
The Journey of Human Transthyretin: Synthesis, Structure Stability, and Catabolism

 , , ,
, , ,
Abstract
:1. Introduction
2. Tissue-Specific Regulation of Transthyretin Expression
2.1. Liver and Choroid Plexus
2.2. Placenta and Visceral Yolk Sac
2.3. Pancreas
2.4. Retinal Pigment Epithelium
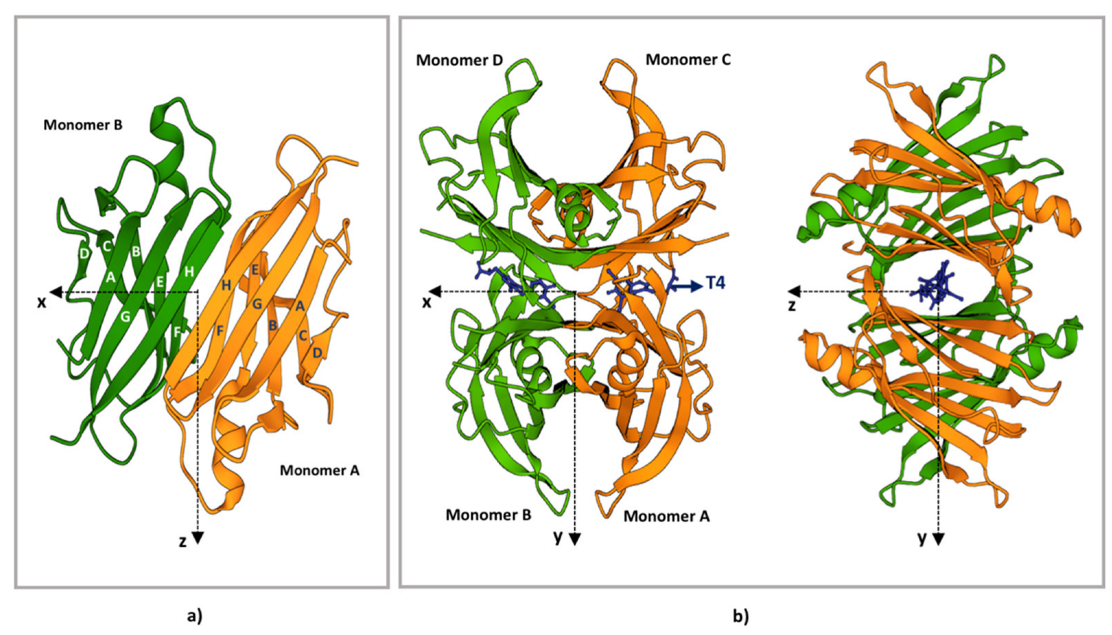
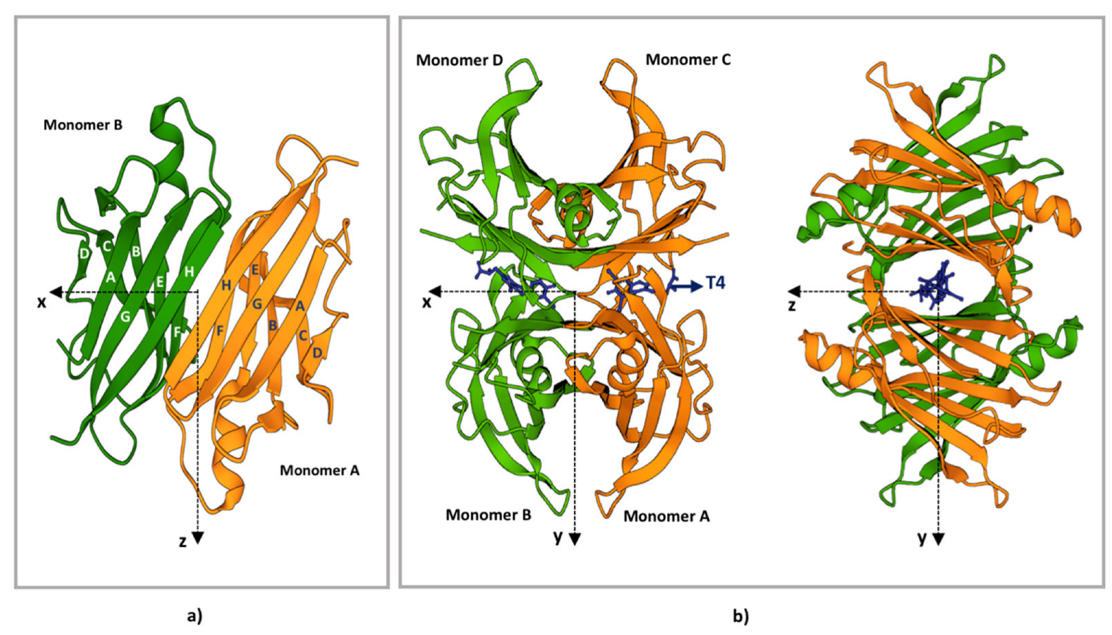
3. TTR Structure
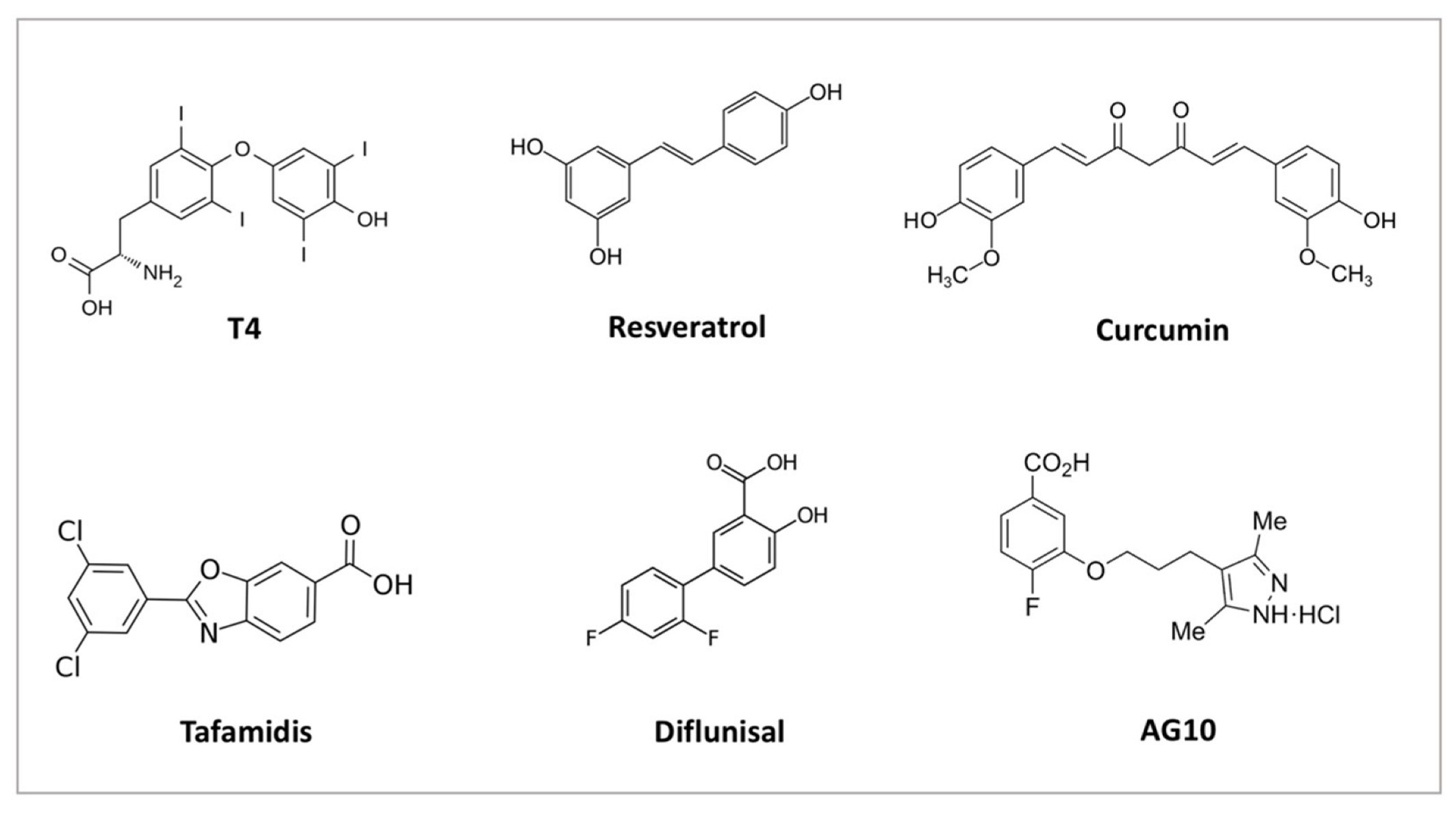
4. TTR Structural Stability
5. TTR Variants and Structural Stability
6. TTR Post-Translational Modifications
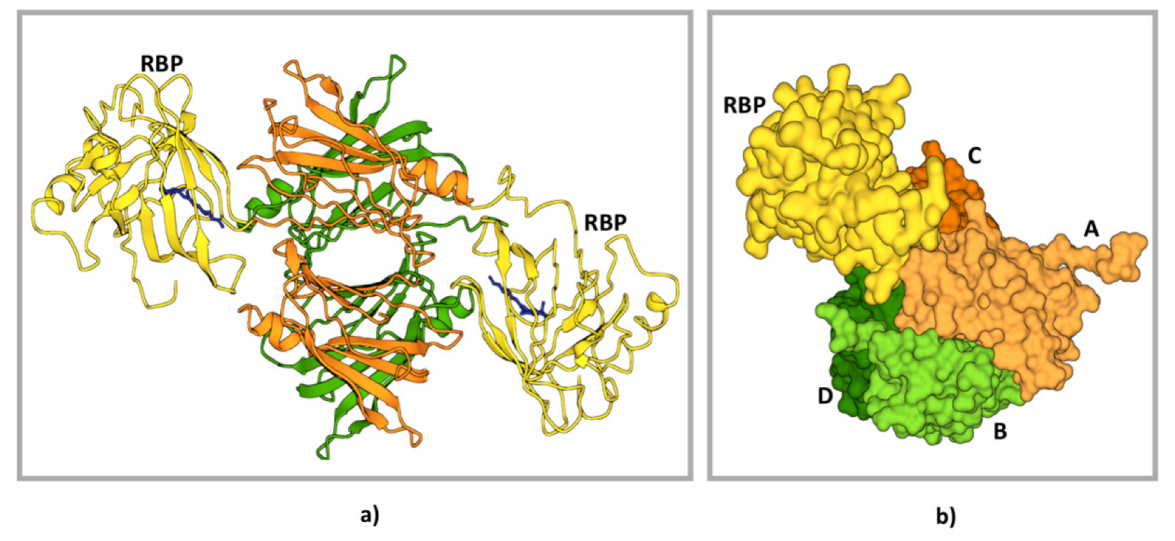
7. TTR–RBP Complex Formation
8. Endoplasmic Reticulum Quality Control in TTRwt and TTRv Cellular Release
9. TTR Catabolism
9.1. Sites of Degradation
9.2. TTR Cellular Internalisation
9.3. Removal of TTR Aggregates
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robbins, J. Transthyretin from discovery to now. Clin. Chem. Lab. Med. (CCLM) 2002, 40, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Nomenclature committee of the International Union of Biochemistry (NC-IUB). Enzyme nomenclature. Recommendations 1978. Supplement 2: Corrections and additions. Eur. J. Biochem. 1981, 116, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Noy, N.; Slosberg, E.; Scarlata, S. Interactions of retinol with binding proteins: Studies with retinol-binding protein and with transthyretin. Biochemistry 1992, 31, 11118–11124. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Rask, L.; Peterson, P. Studies on thyroid hormone-binding proteins. II. Binding of thyroid hormones, retinol-binding protein, and fluorescent probes to prealbumin and effects of thyroxine on prealbumin subunit self association. J. Biol. Chem. 1975, 250, 8554–8563. [Google Scholar] [CrossRef]
- Wei, S.; Episkopou, V.; Piantedosi, R.; Maeda, S.; Shimada, K.; Gottesman, M.E.; Blaner, W.S. Studies on the metabolism of retinol and retinol-binding protein in transthyretin-deficient mice produced by homologous recombination. J. Biol. Chem. 1995, 270, 866–870. [Google Scholar] [CrossRef] [Green Version]
- Hyung, S.J.; Deroo, S.; Robinson, C.V. Retinol and retinol-binding protein stabilize transthyretin via formation of retinol transport complex. ACS Chem. Biol. 2010, 5. [Google Scholar] [CrossRef]
- Goodman, D.S. Plasma retinol-binding protein. Ann. New York Acad. Sci. 1980, 348, 378–390. [Google Scholar] [CrossRef]
- Ronne, H.; Ocklind, C.; Wiman, K.; Rask, L.; Obrink, B.; A Peterson, P. Ligand-dependent regulation of intracellular protein transport: Effect of vitamin a on the secretion of the retinol-binding protein. J. Cell. Biol. 1983, 96, 907–910. [Google Scholar] [CrossRef]
- Stabilini, R.; Vergani, C.; Agostoni, A.; Agostoni, R.V. Influence of age and sex on prealbumin levels. Clin. Chim. Acta 1968, 20, 358–359. [Google Scholar] [CrossRef]
- Ingenbleek, Y.; Bernstein, L.H. Plasma Transthyretin as a Biomarker of Lean Body Mass and Catabolic States. Adv. Nutr. Int. Rev. J. 2015, 6, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Beck, F.K.; Rosenthal, T.C. Prealbumin: A marker for nutritional evaluation. Am. Fam. Physician 2002, 65, 1575–1578. [Google Scholar] [PubMed]
- Dellière, S.; Cynober, L. Is transthyretin a good marker of nutritional status? Clin. Nutr. 2016, 36, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Picken, M.M. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020, 143, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Bergström, J.; Solomon, A.; Murphy, C.; Sletten, K. Transthyretin-derived senile systemic amyloidosis: Clinicopathologic and structural considerations. Amyloid 2003, 10 Suppl 1, 48–54. [Google Scholar] [CrossRef]
- Hanson, J.L.; Arvanitis, M.; Koch, C.M.; Berk, J.L.; Ruberg, F.L.; Prokaeva, T.; Connors, L.H. Use of Serum Transthyretin as a Prognostic Indicator and Predictor of Outcome in Cardiac Amyloid Disease Associated With Wild-Type Transthyretin. Circ. Hear. Fail. 2018, 11, e004000. [Google Scholar] [CrossRef]
- Skinner, M.; Connors, L.; Rubinow, A.; Libbey, C.; Sipe, J.D.; Cohen, A.S. Lowered prealbumin levels in patients with familial amyloid polyneuropathy (FAP) and their non-affected but at risk relatives. Am. J. Med. Sci. 1985, 289, 17–21. [Google Scholar] [CrossRef]
- Buxbaum, J.; Koziol, J.; Connors, L.H. Serum transthyretin levels in senile systemic amyloidosis: Effects of age, gender and ethnicity. Amyloid 2008, 15, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Falk, R.H.; Haddad, M.; Walker, C.R.; Dorbala, S.; Cuddy, S.A. Effect of Tafamidis on Serum Transthyretin Levels in Non-Trial Patients With Transthyretin Amyloid Cardiomyopathy. JACC CardioOncology 2021, 3, 580–586. [Google Scholar] [CrossRef]
- Greene, M.J.; Klimtchuk, E.S.; Seldin, D.C.; Berk, J.L.; Connors, L.H. Cooperative stabilization of transthyretin by clusterin and diflunisal. Biochemistry 2014, 54, 268–278. [Google Scholar] [CrossRef]
- Miroy, G.J.; Lai, Z.; Lashuel, H.A.; Peterson, S.A.; Strang, C.; Kelly, J.W. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc. Natl. Acad. Sci. 1996, 93, 15051–15056. [Google Scholar] [CrossRef] [Green Version]
