
Salivary gland tumors are a heterogeneous group of tumors originating from the major and minor salivary glands. The pleomorphic adenoma (PA), which is the most common subtype, is a benign lesion showing a remarkable morphologic diversity and that, upon recurrence or malignant transformation, can cause significant clinical problems. Cytogenetic studies of >500 PAs have revealed a complex and recurrent pattern of chromosome rearrangements. In this review, we discuss the specificity and frequency of these rearrangements and their molecular/clinical consequences. The genomic hallmark of PA is translocations with breakpoints in 8q12 and 12q13-15 resulting in gene fusions involving the transcription factor genes PLAG1 and HMGA2. Until recently, the association between these two oncogenic drivers was obscure. Studies of the Silver–Russel syndrome, a growth retardation condition infrequently caused by mutations in IGF2/HMGA2/PLAG1, have provided new clues to the understanding of the molecular pathogenesis of PA. These studies have demonstrated that HMGA2 is an upstream regulator of PLAG1 and that HMGA2 regulates the expression of IGF2 via PLAG1. This provides a novel explanation for the 8q12/12q13-15 aberrations in PA and identifies IGF2 as a major oncogenic driver and therapeutic target in PA. These studies have important diagnostic and therapeutic implications for patients with PA.
1. Introduction
Salivary gland tumors are a large and heterogeneous group of neoplasms originating from the major salivary glands (the parotid, submandibular, and sublingual glands) as well as from the numerous minor salivary glands in the oral mucosa and upper aerodigestive tract. The diversity of histologic subtypes originating from these glands is remarkable and there are more than 30 known histological subtypes of benign and malignant salivary gland tumors of which the pleomorphic adenoma (PA) is the most common [1,2]. Mucoepidermoid carcinomas and adenoid cystic carcinomas are the two most common malignant salivary gland tumors.
PA is a benign rather slow-growing epithelial tumor of which approximately three-quarters are located in the parotid gland. They can occur in all age groups but most often in the 5th and 6th decades and with a slight female predominance [3,4]. The etiology is unknown but there is a reported association to radiation exposure [5,6]. PAs are usually encapsulated and are recognized for their morphological diversity [7]. They are composed of ductal epithelial and myoepithelial cells growing in a variety of patterns in a stroma that is often mucoid, myxoid, hyalinized, or chondroid. Some tumors may be predominantly cellular with only scanty stroma. Mitotic figures are rare. PA frequently shows metaplastic changes, of which squamous metaplasia is the most common. Oncocytic and sebaceous metaplasia are other frequent metaplastic changes that can confuse in the diagnostic work up [8]. Because of the broad morphological spectrum of PAs, they may sometimes mimic malignancy and show morphological/architectural overlap with for example adenoid cystic carcinoma and polymorphous adenocarcinoma [8].
PAs may undergo malignant transformation to carcinoma-ex-PA (CXPA). The risk of malignant transformation is greater in long-standing or recurrent tumors and occurs in about 12% of recurrent PAs [9]. CXPA is usually a high-grade tumor with rapid and aggressive growth and frequent recurrences and metastases. The carcinoma component can be of any type, most often salivary duct carcinoma (SDC), myoepithelial carcinoma (MECA), or adenocarcinoma NOS.
PA was the first benign epithelial tumor in which characteristic chromosome translocations and gene fusions were identified [10,11,12,13,14,15]. Recurrent t(3;8)(p21;q12) and t(9;12)(p23-24;q14-15) translocations were shown to be early cytogenetic events [16]. Subsequent molecular cloning of the translocation breakpoints in these and other translocations in PA revealed that they consistently result in gene fusions. The prime molecular targets of these translocations are the transcription factor genes PLAG1 (Pleomorphic Adenoma Gene 1; located in 8q12) and HMGA2 (High Mobility Group AT-Hook 2; located in 12q14-15) [11,12,13,17,18,19]. Notably, the early cytogenetic and molecular genetic studies of translocations in PA during the 80s and 90s paved the way for the discovery of a diagnostically relevant gene fusion network in PA as well as in several other subtypes of salivary gland tumors [18,20,21]. The aim of this paper is to review the comprehensive literature on chromosome translocations/rearrangements and gene fusions in PA and to discuss their molecular consequences and clinical significance.
2. The Cytogenetic Landscape of PA
2.1. Overview of the Chromosomal Pattern in PA
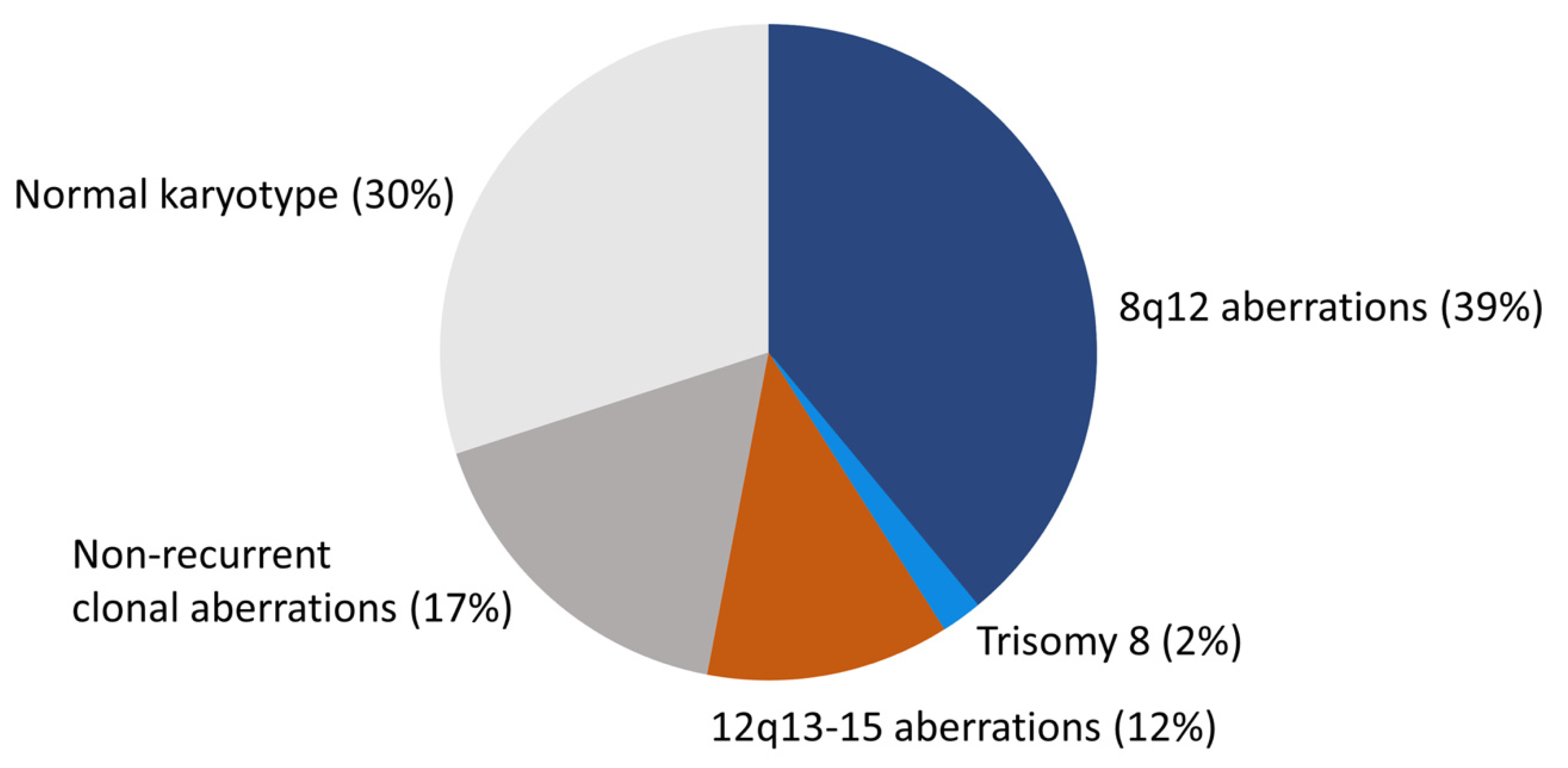
PA is cytogenetically the best-studied benign epithelial neoplasm. Studies by in particular two groups have revealed a consistent pattern of chromosome translocations/rearrangements in about 70% of the cases [10,14,16,19,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. The remaining 30% have shown normal karyotypes without visible chromosome rearrangements (Figure 1). Detailed analysis of high-resolution banded chromosomes have failed to detect cytogenetic alterations in these PAs. However, molecular analyses have unequivocally demonstrated that they have submicroscopic changes, including inversions, small insertions, or deletions resulting in gene fusions (see below) [13,19,37,38].
