
Interaction Interface of Aβ42 with Human Na,K-ATPase Studied by MD and ITC and Inhibitor Screening by MD

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Structure Preparation for Molecular Modeling
2.2. Protein–Protein Docking and Molecular Dynamic Parameters
2.3. The Molecular Modeling Algorithm
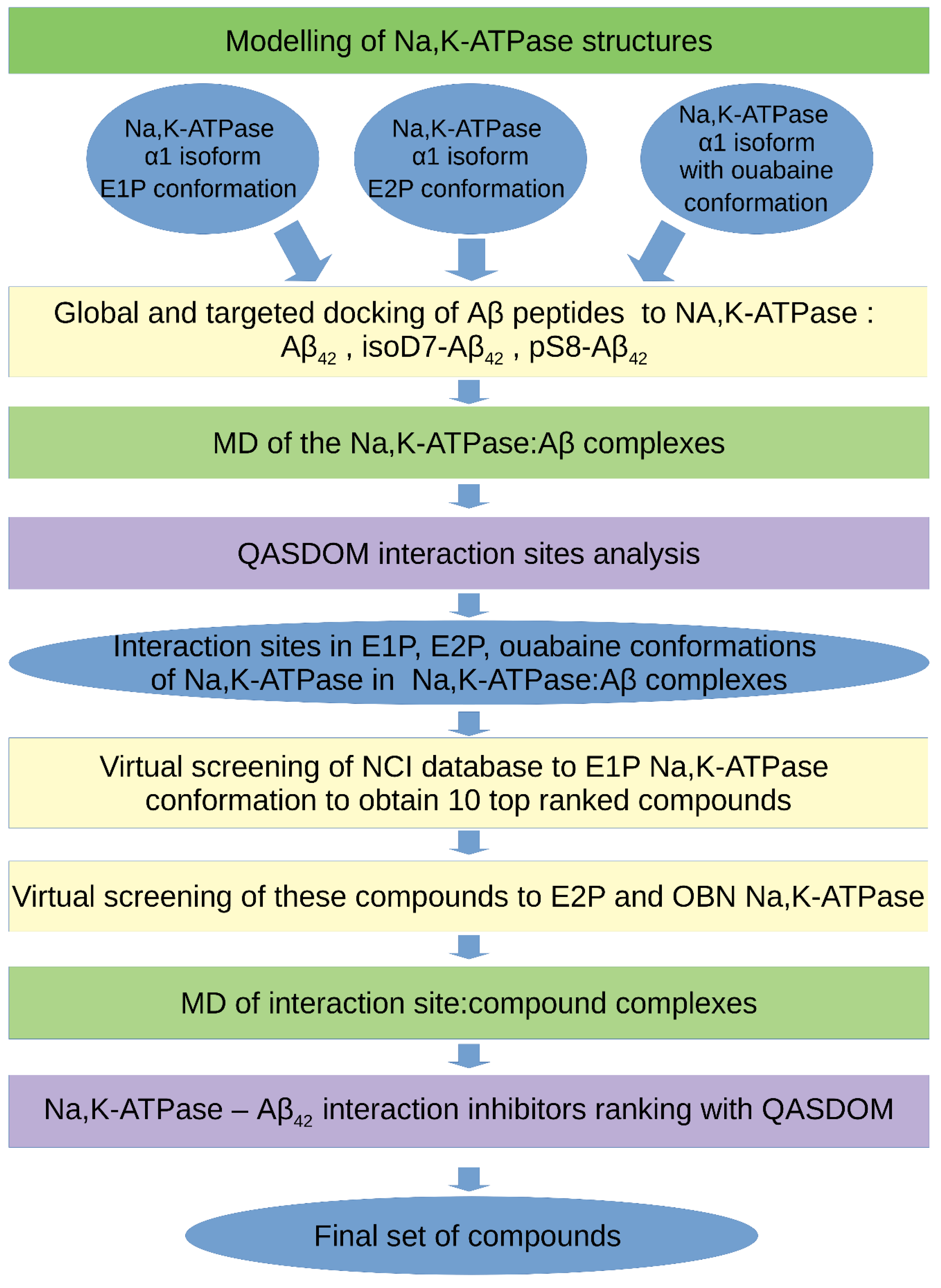
2.4. Na,K-ATPase and Aβ42 Preparations for ITC
2.5. Isothermal Titration Calorimetry
3. Results
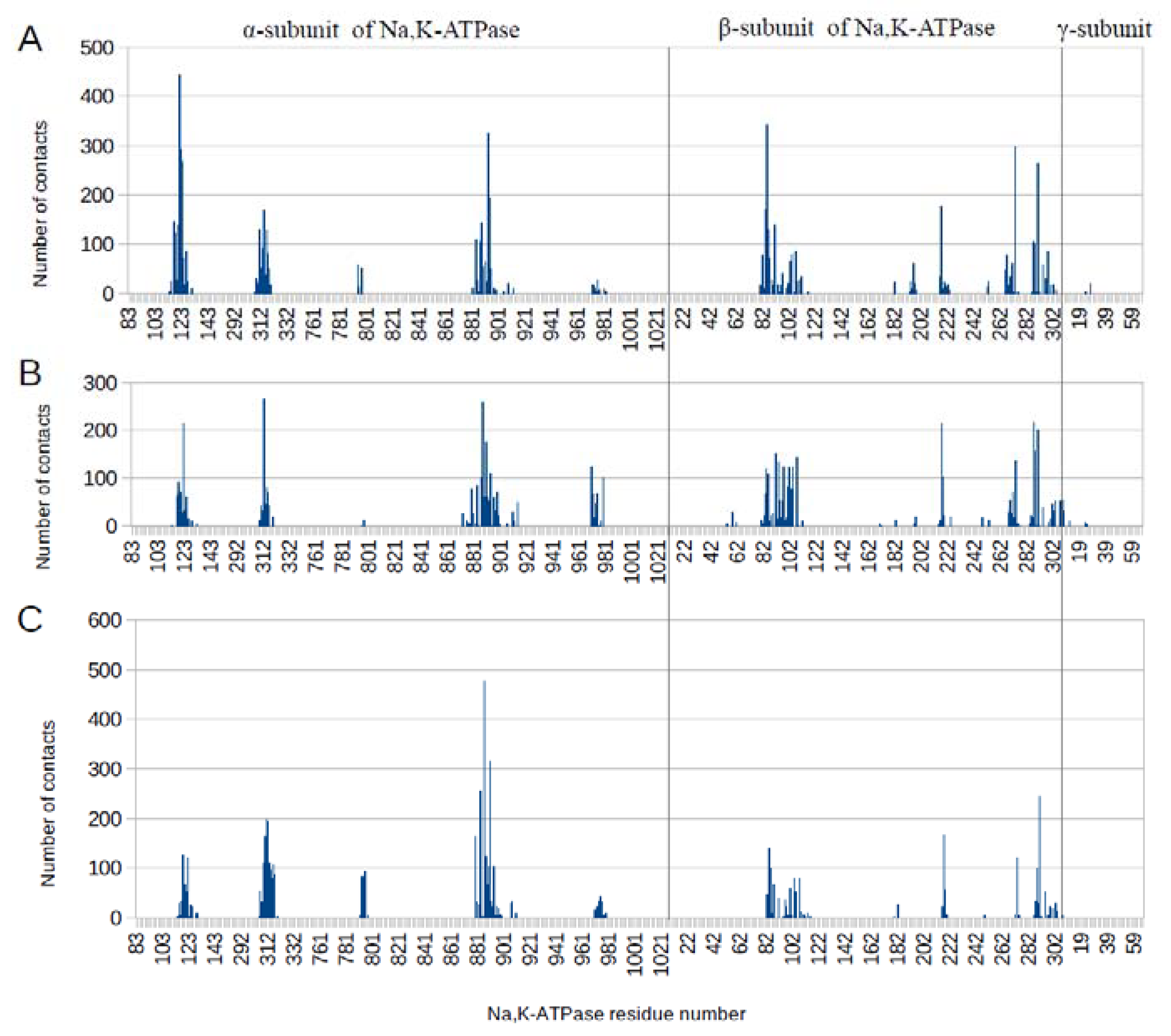
3.1. Interaction Interface of Na,K-ATPase in Various Conformations with Aβ42 and Its Isoforms
3.2. Binding of Na,K-ATPase at Different Conformations with Aβ42
3.3. Virtual Screening of Inhibitors for Na,K-ATPase: Aβ42 Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid β-Protein Assembly and Alzheimer Disease. J. Biol. Chem. 2009, 284, 4749–4753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-Beta: A Crucial Factor in Alzheimer’s Disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Maity, B.K.; Das, A.K.; Dey, S.; Moorthi, U.K.; Kaur, A.; Dey, A.; Surendran, D.; Pandit, R.; Kallianpur, M.; Chandra, B.; et al. Ordered and Disordered Segments of Amyloid-β Drive Sequential Steps of the Toxic Pathway. ACS Chem. Neurosci. 2019, 10, 2498–2509. [Google Scholar] [CrossRef]
- Karisetty, B.C.; Bhatnagar, A.; Armour, E.M.; Beaver, M.; Zhang, H.; Elefant, F. Amyloid-β Peptide Impact on Synaptic Function and Neuroepigenetic Gene Control Reveal New Therapeutic Strategies for Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 577622. [Google Scholar] [CrossRef] [PubMed]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.A.; Makarov, A.A. Amyloid β Modification: A Key to the Sporadic Alzheimer’s Disease? Front. Genet. 2017, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, A.E.; Buneeva, O.A.; Kopylov, A.T.; Mitkevich, V.A.; Kozin, S.A.; Zgoda, V.G.; Makarov, A.A. Chemical Modifications of Amyloid-β(1–42) Have a Significant Impact on the Repertoire of Brain Amyloid-β(1–42) Binding Proteins. Biochimie 2016, 128–129, 55–58. [Google Scholar] [CrossRef]