- Ueda, M.; Okada, M.; Mizuguchi, M.; Kluve-Beckerman, B.; Kanenawa, K.; Isoguchi, A.; Misumi, Y.; Tasaki, M.; Ueda, A.; Kanai, A.; et al. A cell-based high-throughput screening method to directly examine transthyretin amyloid fibril formation at neutral pH. J. Biol. Chem. 2019, 294, 11259–11275. [Google Scholar] [CrossRef] [PubMed]
- Hurshman, A.R.; White, J.T.; Powers, E.T.; Kelly, J.W. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 2004, 43, 7365–7381. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.K.R.; Hughes, R.M.; Wi, S.; Hung, I.; Gan, Z.; Kelly, J.W.; Lim, K.H. Transthyretin Aggregation Pathway toward the Formation of Distinct Cytotoxic Oligomers. Sci. Rep. 2019, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.M.; Brito, R.M.M. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J. Biol. Chem. 2001, 276, 27207–27213. [Google Scholar] [CrossRef] [Green Version]
- Sparkes, R.S.; Sasaki, H.; Mohandas, T.; Yoshioka, K.; Klisak, I.; Sakaki, Y.; Heinzmann, C.; Simon, M.I. Assignment of the prealbumin (PALB) gene (familial amyloidotic polyneuropathy) to human chromosome region 18q11.2-q12.1. Hum. Genet. 1987, 75, 151–154. [Google Scholar] [CrossRef]
- Hiroyuki, S.; Naoko, Y.; Yasuyuki, T.; Yoshiyuki, S. Structure of the chromosomal gene for human serum prealbumin. Gene 1985, 37, 191–197. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Mita, S.; Maeda, S.; Araki, S.; Shimada, K. Structure of the human prealbumin gene. J. Biol. Chem. 1985, 260, 12224–12227. [Google Scholar] [CrossRef]
- Power, D.; Elias, N.; Richardson, S.; Mendes, J.; Soares, C.; Santos, C. Evolution of the thyroid hormone-binding protein, transthyretin. Gen. Comp. Endocrinol. 2000, 119, 241–255. [Google Scholar] [CrossRef] [Green Version]
- Ingenbleek, A.Y.; Young, V. Transthyretin (prealbumin) in health and disease: Nutritional implications. Annu. Rev. Nutr. 1994, 14, 495–533. [Google Scholar] [CrossRef]
- Blake, C.; Geisow, M.; Oatley, S.; Rérat, B.; Rérat, C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J. Mol. Biol. 1978, 121, 339–356. [Google Scholar] [CrossRef]
- Bellovino, D.; Morimoto, T.; Pisaniello, A.; Gaetani, S. In vitro and in vivo studies on transthyretin oligomerization. Exp. Cell Res. 1998, 243, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G.; Richardson, S.J. The evolution of gene expression, structure and function of transthyretin. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997, 116, 137–160. [Google Scholar] [CrossRef]
- Costa, R.H.; Lai, E.; E Darnell, J. Transcriptional control of the mouse prealbumin (transthyretin) gene: Both promoter sequences and a distinct enhancer are cell specific. Mol. Cell. Biol. 1986, 6, 4697–4708. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Burke, P.A. Hepatocyte nuclear factor-4α interacts with other hepatocyte nuclear factors in regulating transthyretin gene expression. FEBS 2010, 277, 4066–4075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, P.W.; Howlett, G.J.; Schreiber, G. Rat transthyretin (prealbumin). Molecular cloning, nucleotide sequence, and gene expression in liver and brain. J. Biol. Chem. 1985, 260, 8214–8219. [Google Scholar] [CrossRef]
- Dickson, P.W.; Aldred, A.R.; Marley, P.D.; Bannister, D.; Schreiber, G. Rat choroid plexus specializes in the synthesis and the secretion of transthyretin (prealbumin). Regulation of transthyretin synthesis in choroid plexus is independent from that in liver. J. Biol. Chem. 1986, 261, 3475–3478. [Google Scholar] [CrossRef]
- Yan, C.; Costa, R.; Darnell, J.; Chen, J.; Van Dyke, T. Distinct positive and negative elements control the limited hepatocyte and choroid plexus expression of transthyretin in transgenic mice. EMBO J. 1990, 9, 869–878. [Google Scholar] [CrossRef]
- Costa, R.H.; A Van Dyke, T.; Yan, C.; Kuo, F.; E Darnell, J. Similarities in transthyretin gene expression and differences in transcription factors: Liver and yolk sac compared to choroid plexus. Proc. Natl. Acad. Sci. USA 1990, 87, 6589–6593. [Google Scholar] [CrossRef] [Green Version]
- SStauder, A.J.; Dickson, P.W.; Aldred, A.R.; Schreiber, G.; A Mendelsohn, F.; Hudson, P. Synthesis of transthyretin (pre-albumin) mRNA in choroid plexus epithelial cells, localized by in situ hybridization in rat brain. J. Histochem. Cytochem. 1986, 34, 949–952. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, G.; Aldred, A.R.; Jaworowski, A.; Nilsson, C.; Achen, M.G.; Segal, M.B. Thyroxine transport from blood to brain via transthyretin synthesis in choroid plexus. Am. J. Physiol. Integr. Comp. Physiol. 1990, 258, R338–R345. [Google Scholar] [CrossRef]