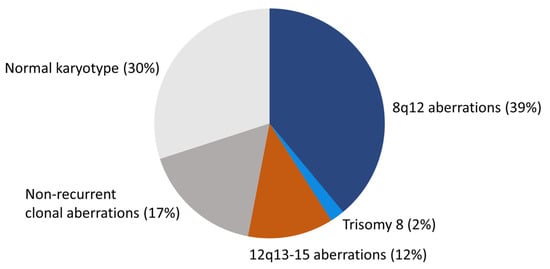
Figure 1.
The cytogenetic landscape of PA. Pie chart showing the different cytogenetic subgroups and their frequencies in PA.
The Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer contains cytogenetic data from 354 salivary gland PAs with abnormal karyotype [36]. Since tumors with normal karyotypes (≈30%) are not included in the database, the total number of PAs that have been cytogenetically characterized is more than 500 [10,14,16,19,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. PAs with an abnormal karyotype form four major subgroups: (i) tumors with translocations/rearrangements involving chromosome 8q12 (≈39%), (ii) tumors with translocations/rearrangements involving chromosome 12q13-15 (≈12%), (iii) tumors with complete or partial trisomy 8 (≈2%), and (iv) tumors with non-recurrent clonal aberrations without involvement of 8q12 or 12q13-15 (≈17%) (Figure 1).
The t(3;8)(p21;q12) translocation is the most common aberration in the 8q12 subgroup (≈47%), followed by t(8;9)(q12;p22-24) or the related ins(9;8)(p23;q11q12) (≈10%), t(5;8)(p13-15;q12) (≈4%), t(6;8)(p21-22;q12) (≈3%), t(8;10)(q12;q22-23) (≈2%), and t(8;15)(q12;q26) (≈2%). There is also a variety of 8q12 variant translocations with other less frequent translocation partners.
The t(9;12)(p21-23;q13-15) and the related ins(9;12)(p23:q14q15) or ins(12;9)(q15;p24p22) (≈16%) are the most common aberrations in the 12q13-15 subgroup, followed by t(3;12)(p12-14;q14-15) (≈5%), t(6;12)(q21-23;q14-15) (≈5%), inv(12)(p11-13q13-14) (≈5%), and inv(12)(q13-15q23-24) (≈5%). Similar to the 8q12 subgroup, there are also several different 12q13-15 variant translocations with other less frequent translocation partners.
Trisomy 8 forms the smallest subgroup of PAs. In 65% of these cases, +8 is seen as the sole aberration. In the remaining cases, it is seen together with other aberrations, in particular 12q13-15 rearrangements. In addition to trisomy 8, there are also several cases reported with partial gain of one to four copies of chromosome 8 in the form of ring chromosomes r(8)(p12q12) (see below) [39]. Complete or partial trisomy 8 is the only major numerical aberration found in PA.
The fourth major subgroup of PAs consists of tumors with non-recurrent clonal aberrations without the involvement of 8q12 or 12q13-15. This is a heterogeneous group of tumors some of which have a single translocation as the sole deviation whereas others have more complex karyotypes with both structural and numerical aberrations. The molecular pathogenesis of these tumors remains to be elucidated. However, previous studies have indicated that at least some of these cases have cryptic rearrangements involving 8q12 [38].
2.2. Double Minute Chromosomes and Homogeneously Staining Regions
A subset of PAs with 12q13-15 aberrations, in particular those with del(12)(q13q15), show cytogenetic evidence of gene amplification in the form of double minute chromosomes (dmin) and homogeneously staining regions (hsr) [35,40,41,42]. Detailed molecular characterization of these aberrations have shown that the prime targets of the amplifications are the HMGA2 and MDM2 genes [42]. Other less frequently co-amplified genes are WIF1, TSPAN31, CDK4, and GLI1. Notably, in several cases a cryptic HMGA2::WIF1 fusion gene was highly amplified and overexpressed [42]. Previous studies have also suggested that PAs with amplification of MDM2 and possibly also other driver genes in 12q have an increased risk of malignant transformation [42,43,44]. However, this hypothesis needs to be confirmed by studies of additional cases of CXPA and by in vitro transformation experiments.
2.3. Ring Chromosomes and Dicentric Chromosomes
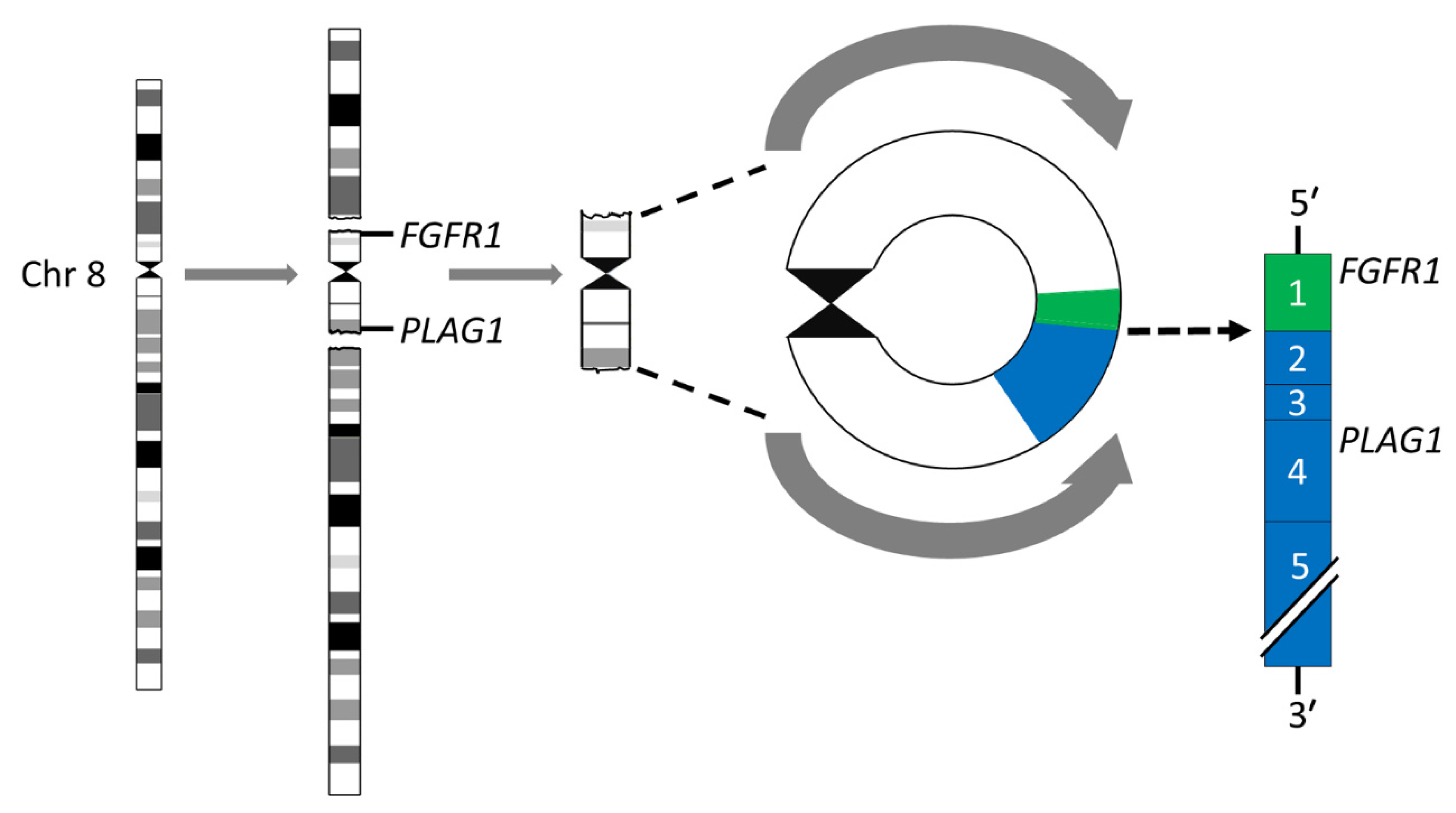
Breakage-fusion-bridge (BFB) cycles [45,46] is a mechanism that generates chromosome variability and mitotically unstable chromosome aberrations such as ring chromosomes and dicentric chromosomes [47]. Ring chromosomes have been found in ≈8% of karyotypically aberrant PAs [24,26,35,36,39,48]. The most common rings are derived from chromosomes 8 and 5, in that order of frequency. The rings may vary in both size and number in a given tumor. Notably, we have previously shown that the r(8)(p12q12.1) consists of a pericentromeric segment with recurrent breakpoints in FGFR1 in 8p12 and PLAG1 in 8q12.1 with amplification and overexpression of an FGFR1::PLAG1 gene fusion (Figure 2) [39]. Importantly, this fusion has also been shown to be enriched 15-fold in myoepithelial carcinoma-ex-PA (MECAXPA) compared to PA, suggesting that amplification and overexpression of the FGFR1::PLAG1 fusion may be a potential biomarker for malignant transformation of PA [49].