- Mitkevich, V.A.; Petrushanko, I.Y.; Yegorov, Y.E.; Simonenko, O.V.; Vishnyakova, K.S.; Kulikova, A.A.; Tsvetkov, P.O.; Makarov, A.A.; Kozin, S.A. Isomerization of Asp7 Leads to Increased Toxic Effect of Amyloid-Β42 on Human Neuronal Cells. Cell Death Dis. 2013, 4, e939. [Google Scholar] [CrossRef] [Green Version]
- Jamasbi, E.; Separovic, F.; Hossain, M.A.; Ciccotosto, G.D. Phosphorylation of a Full Length Amyloid-β Peptide Modulates Its Amyloid Aggregation, Cell Binding and Neurotoxic Properties. Mol. BioSyst. 2017, 13, 1545–1551. [Google Scholar] [CrossRef] [Green Version]
- Barykin, E.P.; Petrushanko, I.Y.; Kozin, S.A.; Telegin, G.B.; Chernov, A.S.; Lopina, O.D.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Phosphorylation of the Amyloid-Beta Peptide Inhibits Zinc-Dependent Aggregation, Prevents Na,K-ATPase Inhibition, and Reduces Cerebral Plaque Deposition. Front. Mol. Neurosci. 2018, 11, 302. [Google Scholar] [CrossRef]
- Deng, L.; Haynes, P.; Wu, Y.; Amirkhani, A.; Kamath, K.; Wu, J.; Pushpitha, K.; Gupta, V.; Graham, S.; Gupta, V.; et al. Amyloid-Beta Peptide Neurotoxicity in Human Neuronal Cells Is Associated with Modulation of Insulin-like Growth Factor Transport, Lysosomal Machinery and Extracellular Matrix Receptor Interactions. Neural Regen. Res. 2020, 15, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Abeysinghe, A.A.D.T.; Deshapriya, R.D.U.S.; Udawatte, C. Alzheimer’s Disease—A Review of the Pathophysiological Basis and Therapeutic Interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef] [PubMed]
- Vidal, C.; Zhang, L. An Analysis of the Neurological and Molecular Alterations Underlying the Pathogenesis of Alzheimer’s Disease. Cells 2021, 10, 546. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Lee, D.H.S.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. β-Amyloid1–42 Binds to A7 Nicotinic Acetylcholine Receptor with High Affinity. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Kuo, Y.