- Lemkine, G.F.; Raji, A.; Alfama, G.; Turque, N.; Hassani, Z.; Alegria-Prévot, O.; Samarut, J.; Levi, G.; Demeneix, B.A. Adult neural stem cell cycling in vivo requires thyroid hormone and its alpha receptor. FASEB J. 2005, 19, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G.; Southwell, B.R.; Richardson, S.J. Hormone delivery systems to the brain - transthyretin. Exp. Clin. Endocrinol. Diabetes 1995, 103, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Palha, J.A. Transthyretin as a thyroid hormone carrier: Function revisited. Clin. Chem. Lab. Med. (CCLM) 2002, 40, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Benson, M.D. Transthyretin: A review from a structural perspective. Cell Mol. Life Sci. 2001, 58, 1491–1521. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, B.; Li, H.; Richard, K.; Mortimer, R. Synthesis of thyroid hormone binding proteins transthyretin and albumin by human trophoblast. J. Clin. Endocrinol. Metab. 2005, 90, 6714–6720. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.; Landers, K.; Li, H.; Mortimer, R.; Richard, K. Ontogenic changes in placental transthyretin. Placenta 2011, 32, 817–822. [Google Scholar] [CrossRef]
- Patel, J.; Landers, K.; Li, H.; Mortimer, R.H.; Richard, K. Delivery of maternal thyroid hormones to the fetus. Trends Endocrinol. Metab. 2011, 22, 164–170. [Google Scholar] [CrossRef]
- Ross, C.; Boroviak, T.E. Origin and function of the yolk sac in primate embryogenesis. Nat. Commun. 2020, 11, 3760. [Google Scholar] [CrossRef]
- Landers, K.; Mortimer, R.; Richard, K. Transthyretin and the human placenta. Placenta 2013, 34, 513–517. [Google Scholar] [CrossRef]
- Costa, R.H.; Grayson, D.R. Site-directed mutagenesis of hepatocyte nuclear factor (HNF) binding sites in the mouse transthyretin (TTR) promoter reveal synergistic interactions with its enhancer region. Nucleic Acids Res. 1991, 19, 4139–4145. [Google Scholar] [CrossRef] [Green Version]
- Westermark, G.T.; Westermark, P. Transthyretin and amyloid in the islets of Langerhans in type-2 diabetes. Exp. Diabetes Res. 2008, 2008, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Jono, H.; Misumi, Y.; Senokuchi, T.; Guo, J.; Ueda, M.; Shinriki, S.; Tasaki, M.; Shono, M.; Obayashi, K.; et al. Novel function of transthyretin in pancreatic alpha cells. FEBS Lett. 2012, 586, 4215–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refai, E.; Dekki, N.; Yang, S.N.; Imreh, G.; Cabrera, O.; Yu, L.; Yang, G.; Norgren, S.; Rössner, S.M.; Inverardi, L.; et al. Transthyretin constitutes a functional component in pancreatic β-cell stimulus-secretion coupling. Proc. Natl. Acad. Sci. USA 2005, 102, 17020–17025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N.; Hanafusa, T.; Miyagawa, J.; Tamura, S.; Inada, M.; Kawata, S.; Kono, N.; Tarui, S. Transthyretin (prealbumin) in the pancreas and sera of newly diagnosed type I (insulin-dependent) diabetic patients. J. Clin. Endocrinol. Metab. 1992, 74, 1372–1377. [Google Scholar] [CrossRef]
- Cavallaro, T.; Martone, R.; Dwork, A.J.; A Schon, E.; Herbert, J. The retinal pigment epithelium is the unique site of transthyretin synthesis in the rat eye. Investig. Ophthalmol. Vis. Sci. 1990, 31, 497–501. [Google Scholar]
- Martone, R.L.; Herbert, J.; Dwork, A.; Schon, E.A. Transthyretin is synthesized in the mammalian eye. Biochem. Biophys. Res. Commun. 1988, 151, 905–912. [Google Scholar] [CrossRef]
- Ong, D.E.; Davis, J.T.; O’Day, W.T.; Bok, D. Synthesis and secretion of retinol-binding protein and transthyretin by cultured retinal pigment epithelium. Biochemistry 1994, 33, 1835–1842. [Google Scholar] [CrossRef]
- Eichenbaum, J.W.; Zheng, W. Distribution of lead and transthyretin in human eyes. J. Toxicol. Clin. Toxicol. 2000, 38, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Liepnieks, J.J.; Wilson, D.L.; Benson, M.D. Biochemical characterization of vitreous and cardiac amyloid in Ile84Ser transthyretin amyloidosis. Amyloid 2006, 13, 170–177. [Google Scholar] [CrossRef]
- Iakovleva, I.; Hall, M.; Oelker, M.; Sandblad, L.; Anan, I.; Sauer-Eriksson, A.E. Structural basis for transthyretin amyloid formation in vitreous body of the eye. Nat. Commun. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Blake, C.; Swan, I.; Rerat, C.; Berthou, J.; Laurent, A.; Rerat, B. An x-ray study of the subunit structure of prealbumin. J. Mol. Biol. 1971, 61, 217–224. [Google Scholar] [CrossRef]
- Kanda, Y.; Goodman, D.S.; Canfield, R.E.; Morgan, F.J. The amino acid sequence of human plasma prealbumin. J. Biol. Chem. 1974, 249, 6796–6805. [Google Scholar] [CrossRef]
- He, S.; He, X.; Liu, L.; Zhang, W.; Yu, L.; Deng, Z.; Feiyi, Z.; Mo, S.; Fan, Y.; Zhao, X.; et al. The Structural Understanding of Transthyretin Misfolding and the Inspired Drug Approaches for the Treatment of Heart Failure Associated With Transthyretin Amyloidosis. Front. Pharmacol. 2021, 12, 628184. [Google Scholar] [CrossRef] [PubMed]
- Park, G.Y.; Jamerlan, A.; Shim, K.H.; An, S.S.A. Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. Int. J. Mol. Sci. 2019, 20, 2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, F.; Hammarstrom, P.; Kelly, J.W. Transthyretin slowly exchanges subunits under physiological conditions: A convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein. Sci. 2001, 10, 1606–1613. [Google Scholar] [CrossRef] [Green Version]
