
Small Molecules and Immunotherapy Agents for Enhancing Radiotherapy in Glioblastoma

 , ,
, , 

Abstract
:1. Introduction
2. Modern Treatment Strategies for Glioblastoma
2.1. Surgical Resection
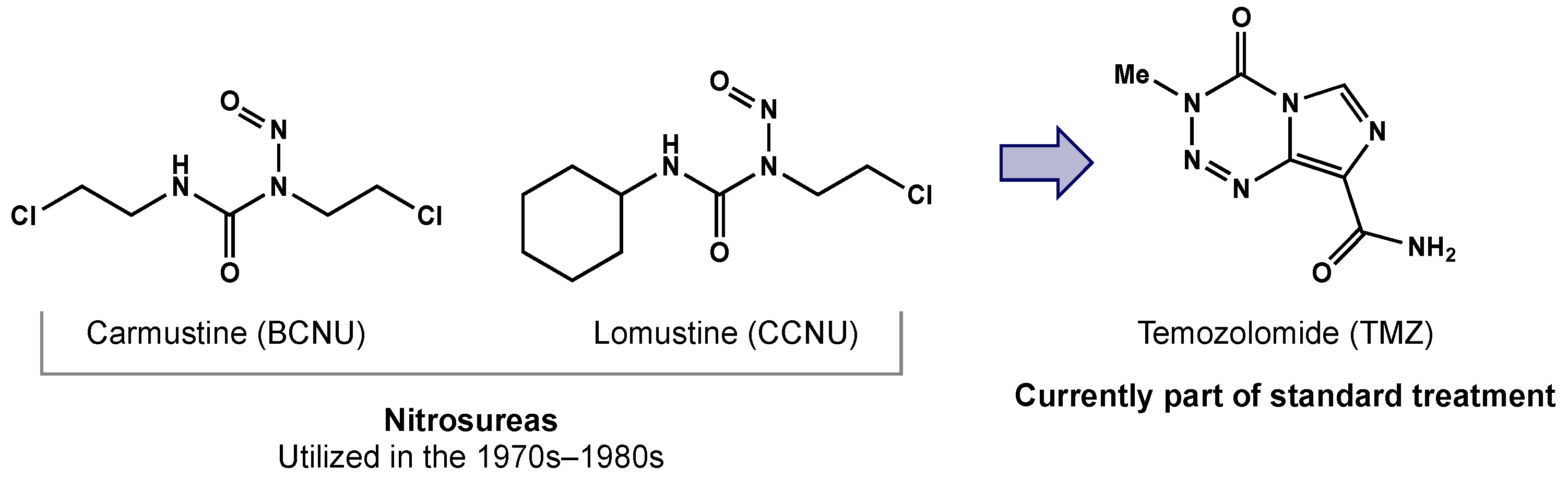
2.2. Chemotherapy
2.3. Radiotherapy
3. Mechanisms of Radiation-Induced Cancer Cell Death
4. Radioresistance
4.1. Glioma Stem Cells
4.2. Hypoxia
5. Radiosensitizers
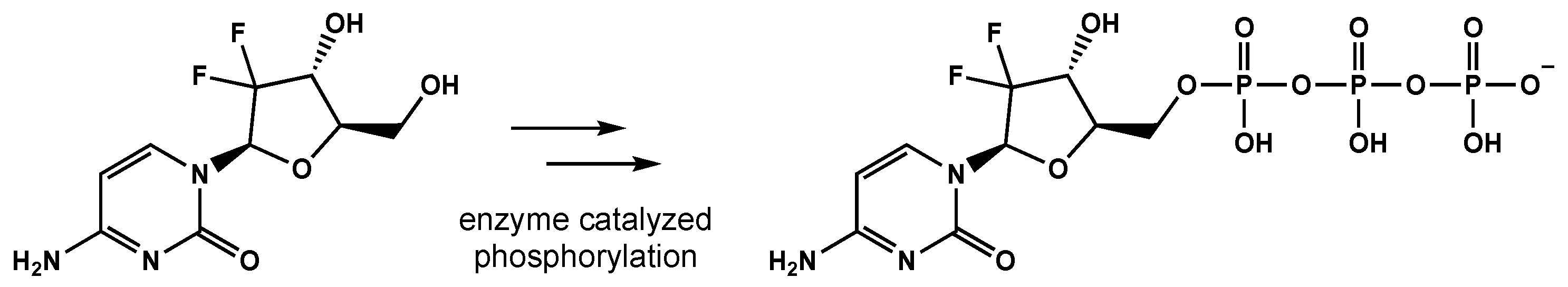
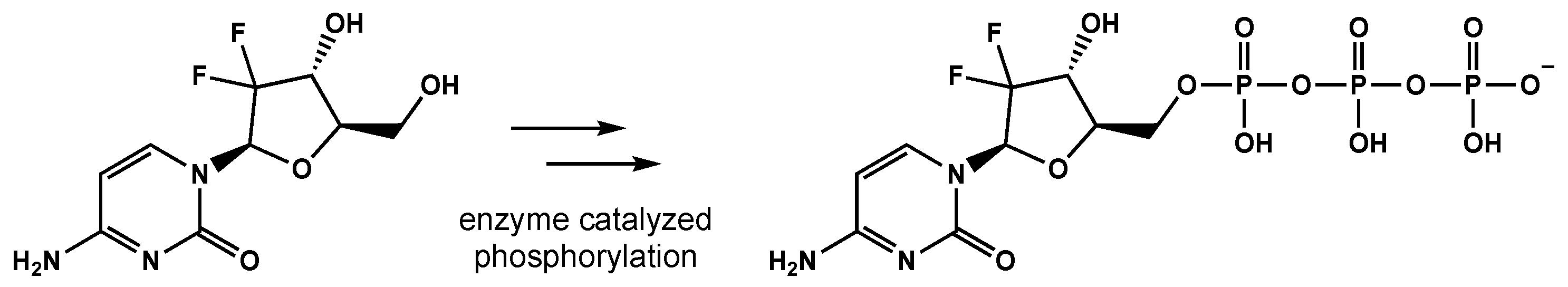
5.1. Pyrmidine Analogues
5.2. Kinase Inhibitors
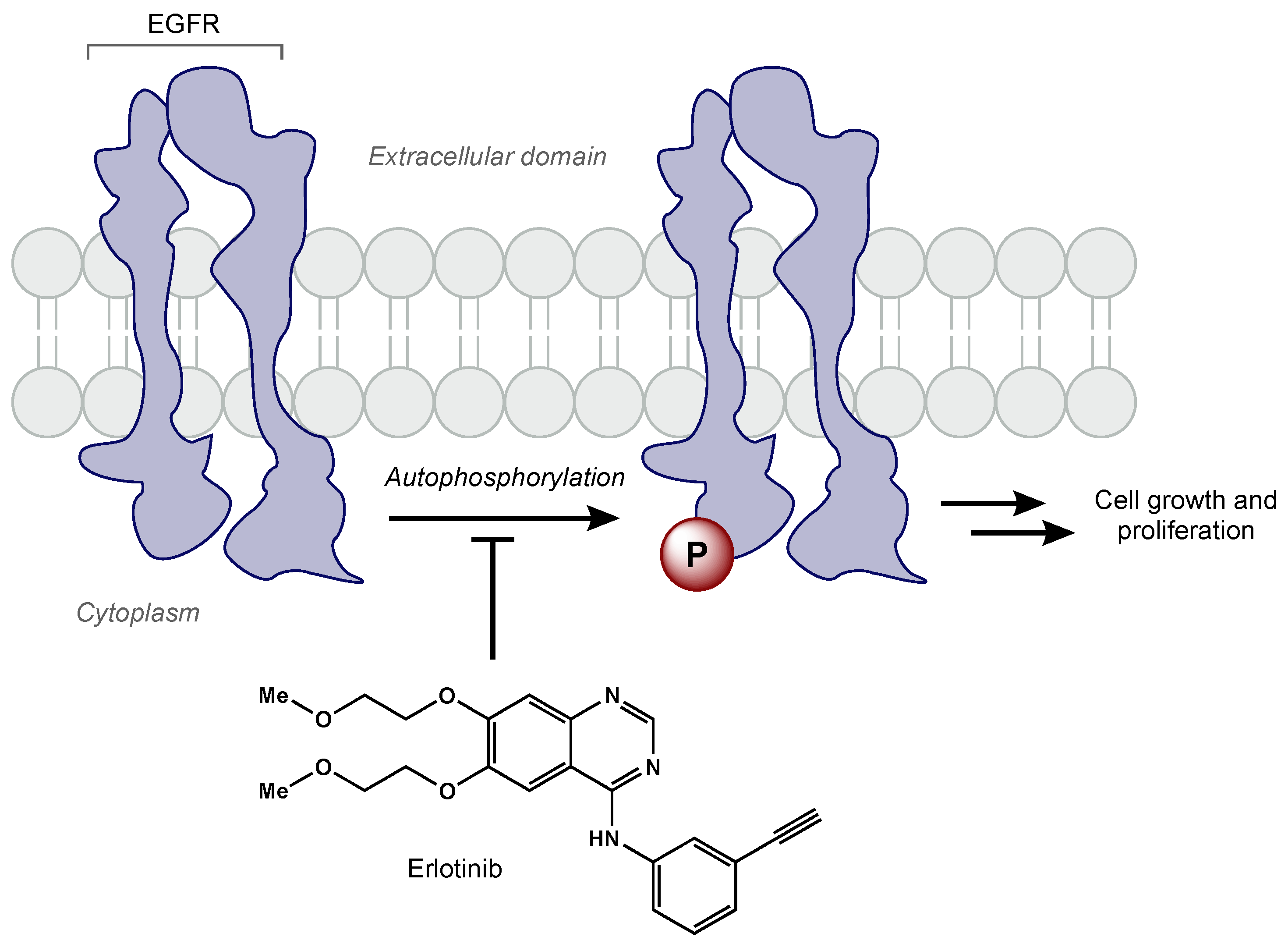
5.2.1. Tyrosine Kinase Inhibitors
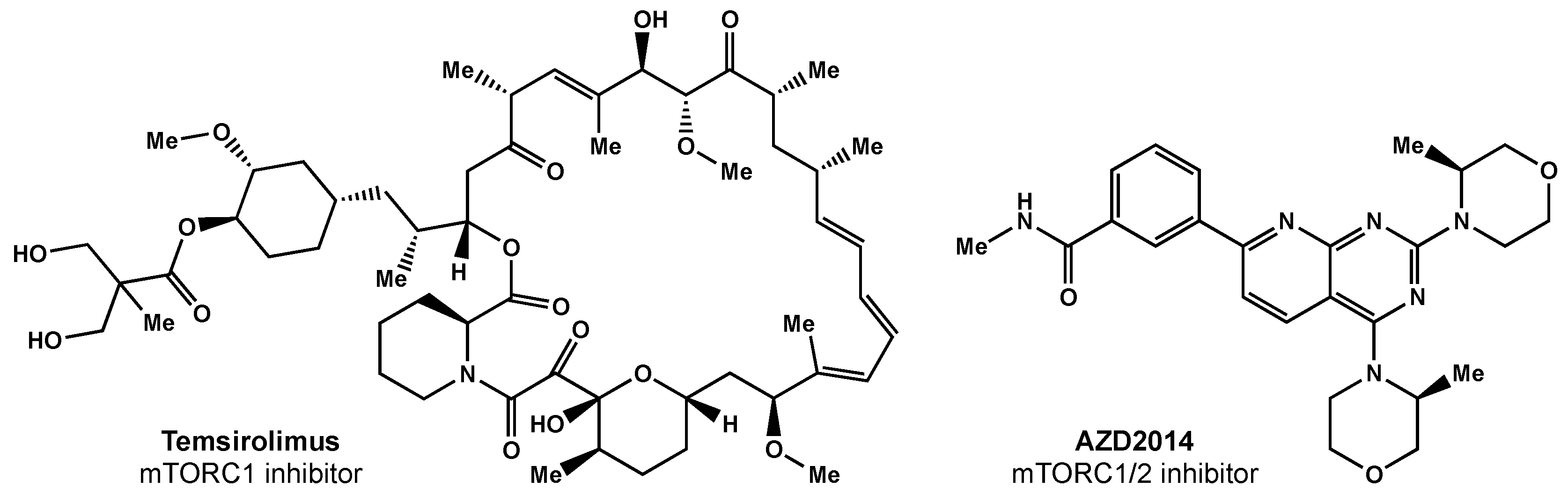
5.2.2. mTOR Inhibitors
5.3. Oxygen Mimetics
5.4. Reductive Agents
5.5. Histone Deactylase Inhibitors
5.6. Targeting DNA Repair Pathways
5.7. Allosteric Modifiers of Hemoglobin
5.8. Immunotherapy
5.8.1. Anti-Angiogenic Therapy
5.8.2. Immune Checkpoint Inhibitors
6. Recent Preclinical Studies
6.1. Purine Metabolism
6.2. Metabolic Targeting
6.3. Curcumin
6.4. Hsp90 Inhibitors
6.5. MDM2 Inhibitors
6.6. Chimeric Antigen Receptor (CAR) T Cell Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-Oncology 2014, 16, iv1–iv63. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef]
- Sanai, N.; Polley, M.-Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An Extent of Resection Threshold for Newly Diagnosed Glioblastomas: Clinical Article. J. Neurosurg. JNS 2011, 115, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.M.; Suki, D.; Hess, K.; Sawaya, R. The Influence of Maximum Safe Resection of Glioblastoma on Survival in 1229 Patients: Can We Do Better than Gross-Total Resection? J. Neurosurg. JNS 2016, 124, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Duffau, H. Long-Term Outcomes after Supratotal Resection of Diffuse Low-Grade Gliomas: A Consecutive Series with 11-Year Follow-Up. Acta Neurochir. 2016, 158, 51–58. [Google Scholar] [CrossRef]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2021, 10, 574012. [Google Scholar] [CrossRef]
- Rong, L.; Li, N.; Zhang, Z. Emerging Therapies for Glioblastoma: Current State and Future Directions. J. Exp. Clin. Cancer Res. 2022, 41, 142. [Google Scholar] [CrossRef]
- Fan, C.-H.; Liu, W.-L.; Cao, H.; Wen, C.; Chen, L.; Jiang, G. O6-Methylguanine DNA Methyltransferase as a Promising Target for the Treatment of Temozolomide-Resistant Gliomas. Cell Death Dis. 2013, 4, e876. [Google Scholar] [CrossRef] [Green Version]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. P53 Effects Both the Duration of G2/M Arrest and the Fate of Temozolomide-Treated Human Glioblastoma Cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laperriere, N.; Zuraw, L.; Cairncross, G. Radiotherapy for Newly Diagnosed Malignant Glioma in Adults: A Systematic Review. Radiother. Oncol. 2002, 64, 259–273. [Google Scholar] [CrossRef]
- Davies, A.M.; Weinberg, U.; Palti, Y. Tumor Treating Fields: A New Frontier in Cancer Therapy. Ann. N. Y. Acad. Sci. 2013, 1291, 86–95. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Trone, J.-C.; Vallard, A.; Sotton, S.; Ben Mrad, M.; Jmour, O.; Magné, N.; Pommier, B.; Laporte, S.; Ollier, E. Survival after Hypofractionation in Glioblastoma: A Systematic Review and Meta-Analysis. Radiat. Oncol. 2020, 15, 145. [Google Scholar] [CrossRef]
- Liao, G.; Zhao, Z.; Yang, H.; Li, X. Efficacy and Safety of Hypofractionated Radiotherapy for the Treatment of Newly Diagnosed Glioblastoma Multiforme: A Systematic Review and Meta-Analysis. Front. Oncol. 2019, 9, 1017. [Google Scholar] [CrossRef]
- Perlow, H.K.; Yaney, A.; Yang, M.; Klamer, B.; Matsui, J.; Raval, R.R.; Blakaj, D.M.; Arnett, A.; Beyer, S.; Elder, J.B.; et al. Dose-Escalated Accelerated Hypofractionation for Elderly or Frail Patients with a Newly Diagnosed Glioblastoma. J. Neurooncol. 2022, 156, 399–406. [Google Scholar] [CrossRef]
- Perlow, H.K.; Prasad, R.N.; Yang, M.; Klamer, B.; Matsui, J.; Marrazzo, L.; Detti, B.; Scorsetti, M.; Clerici, E.; Arnett, A.; et al. Accelerated Hypofractionated Radiation for Elderly or Frail Patients with a Newly Diagnosed Glioblastoma: A Pooled Analysis of Patient-Level Data from 4 Prospective Trials. Cancer 2022, 128, 2367–2374. [Google Scholar] [CrossRef]
- Cho, K.H.; Hall, W.A.; Gerbi, B.J.; Higgins, P.D.; McGuire, W.A.; Clark, H.B. Single Dose versus Fractionated Stereotactic Radiotherapy for Recurrent High-Grade Gliomas. Int. J. Radiat. Oncol. 1999, 45, 1133–1141. [Google Scholar] [CrossRef]
- Combs, S.E.; Thilmann, C.; Edler, L.; Debus, J.; Schulz-Ertner, D. Efficacy of Fractionated Stereotactic Reirradiation in Recurrent Gliomas: Long-Term Results in 172 Patients Treated in a Single Institution. J. Clin. Oncol. 2005, 23, 8863–8869. [Google Scholar] [CrossRef]
- Fogh, S.E.; Andrews, D.W.; Glass, J.; Curran, W.; Glass, C.; Champ, C.; Evans, J.J.; Hyslop, T.; Pequignot, E.; Downes, B.; et al. Hypofractionated Stereotactic Radiation Therapy: An Effective Therapy for Recurrent High-Grade Gliomas. J. Clin. Oncol. 2010, 28, 3048–3053. [Google Scholar] [CrossRef]