-P.; George, A.A.; Xu, L.; Hu, J.; Lukas, R.J. β-Amyloid Directly Inhibits Human A4β2-Nicotinic Acetylcholine Receptors Heterologously Expressed in Human SH-EP1 Cells. J. Biol. Chem. 2004, 279, 37842–37851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and Amyloid-β Peptide Neurotoxicity in Alzheimer’s Disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Nakanishi, A.; Kaneko, N.; Takeda, H.; Sawasaki, T.; Morikawa, S.; Zhou, W.; Kurata, M.; Yamamoto, T.; Akbar, S.M.F.; Zako, T.; et al. Amyloid β Directly Interacts with NLRP3 to Initiate Inflammasome Activation: Identification of an Intrinsic NLRP3 Ligand in a Cell-Free System. Inflamm. Regen. 2018, 38, 27. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Mitkevich, V.A.; Anashkina, A.A.; Adzhubei, A.A.; Burnysheva, K.M.; Lakunina, V.A.; Kamanina, Y.V.; Dergousova, E.A.; Lopina, O.D.; Ogunshola, O.O.; et al. Direct Interaction of Beta-Amyloid with Na,K-ATPase as a Putative Regulator of the Enzyme Function. Sci. Rep. 2016, 6, 27738. [Google Scholar] [CrossRef] [Green Version]
- Dickey, C.A.; Gordon, M.N.; Wilcock, D.M.; Herber, D.L.; Freeman, M.J.; Morgan, D. Dysregulation of Na+/K+ ATPase by Amyloid in APP+PS1 Transgenic Mice. BMC Neurosci. 2005, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-N.; Sun, Y.-J.; Pan, S.; Li, J.-X.; Qu, Y.-E.; Li, Y.; Wang, Y.-L.; Gao, Z.-B. Na+-K+-ATPase, a Potent Neuroprotective Modulator against Alzheimer Disease. Fundam. Clin. Pharmacol. 2013, 27, 96–103. [Google Scholar] [CrossRef]
- Hattori, N.; Kitagawa, K.; Higashida, T.; Yagyu, K.; Shimohama, S.; Wataya, T.; Perry, G.; Smith, M.A.; Inagaki, C. Cl−-ATPase and Na+/K+-ATPase Activities in Alzheimer’s Disease Brains. Neurosci. Lett. 1998, 254, 141–144. [Google Scholar] [CrossRef]