- Tomar, D.; Khan, T.; Singh, R.R.; Mishra, S.; Gupta, S.; Surolia, A.; Salunke, D.M. Crystallographic study of novel transthyretin ligands exhibiting negative-cooperativity between two thyroxine binding sites. PLoS ONE 2012, 7, e43522. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, R.N.; Edelhoch, H.; Saroff, H.A.; Robbins, J.; Cahnmann, H.J. Negative cooperativity in the binding of thyroxine to human serum prealbumin. Biochemistry 1975, 14, 282–289. [Google Scholar] [CrossRef]
- Johnson, S.M.; Wiseman, L.; Sekijima, Y.; Green, N.S.; Adamski-Werner, S.L.; Kelly, J.W. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: A focus on the transthyretin amyloidoses. Accounts Chem. Res. 2005, 38, 911–921. [Google Scholar] [CrossRef]
- Foss, T.R.; Kelker, M.S.; Wiseman, R.L.; Wilson, I.A.; Kelly, J.W. Kinetic stabilization of the native state by protein engineering: Implications for inhibition of transthyretin amyloidogenesis. J. Mol. Biol. 2005, 347, 841–854. [Google Scholar] [CrossRef]
- Johnson, S.M.; Connelly, S.; Fearns, C.; Powers, E.T.; Kelly, J.W. The transthyretin amyloidoses: From delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J. Mol. Biol. 2012, 421, 185–203. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. J. Hear. Fail. 2021, 23, 512–526. [Google Scholar] [CrossRef] [PubMed]
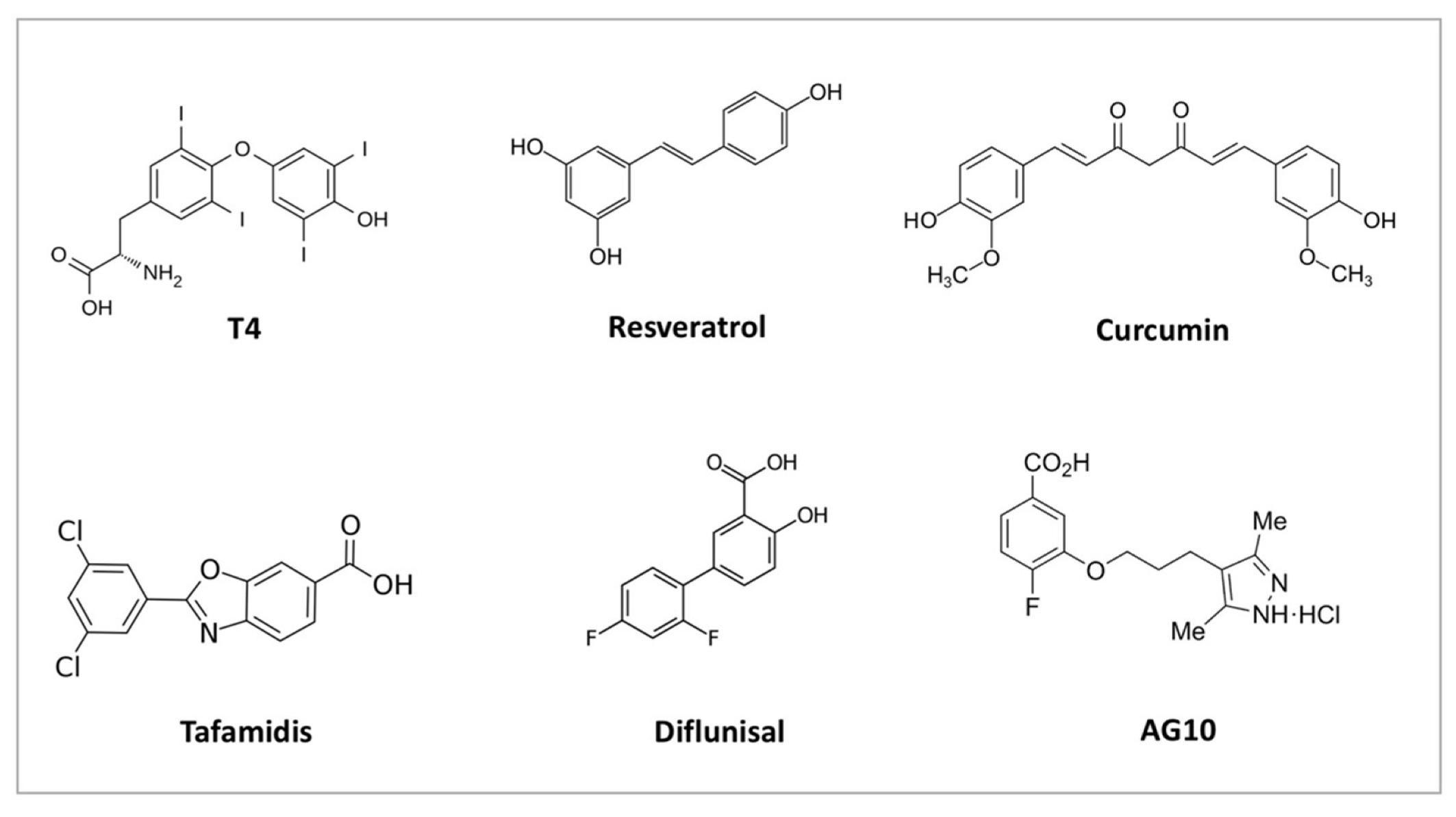
- Coelho, T.; Merlini, G.; Bulawa, C.E.; Fleming, J.A.; Judge, D.; Kelly, J.W.; Maurer, M.S.; Planté-Bordeneuve, V.; Labaudinière, R.; Mundayat, R.; et al. Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2016, 5, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekijima, Y.; Dendle, M.A.; Kelly, J.W. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006, 13, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Heitner, S.B.; Falk, R.H.; Maurer, M.S.; Shah, S.J.; Witteles, R.M.; Grogan, M.; Selby, V.N.; Jacoby, D.; Hanna, M.; et al. Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Pullakhandam, R.; Srinivas, P.; Nair, M.K.; Reddy, G.B. Binding and stabilization of transthyretin by curcumin. Arch. Biochem. Biophys. 2009, 485, 115–119. [Google Scholar] [CrossRef]
- Santos, L.M.; Rodrigues, D.; Alemi, M.; Silva, S.C.; Ribeiro, C.A.; Cardoso, I. Resveratrol administration increases Transthyretin protein levels ameliorating AD features- importance of transthyretin tetrameric stability. Mol. Med. 2016, 22, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, T.; Takaki, S.; Chosa, K.; Sato, T.; Suico, M.A.; Teranishi, Y.; Shuto, T.; Mizuguchi, M.; Kai, H. Structural stabilization of transthyretin by a new compound, 6-benzoyl-2-hydroxy-1H-benzo[de]isoquinoline-1,3(2H)-dione. J. Pharmacol. Sci. 2015, 129, 240–243. [Google Scholar] [CrossRef] [Green Version]
- Ciccone, L.; Tonali, N.; Nencetti, S.; Orlandini, E. Natural compounds as inhibitors of transthyretin amyloidosis and neuroprotective agents: Analysis of structural data for future drug design. J. Enzym. Inhib. Med. Chem. 2020, 35, 1145–1162. [Google Scholar] [CrossRef]
- Almeida, M.; Gales, L.; Damas, A.M.; Cardoso, I.; Saraiva, M.J. Small transthyretin (TTR) ligands as possible therapeutic agents in TTR amyloidoses. Curr. Drug Target.-CNS Neurol. Disord. 2005, 4, 587–596. [Google Scholar] [CrossRef]
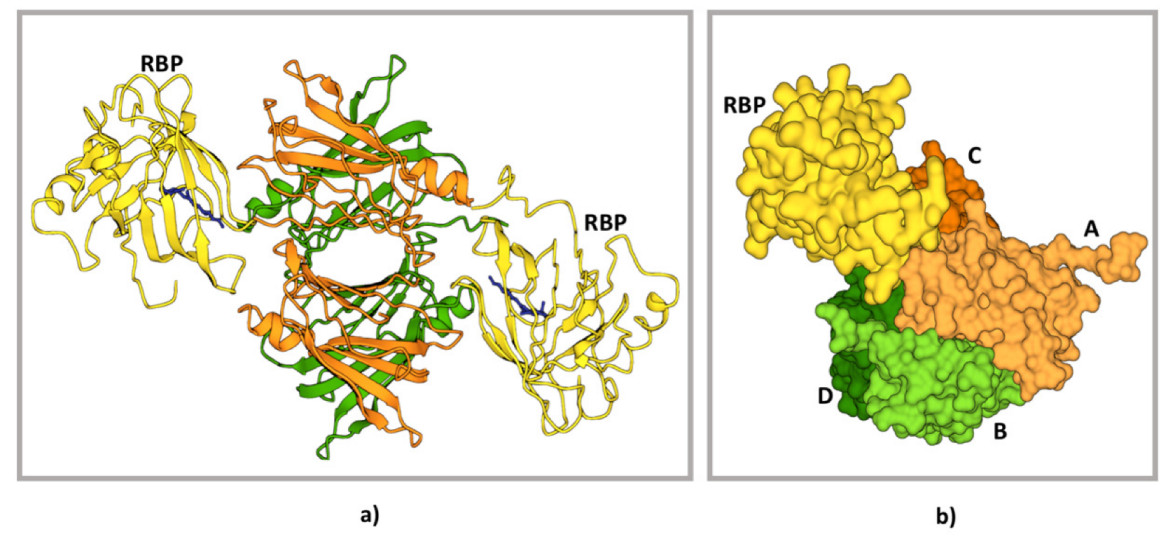
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef]
- Naylor, H.M.; Newcomer, M.E. The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry 1999, 38, 2647–2653. [Google Scholar] [CrossRef] [PubMed]
- Berni, R.; Malpeli, G.; Folli, C.; Murrell, J.; Liepnieks, J.; Benson, M. The Ile-84-->Ser amino acid substitution in transthyretin interferes with the interaction with plasma retinol-binding protein. J. Biol. Chem. 1994, 269, 23395–23398. [Google Scholar] [CrossRef]
- TTrägårdh, L.; Anundi, H.; Rask, L.; Sege, K.; A Peterson, P. On the stoichiometry of the interaction between prealbumin and retinol-binding protein. J. Biol. Chem. 1980, 255, 9243–9248. [Google Scholar] [CrossRef]
- Prapunpoj, P.; Leelawatwattana, L. Evolutionary changes to transthyretin: Structure-function relationships. FEBS J. 2009, 276, 5330–5341. [Google Scholar] [CrossRef]
- Zanotti, G.; Berni, R. Plasma retinol-binding protein: Structure and interactions with retinol, retinoids, and transthyretin. Vitam. & Horm. 2004, 69, 271–295. [Google Scholar] [CrossRef]
- Malpeli, G.; Folli, C.; Berni, R. Retinoid binding to retinol-binding protein and the interference with the interaction with transthyretin. Biochim. Biophys. Acta. 1996, 1294, 48–54. [Google Scholar] [CrossRef]
- White, J.T.; Kelly, J.W. Support for the multigenic hypothesis of amyloidosis: The binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 13019–13024. [Google Scholar] [CrossRef] [Green Version]
- Heller, J.; Horwitz, J. Conformational changes following interaction between retinol isomers and human retinol-binding protein and between the retinol-binding protein and prealbumin. J. Biol. Chem. 1973, 248, 6308–6316. [Google Scholar] [CrossRef]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Uncovering the Neuroprotective Mechanisms of Curcumin on Transthyretin Amyloidosis. Int. J. Mol. Sci. 2019, 20, 1287. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, E.; Ożyhar, A. Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases. Cells 2021, 10, 1768. [Google Scholar] [CrossRef]