- Brown, J.M.; Koong, A.C. High-Dose Single-Fraction Radiotherapy: Exploiting a New Biology? Int. J. Radiat. Oncol. 2008, 71, 324–325. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using Immunotherapy to Boost the Abscopal Effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Tharmalingam, H.; Hoskin, P.J. The Optimism Surrounding Stereotactic Body Radiation Therapy and Immunomodulation. Chin. Clin. Oncol. 2017, 6, S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotera, Y.; Shimizu, K.; Mulé, J.J. Comparative Analysis of Necrotic and Apoptotic Tumor Cells As a Source of Antigen(s) in Dendritic Cell-Based Immunization. Cancer Res. 2001, 61, 8105–8109. [Google Scholar]
- Pouessel, D.; Mervoyer, A.; Larrieu-Ciron, D.; Cabarrou, B.; Attal, J.; Robert, M.; Frenel, J.-S.; Olivier, P.; Poublanc, M.; Mounier, M.; et al. Hypofractionnated Stereotactic Radiotherapy and Anti-PDL1 Durvalumab Combination in Recurrent Glioblastoma: Results of the Phase I Part of the Phase I/II STERIMGLI Trial. J. Clin. Oncol. 2018, 36, 2046. [Google Scholar] [CrossRef]
- Nguyen, N.; Nguyen, M.; Vock, J.; Lemanski, C.; Kerr, C.; Vinh-Hung, V.; Chi, A.; Khan, R.; Woods, W.; Altdorfer, G.; et al. Potential Applications of Imaging and Image-Guided Radiotherapy for Brain Metastases and Glioblastoma to Improve Patient Quality of Life. Front. Oncol. 2013, 3, 284. [Google Scholar] [CrossRef] [Green Version]
- Mizumoto, M.; Yamamoto, T.; Takano, S.; Ishikawa, E.; Matsumura, A.; Ishikawa, H.; Okumura, T.; Sakurai, H.; Miyatake, S.-I.; Tsuboi, K. Long-Term Survival after Treatment of Glioblastoma Multiforme with Hyperfractionated Concomitant Boost Proton Beam Therapy. Pract. Radiat. Oncol. 2015, 5, e9–e16. [Google Scholar] [CrossRef]
- Fitzek, M.M.; Thornton, A.F.; Rabinov, J.D.; Lev, M.H.; Pardo, F.S.; Munzenrider, J.E.; Okunieff, P.; Bussière, M.; Braun, I.; Hochberg, F.H.; et al. Accelerated Fractionated Proton/Photon Irradiation to 90 Cobalt Gray Equivalent for Glioblastoma Multiforme: Results of a Phase II Prospective Trial. J. Neurosurg. 1999, 91, 251–260. [Google Scholar] [CrossRef]
- Combs, S.E.; Kieser, M.; Rieken, S.; Habermehl, D.; Jäkel, O.; Haberer, T.; Nikoghosyan, A.; Haselmann, R.; Unterberg, A.; Wick, W.; et al. Randomized Phase II Study Evaluating a Carbon Ion Boost Applied after Combined Radiochemotherapy with Temozolomide versus a Proton Boost after Radiochemotherapy with Temozolomide in Patients with Primary Glioblastoma: The CLEOPATRA Trial. BMC Cancer 2010, 10, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chédeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef]
- Riballo, E.; Kühne, M.; Rief, N.; Doherty, A.; Smith, G.C.M.; Recio, M.-J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A Pathway of Double-Strand Break Rejoining Dependent upon ATM, Artemis, and Proteins Locating to γ-H2AX Foci. Mol. Cell 2004, 16, 715–724. [Google Scholar] [CrossRef] [PubMed]
- van der Schans, G.P. Gamma-Ray Induced Double-Strand Breaks in DNA Resulting from Randomly-Inflicted Single-Strand Breaks: Temporal Local Denaturation, a New Radiation Phenomenon? Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1978, 33, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes 2020, 11, 99. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Leidal, A.M.; Madureira, P.A.; Gillis, L.D.; Waisman, D.M.; Chiu, A.; Lee, P.W.K. Chromium-Mediated Apoptosis: Involvement of DNA-Dependent Protein Kinase (DNA-PK) and Differential Induction of P53 Target Genes. DNA Repair 2008, 7, 1484–1499. [Google Scholar] [CrossRef]
- Corre, I.; Niaudet, C.; Paris, F. Plasma Membrane Signaling Induced by Ionizing Radiation. Mutat. Res. /Rev. Mutat. Res. 2010, 704, 61–67. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The Role of Ferroptosis in Ionizing Radiation-Induced Cell Death and Tumor Suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Carlos-Reyes, A.; Muñiz-Lino, M.A.; Romero-Garcia, S.; López-Camarillo, C.; Hernández-de la Cruz, O.N. Biological Adaptations of Tumor Cells to Radiation Therapy. Front. Oncol. 2021, 11, 718636. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells in Solid Tumours: Accumulating Evidence and Unresolved Questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma Stem Cells Microenvironment: The Paracrine Roles of the Niche in Drug and Radioresistance. Stem Cells Int. 2016, 2016, 6809105. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Thomlinson, R.H.; Gray, L.H. The Histological Structure of Some Human Lung Cancers and the Possible Implications for Radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C.A. The Concentration of Oxygen Dissolved in Tissues at the Time of Irradiation as a Factor in Radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Ewing, D. The Oxygen Fixation Hypothesis: A Reevaluation. Am. J. Clin. Oncol. 