- Scheiner-Bobis, G. The Sodium Pump: Its Molecular Properties and Mechanics of Ion Transport. Eur. J. Biochem. 2002, 269, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Kanai, R.; Ogawa, H.; Vilsen, B.; Cornelius, F.; Toyoshima, C. Crystal Structure of a Na+-Bound Na+,K+-ATPase Preceding the E1P State. Nature 2013, 502, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, T.; Ogawa, H.; Cornelius, F.; Toyoshima, C. Crystal Structure of the Sodium–Potassium Pump at 2.4 Å Resolution. Nature 2009, 459, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Laursen, M.; Yatime, L.; Nissen, P.; Fedosova, N.U. Crystal Structure of the High-Affinity Na+,K+-ATPase-Ouabain Complex with Mg2+ Bound in the Cation Binding Site. Proc. Natl. Acad. Sci. USA 2013, 110, 10958–10963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Adzhubei, A.A.; Anashkina, A.A.; Makarov, A.A. Left-Handed Polyproline-II Helix Revisited: Proteins Causing Proteopathies. J. Biomol. Struct. Dyn. 2017, 35, 2701–2713. [Google Scholar] [CrossRef]
- Barykin, E.P.; Garifulina, A.I.; Kruykova, E.V.; Spirova, E.N.; Anashkina, A.A.; Adzhubei, A.A.; Shelukhina, I.V.; Kasheverov, I.E.; Mitkevich, V.A.; Kozin, S.A.; et al. Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the A7 Nicotinic Receptor and Promotes Neurotoxicity. Cells 2019, 8, 771. [Google Scholar] [CrossRef] [Green Version]
- Moal, I.H.; Chaleil, R.A.G.; Bates, P.A. Flexible Protein-Protein Docking with SwarmDock. In Protein Complex Assembly; Methods in Molecular, Biology; Marsh, J.A., Ed.; Springer: New York, NY, USA, 2018; Volume 1764, pp. 413–428. ISBN 978-1-4939-7758-1. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anashkina, A.A.; Kravatsky, Y.; Kuznetsov, E.; Makarov, A.A.; Adzhubei, A.A. Meta-Server for Automatic Analysis, Scoring and Ranking of Docking Models. Bioinformatics 2018, 34, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, W.L. Aβ Toxicity in Alzheimer’s Disease: Globular Oligomers (ADDLs) as New Vaccine and Drug Targets. Neurochem. Int. 2002, 41, 345–352. [Google Scholar] [CrossRef]