- Si, J.B.; Kim, B.; Kim, J.H. Transthyretin Misfolding, A Fatal Structural Pathogenesis Mechanism. Int. J. Mol. Sci. 2021, 22, 4429. [Google Scholar] [CrossRef] [PubMed]
- Pires, R.H.; Karsai, A.; Saraiva, M.J.; Damas, A.M.; Kellermayer, M.S.Z. Distinct annular oligomers captured along the assembly and disassembly pathways of transthyretin amyloid protofibrils. PLoS ONE 2012, 7, e44992. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Katsuno, M. Transthyretin Amyloidosis: Update on the Clinical Spectrum, Pathogenesis, and Disease-Modifying Therapies. Neurol. Ther. 2020, 9, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Leach, B.I.; Zhang, X.; Kelly, J.W.; Dyson, H.J.; Wright, P.E. NMR Measurements Reveal the Structural Basis of Transthyretin Destabilization by Pathogenic Mutations. Biochemistry 2018, 57, 4421–4430. [Google Scholar] [CrossRef]
- Conceição, I.; Coelho, T.; Rapezzi, C.; Parman, Y.; Obici, L.; Galán, L.; Rousseau, A. Assessment of patients with hereditary transthyretin amyloidosis - understanding the impact of management and disease progression. Amyloid 2019, 26, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Rowczenio, D.M.; Noor, I.; Gillmore, J.D.; Lachmann, H.J.; Whelan, C.; Hawkins, P.N.; Obici, L.; Westermark, P.; Grateau, G.; Wechalekar, A.D. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum. Mutat. 2014, 35, E2403–E2412. [Google Scholar] [CrossRef]
- Sant’Anna, R.; Almeida, M.; Varejao, N.; Gallego, P.; Esperante, S.; Ferreira, P.; Pereira-Henriques, A.; Palhano, F.; De Carvalho, M.; Foguel, D.; et al. Cavity filling mutations at the thyroxine-binding site dramatically increase transthyretin stability and prevent its aggregation. Sci. Rep. 2017, 7, srep44709. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.R.; Alves, I.L.; Terazaki, H.; Ando, Y.; Saraiva, M.J. Comparative studies of two transthyretin variants with protective effects on familial amyloidotic polyneuropathy: TTR R104H and TTR T119M. Biochem. Biophys. Res. Commun. 2000, 270, 1024–1028. [Google Scholar] [CrossRef]
- Sekijima, Y.; Dendle, M.T.; Wiseman, R.L.; White, J.T.; D’Haeze, W.; Kelly, J.W. R104H may suppress transthyretin amyloidogenesis by thermodynamic stabilization, but not by the kinetic mechanism characterizing T119 interallelic trans-suppression. Amyloid 2006, 13, 57–66. [Google Scholar] [CrossRef]
- Palaninathan, S.K. Nearly 200 X-ray crystal structures of transthyretin: What do they tell us about this protein and the design of drugs for TTR amyloidoses? Curr. Med. Chem. 2012, 19, 2324–2342. [Google Scholar] [CrossRef]
- Hörnberg, A.; Eneqvist, T.; Olofsson, A.; Lundgren, E.; Sauer-Eriksson, E. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J. Mol. Biol. 2000, 302, 649–669. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.A.; Sun, X.; Dyson, H.J.; Wright, P.E. Thermodynamic Stability and Aggregation Kinetics of EF Helix and EF Loop Variants of Transthyretin. Biochemistry 2021, 60, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Saldaño, T.E.; Zanotti, G.; Parisi, G.; Fernandez-Alberti, S. Evaluating the effect of mutations and ligand binding on transthyretin homotetramer dynamics. PLoS ONE 2017, 12, e0181019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.; Steinrauf, L.; Braden, B.; Liepnieks, J.; Benson, M.; Holmgren, G.; Sandgren, O.; Steen, L. The X-ray crystal structure refinements of normal human transthyretin and the amyloidogenic Val-30-->Met variant to 1.7-A resolution. J. Biol. Chem. 1993, 268, 2416–2424. [Google Scholar] [CrossRef]
- Terry, C.; Damas, A.; Oliveira, P.; Saraiva, M.J.; Alves, I.; Costa, P.; Matias, P.; Sakaki, Y.; Blake, C. Structure of Met30 variant of transthyretin and its amyloidogenic implications. EMBO J. 1993, 12, 735–741. [Google Scholar] [CrossRef]
- Yee, A.W.; Aldeghi, M.; Blakeley, M.P.; Ostermann, A.; Mas, P.J.; Moulin, M.; de Sanctis, D.; Bowler, M.W.; Mueller-Dieckmann, C.; Mitchell, E.P.; et al. A molecular mechanism for transthyretin amyloidogenesis. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Miyata, M.; Sato, T.; Mizuguchi, M.; Nakamura, T.; Ikemizu, S.; Nabeshima, Y.; Susuki, S.; Suwa, Y.; Morioka, H.; Ando, Y.; et al. Role of the glutamic acid 54 residue in transthyretin stability and thyroxine binding. Biochemistry 2009, 49, 114–123. [Google Scholar] [CrossRef]
- Sebastião, P.; Dauter, Z.; Saraiva, M.J.; Damas, A.M. Crystallization and preliminary X-ray diffraction studies of Leu55Pro variant transthyretin. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 566–568. [Google Scholar] [CrossRef]
- Sebastião, M.P.; Saraiva, M.J.; Damas, A.M. The crystal structure of amyloidogenic Leu55 --> Pro transthyretin variant reveals a possible pathway for transthyretin polymerization into amyloid fibrils. J. Biol. Chem. 1998, 273, 24715–24722. [Google Scholar] [CrossRef] [Green Version]