1998, 21, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Farace, C.; Oliver, J.A.; Melguizo, C.; Alvarez, P.; Bandiera, P.; Rama, A.R.; Malaguarnera, G.; Ortiz, R.; Madeddu, R.; Prados, J. Microenvironmental Modulation of Decorin and Lumican in Temozolomide-Resistant Glioblastoma and Neuroblastoma Cancer Stem-Like Cells. PLOS ONE 2015, 10, e0134111. [Google Scholar] [CrossRef] [Green Version]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; De Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood Flow, Oxygen and Nutrient Supply, and Metabolic Microenvironment of Human Tumors: A Review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [PubMed]
- Gordon Steel, G.; Peckham, M.J. Exploitable Mechanisms in Combined Radiotherapy-Chemotherapy: The Concept of Additivity. Int. J. Radiat. Oncol. 1979, 5, 85–91. [Google Scholar] [CrossRef]
- Gill, M.R.; Vallis, K.A. Transition Metal Compounds as Cancer Radiosensitizers. Chem. Soc. Rev. 2019, 48, 540–557. [Google Scholar] [CrossRef]
- Fowler, J.F.; Adams, G.E.; Denekamp, J. Radiosensitizers of Hypoxic Cells in Solid Tumours. Cancer Treat. Rev. 1976, 3, 227–256. [Google Scholar] [CrossRef]
- Adams, G.E. Chemical radiosensitization of hypoxic cells. Br. Med. Bull. 1973, 29, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Bastiancich, C.; Bastiat, G.; Lagarce, F. Gemcitabine and Glioblastoma: Challenges and Current Perspectives. Drug Discov. Today 2018, 23, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Mrugala, M.M.; Chamberlain, M.C. Mechanisms of Disease: Temozolomide and Glioblastoma—Look to the Future. Nat. Clin. Pract. Oncol. 2008, 5, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Erkkinen, M.G.; Nestler, U.; Stupp, R.; Mehta, M.; Aldape, K.; Gilbert, M.R.; Black, P.M.L.; Loeffler, J.S. Temozolomide-Mediated Radiation Enhancement in Glioblastoma: A Report on Underlying Mechanisms. Clin. Cancer Res. 2006, 12, 4738–4746. [Google Scholar] [CrossRef] [Green Version]
- Lund, B.; Kristjansen, P.E.G.; Hansen, H.H. Clinical and Preclinical Activity of 2′,2′- Difluorodeoxycytidine (Gemcitabine). Cancer Treat. Rev. 1993, 19, 45–55. [Google Scholar] [CrossRef]
- Lawrence, T.S.; Blackstock, A.W.; McGinn, C. The Mechanism of Action of Radiosensitization of Conventional Chemotherapeutic Agents. Semin. Radiat. Oncol. 2003, 13, 13–21. [Google Scholar] [CrossRef]
- Pauwels, B.; Korst, A.E.C.; Pattyn, G.G.O.; Lambrechts, H.A.J.; Van Bockstaele, D.R.; Vermeulen, K.; Lenjou, M.; de Pooter, C.M.J.; Vermorken, J.B.; Lardon, F. Cell Cycle Effect of Gemcitabine and Its Role in the Radiosensitizing Mechanism in Vitro. Int. J. Radiat. Oncol. 2003, 57, 1075–1083. [Google Scholar] [CrossRef]
- Latz, D.; Fleckenstein, K.; Eble, M.; Blatter, J.; Wannenmacher, M.; Weber, K.J. Radiosensitizing Potential of Gemcitabine (2′,2′-Difluoro-2′-Deoxycytidine) within the Cell Cycle in Vitro. Int. J. Radiat. Oncol. 1998, 41, 875–882. [Google Scholar] [CrossRef]
- Ciccolini, J.; Serdjebi, C.; Peters, G.J.; Giovannetti, E. Pharmacokinetics and Pharmacogenetics of Gemcitabine as a Mainstay in Adult and Pediatric Oncology: An EORTC-PAMM Perspective. Cancer Chemother. Pharmacol. 2016, 78, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T.; Eisbruch, A.; Shewach, D. Gemcitabine-Mediated Radiosensitization. Semin. Oncol. 1997, 24 (Suppl. 7), 24–28. [Google Scholar]
- Maraveyas, A.; Sgouros, J.; Upadhyay, S.; Abdel-Hamid, A.-H.; Holmes, M.; Lind, M. Gemcitabine Twice Weekly as a Radiosensitiser for the Treatment of Brain Metastases in Patients with Carcinoma: A Phase I Study. Br. J. Cancer 2005, 92, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabi, A.; Mirri, A.; Felici, A.; Vidiri, A.; Pace, A.; Occhipinti, E.; Cognetti, F.; Arcangeli, G.; Iandolo, B.; Carosi, M.A.; et al. Fixed Dose-Rate Gemcitabine as Radiosensitizer for Newly Diagnosed Glioblastoma: A Dose-Finding Study. J. Neurooncol. 2008, 87, 79–84. [Google Scholar] [CrossRef]
- Metro, G.; Fabi, A.; Mirri, M.A.; Vidiri, A.; Pace, A.; Carosi, M.; Russillo, M.; Maschio, M.; Giannarelli, D.; Pellegrini, D.; et al. Phase II Study of Fixed Dose Rate Gemcitabine as Radiosensitizer for Newly Diagnosed Glioblastoma Multiforme. Cancer Chemother. Pharmacol. 2009, 65, 391. [Google Scholar] [CrossRef] [PubMed]
- Sigmond, J.; Honeywell, R.J.; Postma, T.J.; Dirven, C.M.F.; de Lange, S.M.; van der Born, K.; Laan, A.C.; Baayen, J.C.A.; Van Groeningen, C.J.; Bergman, A.M.; et al. Gemcitabine Uptake in Glioblastoma Multiforme: Potential as a Radiosensitizer. Ann. Oncol. 2009, 20, 182–187. [Google Scholar] [CrossRef]
- Degen, J.W.; Walbridge, S.; Vortmeyer, A.O.; Oldfield, E.H.; Lonser, R.R. Safety and Efficacy of Convection-Enhanced Delivery of Gemcitabine or Carboplatin in a Malignant Glioma Model in Rats. J. Neurosurg. 2003, 99, 893–898. [Google Scholar] [CrossRef]