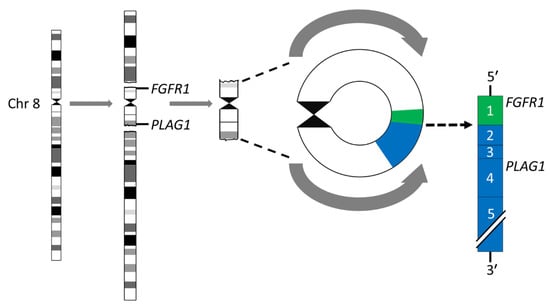
Figure 2.
Ring chromosome 8 in PA. Schematic illustration of the formation of the ring chromosome r(8)(p12q12.1) and the resulting FGR1::PLAG1 gene fusion in which exon 1 of FGR1 (green) is fused to exon 2 of PLAG1 (blue).
Rings derived from other chromosomes have so far not resulted in gene fusions, but instead losses of segments of for example 8p, 5p, 5q, and/or 6q [39], indicating the presence of putative tumor suppressor genes in these regions. Dicentric chromosomes are rarer than rings and are seen in only 1.5% of cytogenetically abnormal PAs. The majority of these involve chromosome 8 with breakpoints in 8q12.
2.4. Polyclonal Aberrations in Radiation-Associated PAs
Cytogenetic studies on radiation-associated PAs are rare. To the best of our knowledge, there are only two such cases reported in which PA developed in patients previously treated with radiotherapy for tuberculous lymphadenitis in the neck [5,50]. Both cases were cytogenetically polyclonal with a variety of mainly unique structural aberrations, but without involvement of the PA-specific breakpoints 8q12 and 12q13-15. Subsequent molecular analyses revealed that both tumors had activation of PLAG1 and HMGA2 and one of the cases had a cryptic CTNNB1::PLAG1 fusion [6]. Studies of these two unique cases demonstrate that polyclonal, radiation-associated PAs develop as a result of very similar basic molecular mechanisms as sporadic PAs with monoclonal karyotypes.
3. The Gene Fusion Landscape of PA
3.1. The Transcription Factor Gene PLAG1 Is the Target of Translocations and Rearrangements of 8q12 in PA
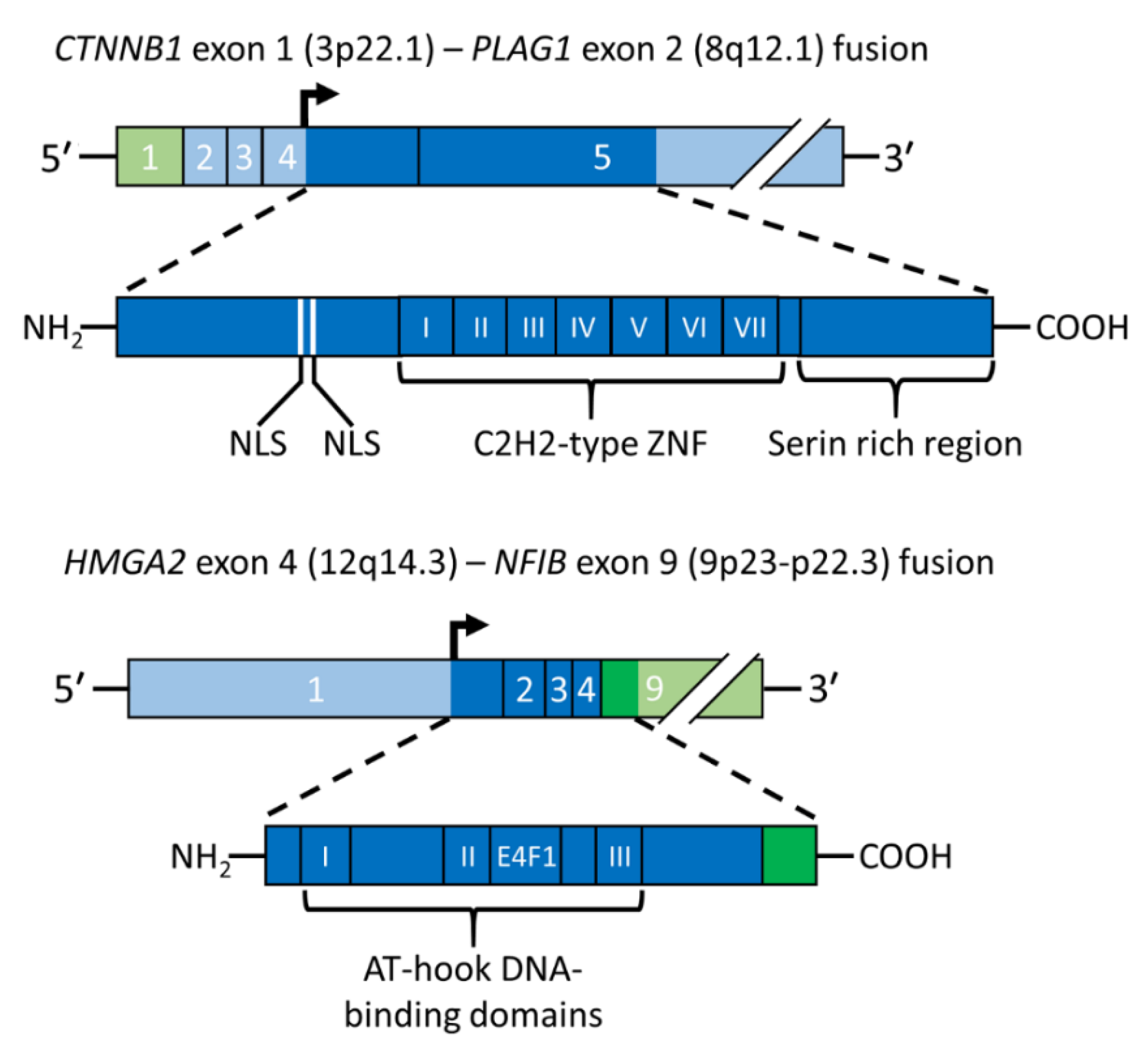
PA was the first benign epithelial tumor in which a tumor type-specific translocation was shown to result in a gene fusion. Positional cloning of the breakpoints in the t(3;8)(p21;q12) translocation revealed that it generates a fusion between the PLAG1 gene in 8q12 and CTNNB1 gene in 3p21 [11] (Figure 3, upper panel). The PLAG1 oncogene encodes a developmentally regulated transcription factor that is highly expressed in various fetal tissues, but whose expression is low or absent in adult tissues, including normal salivary gland [11,37]. PLAG1 has seven canonical C2H2 zinc finger domains in the N-terminal part of the protein and a serine-rich transcriptional activation domain in its C-terminal part [11,51]. The PLAG1 oncoprotein is involved in the regulation of crucial cellular processes such as transcriptional regulation, growth control, cell proliferation, and apoptosis [52,53]. Its fundamental role in growth control is further emphasized by studies of knockout mice showing that the pre- and postnatal growth of Plag1−/− mice is significantly retarded [54]. The finding that the growth factor IGF2 is a major downstream target of PLAG1 [55] also agrees with this finding. PLAG1 binds the IGF2 P3 promoter and aberrantly activates its expression in PAs overexpressing PLAG1 [55]. Other known PLAG1 targets are BCL2, CDKN1C, CRABP2, CYTL1, EFNB1, SMARCD3, and TSPAN4 [53].
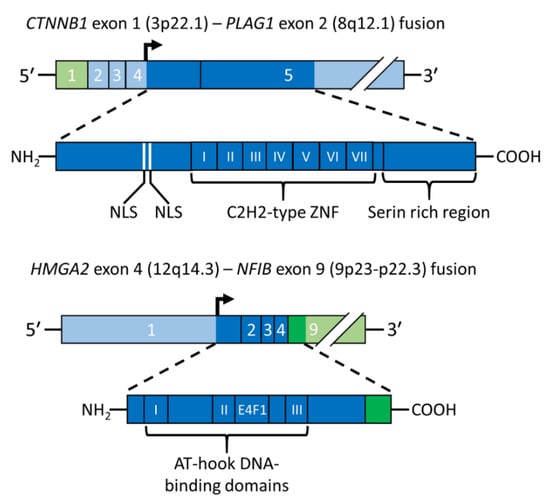
Figure 3.