- Damas, A.M.; Ribeiro, S.; Lamzin, V.; Palha, J.A.; Saraiva, M.J. Structure of the Val122Ile variant transthyretin - a cardiomyopathic mutant. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 966–972. [Google Scholar] [CrossRef]
- Vugmeyster, L.; Au, D.F.; Ostrovsky, D.; Kierl, B.; Fu, R.; Hu, Z.W.; Qiang, W. Effect of Post-Translational Modifications and Mutations on Amyloid-β Fibrils Dynamics at N Terminus. Biophys. J. 2019, 117, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Dorval, V.; Fraser, P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and and α-Synuclein. J. Biol. Chem. 2006, 281, 9919–9924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.; Makarov, A.A. Amyloid β Modification: A Key to the Sporadic Alzheimer’s Disease? Front. Genet. 2017, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Radamaker, L.; Karimi-Farsijani, S.; Andreotti, G.; Baur, J.; Neumann, M.; Schreiner, S.; Berghaus, N.; Motika, R.; Haupt, C.; Walther, P.; et al. Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
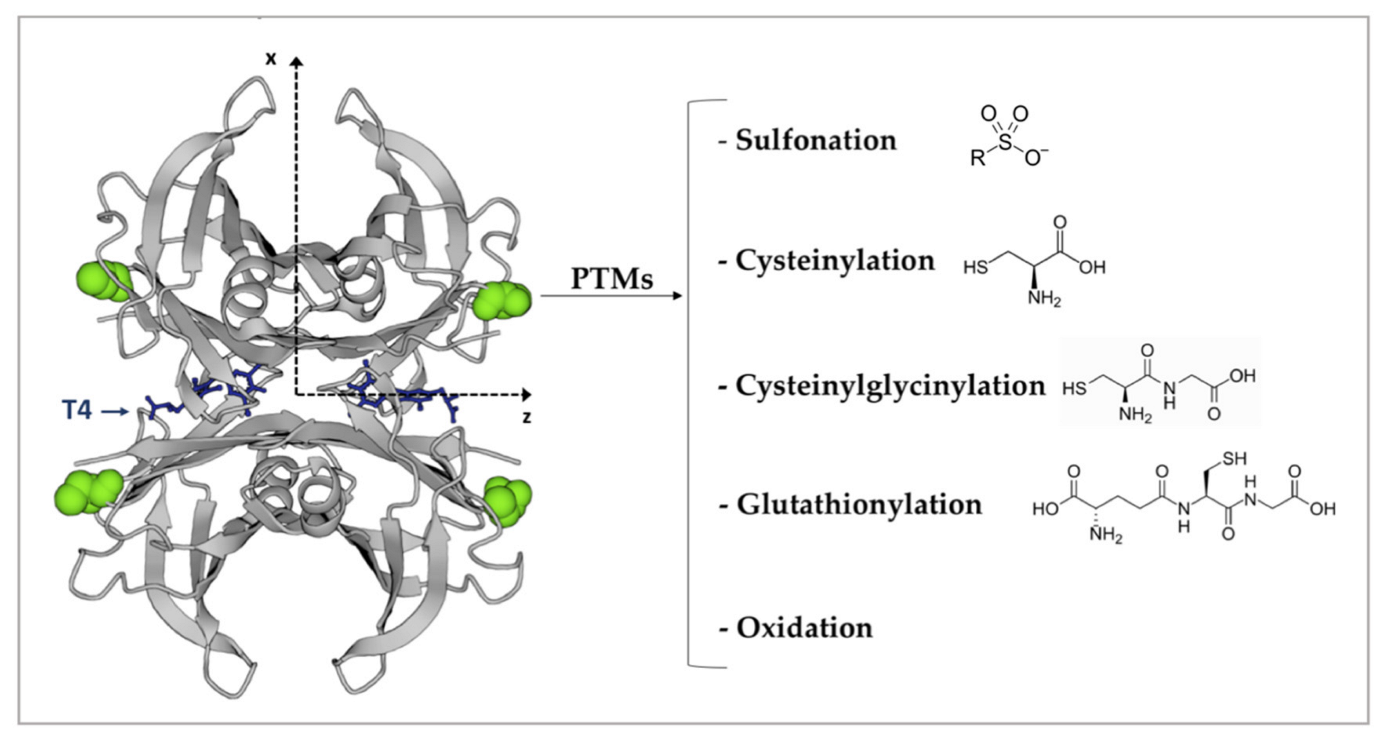
- Henze, A.; Homann, T.; Serteser, M.; Can, O.; Sezgin, O.; Coskun, A.; Unsal, I.; Schweigert, F.; Ozpinar, A. Post-translational modifications of transthyretin affect the triiodonine-binding potential. J. Cell. Mol. Med. 2015, 19, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, T.; Carlstroem, A.; Joernvall, H. Different types of microheterogeneity of human thyroxine-binding prealbumin. Biochemistry 1987, 26, 4572–4583. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, K.; Bahl, J.M.; Tanassi, J.T.; Simonsen, A.H.; Heegaard, N.H. Characterization and stability of transthyretin isoforms in cerebrospinal fluid examined by immunoprecipitation and high-resolution mass spectrometry of intact protein. Methods 2012, 56, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Henze, A.; Homann, T.; Rohn, I.; Aschner, M.; Link, C.D.; Kleuser, B.; Schweigert, F.; Schwerdtle, T.; Bornhorst, J. Caenorhabditis elegans as a model system to study post-translational modifications of human transthyretin. Sci. Rep. 2016, 6, 37346. [Google Scholar] [CrossRef] [Green Version]
- Gales, L.; Saraiva, M.J.; Damas, A.M. Structural basis for the protective role of sulfite against transthyretin amyloid formation. Biochim. et Biophys. Acta (BBA)-Proteins Proteom. 2007, 1774, 59–64. [Google Scholar] [CrossRef]
- Altland, K.; Winter, P. Potential treatment of transthyretin-type amyloidoses by sulfite. Neurogenetics 1999, 2, 183–188. [Google Scholar] [CrossRef]
- Kingsbury, J.; Laue, T.M.; Klimtchuk, E.S.; Théberge, R.; Costello, C.; Connors, L.H. The modulation of transthyretin tetramer stability by cysteine 10 adducts and the drug diflunisal: Direct analysis by fluorescence-detected analytical ultracentrifugation. J. Biol. Chem. 2008, 283, 11887–11896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaoka, Y.; Ohta, M.; Miyakawa, K.; Nakamura, O.; Suzuki, M.; Takahashi, K.; Yamamura, K.; Sakaki, Y. Cysteine 10 is a key residue in amyloidogenesis of human transthyretin Val30Met. Am. J. Pathol. 2004, 164, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Kelly, J.W. Cys10 mixed disulfides make transthyretin more amyloidogenic under mildly acidic conditions. Biochemistry 2003, 42, 8756–8761. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Buxbaum, J.N.; Reixach, N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 2013, 52, 1913–1926. [Google Scholar] [CrossRef] [Green Version]
- Navab, M.; E Smith, J.; Goodman, D.S. Rat plasma prealbumin. Metabolic studies on effects of vitamin A status and on tissue distribution. J. Biol. Chem. 1977, 252, 5107–5114. [Google Scholar] [CrossRef]
- Rask, L.; Valtersson, C.; Anundi, H.; Kvist, S.; Eriksson, U.; Dallner, G.; A Peterson, P. Subcellular localization in normal and vitamin A-deficient rat liver of vitamin A serum transport proteins, albumin, ceruloplasmin and class I major histocompatibility antigens. Exp. Cell Res. 1983, 143, 91–102. [Google Scholar] [CrossRef]
- Melhus, H.; Nilsson, T.; Peterson, P.A.; Rask, L. Retinol-binding protein and transthyretin expressed in HeLa cells form a complex in the endoplasmic reticulum in both the absence and the presence of retinol. Exp. Cell Res. 1991, 197, 119–124. [Google Scholar] [CrossRef]
- Episkopou, V.; Maeda, S.; Nishiguchi, S.; Shimada, K.; Gaitanaris, G.A.; Gottesman, M.E.; Robertson, E.J. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc. Natl. Acad. Sci. USA 1993, 90, 2375–2379. [Google Scholar] [CrossRef] [Green Version]
- E Smith, J.; Deen, D.D.; Sklan, D.; Goodman, D.S. Colchicine inhibition of retinol-binding protein secretion by rat liver. J. Lipid Res. 1980, 21, 229–237. [Google Scholar] [CrossRef]
- Berni, R.; Clerici, M.; Malpeli, G.; Cleris, L.; Formelli, F. Retinoids: In vitro interaction with retinol-binding protein and influence on plasma retinol. FASEB J. 1993, 7, 1179–1184. [Google Scholar] [CrossRef]