- Guo, P.; Ma, J.; Li, S.; Guo, Z.; Adams, A.L.; Gallo, J.M. Targeted Delivery of a Peripheral Benzodiazepine Receptor Ligand-Gemcitabine Conjugate to Brain Tumors in a Xenograft Model. Cancer Chemother. Pharmacol. 2001, 48, 169–176. [Google Scholar] [CrossRef]
- Wang, C.-X.; Huang, L.-S.; Hou, L.-B.; Jiang, L.; Yan, Z.-T.; Wang, Y.-L.; Chen, Z.-L. Antitumor Effects of Polysorbate-80 Coated Gemcitabine Polybutylcyanoacrylate Nanoparticles in Vitro and Its Pharmacodynamics in Vivo on C6 Glioma Cells of a Brain Tumor Model. Brain Res. 2009, 1261, 91–99. [Google Scholar] [CrossRef] [PubMed]
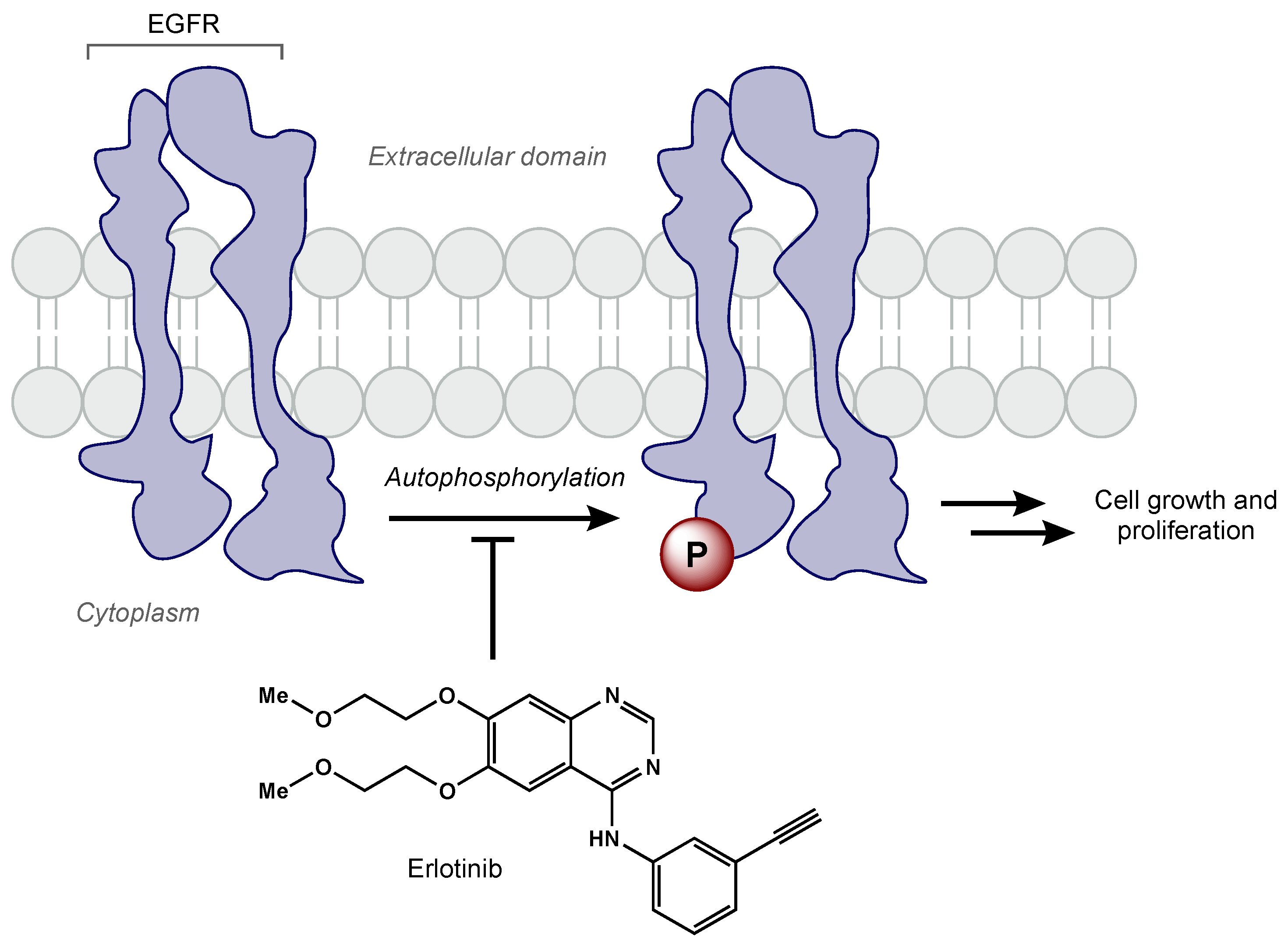
- Kim, G.; Ko, Y.T. Small Molecule Tyrosine Kinase Inhibitors in Glioblastoma. Arch. Pharm. Res. 2020, 43, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification, Enhanced Expression and Possible Rearrangement of EGF Receptor Gene in Primary Human Brain Tumours of Glial Origin. Nature 1985, 313, 144–147. [Google Scholar] [CrossRef]
- Chakravarti, A.; Chakladar, A.; Delaney, M.A.; Latham, D.E.; Loeffler, J.S. The Epidermal Growth Factor Receptor Pathway Mediates Resistance to Sequential Administration of Radiation and Chemotherapy in Primary Human Glioblastoma Cells in a RAS-Dependent Manner. Cancer Res. 2002, 62, 4307–4315. [Google Scholar] [PubMed]
- Barker, F.G.; Simmons, M.L.; Chang, S.M.; Prados, M.D.; Larson, D.A.; Sneed, P.K.; Wara, W.M.; Berger, M.S.; Chen, P.; Israel, M.A.; et al. EGFR Overexpression and Radiation Response in Glioblastoma Multiforme. Int. J. Radiat. Oncol. 2001, 51, 410–418. [Google Scholar] [CrossRef]
- Prados, M.D.; Lamborn, K.R.; Chang, S.; Burton, E.; Butowski, N.; Malec, M.; Kapadia, A.; Rabbitt, J.; Page, M.S.; Fedoroff, A.; et al. Phase 1 Study of Erlotinib HCl Alone and Combined with Temozolomide in Patients with Stable or Recurrent Malignant Glioma. Neuro-Oncology 2006, 8, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Schettino, C.; Bareschino, M.A.; Ricci, V.; Ciardiello, F. Erlotinib: An EGF Receptor Tyrosine Kinase Inhibitor in Non-Small-Cell Lung Cancer Treatment. Expert Rev. Respir. Med. 2008, 2, 167–178. [Google Scholar] [CrossRef]
- Brown, P.D.; Krishnan, S.; Sarkaria, J.N.; Wu, W.; Jaeckle, K.A.; Uhm, J.H.; Geoffroy, F.J.; Arusell, R.; Kitange, G.; Jenkins, R.B.; et al. Phase I/II Trial of Erlotinib and Temozolomide With Radiation Therapy in the Treatment of Newly Diagnosed Glioblastoma Multiforme: North Central Cancer Treatment Group Study N0177. J. Clin. Oncol. 2008, 26, 5603–5609. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Shepard, D.R.; Ahluwalia, M.S.; Brewer, C.J.; Agarwal, N.; Stevens, G.H.J.; Suh, J.H.; Toms, S.A.; Vogelbaum, M.A.; Weil, R.J.; et al. Phase II Trial of Erlotinib with Temozolomide and Radiation in Patients with Newly Diagnosed Glioblastoma Multiforme. J. Neurooncol. 2010, 98, 93–99. [Google Scholar] [CrossRef]
- Uhm, J.H.; Ballman, K.V.; Wu, W.; Giannini, C.; Krauss, J.C.; Buckner, J.C.; James, C.D.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J.; et al. Phase II Evaluation of Gefitinib in Patients With Newly Diagnosed Grade 4 Astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. 2011, 80, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y.; et al. Phase I/Randomized Phase II Study of Afatinib, an Irreversible ErbB Family Blocker, with or without Protracted Temozolomide in Adults with Recurrent Glioblastoma. Neuro-Oncology 2015, 17, 430–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.; McBain, C.; Nash, S.; Hopkins, K.; Sanghera, P.; Saran, F.; Phillips, M.; Dungey, F.; Clifton-Hadley, L.; Wanek, K.; et al. Multi-Center Randomized Phase II Study Comparing Cediranib plus Gefitinib with Cediranib plus Placebo in Subjects with Recurrent/Progressive Glioblastoma. PLoS ONE 2016, 11, e0156369. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, X.; Shi, L.; Shan, Q.; Cao, Q.; Yue, C.; Li, H.; Li, S.; Wang, J.; Gao, S.; et al. The Third-Generation EGFR Inhibitor AZD9291 Overcomes Primary Resistance by Continuously Blocking ERK Signaling in Glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 219. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/MTOR Signaling Pathway and Targeted Therapy for Glioblastoma. Oncotarget 2016, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Jhanwar-Uniyal, M.; Gillick, J.L.; Neil, J.; Tobias, M.; Thwing, Z.E.; Murali, R. Distinct Signaling Mechanisms of MTORC1 and MTORC2 in Glioblastoma Multiforme: A Tale of Two Complexes. Adv. Biol. Regul. 2015, 57, 64–74. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse Signaling Mechanisms of MTOR Complexes: MTORC1 and MTORC2 in Forming a Formidable Relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia Promote Glioblastoma via MTOR-Mediated Immunosuppression of the Tumour Microenvironment. EMBO J. 2020, 39, e103790. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; van den Bent, M.J.; Taphoorn, M.J.B.; Steuve, J.; Brandes, A.A.; Hamou, M.-F.; Wick, A.; et al. Phase II Study of Radiotherapy and Temsirolimus versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Gururangan, S.; Friedman, A.H.; Herndon, J.E.; Marcello, J.; Norfleet, J.A.; McLendon, R.E.; Sampson, J.H.; et al. Phase 2 Trial of Erlotinib plus Sirolimus in Adults with Recurrent Glioblastoma. J. Neurooncol. 2010, 96, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.D.; Mladek, A.C.; Lamont, J.D.; Goble, J.M.; Erlichman, C.; James, C.D.; Sarkaria, J.N. Disruption of Parallel and Converging Signaling Pathways Contributes to the Synergistic Antitumor Effects of Simultaneous MTOR and EGFR Inhibition in GBM Cells. Neoplasia 2005, 7, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A Phase II Trial of Everolimus, Temozolomide, and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: NCCTG N057K. Neuro-Oncology 2015, 17, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Michael Yu, H.-H.; Stieber, V.W.; Malone, S.C.; et al. A Randomized Phase II Study of Everolimus in Combination with Chemoradiation in Newly Diagnosed Glioblastoma: Results of NRG Oncology RTOG 0913. Neuro-Oncology 2018, 20, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. MTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
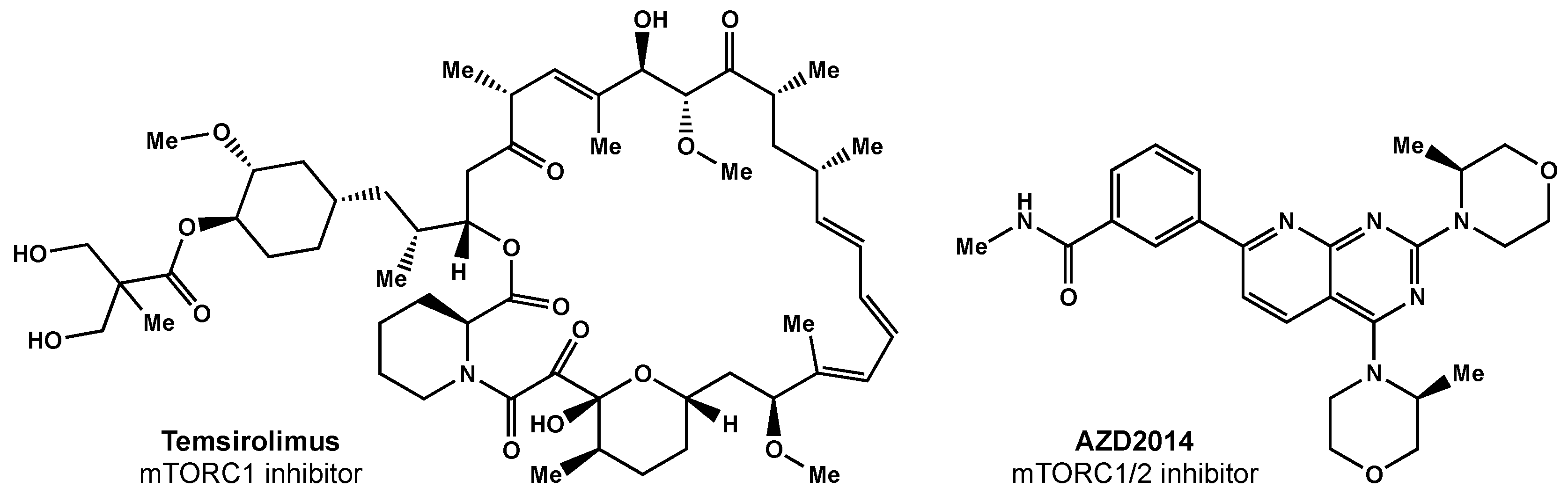
- Kahn, J.; Hayman, T.J.; Jamal, M.; Rath, B.H.; Kramp, T.; Camphausen, K.; Tofilon, P.J. The MTORC1/MTORC2 Inhibitor AZD2014 Enhances the Radiosensitivity of Glioblastoma Stem-like Cells. Neuro-Oncology 2014, 16, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Mu, X.; He, H.; Zhang, X.-D. Cancer Radiosensitizers. Trends Pharmacol. Sci. 2018, 39, 24–48. [Google Scholar] [CrossRef]
- Adams, G.E.; Asquith, J.C.; Dewey, D.L.; Foster, J.L.; Michael, B.D.; Willson, R.L. Electron Affinic Sensitization. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1971, 19, 575–585. [Google Scholar] [CrossRef]
- De, P.; Roy, K. Nitroaromatics as Hypoxic Cell Radiosensitizers: A 2D-QSAR Approach to Explore Structural Features Contributing to Radiosensitization Effectiveness. Eur. J. Med. Chem. Rep. 2022, 4, 100035. [Google Scholar] [CrossRef]
- Overgaard, J.; Sand Hansen, H.; Andersen, A.P.; Hjelm-Hansen, M.; Jørgensen, K.; Sandberg, E.; Berthelsen, A.; Hammer, R.; Pedersen, M. Misonidazole Combined with Split-Course Radiotherapy in the Treatment of Invasive Carcinoma of Larynx and Pharynx: Report from the DAHANCA 2 Study. Int. J. Radiat. Oncol. 1989, 16, 1065–1068. [Google Scholar] [CrossRef]
- Coleman, C.N.; Wasserman, T.H.; Urtasun, R.C.; Halsey, J.; Noll, L.; Hancock, S.; Phillips, T.L. Final Report of the Phase i Trial of the Hypoxic Cell Radiosensitizer SR 2508 (Etanidazole) Radiation Therapy Oncology Group 83-03. Int. J. Radiat. Oncol. 1990, 18, 389–393. [Google Scholar] [CrossRef]
- Oronsky, B.; Scicinski, J.; Ning, S.; Peehl, D.; Oronsky, A.; Cabrales, P.; Bednarski, M.; Knox, S. RRx-001, A Novel Dinitroazetidine Radiosensitizer. Investig. New Drugs 2016, 34, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaoka, T.; Shibamoto, Y.; Matsuo, M.; Sugie, C.; Murai, T.; Ogawa, Y.; Miyakawa, A.; Manabe, Y.; Kondo, T.; Nakajima, K.; et al. Biological Effects of Hydrogen Peroxide Administered Intratumorally with or without Irradiation in Murine Tumors. Cancer Sci. 2017, 108, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Janssens, G.O.; Rademakers, S.E.; Terhaard, C.H.; Doornaert, P.A.; Bijl, H.P.; van den Ende, P.; Chin, A.; Marres, H.A.; de Bree, R.; van der Kogel, A.J.; et al. Accelerated Radiotherapy With Carbogen and Nicotinamide for Laryngeal Cancer: Results of a Phase III Randomized Trial. J. Clin. Oncol. 2012, 30, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Taylor, J.; Eustace, A.; Irlam, J.J.; Denley, H.; Hoskin, P.J.; Alsner, J.; Buffa, F.M.; Harris, A.L.; Choudhury, A.; et al. A Gene Signature for Selecting Benefit from Hypoxia Modification of Radiotherapy for High-Risk Bladder Cancer Patients. Clin. Cancer Res. 2017, 23, 4761–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rademakers, S.E.; Hoogsteen, I.J.; Rijken, P.F.; Terhaard, C.H.; Doornaert, P.A.; Langendijk, J.A.; van den Ende, P.; van der Kogel, A.J.; Bussink, J.; Kaanders, J.H. Prognostic Value of the Proliferation Marker Ki-67 in Laryngeal Carcinoma: Results of the Accelerated Radiotherapy with Carbogen Breathing and Nicotinamide Phase III Randomized Trial. Head Neck 2015, 37, 171–176. [Google Scholar] [CrossRef]
- Hoskin, P.J.; Rojas, A.M.; Saunders, M.I.; Bentzen, S.M.; Motohashi, K.J. Carbogen and Nicotinamide in Locally Advanced Bladder Cancer: Early Results of a Phase-III Randomized Trial. Radiother. Oncol. 2009, 91, 120–125. [Google Scholar] [CrossRef]
- Horsman, M.R.; Overgaard, J.; Chaplin, D.J. Combination of Nicotinamide and Hyperthermia to Eliminate Radioresistant Chronically and Acutely Hypoxic Tumor Cells. Cancer Res. 1990, 50, 7430–7436. [Google Scholar]