Schematic illustration of gene fusions in PA. The CTNNB1::PLAG1 fusion gene and the encoded fusion protein is depicted in the upper panel and the HMGA2::NFIB fusion gene and the encoded fusion protein in the lower panel. Coding exons are shown in darker green and blue and the translation start site is indicated by an arrow. AT-hook, DNA binding motif; E4F1, E4F1-domain; NLS, nuclear localization signal; ZNF, zinc finger.
CTNNB1 is the most common fusion partner gene to PLAG1 in PA. CTNNB1 encodes β-catenin, a ubiquitously expressed protein involved in the regulation of cell–cell adhesion, cell proliferation, and differentiation. β-catenin acts as an intracellular signal transducer in the Wnt signaling pathway and is inhibited by the tumor suppressor protein APC [56]. CTNNB1 is mutated in a variety of solid tumors [56,57].
The t(3;8) translocation in PA results in promoter swapping between PLAG1 and CTNNB1 leading to activation of PLAG1 expression [11] (Figure 3, upper panel). The CTNNB1::PLAG1 fusion was the first example of promoter swapping in solid tumors. The breakpoints in PLAG1 and CTNNB1 occur in the non-coding regions of both genes, leading to exchange of regulatory control elements with the coding sequences preserved. Subsequent studies of other translocations have confirmed that ectopic overexpression of a normal PLAG1 oncoprotein due to promoter swapping is a recurrent theme in PA. Thus, also the t(8;9)/ins(9;8) and t(5;8), which are the second and third most common aberrations in the 8q12 subgroup of PAs, result in exchange of regulatory sequences between PLAG1 and the NFIB and LIFR genes, respectively [17,37]. Notably, NFIB is also a fusion partner of HMGA2 in PA and of MYB in salivary gland adenoid cystic carcinomas (see below) [13,37,58,59].
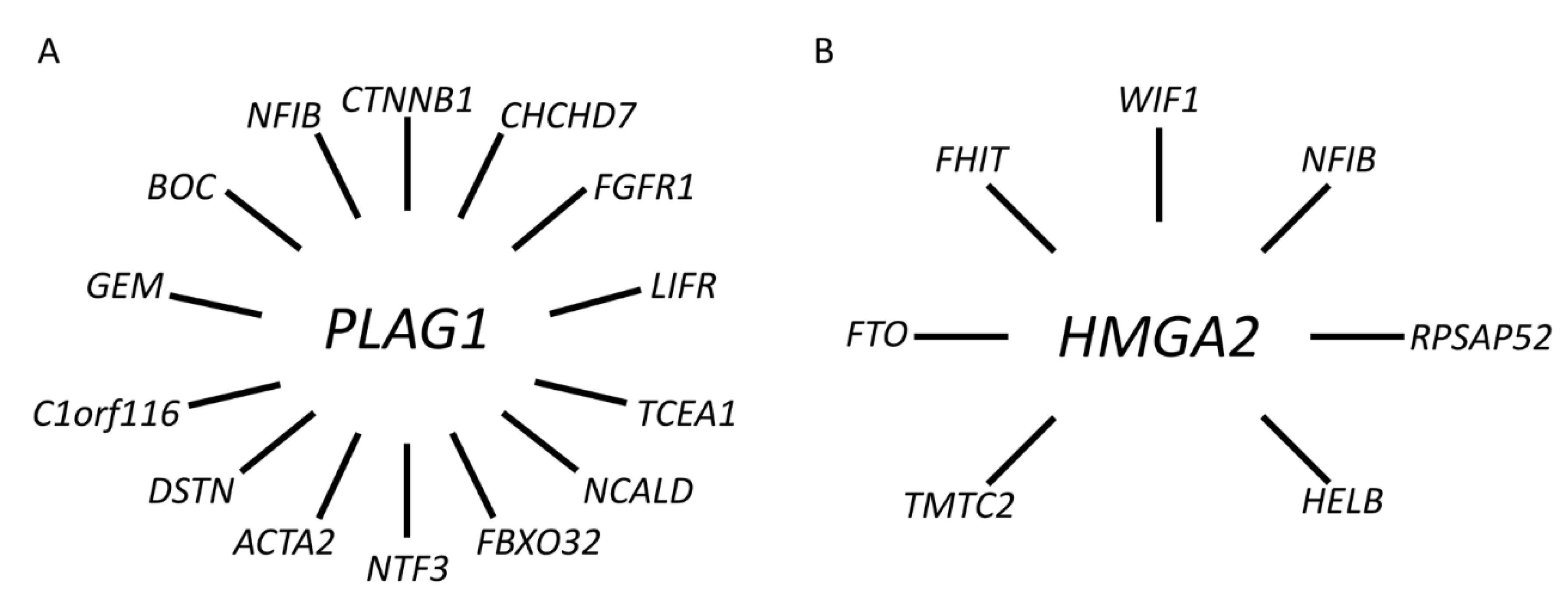
To date, we and others have identified a network of PLAG1 fusions in PA involving at least 11 additional fusion partner genes, including CHCHD7, FGFR1, TCEA1, NCALD, FBXO32, NTF3, ACTA2, DSTN, C1orf116, GEM, and BOC [38,39,60,61,62] (Figure 4A and Supplementary Table S1).
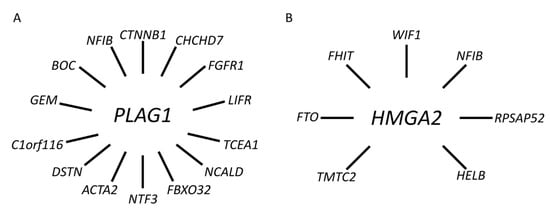
Figure 4.
Gene fusion network in PA. (A) PLAG1 and its 14 known fusion partner genes. (B) HMGA2 and its seven known fusion partner genes.
It should be emphasized that several of these fusions, including e.g., CHCHD7::PLAG1 and TCEA1::PLAG1, are cryptic fusions generated by intrachromosomal rearrangements [38]. For example, CHCHD7 and PLAG1 are located head-to-head only 500 bp apart and due to a paracentric inversion PLAG1 is activated by promoter swapping. The common denominator for all fusion partners is that they are either ubiquitously expressed or highly expressed in normal salivary gland. Importantly, the fusions are generated both by cytogenetically visible translocations/aberrations and cryptic intra- and interchromosomal rearrangements [38]. The latter are often, but not exclusively, found in PAs with an apparently normal karyotype [19]. The diversity of chromosome 8q12 aberrations in PA suggest that additional PLAG1 fusion partners are likely to be found.
In summary, the 8q12 translocations/aberrations in PA result in ectopic activation of PLAG1 expression due to promoter swapping with a variety of different, mostly ubiquitously expressed, fusion partner genes. The oncogenic activity of PLAG1 is partly due to activation of IGF2-signaling, suggesting that the IGF2-pathway is a potential therapeutic target in PA.
3.2. HMGA2 Is the Target Gene of Translocations and Rearrangements of 12q13-15 in PA
Parallel to the discovery of the PLAG1 gene as the target of the 8q12 aberrations in PA [11] we could demonstrate by positional cloning that the target of the 12q13-15 rearrangements in PA was the HMGA2 gene (previously known as HMGIC) [12,13]. HMGA2 is a member of the non-histone high-mobility group (HMG) protein gene family and encodes an architectural transcription factor [63,64,65,66]. HMG-proteins are essential components of enhanceosomes and are involved in the regulation of gene transcription, recombination, and chromatin structure. The HMGA2 protein has three AT-hook DNA-binding domains in its N-terminus, a spacer domain, and an acidic C-terminus. Examples of known down-stream targets of HMGA2 are PLAG1 and the cell cycle regulator CCNA1 [67,68].