- Bellovino, D.; Morimoto, T.; Tosetti, F.; Gaetani, S. Retinol binding protein and transthyretin are secreted as a complex formed in the endoplasmic reticulum in HepG2 human hepatocarcinoma cells. Exp. Cell Res. 1996, 222, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, P.; Sekijima, Y.; White, J.T.; Wiseman, R.L.; Lim, A.; Costello, C.E.; Altland, K.; Garzuly, F.; Budka, H.; Kelly, J.W. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: A prescription for central nervous system amyloidosis? Biochemistry 2003, 42, 6656–6663. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Hammarström, P.; Matsumura, M.; Shimizu, Y.; Iwata, M.; Tokuda, T.; Ikeda, S.; Kelly, J.W. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab. Investig. 2003, 83, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sörgjerd, K.; Ghafouri, B.; Jonsson, B.H.; Kelly, J.W.; Blond, S.Y.; Hammarström, P. Retention of misfolded mutant transthyretin by the chaperone BiP/GRP78 mitigates amyloidogenesis. J. Mol. Biol. 2006, 356, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Susuki, S.; Sato, T.; Miyata, M.; Momohara, M.; Suico, M.A.; Shuto, T.; Ando, Y.; Kai, H. The Endoplasmic Reticulum-associated Degradation of Transthyretin Variants Is Negatively Regulated by BiP in Mammalian Cells. J. Biol. Chem. 2009, 284, 8312–8321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Costa, G.; Ribeiro-Silva, C.; Ribeiro, R.; Gilberto, S.; Gomes, R.A.; Ferreira, A.; Mateus, É.; Barroso, E.; Coelho, A.V.; Freire, A.P.; et al. Transthyretin Amyloidosis: Chaperone Concentration Changes and Increased Proteolysis in the Pathway to Disease. PLoS ONE 2015, 10, e0125392. [Google Scholar] [CrossRef]
- Mesgarzadeh, J.S.; Romine, I.C.; Smith-Cohen, E.M.; Grandjean, J.M.D.; Kelly, J.W.; Genereux, J.C.; Wiseman, R.L. ATF6 Activation Reduces Amyloidogenic Transthyretin Secretion through Increased Interactions with Endoplasmic Reticulum Proteostasis Factors. Cells 2022, 11, 1661. [Google Scholar] [CrossRef]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarström, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Cohen, F.E.; Kelly, J.W. Therapeutic approaches to protein-misfolding diseases. Nature 2003, 426, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, A.R.; Yerbury, J.J.; Ecroyd, H.; Wilson, M.R. Extracellular chaperones and proteostasis. Annu. Rev. Biochem. 2013, 82, 295–322. [Google Scholar] [CrossRef] [Green Version]
- Dabbs, R.A.; Wyatt, A.R.; Yerbury, J.J.; Ecroyd, H.; Wilson, M.R. Extracellular chaperones. Top. Curr. Chem. 2013, 328, 241–268. [Google Scholar] [CrossRef] [Green Version]
- Nilselid, A.M.; Davidsson, P.; Nägga, K.; Andreasen, N.; Fredman, P.; Blennow, K. Clusterin in cerebrospinal fluid: Analysis of carbohydrates and quantification of native and glycosylated forms. Neurochem. Int. 2006, 48, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Gollin, P.A.; Kalaria, R.N.; Eikelenboom, P.; Rozemuller, A.; Perry, G. Alpha 1-antitrypsin and alpha 1-antichymotrypsin are in the lesions of Alzheimer’s disease. Neuroreport 1992, 3, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Poon, S.; Meehan, S.; Thompson, B.; Kumita, J.R.; Dobson, C.M.; Wilson, M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007, 21, 2312–2322. [Google Scholar] [CrossRef]
- Makover, A.; Moriwaki, H.; Ramakrishnan, R.; Saraiva, M.J.; Blaner, W.S.; Goodman, D.S. Plasma transthyretin. Tissue sites of degradation and turnover in the rat. J. Biol. Chem. 1988, 263, 8598–8603. [Google Scholar] [CrossRef]
- Gjøen, T.; Bjerkelund, T.; Blomhoff, H.K.; Norum, K.R.; Berg, T.; Blomhoff, R. Liver takes up retinol-binding protein from plasma. J. Biol. Chem. 1987, 262, 10926–10930. [Google Scholar] [CrossRef]
- Sousa, M.M.; Norden, A.G.; Jacobsen, C.; Willnow, T.E.; Christensen, E.I.; Thakker, R.V.; Verroust, P.J.; Moestrup, S.K.; Saraiva, M.J. Evidence for the role of megalin in renal uptake of transthyretin. J. Biol. Chem. 2000, 275, 38176–38181. [Google Scholar] [CrossRef] [Green Version]
- May, P.; Woldt, E.; Matz, R.L.; Boucher, P. The LDL receptor-related protein (LRP) family: An old family of proteins with new physiological functions. Ann. Med. 2007, 39, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.I.; Moskaug, J.O.; Vorum, H.; Jacobsen, C.; Gundersen, T.E.; Nykjaer, A.; Blomhoff, R.; Willnow, T.E.; Moestrup, S.K. Evidence for an essential role of megalin in transepithelial transport of retinol. J. Am. Soc. Nephrol. 1999, 10, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Fleming, C.E.; Mar, F.M.; Franquinho, F.; Saraiva, M.J.; Sousa, M.M. Transthyretin internalization by sensory neurons is megalin mediated and necessary for its neuritogenic activity. J. Neurosci. 2009, 29, 3220–3232. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; Saraiva, M.J. Internalization of transthyretin:. Evidence of a novel yet unidentified receptor-associated protein (RAP)-sensitive receptor. J. Biol. Chem. 2001, 276, 14420–14425. [Google Scholar] [CrossRef] [Green Version]
- Divino, C.M.; Schussler, G.C. Receptor-mediated uptake and internalization of transthyretin. J. Biol. Chem. 1990, 265, 1425–1429. [Google Scholar] [CrossRef]
- Sousa, M.M.; Berglund, L.; Saraiva, M.J. Transthyretin in high density lipoproteins: Association with apolipoprotein A-I. J. Lipid Res. 2000, 41, 58–65. [Google Scholar] [CrossRef]