- Boldyrev, A.A.; Lopina, O.D.; Kenney, M.; Johnson, P. Characterization of the Subunit Isoforms of Duck Salt Gland Na/K Adenosine Triphosphatase. Biochem. Biophys. Res. Commun. 1995, 216, 1048–1053. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-Glutathionylation of the Na,K-ATPase Catalytic α Subunit Is a Determinant of the Enzyme Redox Sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.W. Purification of Na+,K+-ATPase from the Supraorbital Salt Gland of the Duck. Methods Enzymol. 1988, 156, 46–48. [Google Scholar]
- Klimanova, E.A.; Petrushanko, I.Y.; Mitkevich, V.A.; Anashkina, A.A.; Orlov, S.N.; Makarov, A.A.; Lopina, O.D. Binding of Ouabain and Marinobufagenin Leads to Different Structural Changes in Na,K-ATPase and Depends on the Enzyme Conformation. FEBS Lett. 2015, 589, 2668–2674. [Google Scholar] [CrossRef] [Green Version]
- Kairane, C.; Mahlapuu, R.; Ehrlich, K.; Zilmer, M.; Soomets, U. The Effects of Different Antioxidants on the Activity of Cerebrocortical MnSOD and Na,K-ATPase from Post Mortem Alzheimer’s Disease and Age-Matched Normal Brains. Curr. Alzheimer Res. 2014, 11, 79–85. [Google Scholar] [CrossRef]
- Tverskoi, A.M.; Poluektov, Y.M.; Klimanova, E.A.; Mitkevich, V.A.; Makarov, A.A.; Orlov, S.N.; Petrushanko, I.Y.; Lopina, O.D. Depth of the Steroid Core Location Determines the Mode of Na,K-ATPase Inhibition by Cardiotonic Steroids. Int. J. Mol. Sci. 2021, 22, 13268. [Google Scholar] [CrossRef]
- Xie, Z.; Askari, A. Na+/K+-ATPase as a Signal Transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlov, S.N.; Tverskoi, A.M.; Sidorenko, S.V.; Smolyaninova, L.V.; Lopina, O.D.; Dulin, N.O.; Klimanova, E.A. Na,K-ATPase as a Target for Endogenous Cardiotonic Steroids: What’s the Evidence? Genes Dis. 2021, 8, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Kozin, S.A.; Barykin, E.P.; Telegin, G.B.; Chernov, A.S.; Adzhubei, A.A.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Intravenously Injected Amyloid-β Peptide with Isomerized Asp7 and Phosphorylated Ser8 Residues Inhibits Cerebral β-Amyloidosis in AβPP/PS1 Transgenic Mice Model of Alzheimer’s Disease. Front. Neurosci. 2018, 12, 518. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Conformation of the Na, K-ATPase | Interaction Sites with Aβ42 after Docking, a.a. Residues | Interaction Sites with Aβ42 after MD, a.a. Residues |