Detailed genomic analyses of the 3p and 12q breakpoints in a PA with a complex karyotype, including an ins(3;12)(p14.2;q14q15), revealed an HMGA2-fusion linking the first three exons (encoding the DNA-binding domains) of HMGA2 to the two last coding exons of the FHIT gene in 3p14.2 [12]. The deduced HMGA2::FHIT fusion protein consists of the N-terminal part of HMGA2, including the three DNA-binding domains, fused to the C-terminal part of FHIT which encodes the last 31 amino acids of the protein. Subsequent molecular analysis of a PA with an ins(9;12) confirmed that also this rearrangement involves HMGA2 and results in a fusion of the 3′-part of the gene linked to the last coding exon of the NFIB gene which only encodes five amino acids [13]. Notably, it was also shown that a PA with an apparently normal karyotype had a cryptic ins(9;12) generating an HMGA2::NFIB fusion. Later studies confirmed that also the t(9;12)(p21-23;q13-15), which together with the ins(9;12) variant are the most common aberrations in the 12q13-15 subgroup of PAs, result in HMGA2::NFIB gene fusions [37] (Figure 3, lower panel). Notably, NFIB is so far the only fusion partner gene that is shared between PLAG1 and HMGA2. NFIB is also a fusion partner to the MYB oncogene in adenoid cystic carcinoma [58,69]. In addition to being highly expressed in normal salivary gland, NFIB also contains several super-enhancers in the 5′- and 3′-parts of the gene and its flanking sequences [37,70], suggesting that enhancer-hijacking events may also contribute to the activation of PLAG1 and HMGA2 in PAs [37].
In addition to NFIB and FHIT, there are at least five additional known fusion partner genes to HMGA2 in PA, including FTO, HELB, TMTC2, RPSAP52, and WIF1 [71,72,73,74] (Figure 4B and Supplementary Table S1). Available data indicate that the latter, together with NFIB, are the two most common fusion partners [37,42,72,74]. WIF1 is located only 0.7 Mb centromeric to HMGA2 and the HMGA2::WIF1 fusion is thus generated by a cryptic intrachromosomal rearrangement [42]. Previous studies have shown that the HMGA2::WIF1 fusion is also amplified together with several other closely linked genes in 12q. Interestingly, Agaimy and co-workers recently suggested that PAs with HMGA2::WIF1 fusions are characterized by a prominent trabecular and canalicular adenoma-like morphology [72]. The molecular mechanism by which the fusions activate HMGA2 is only partly known. In addition to enhancer-hijacking, it has been suggested that binding sites for negatively regulating microRNAs in the 3′-UTR of HMGA2 are lost in the fusions that may contribute to activation of the gene [18,75]. However, additional studies are needed to fully understand the mechanisms by which HMGA2 is activated in PA.
4. The 8q12 and 12q13-15 Rearrangements in PA Activate the HMGA2-PLAG1-IGF2 Pathway
As discussed in this review, the genomic hallmarks of PA are translocations/rearrangements, with consistent breakpoints in 8q12 and 12q13-15, leading to gene fusions with the oncogenic drivers PLAG1 and HMGA2. However, the molecular association between these two seemingly different aberrations have been unclear. There is now convincing evidence to suggest that HMGA2 is an upstream regulator of PLAG1 expression and that HMGA2 regulates the expression of IGF2 via PLAG1 in a PLAG1-independent manner [68,76]. This new information emanates mainly from recent studies of the Silver–Russell syndrome (SRS) [76], a condition characterized by pre- and postnatal growth retardation associated with IGF2-deficiency [77,78]. Notably, it was recently shown that a subset of SRSs is caused by mutations in HMGA2 or in the HMGA2 regulated gene PLAG1 [76,79,80]. Inactivation of either of these genes leads to downregulation of IGF2 and a growth retardation phenotype. We now propose that activation of HMGA2 or PLAG1 expression due to chromosome 12q13-15 and 8q12 rearrangements in PA result in upregulation of IGF2 expression and tumorigenesis. This conclusion is also supported by previous studies showing co-expression of HMGA2 and PLAG1 in PAs with 12q13-15 rearrangements [19,68]. Taken together, these studies for the first time provide a common explanation for the 8q12 and 12q13-15 aberrations in PA and identifies IGF2 as a major oncogenic driver in PA.
5. Clinical Significance of Genomic Alterations in PA
Recurrent PAs may pose a significant clinical problem [81,82,83]. With current management of parotid PAs, the recurrence rate is around 3% [3,83]. Little, if anything, is known about the genomic profile of recurrent PAs. Preliminary cytogenetic analysis of 19 recurrent PAs have shown a similar distribution of 8q12, 12q13-15, and non-recurrent clonal aberrations as in primary non-recurrent tumors after more than 20 years follow-up (unpublished data). Similar observations were made also from an unpublished smaller series of cytogenetically analyzed primary PAs that recurred after 6–36 years. Taken together, these preliminary studies demonstrate that recurrent PAs are genetically stable even after long growth periods in vivo and that they do not deviate from the general pattern of chromosome aberrations in PA.
Malignant transformation of PA may occur in up to 10% of the cases and is increased in recurrent tumors and in tumors with long growth periods in vivo [1]. Studies of the cytogenetic and gene fusion landscape of CXPA is very limited. CXPAs show the PA specific PLAG1- or HMGA2-fusions as well as a number of other both recurrent and non-recurrent aberrations [42,43,60,84,85]. Previous studies have suggested that PAs with amplification of genes in 12q13-21, including MDM2, HMGA2 or an HMGA2-fusion, have an increased risk of malignant transformation [42,43,44]. In addition, amplification and overexpression of the FGFR1::PLAG1 fusion has been suggested as a potential biomarker for malignant transformation of PA [49].
PAs are characterized by histopathological diversity and there are several other benign and malignant salivary gland tumors that can mimic PA (reviewed in [8]). There is thus a need for diagnostically useful molecular markers to help distinguish PA from its mimics. Importantly, previous studies have shown that aberrations of 8q12 and 12q13-15 involving PLAG1 and HMGA2 are specific to PA and CXPA (cf. above) among salivary gland tumors [18,27,84,86,87,88,89]. Aberrations of these genes may be detected by FISH, RT-PCR, immunohistochemistry (IHC), and other molecular techniques such as for example RT-PCR and next generation sequencing. For routine histopathological purposes, FISH and IHC are perhaps most useful and there are several studies showing that PLAG1 IHC is a sensitive marker for PA and CXPA (cf. above).
Today the mainstay treatment of PA is surgery. However, surgery in the parotid and other regions of the head and neck is not uncomplicated with risk of damage to the facial nerve and a number of other complications. Therefore, especially in the recurrent setting with substantial risk for the VIIth nerve, non-surgical treatments are needed, and the recent discovery that the 8q12 and 12q13-15 aberrations lead to activation of the HMGA2-PLAG1-IGF2 pathway opens up new avenues for future medical treatment of PAs with for example IGF2-inhibitors.
6. Conclusions
The genomic hallmark of PA is translocations with consistent breakpoints in 8q12 and 12q13-15 resulting in gene fusions involving the transcription factor genes PLAG1 and HMGA2. PLAG1 is activated by promoter swapping/enhancer hijacking whereas HMGA2 is activated by gene truncation/enhancer hijacking. Importantly, recent studies have shown that HMGA2 is an upstream regulator of PLAG1 expression and that HMGA2 regulates the expression of IGF2 via PLAG1. This provides a novel explanation for the 8q12 and 12q13-15 aberrations in PA and identifies IGF2 as a major oncogenic driver and therapeutic target in PA. Taken together, these studies have important diagnostic and possible future therapeutic implications for patients with PA.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biomedicines10081970/s1. Table S1: Overview of all PLAG1- and HMGA2-fusions so far described in pleomorphic salivary gland adenomas in the literature. References [11,12,13,17,19,37,38,39,42,60,61,62,71,72,73,74,85,89,90,91] are cited in the supplementary materials.
Author Contributions
Conceptualization and writing—original draft: G.S.; data retrieval: G.S. and A.F. (André Fehr); visualization: A.F. (André Fehr), A.S. and G.S.; writing—review and editing: G.S., A.F. (André Fehr), A.S., V.V.P., H.H., L.H.M., N.F.S., O.G.-L., C.M.C.-E., M.K.A. and A.F. (Alfio Ferlito). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors wish to dedicate this review to the late Joachim Mark, a pathologist and pioneer in cancer cytogenetics and a great scientist, who in the late 70s together with GS initiated the cytogenetic studies of salivary gland tumors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO Classification of Tumours Editorial Board. International Agency for Research on Cancer, 5th ed.; WHO Classification of Tumours Series; Head and Neck Tumours [Internet; Beta Version Ahead of Print]; WHO Classification of Tumours Editorial Board: Lyon, France, 2022; Volume 9, Available online: https://tumourclassification.iarc.who.int/chapters/52 (accessed on 14 June 2022).