- Welsh, L.; Panek, R.; Riddell, A.; Wong, K.; Leach, M.O.; Tavassoli, M.; Rahman, D.; Schmidt, M.; Hurley, T.; Grove, L.; et al. Blood Transfusion during Radical Chemo-Radiotherapy Does Not Reduce Tumour Hypoxia in Squamous Cell Cancer of the Head and Neck. Br. J. Cancer 2017, 116, 28–35. [Google Scholar] [CrossRef] [Green Version]
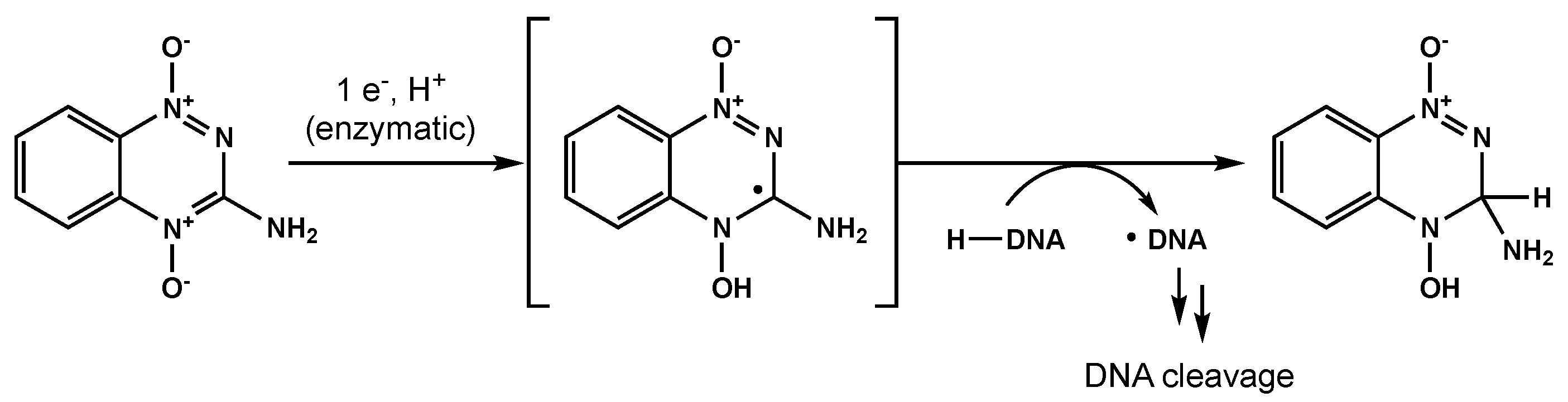
- Daniels, J.S.; Gates, K.S. DNA Cleavage by the Antitumor Agent 3-Amino-1,2,4-Benzotriazine 1,4-Dioxide (SR4233): Evidence for Involvement of Hydroxyl Radical. J. Am. Chem. Soc. 1996, 118, 3380–3385. [Google Scholar] [CrossRef]
- Del Rowe, J.; Scott, C.; Werner-Wasik, M.; Bahary, J.P.; Curran, W.J.; Urtasun, R.C.; Fisher, B. Single-Arm, Open-Label Phase II Study of Intravenously Administered Tirapazamine and Radiation Therapy for Glioblastoma Multiforme. J. Clin. Oncol. 2000, 18, 1254–1259. [Google Scholar] [CrossRef]
- Wang, J.; Guise, C.P.; Dachs, G.U.; Phung, Y.; Hsu, A.H.-L.; Lambie, N.K.; Patterson, A.V.; Wilson, W.R. Identification of One-Electron Reductases That Activate Both the Hypoxia Prodrug SN30000 and Diagnostic Probe EF5. Biochem. Pharmacol. 2014, 91, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Conway, S.J.; Hammond, E.M. Clinical Advances of Hypoxia-Activated Prodrugs in Combination with Radiation Therapy. Int. J. Radiat. Oncol. 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overgaard, J.; Sand Hansen, H.; Overgaard, M.; Bastholt, L.; Berthelsen, A.; Specht, L.; Lindeløv, B.; Jørgensen, K. A Randomized Double-Blind Phase III Study of Nimorazole as a Hypoxic Radiosensitizer of Primary Radiotherapy in Supraglottic Larynx and Pharynx Carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother. Oncol. 1998, 46, 135–146. [Google Scholar] [CrossRef]
- Cerna, D.; Camphausen, K.; Tofilon, P.J. Histone Deacetylation as a Target for Radiosensitization. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2006; Volume 73, pp. 173–204. ISBN 0070-2153. [Google Scholar]
- Chinnaiyan, P.; Cerna, D.; Burgan, W.E.; Beam, K.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. Postradiation Sensitization of the Histone Deacetylase Inhibitor Valproic Acid. Clin. Cancer Res. 2008, 14, 5410–5415. [Google Scholar] [CrossRef] [Green Version]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. 2015, 92, 986–992. [Google Scholar] [CrossRef] [Green Version]
- Happold, C.; Gorlia, T.; Chinot, O.; Gilbert, M.R.; Nabors, L.B.; Wick, W.; Pugh, S.L.; Hegi, M.; Cloughesy, T.; Roth, P.; et al. Does Valproic Acid or Levetiracetam Improve Survival in Glioblastoma? A Pooled Analysis of Prospective Clinical Trials in Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2016, 34, 731–739. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II Trial of Vorinostat Combined with Temozolomide and Radiation Therapy for Newly Diagnosed Glioblastoma: Results of Alliance N0874/ABTC 02. Neuro-Oncology 2018, 20, 546–556. [Google Scholar] [CrossRef] [Green Version]
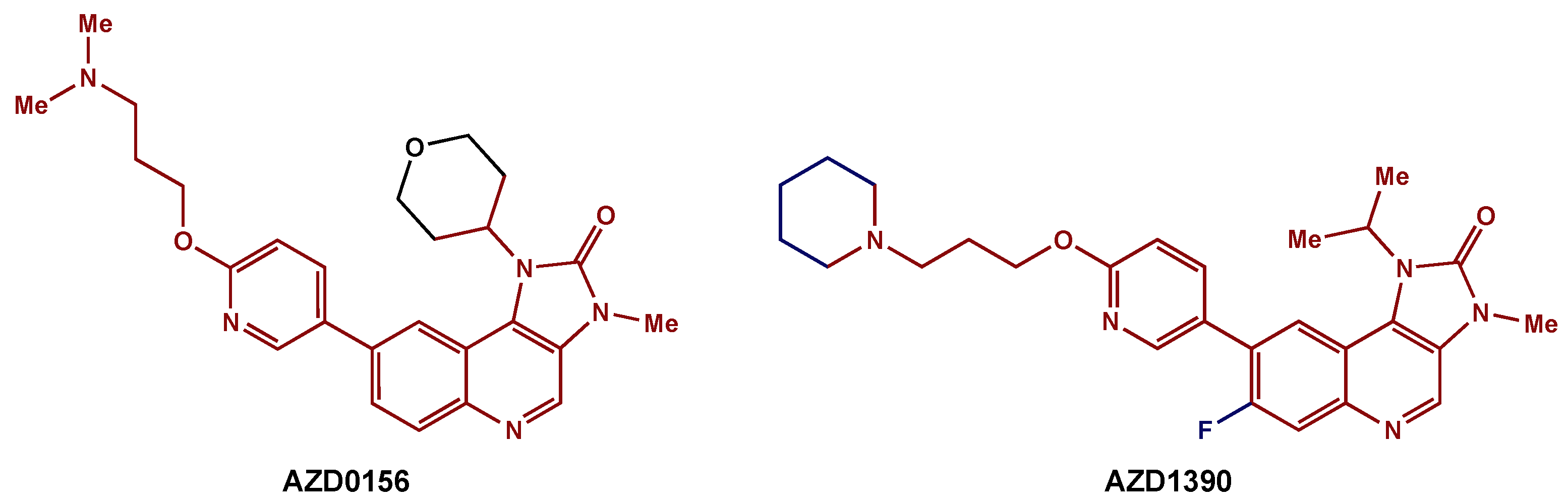
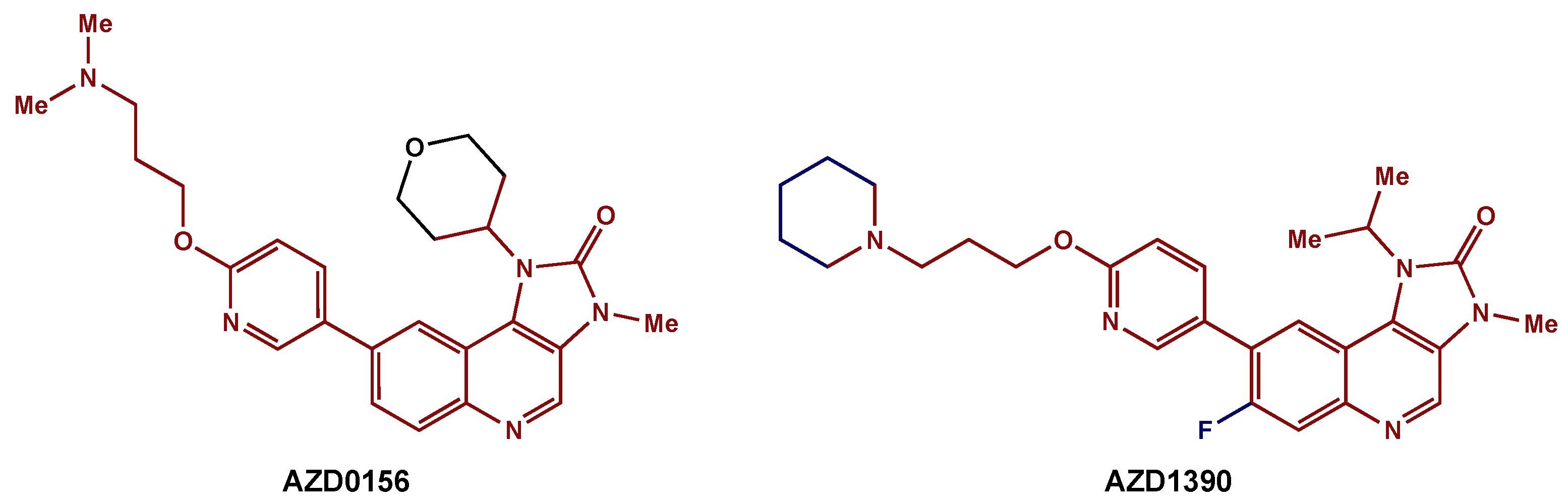
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.; et al. The Brain-Penetrant Clinical ATM Inhibitor AZD1390 Radiosensitizes and Improves Survival of Preclinical Brain Tumor Models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA Damage Activates ATM through Intermolecular Autophosphorylation and Dimer Dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of Radioresistance in Glioblastoma Stem-like Cells by Inhibition of ATM Kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM Kinase Inhibitor KU-60019 Radiosensitizes Glioma Cells, Compromises Insulin, AKT and ERK Prosurvival Signaling, and Inhibits Migration and Invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pike, K.G.; Barlaam, B.; Cadogan, E.; Campbell, A.; Chen, Y.; Colclough, N.; Davies, N.L.; de-Almeida, C.; Degorce, S.L.; Didelot, M.; et al. The Identification of Potent, Selective, and Orally Available Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase: The Discovery of AZD0156 (8-{6-[3-(Dimethylamino)Propoxy]Pyridin-3-Yl}-3-Methyl-1-(Tetrahydro-2H-Pyran-4-Yl)-1,3-Dihydro-2H-Imidazo[4,5-c]Quinolin-2-One). J. Med. Chem. 2018, 61, 3823–3841. [Google Scholar] [CrossRef]
- Abraham, D.J.; Kennedy, P.E.; Mehanna, A.S.; Patwa, D.C.; Williams, F.L. Design, Synthesis, and Testing of Potential Antisickling Agents. 4. Structure-Activity Relationships of Benzyloxy and Phenoxy Acids. J. Med. Chem. 1984, 27, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Stea, B.; Nabid, A.; Kresl, J.J.; Fortin, A.; Mercier, J.-P.; Senzer, N.; Chang, E.L.; Boyd, A.P.; Cagnoni, P.J.; et al. Phase III Study of Efaproxiral As an Adjunct to Whole-Brain Radiation Therapy for Brain Metastases. J. Clin. Oncol. 2006, 24, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Rampling, R.; Cruickshank, G.; Lewis, A.D.; Fitzsimmons, S.A.; Workman, P. Direct Measurement of PO2 Distribution and Bioreductive Enzymes in Human Malignant Brain Tumors. Int. J. Radiat. Oncol. 1994, 29, 427–431. [Google Scholar] [CrossRef]