- Misumi, Y.; Ando, Y.; Gonçalves, N.P.; Saraiva, M.J. Fibroblasts endocytose and degrade transthyretin aggregates in transthyretin-related amyloidosis. Lab. Investig. 2013, 93, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Misumi, Y.; Ueda, M.; Obayashi, K.; Jono, H.; Yamashita, T.; Ando, Y. Interaction between amyloid fibril formation and extracellular matrix in the proceedings of VIIIth International Symposium on Familial Amyloidotic Polyneuropathy. Amyloid 2012, 19, 8–10. [Google Scholar] [CrossRef]
- Bourgault, S.; Solomon, J.P.; Reixach, N.; Kelly, J.W. Sulfated glycosaminoglycans accelerate transthyretin amyloidogenesis by quaternary structural conversion. Biochemistry 2011, 50, 1001–1015. [Google Scholar] [CrossRef] [Green Version]
- Madine, J.; Davies, H.A.; Hughes, E.; Middleton, D.A. Heparin promotes the rapid fibrillization of a peptide with low intrinsic amyloidogenicity. Biochemistry 2013, 52, 8984–8992. [Google Scholar] [CrossRef]
- Fella, E.; Sokratous, K.; Papacharalambous, R.; Kyriacou, K.; Phillips, J.; Sanderson, S.; Panayiotou, E.; Kyriakides, T. Pharmacological Stimulation of Phagocytosis Enhances Amyloid Plaque Clearance; Evidence from a Transgenic Mouse Model of ATTR Neuropathy. Front. Mol. Neurosci. 2017, 10, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoi, A.; Su, Y.; Torikai, M.; Jono, H.; Ishikawa, D.; Soejima, K.; Higuchi, H.; Guo, J.; Ueda, M.; Suenaga, G.; et al. Novel Antibody for the Treatment of Transthyretin Amyloidosis. J. Biol. Chem. 2016, 291, 25096–25105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalon, A.; Hagenbuch, A.; Huy, C.; Varela, E.; Combaluzier, B.; Damy, T.; Suhr, O.B.; Saraiva, M.J.; Hock, C.; Nitsch, R.M.; et al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat. Commun. 2021, 12, 3142. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, A.; Yazaki, M.; Kametani, F.; Takei, Y.; Ikeda, S. Marked regression of abdominal fat amyloid in patients with familial amyloid polyneuropathy during long-term follow-up after liver transplantation. Liver Transpl. 2008, 14, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Morfino, P.; Aimo, A.; Panichella, G.; Rapezzi, C.; Emdin, M. Amyloid seeding as a disease mechanism and treatment target in transthyretin cardiac amyloidosis. Hear. Fail. Rev. 2022. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
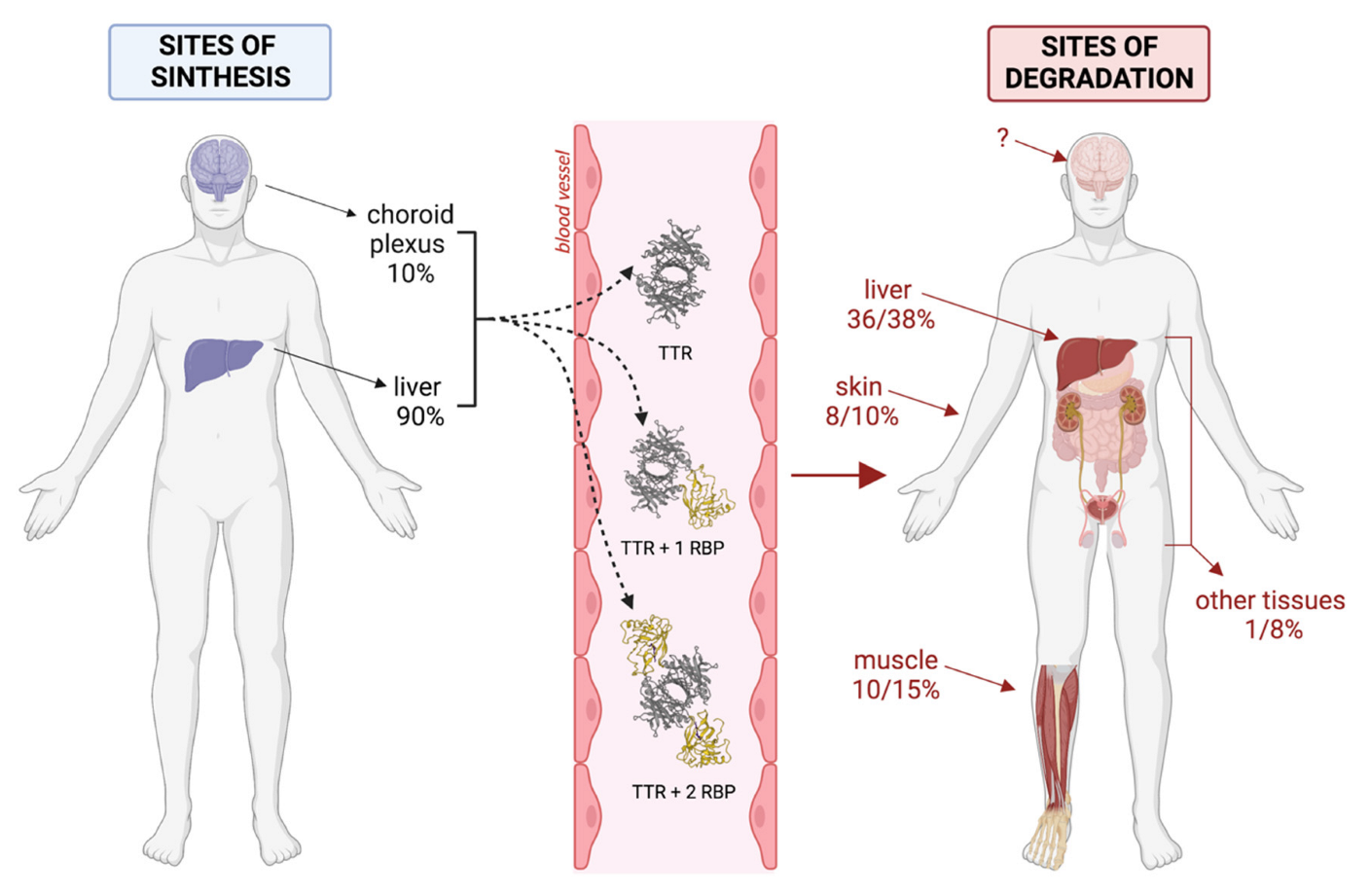{kind=link}
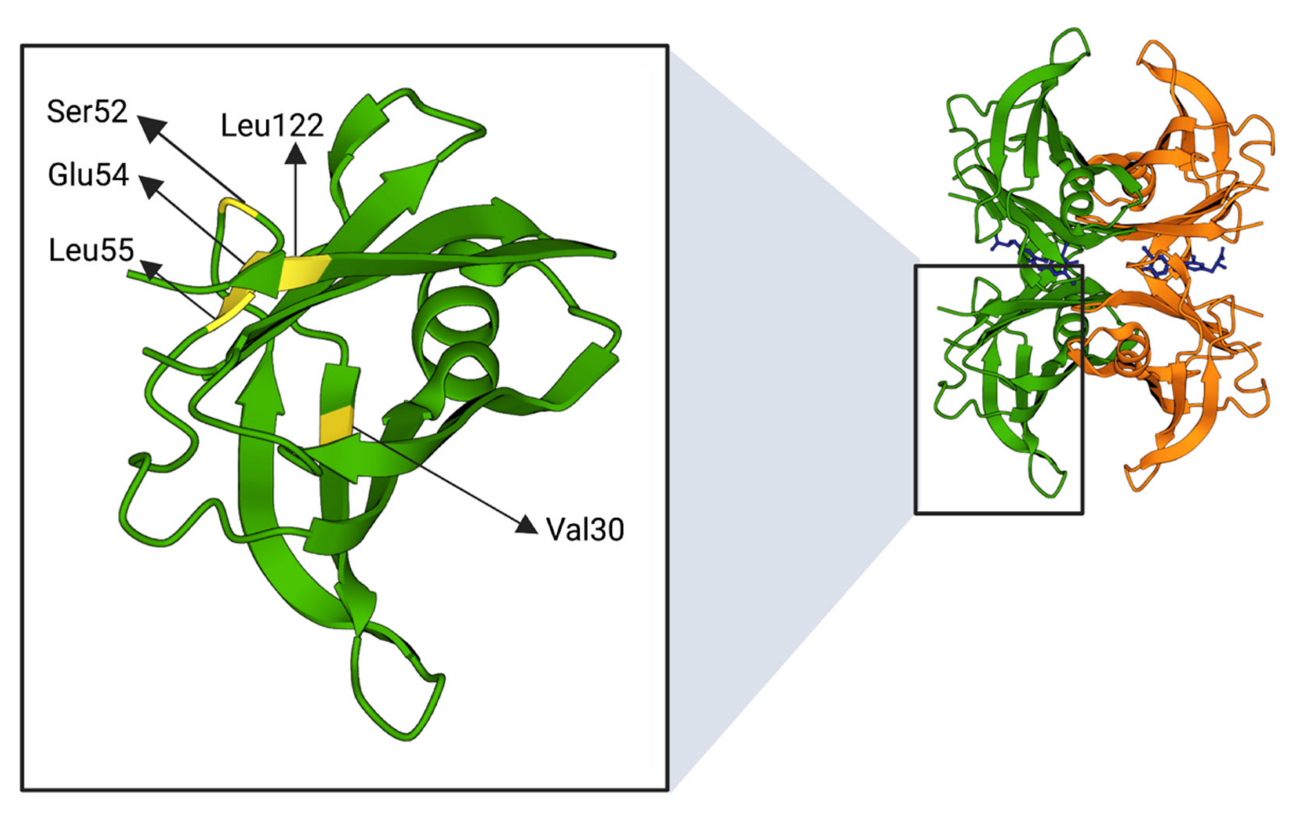
| TTRv (Other Name) * | Mutated Nucleotide (mRNA) | Clinical Phenotype | Involved Secondary Structure [30] | Overall Structural Alteration | Ref. |
|---|---|---|---|---|---|
| Val30Met (p.Val50Met) | c.148G>A | AN, E, LM, PN | β-strand B | Destabilisation of B and E strands resulting in a distortion of T4 binding channel. Causes a lower affinity for T4 | [104,105] |
| Ser52Pro (p.Ser72Pro) | c.214T>C | AN, H, K, PN | β-bend | Stability alteration of C-D loop in protein monomer | [106] |
| Glu54Lys (p.Glu74Lys) | c.220G>A | AN, H, PN | β-strand D | Lys54 destabilises tetramer structure due to increased electrostatic repulsion between Lys15 of two monomers. The T4 binding pocket is markedly narrower in Glu54Lys compared with wtTTR, suggesting a decrease of affinity for T4 | [107] |
| Leu55Pro (p.Leu75Pro) | c.224T>C | AN, E, H, PN | β-strand D | Disruption of hydrogen bond interaction between β-strands A and D leads a β-strand D structure highly disordered with different contacts between the subunits as well as a significant variation in the CE region of the monomer | [108,109] |
| Val122Ile (p.Val142Ile) | c.424G>A | H | 122-127 AAs terminal loop | small changes in the region associated with the intra- and inter dimer interactions | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanguinetti, C.; Minniti, M.; Susini, V.; Caponi, L.; Panichella, G.; Castiglione, V.; Aimo, A.; Emdin, M.; Vergaro, G.; Franzini, M. The Journey of Human Transthyretin: Synthesis, Structure Stability, and Catabolism. Biomedicines 2022, 10, 1906. https://doi.org/10.3390/biomedicines10081906
Sanguinetti C, Minniti M, Susini V, Caponi L, Panichella G, Castiglione V, Aimo A, Emdin M, Vergaro G, Franzini M. The Journey of Human Transthyretin: Synthesis, Structure Stability, and Catabolism. Biomedicines. 2022; 10(8):1906. https://doi.org/10.3390/biomedicines10081906
Chicago/Turabian StyleSanguinetti, Chiara, Marianna Minniti, Vanessa Susini, Laura Caponi, Giorgia Panichella, Vincenzo Castiglione, Alberto Aimo, Michele Emdin, Giuseppe Vergaro, and Maria Franzini. 2022. "The Journey of Human Transthyretin: Synthesis, Structure Stability, and Catabolism" Biomedicines 10, no. 8: 1906. https://doi.org/10.3390/biomedicines10081906
APA StyleSanguinetti, C., Minniti, M., Susini, V., Caponi, L., Panichella, G., Castiglione, V., Aimo, A., Emdin, M., Vergaro, G., & Franzini, M. (2022). The Journey of Human Transthyretin: Synthesis, Structure Stability, and Catabolism. Biomedicines, 10(8), 1906. https://doi.org/10.3390/biomedicines10081906