- Skalova, A.; Hyrcza, M.D.; Leivo, I. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Salivary Glands. Head Neck Pathol. 2022, 16, 40–53. [Google Scholar] [CrossRef]
- Andreasen, S.; Therkildsen, M.H.; Bjorndal, K.; Homoe, P. Pleomorphic adenoma of the parotid gland 1985-2010: A Danish nationwide study of incidence, recurrence rate, and malignant transformation. Head Neck 2016, 38 (Suppl. S1), E1364–E1369. [Google Scholar] [CrossRef]
- Valstar, M.H.; de Ridder, M.; van den Broek, E.C.; Stuiver, M.M.; van Dijk, B.A.C.; van Velthuysen, M.L.F.; Balm, A.J.M.; Smeele, L.E. Salivary gland pleomorphic adenoma in the Netherlands: A nationwide observational study of primary tumor incidence, malignant transformation, recurrence, and risk factors for recurrence. Oral Oncol. 2017, 66, 93–99. [Google Scholar] [CrossRef]
- Mark, J.; Ekedahl, C. Polyclonal chromosomal evolution in a benign mixed salivary gland tumor. Cancer Genet. Cytogenet. 1987, 28, 237–243. [Google Scholar] [CrossRef]
- Enlund, F.; Nordkvist, A.; Sahlin, P.; Mark, J.; Stenman, G. Expression of PLAG1 and HMGIC proteins and fusion transcripts in radiation-associated pleomorphic adenomas. Int. J. Oncol. 2002, 20, 713–716. [Google Scholar] [CrossRef]
- Hernandez-Prera, J.C.; Faquin, W.C.; Ihrler, S.; Katabi, N.; Weinreb, I.; Altemani, A.; Machado de Sousa, S.O.; Wasserman, J.K. Pleomorphic Adenoma. In WHO Classification of Tumours Editorial Board, 5th ed.; WHO Classification of Tumours Series; Head and Neck Tumours [Internet; Beta Version Ahead of Print]; International Agency for Research on Cancer: Lyon, France, 2022; Volume 9, Available online: https://tumourclassification.iarc.who.int/chapters/52 (accessed on 14 June 2022).
- Hernandez-Prera, J.C.; Skalova, A.; Franchi, A.; Rinaldo, A.; Vander Poorten, V.; Zbaren, P.; Ferlito, A.; Wenig, B.M. Pleomorphic adenoma: The great mimicker of malignancy. Histopathology 2021, 79, 279–290. [Google Scholar] [CrossRef]
- Katabi, N.; Chiosea, S.; Fonseca, I.; Ihrler, S.; Klijanienko, J.; Altemani, A. Carcinoma ex Pleomorphic Adenoma. In WHO Classification of Tumours Editorial Board, 5th ed.; WHO Classification of Tumours Series; Head and Neck Tumours [Internet; Beta Version Ahead of Print]; International Agency for Research on Cancer: Lyon, France, 2022; Volume 9, Available online: https://tumourclassification.iarc.who.int/chapters/52 (accessed on 14 June 2022).
- Mark, J.; Dahlenfors, R.; Ekedahl, C.; Stenman, G. The mixed salivary gland tumor—A normally benign human neoplasm frequently showing specific chromosomal abnormalities. Cancer Genet. Cytogenet. 1980, 2, 231–241. [Google Scholar] [CrossRef]
- Kas, K.; Voz, M.L.; Röijer, E.; Aström, A.K.; Meyen, E.; Stenman, G.; Van de Ven, W.J. Promoter swapping between the genes for a novel zinc finger protein and beta-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat. Genet. 1997, 15, 170–174. [Google Scholar] [CrossRef]
- Geurts, J.M.; Schoenmakers, E.F.; Röijer, E.; Stenman, G.; Van de Ven, W.J. Expression of reciprocal hybrid transcripts of HMGIC and FHIT in a pleomorphic adenoma of the parotid gland. Cancer Res. 1997, 57, 13–17. [Google Scholar]
- Geurts, J.M.; Schoenmakers, E.F.; Röijer, E.; Aström, A.K.; Stenman, G.; van de Ven, W.J. Identification of NFIB as recurrent translocation partner gene of HMGIC in pleomorphic adenomas. Oncogene 1998, 16, 865–872. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Ekedahl, C.; Stenman, G. Chromosomal patterns in a benign human neoplasm, the mixed salivary gland tumour. Hereditas 1982, 96, 141–148. [Google Scholar] [CrossRef]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Stenman, G.; Mark, J. Specificity of the involvement of chromosomes 8 and 12 in human mixed salivary-gland tumours. J. Oral. Pathol. 1983, 12, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Voz, M.L.; Aström, A.K.; Kas, K.; Mark, J.; Stenman, G.; Van de Ven, W.J. The recurrent translocation t(5;8)(p13;q12) in pleomorphic adenomas results in upregulation of PLAG1 gene expression under control of the LIFR promoter. Oncogene 1998, 16, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Stenman, G. Fusion oncogenes in salivary gland tumors: Molecular and clinical consequences. Head Neck Pathol. 2013, 7 (Suppl. S1), S12–S19. [Google Scholar] [CrossRef]
- Astrom, A.K.; Voz, M.L.; Kas, K.; Roijer, E.; Wedell, B.; Mandahl, N.; Van de Ven, W.; Mark, J.; Stenman, G. Conserved mechanism of PLAG1 activation in salivary gland tumors with and without chromosome 8q12 abnormalities: Identification of SII as a new fusion partner gene. Cancer Res. 1999, 59, 918–923. [Google Scholar] [PubMed]
- Skalova, A.; Hyrcza, M.D.; Vanecek, T.; Baneckova, M.; Leivo, I. Fusion-positive salivary gland carcinomas. Genes Chromosomes Cancer 2022, 61, 228–243. [Google Scholar] [CrossRef]
- Andersson, M.K.; Stenman, G. The landscape of gene fusions and somatic mutations in salivary gland neoplasms—Implications for diagnosis and therapy. Oral Oncol. 2016, 57, 63–69. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Ekedahl, C. Cytogenetics of the human benign mixed salivary gland tumour. Hereditas 1983, 99, 115–129. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R. Cytogenetical observations in 100 human benign pleomorphic adenomas: Specificity of the chromosomal aberrations and their relationship to sites of localized oncogenes. Anticancer Res. 1986, 6, 299–308. [Google Scholar]
- Mark, J.; Sandros, J.; Wedell, B.; Stenman, G.; Ekedahl, C. Significance of the choice of tissue culture technique on the chromosomal patterns in human mixed salivary gland tumors. Cancer Genet. Cytogenet. 1988, 33, 229–244. [Google Scholar] [CrossRef]
- Sandros, J.; Stenman, G.; Mark, J. Cytogenetic and molecular observations in human and experimental salivary gland tumors. Cancer Genet. Cytogenet. 1990, 44, 153–167. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Wedell, B. Impact of the in vitro technique used on the cytogenetic patterns in pleomorphic adenomas. Cancer Genet. Cytogenet. 1997, 95, 9–15. [Google Scholar] [CrossRef]
- Stenman, G. Fusion oncogenes and tumor type specificity—Insights from salivary gland tumors. Semin. Cancer Biol. 2005, 15, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Bullerdiek, J.; Bartnitzke, S.; Weinberg, M.; Chilla, R.; Haubrich, J.; Schloot, W. Rearrangements of chromosome region 12q13----q15 in pleomorphic adenomas of the human salivary gland (PSA). Cytogenet Cell Genet. 1987, 45, 187–190. [Google Scholar] [CrossRef]
- Bullerdiek, J.; Boschen, C.; Bartnitzke, S. Aberrations of chromosome 8 in mixed salivary gland tumors--cytogenetic findings on seven cases. Cancer Genet. Cytogenet. 1987, 24, 205–212. [Google Scholar] [CrossRef]
- Bullerdiek, J.; Chilla, R.; Haubrich, J.; Meyer, K.; Bartnitzke, S. A causal relationship between chromosomal rearrangements and the genesis of salivary gland pleomorphic adenomas. Arch. Otorhinolaryngol. 1988, 245, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Bullerdiek, J.; Wobst, G.; Meyer-Bolte, K.; Chilla, R.; Haubrich, J.; Thode, B.; Bartnitzke, S. Cytogenetic subtyping of 220 salivary gland pleomorphic adenomas: Correlation to occurrence, histological subtype, and in vitro cellular behavior. Cancer Genet. Cytogenet. 1993, 65, 27–31. [Google Scholar] [CrossRef]
- Martins, C.; Fonseca, I.; Felix, A.; Roque, L.; Soares, J. Benign salivary gland tumors: A cytogenetic study of 21 cases. J. Surg. Oncol. 1995, 60, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Hrynchak, M.; White, V.; Berean, K.; Horsman, D. Cytogenetic findings in seven lacrimal gland neoplasms. Cancer Genet. Cytogenet. 1994, 75, 133–138. [Google Scholar] [CrossRef]
- Mark, H.F.; Hanna, I.; Gnepp, D.R. Cytogenetic analysis of salivary gland type tumors. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1996, 82, 187–192. [Google Scholar] [CrossRef]
- Jin, C.; Martins, C.; Jin, Y.; Wiegant, J.; Wennerberg, J.; Dictor, M.; Gisselsson, D.; Strombeck, B.; Fonseca, I.; Mitelman, F.; et al. Characterization of chromosome aberrations in salivary gland tumors by FISH, including multicolor COBRA-FISH. Genes Chromosomes Cancer 2001, 30, 161–167. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. (Eds.) Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer: 2022. Available online: https://mitelmandatabase.isb-cgc.org (accessed on 21 June 2022).