- Taghian, A.; duBois, W.; Budach, W.; Baumann, M.; Freeman, J.; Suit, H. In Vivo Radiation Sensitivity of Glioblastoma Multiforme. Int. J. Radiat. Oncol. 1995, 32, 99–104. [Google Scholar] [CrossRef]
- Kleinberg, L.; Grossman, S.A.; Carson, K.; Lesser, G.; O’Neill, A.; Pearlman, J.; Phillips, P.; Herman, T.; Gerber, M. Survival of Patients With Newly Diagnosed Glioblastoma Multiforme Treated With RSR13 and Radiotherapy: Results of a Phase II New Approaches to Brain Tumor Therapy CNS Consortium Safety and Efficacy Study. J. Clin. Oncol. 2002, 20, 3149–3155. [Google Scholar] [CrossRef]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in Brain Tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Schreiber, R.D. Cancer Immunoediting: Antigens, Mechanisms, and Implications to Cancer Immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A Review of Glioblastoma Immunotherapy. J. Neurooncol. 2021, 151, 41–53. [Google Scholar] [CrossRef]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Sharabi, A.B.; Lim, M.; DeWeese, T.L.; Drake, C.G. Radiation and Checkpoint Blockade Immunotherapy: Radiosensitisation and Potential Mechanisms of Synergy. Lancet Oncol. 2015, 16, e498–e509. [Google Scholar] [CrossRef]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic Effects of Ablative Radiation on Local Tumor Require CD8+ T Cells: Changing Strategies for Cancer Treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local Radiation Therapy of B16 Melanoma Tumors Increases the Generation of Tumor Antigen-Specific Effector Cells That Traffic to the Tumor. J. Immunol. 2005, 174, 7516. [Google Scholar] [CrossRef] [Green Version]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and Genomic Correlates of Response to Anti-PD-1 Immunotherapy in Glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Vlahovic, G.; Sahebjam, S.; Omuro, A.M.P.; Baehring, J.M.; Hafler, D.A.; Voloschin, A.D.; Paliwal, P.; Grosso, J.; Coric, V.; et al. Preliminary Safety and Activity of Nivolumab and Its Combination with Ipilimumab in Recurrent Glioblastoma (GBM): CHECKMATE-143. J. Clin. Oncol. 2015, 33, 3010. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant Anti-PD-1 Immunotherapy Promotes a Survival Benefit with Intratumoral and Systemic Immune Responses in Recurrent Glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of Immunotherapy Resistance: Lessons from Glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current State of Immunotherapy for Glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Hu, J.; Locasale, J.W.; Bielas, J.H.; O’Sullivan, J.; Sheahan, K.; Cantley, L.C.; Heiden, M.G.V.; Vitkup, D. Heterogeneity of Tumor-Induced Gene Expression Changes in the Human Metabolic Network. Nat. Biotechnol. 2013, 31, 522–529. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine Synthesis Promotes Maintenance of Brain Tumor Initiating Cells in Glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Kofuji, S.; Hirayama, A.; Eberhardt, A.O.; Kawaguchi, R.; Sugiura, Y.; Sampetrean, O.; Ikeda, Y.; Warren, M.; Sakamoto, N.; Kitahara, S.; et al. IMP Dehydrogenase-2 Drives Aberrant Nucleolar Activity and Promotes Tumorigenesis in Glioblastoma. Nat. Cell Biol. 2019, 21, 1003–1014. [Google Scholar] [CrossRef]
- Zhou, W.; Yao, Y.; Scott, A.J.; Wilder-Romans, K.; Dresser, J.J.; Werner, C.K.; Sun, H.; Pratt, D.; Sajjakulnukit, P.; Zhao, S.G.; et al. Purine Metabolism Regulates DNA Repair and Therapy Resistance in Glioblastoma. Nat. Commun. 2020, 11, 3811. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitroda, S.P.; Wakim, B.T.; Sood, R.F.; Beveridge, M.G.; Beckett, M.A.; MacDermed, D.M.; Weichselbaum, R.R.; Khodarev, N.N. STAT1-Dependent Expression of Energy Metabolic Pathways Links Tumour Growth and Radioresistance to the Warburg Effect. BMC Med. 2009, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Zoi, V.; Galani, V.; Tsekeris, P.; Kyritsis, A.P.; Alexiou, G.A. Radiosensitization and Radioprotection by Curcumin in Glioblastoma and Other Cancers. Biomedicines 2022, 10, 312. [Google Scholar] [CrossRef] [PubMed]
- Nakamae, I.; Morimoto, T.; Shima, H.; Shionyu, M.; Fujiki, H.; Yoneda-Kato, N.; Yokoyama, T.; Kanaya, S.; Kakiuchi, K.; Shirai, T.; et al. Curcumin Derivatives Verify the Essentiality of ROS Upregulation in Tumor Suppression. Molecules 2019, 24, 4067. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Du, Q.; Zhang, Z.; Ma, J.; Han, L.; Wang, Y.; Yang, L.; Tao, N.; Qin, Z. Curcumin Promotes Cancer-Associated Fibroblasts Apoptosis via ROS-Mediated Endoplasmic Reticulum Stress. Arch. Biochem. Biophys. 2020, 694, 108613. [Google Scholar] [CrossRef]