- Afshari, M.K.; Fehr, A.; Nevado, P.T.; Andersson, M.K.; Stenman, G. Activation of PLAG1 and HMGA2 by gene fusions involving the transcriptional regulator gene NFIB. Genes Chromosomes Cancer 2020, 59, 652–660. [Google Scholar] [CrossRef]
- Asp, J.; Persson, F.; Kost-Alimova, M.; Stenman, G. CHCHD7-PLAG1 and TCEA1-PLAG1 gene fusions resulting from cryptic, intrachromosomal 8q rearrangements in pleomorphic salivary gland adenomas. Genes Chromosomes Cancer 2006, 45, 820–828. [Google Scholar] [CrossRef]
- Persson, F.; Winnes, M.; Andrén, Y.; Wedell, B.; Dahlenfors, R.; Asp, J.; Mark, J.; Enlund, F.; Stenman, G. High-resolution array CGH analysis of salivary gland tumors reveals fusion and amplification of the FGFR1 and PLAG1 genes in ring chromosomes. Oncogene 2008, 27, 3072–3080. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Ekedahl, C. On double-minutes and their origin in a benign human neoplasm, a mixed salivary gland tumour. Anticancer Res. 1982, 2, 261–264. [Google Scholar] [PubMed]
- Di Palma, S.; Lambros, M.B.; Savage, K.; Jones, C.; Mackay, A.; Dexter, T.; Iravani, M.; Fenwick, K.; Ashworth, A.; Reis-Filho, J.S. Oncocytic change in pleomorphic adenoma: Molecular evidence in support of an origin in neoplastic cells. J. Clin. Pathol. 2007, 60, 492–499. [Google Scholar] [CrossRef]
- Persson, F.; Andrén, Y.; Winnes, M.; Wedell, B.; Nordkvist, A.; Gudnadottir, G.; Dahlenfors, R.; Sjögren, H.; Mark, J.; Stenman, G. High-resolution genomic profiling of adenomas and carcinomas of the salivary glands reveals amplification, rearrangement, and fusion of HMGA2. Genes Chromosomes Cancer 2009, 48, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Roijer, E.; Nordkvist, A.; Strom, A.K.; Ryd, W.; Behrendt, M.; Bullerdiek, J.; Mark, J.; Stenman, G. Translocation, deletion/amplification, and expression of HMGIC and MDM2 in a carcinoma ex pleomorphic adenoma. Am. J. Pathol. 2002, 160, 433–440. [Google Scholar] [CrossRef]
- Rao, P.H.; Murty, V.V.; Louie, D.C.; Chaganti, R.S. Nonsyntenic amplification of MYC with CDK4 and MDM2 in a malignant mixed tumor of salivary gland. Cancer Genet. Cytogenet. 1998, 105, 160–163. [Google Scholar] [CrossRef]
- McClintock, B. The Production of Homozygous Deficient Tissues with Mutant Characteristics by Means of the Aberrant Mitotic Behavior of Ring-Shaped Chromosomes. Genetics 1938, 23, 315–376. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Pettersson, L.; Hoglund, M.; Heidenblad, M.; Gorunova, L.; Wiegant, J.; Mertens, F.; Dal Cin, P.; Mitelman, F.; Mandahl, N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc. Natl. Acad. Sci. USA 2000, 97, 5357–5362. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Ekedahl, C. Specificity and implications of ring chromosomes and dicentrics in benign mixed salivary gland tumours. Acta Pathol. Microbiol. Immunol. Scand. A 1983, 91, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Dalin, M.G.; Katabi, N.; Persson, M.; Lee, K.W.; Makarov, V.; Desrichard, A.; Walsh, L.A.; West, L.; Nadeem, Z.; Ramaswami, D.; et al. Multi-dimensional genomic analysis of myoepithelial carcinoma identifies prevalent oncogenic gene fusions. Nat. Commun. 2017, 8, 1197. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Wedell, B. Is pleomorphic adenoma of the salivary glands a tumor of congenital or very early origin? Oncol. Rep. 1996, 3, 1075–1077. [Google Scholar] [CrossRef]
- Kas, K.; Voz, M.L.; Hensen, K.; Meyen, E.; Van de Ven, W.J. Transcriptional activation capacity of the novel PLAG family of zinc finger proteins. J. Biol. Chem. 1998, 273, 23026–23032. [Google Scholar] [CrossRef]
- Van Dyck, F.; Declercq, J.; Braem, C.V.; Van de Ven, W.J. PLAG1, the prototype of the PLAG gene family: Versatility in tumour development (review). Int. J. Oncol. 2007, 30, 765–774. [Google Scholar] [CrossRef]
- Juma, A.R.; Damdimopoulou, P.E.; Grommen, S.V.; Van de Ven, W.J.; De Groef, B. Emerging role of PLAG1 as a regulator of growth and reproduction. J. Endocrinol. 2016, 228, R45–R56. [Google Scholar] [CrossRef]
- Hensen, K.; Braem, C.; Declercq, J.; Van Dyck, F.; Dewerchin, M.; Fiette, L.; Denef, C.; Van de Ven, W.J. Targeted disruption of the murine Plag1 proto-oncogene causes growth retardation and reduced fertility. Dev. Growth Differ. 2004, 46, 459–470. [Google Scholar] [CrossRef]
- Voz, M.L.; Agten, N.S.; Van de Ven, W.J.; Kas, K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000, 60, 106–113. [Google Scholar]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, S. Mutation Hotspots in the beta-Catenin Gene: Lessons from the Human Cancer Genome Databases. Mol. Cells 2019, 42, 8–16. [Google Scholar] [CrossRef]
- Persson, M.; Andren, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef]
- Persson, M.; Andren, Y.; Moskaluk, C.A.; Frierson, H.F., Jr.; Cooke, S.L.; Futreal, P.A.; Kling, T.; Nelander, S.; Nordkvist, A.; Persson, F.; et al. Clinically significant copy number alterations and complex rearrangements of MYB and NFIB in head and neck adenoid cystic carcinoma. Genes Chromosomes Cancer 2012, 51, 805–817. [Google Scholar] [CrossRef]
- Bubola, J.; MacMillan, C.M.; Demicco, E.G.; Chami, R.A.; Chung, C.T.; Leong, I.; Marrano, P.; Onkal, Z.; Swanson, D.; Veremis, B.M.; et al. Targeted RNA sequencing in the routine clinical detection of fusion genes in salivary gland tumors. Genes Chromosomes Cancer 2021, 60, 695–708. [Google Scholar] [CrossRef]
- Baněčková, M.; Uro-Coste, E.; Ptáková, N.; Šteiner, P.; Stanowska, O.; Benincasa, G.; Colella, G.; Vondrák, J., Jr.; Michal, M.; Leivo, I.; et al. What is hiding behind S100 protein and SOX10 positive oncocytomas? Oncocytic pleomorphic adenoma and myoepithelioma with novel gene fusions in a subset of cases. Hum. Pathol. 2020, 103, 52–62. [Google Scholar] [CrossRef]
- Pei, J.; Liu, J.C.; Ehya, H.; Wei, S. BOC-PLAG1, a new fusion gene of pleomorphic adenoma: Identified in a fine-needle aspirate by RNA next-generation sequencing. Diagn. Cytopathol. 2021, 49, 790–792. [Google Scholar] [CrossRef]
- Ashar, H.R.; Fejzo, M.S.; Tkachenko, A.; Zhou, X.; Fletcher, J.A.; Weremowicz, S.; Morton, C.C.; Chada, K. Disruption of the architectural factor HMGI-C: DNA-binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell 1995, 82, 57–65. [Google Scholar] [CrossRef]
- Tkachenko, A.; Ashar, H.R.; Meloni, A.M.; Sandberg, A.A.; Chada, K.K. Misexpression of disrupted HMGI architectural factors activates alternative pathways of tumorigenesis. Cancer Res. 1997, 57, 2276–2280. [Google Scholar]