- Zoi, V.; Galani, V.; Vartholomatos, E.; Zacharopoulou, N.; Tsoumeleka, E.; Gkizas, G.; Bozios, G.; Tsekeris, P.; Chousidis, I.; Leonardos, I.; et al. Curcumin and Radiotherapy Exert Synergistic Anti-Glioma Effect In Vitro. Biomedicines 2021, 9, 1562. [Google Scholar] [CrossRef]
- Dote, H.; Burgan, W.E.; Camphausen, K.; Tofilon, P.J. Inhibition of Hsp90 Compromises the DNA Damage Response to Radiation. Cancer Res. 2006, 66, 9211–9220. [Google Scholar] [CrossRef] [Green Version]
- Bisht, K.S.; Bradbury, C.M.; Mattson, D.; Kaushal, A.; Sowers, A.; Markovina, S.; Ortiz, K.L.; Sieck, L.K.; Isaacs, J.S.; Brechbiel, M.W.; et al. Geldanamycin and 17-Allylamino-17-Demethoxygeldanamycin Potentiate the in Vitro and in Vivo Radiation Response of Cervical Tumor Cells via the Heat Shock Protein 90-Mediated Intracellular Signaling and Cytotoxicity. Cancer Res. 2003, 63, 8984–8995. [Google Scholar]
- Machida, H.; Matsumoto, Y.; Shirai, M.; Kubota, N. Geldanamycin, an Inhibitor of Hsp90, Sensitizes Human Tumour Cells to Radiation. Int. J. Radiat. Biol. 2003, 79, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Tani, T.; Tojo, N.; Ohnishi, K. Preferential Radiosensitization to Glioblastoma Cancer Stem Cell-like Cells by a Hsp90 Inhibitor, N-vinylpyrrolidone-AUY922. Oncol. Lett. 2022, 23, 102. [Google Scholar] [CrossRef] [PubMed]
- Miles, X.; Vandevoorde, C.; Hunter, A.; Bolcaen, J. MDM2/X Inhibitors as Radiosensitizers for Glioblastoma Targeted Therapy. Front. Oncol. 2021, 11, 703442. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Ornstein, J.; Iwamoto, Y.; Miller, M.A.; Prytyskach, M.A.; Ferretti, S.; Holzer, P.; Kallen, J.; Furet, P.; Jambhekar, A.; Forrester, W.C.; et al. P53 Dynamics Vary between Tissues and Are Linked with Radiation Sensitivity. Nat. Commun. 2021, 12, 898. [Google Scholar] [CrossRef]
- Spiegelberg, D.; Mortensen, A.C.; Lundsten, S.; Brown, C.J.; Lane, D.P.; Nestor, M. The MDM2/MDMX-P53 Antagonist PM2 Radiosensitizes Wild-Type P53 Tumors. Cancer Res. 2018, 78, 5084–5093. [Google Scholar] [CrossRef] [Green Version]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussière, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-Type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.P.; O’Hara, M.H.; Gandhi, S.J. Preclinical Rationale for Combining Radiation Therapy and Immunotherapy beyond Checkpoint Inhibitors (i.e., CART). Transl. Lung Cancer Res. 2007, 6, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Ge, H.; Long, Y.; Yang, C.; Chang, Y.; Mu, L.; Sayour, E.J.; De Leon, G.; Wang, Q.J.; Yang, J.C.; et al. CD70, a Novel Target of CAR T-Cell Therapy for Gliomas. Neuro-Oncology 2018, 20, 55–65. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Agent(s) | Proposed Mechanism |
|---|---|---|
| Purine synthesis inhibitor | Mycophenolate mofetil | GBM upregulates GTP synthesis and mycophenolate mofetil inhibits GTP synthesis |
| PDK inhibitor | Dichloroacetate | PDK inhibitor that sensitizes GBM cells to RT via G2/M phase cell-cycle arrest. |
| DNA repair inhibitor | Curcumin | Curcumin radiosensitizes tumor cells and leads to greater G2/M cell-cycle arrest. |
| Hsp90 inhibitor | Geldanamycin, 17DMAG, radicicol, NVP-AUY922 | Targets Hsp90, a chaperone involved in protecting cells against radiation-induced death. |
| MDM2 inhibitor | RG7112 | MDM2 inhibitors increase expression of p53 and may be beneficial in patients with TP53 wildtype and MDM2 amplification. |
| CAR T cell therapy | CD70 CAR T cells | Targets CD70-expressing GBM tumors and may offset the immunosuppressive effects. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsui, J.K.; Perlow, H.K.; Ritter, A.R.; Upadhyay, R.; Raval, R.R.; Thomas, E.M.; Beyer, S.J.; Pillainayagam, C.; Goranovich, J.; Ong, S.; et al. Small Molecules and Immunotherapy Agents for Enhancing Radiotherapy in Glioblastoma. Biomedicines 2022, 10, 1763. https://doi.org/10.3390/biomedicines10071763
Matsui JK, Perlow HK, Ritter AR, Upadhyay R, Raval RR, Thomas EM, Beyer SJ, Pillainayagam C, Goranovich J, Ong S, et al. Small Molecules and Immunotherapy Agents for Enhancing Radiotherapy in Glioblastoma. Biomedicines. 2022; 10(7):1763. https://doi.org/10.3390/biomedicines10071763
Chicago/Turabian StyleMatsui, Jennifer K., Haley K. Perlow, Alex R. Ritter, Rituraj Upadhyay, Raju R. Raval, Evan M. Thomas, Sasha J. Beyer, Clement Pillainayagam, Justin Goranovich, Shirley Ong, and et al. 2022. "Small Molecules and Immunotherapy Agents for Enhancing Radiotherapy in Glioblastoma" Biomedicines 10, no. 7: 1763. https://doi.org/10.3390/biomedicines10071763
APA StyleMatsui, J. K., Perlow, H. K., Ritter, A. R., Upadhyay, R., Raval, R. R., Thomas, E. M., Beyer, S. J., Pillainayagam, C., Goranovich, J., Ong, S., Giglio, P., & Palmer, J. D. (2022). Small Molecules and Immunotherapy Agents for Enhancing Radiotherapy in Glioblastoma. Biomedicines, 10(7), 1763. https://doi.org/10.3390/biomedicines10071763