- Mansoori, B.; Mohammadi, A.; Ditzel, H.J.; Duijf, P.H.G.; Khaze, V.; Gjerstorff, M.F.; Baradaran, B. HMGA2 as a Critical Regulator in Cancer Development. Genes 2021, 12, 269. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Fusco, A.; Esposito, F. HMGA and Cancer: A Review on Patent Literatures. Recent Pat. Anti-Cancer Drug Discov. 2019, 14, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Tessari, M.A.; Gostissa, M.; Altamura, S.; Sgarra, R.; Rustighi, A.; Salvagno, C.; Caretti, G.; Imbriano, C.; Mantovani, R.; Del Sal, G.; et al. Transcriptional activation of the cyclin A gene by the architectural transcription factor HMGA2. Mol. Cell. Biol. 2003, 23, 9104–9116. [Google Scholar] [CrossRef]
- Klemke, M.; Muller, M.H.; Wosniok, W.; Markowski, D.N.; Nimzyk, R.; Helmke, B.M.; Bullerdiek, J. Correlated expression of HMGA2 and PLAG1 in thyroid tumors, uterine leiomyomas and experimental models. PLoS ONE 2014, 9, e88126. [Google Scholar] [CrossRef]
- Andersson, M.K.; Aman, P.; Stenman, G. IGF2/IGF1R Signaling as a Therapeutic Target in MYB-Positive Adenoid Cystic Carcinomas and Other Fusion Gen.ne-Driven Tumors. Cells 2019, 8, 913. [Google Scholar] [CrossRef]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef]
- Sun, L.; Petrone, J.S.; McNulty, S.N.; Evenson, M.J.; Zhu, X.; Robinson, J.A.; Chernock, R.D.; Duncavage, E.J.; Pfeifer, J.D. Comparison of gene fusion detection methods in salivary gland tumors. Hum. Pathol. 2022, 123, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Agaimy, A.; Ihrler, S.; Baněčková, M.; Costés Martineau, V.; Mantsopoulos, K.; Hartmann, A.; Iro, H.; Stoehr, R.; Skálová, A. HMGA2-WIF1 Rearrangements Characterize a Distinctive Subset of Salivary Pleomorphic Adenomas With Prominent Trabecular (Canalicular Adenoma-like) Morphology. Am. J. Surg. Pathol. 2022, 46, 190–199. [Google Scholar] [CrossRef]
- Wasserman, J.K.; Dickson, B.C.; Smith, A.; Swanson, D.; Purgina, B.M.; Weinreb, I. Metastasizing Pleomorphic Adenoma: Recurrent PLAG1/HMGA2 Rearrangements and Identification of a Novel HMGA2-TMTC2 Fusion. Am. J. Surg. Pathol. 2019, 43, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Queimado, L.; Lopes, C.S.; Reis, A.M. WIF1, an inhibitor of the Wnt pathway, is rearranged in salivary gland tumors. Genes Chromosomes Cancer 2007, 46, 215–225. [Google Scholar] [CrossRef]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Abi Habib, W.; Brioude, F.; Edouard, T.; Bennett, J.T.; Lienhardt-Roussie, A.; Tixier, F.; Salem, J.; Yuen, T.; Azzi, S.; Le Bouc, Y.; et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet. Med. 2018, 20, 250–258. [Google Scholar] [CrossRef]
- Gicquel, C.; Rossignol, S.; Cabrol, S.; Houang, M.; Steunou, V.; Barbu, V.; Danton, F.; Thibaud, N.; Le Merrer, M.; Burglen, L.; et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat. Genet. 2005, 37, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Azzi, S.; Abi Habib, W.; Netchine, I. Beckwith-Wiedemann and Russell-Silver Syndromes: From new molecular insights to the comprehension of imprinting regulation. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 30–38. [Google Scholar] [CrossRef]
- Leszinski, G.S.; Warncke, K.; Hoefele, J.; Wagner, M. A case report and review of the literature indicate that HMGA2 should be added as a disease gene for Silver-Russell syndrome. Gene 2018, 663, 110–114. [Google Scholar] [CrossRef]
- Vado, Y.; Pereda, A.; Llano-Rivas, I.; Gorria-Redondo, N.; Diez, I.; Perez de Nanclares, G. Novel Variant in PLAG1 in a Familial Case with Silver-Russell Syndrome Suspicion. Genes 2020, 11, 1461. [Google Scholar] [CrossRef]
- Psychogios, G.; Bohr, C.; Constantinidis, J.; Canis, M.; Vander Poorten, V.; Plzak, J.; Knopf, A.; Betz, C.; Guntinas-Lichius, O.; Zenk, J. Review of surgical techniques and guide for decision making in the treatment of benign parotid tumors. Eur. Arch. Otorhinolaryngol. 2021, 278, 15–29. [Google Scholar] [CrossRef]
- Rooker, S.A.; Van Abel, K.M.; Yin, L.X.; Nagelschneider, A.A.; Price, D.L.; Olsen, K.D.; Janus, J.R.; Kasperbauer, J.L.; Moore, E.J. Risk factors for subsequent recurrence after surgical treatment of recurrent pleomorphic adenoma of the parotid gland. Head Neck 2021, 43, 1088–1096. [Google Scholar] [CrossRef]
- Alzumaili, B.; Xu, B.; Saliba, M.; Abuhashem, A.; Ganly, I.; Ghossein, R.; Katabi, N. Clinicopathologic Characteristics and Prognostic Factors of Primary and Recurrent Pleomorphic Adenoma: A Single Institution Retrospective Study of 705 Cases. Am. J. Surg. Pathol. 2022, 46, 854–862. [Google Scholar] [CrossRef] [PubMed]
- von Holstein, S.L.; Fehr, A.; Persson, M.; Nickelsen, M.; Therkildsen, M.H.; Prause, J.U.; Heegaard, S.; Stenman, G. Lacrimal gland pleomorphic adenoma and carcinoma ex pleomorphic adenoma: Genomic profiles, gene fusions, and clinical characteristics. Ophthalmology 2014, 121, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Asahina, M.; Saito, T.; Hayashi, T.; Fukumura, Y.; Mitani, K.; Yao, T. Clinicopathological effect of PLAG1 fusion genes in pleomorphic adenoma and carcinoma ex pleomorphic adenoma with special emphasis on histological features. Histopathology 2019, 74, 514–525. [Google Scholar] [CrossRef]
- Katabi, N.; Xu, B.; Jungbluth, A.A.; Zhang, L.; Shao, S.Y.; Lane, J.; Ghossein, R.; Antonescu, C.R. PLAG1 immunohistochemistry is a sensitive marker for pleomorphic adenoma: A comparative study with PLAG1 genetic abnormalities. Histopathology 2018, 72, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, A.; Dalton, J.D.; Shivakumar, B.; Krane, J.F. PLAG1 alteration in carcinoma ex pleomorphic adenoma: Immunohistochemical and fluorescence in situ hybridization studies of 22 cases. Head Neck Pathol. 2012, 6, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Rotellini, M.; Palomba, A.; Baroni, G.; Franchi, A. Diagnostic utility of PLAG1 immunohistochemical determination in salivary gland tumors. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, A.; Hisaoka, M.; Nagao, Y.; Hashimoto, H. Aberrant PLAG1 expression in pleomorphic adenomas of the salivary gland: A molecular genetic and immunohistochemical study. Virchows Arch. Int. J. Pathol. 2011, 458, 583–592. [Google Scholar] [CrossRef]
- Katabi, N.; Ghossein, R.; Ho, A.; Dogan, S.; Zhang, L.; Sung, Y.S.; Antonescu, C.R. Consistent PLAG1 and HMGA2 abnormalities distinguish carcinoma ex-pleomorphic adenoma from its de novo counterparts. Hum. Pathol. 2015, 46, 26–33. [Google Scholar] [CrossRef]
- Pareja, F.; Da Cruz Paula, A.; Gularte-Mérida, R.; Vahdatinia, M.; Li, A.; Geyer, F.C.; da Silva, E.M.; Nanjangud, G.; Wen, H.Y.; Varga, Z.; et al. Pleomorphic adenomas and mucoepidermoid carcinomas of the breast are underpinned by fusion genes. NPJ Breast Cancer 2020, 6, 20. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).