|---|---|---|
| E1P | α-subunit: 121–124 312–317 887–893 β-subunit: 84–87 216–222 270–273 287–290 | α-subunit: 117–126 311–318 794 884–894 β-subunit: 82–87 91–111 196 217 266–273 287–298 |
| E2P | α-subunit: 113–128 309–316 883–900 970–979 β-subunit: 82–107 197 216–226 267–273 285–290 294–303 | α-subunit: 117–124 310–316 879 883 886–893 970–979 β-subunit: 83–85 91–107 217–218 268–273 287–290 |
| OBN | α-subunit: 117–125 309–317 886–893 969–979 β-subunit: 82–87 96–107 216–221 271–273 287–290 | α-subunit: 115–125 306–317 792–795 879 883 886–893 974 β-subunit: 2–87 91 100 103–107 216–218 273 287–290 294 |
| Na,K-ATPase State a | Ka,b M−1 | Kd c µM | ∆H d kcal/mol | T∆S e kcal/mol | ∆G f kcal/mol |
|---|---|---|---|---|---|
| E1/E2 | 7.7 × 105 | 1.3 | −2.54 | 5.48 | −8.02 |
| E1 | 3.7 × 105 | 2.7 | −1.54 | 6.02 | −7.56 |
| E2 | 4.9 × 105 | 2.0 | −2.21 | 5.55 | −7.76 |
| E2P | 5.1 × 105 | 2.0 | −1.59 | 6.19 | −7.78 |
| OBN | 8.3 × 105 | 1.2 | −1.21 | 6.87 | −8.08 |
| Compound Name in the NCI and ZINC15 Databases | Rating Number and Affinity to E1P, kcal/mol | Rating Number and Affinity to E2P, kcal/mol | Rating Number and Affinity to OBN, kcal/mol | Compound Chemical Formula |
|---|---|---|---|---|
| NCI39918 ZINC4430655 | 1 –9.4 | 4 –8.0 | 3 –8.6 | 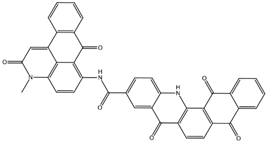 |
| NCI610512 iodine (I) atom changed to hydrogen | 2 –9.3 | 7 –7.4 | 5 –7.6 | 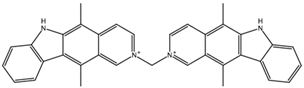 |
| NCI686480 ZINC5542961 | 3 –9.1 | 3 –8.2 | 4 –8.1 |  |
| NCI39921 ZINC150471868 | 4 –8.9 | 1 –8.7 | 2 –8.7 | 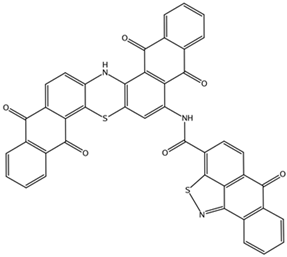 |
| NCI617551 ZINC153412538 ZINC153412622 ZINC153412744 ZINC153412881 ZINC160342695 ZINC160342850 | 5 –8.9 | 2 –8.3 | 7 –7.4 | 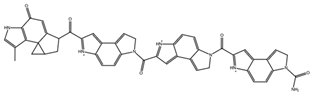 |
| NCI84171 ZINC95857648 | 6 –8.7 | 5 –7.9 | 1 –8.9 |  |
| NCI298806 ZINC5390003 | 7 –8.7 | 10 –5.6 | 9 –6.7 | 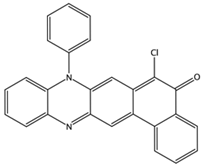 |
| NCI23128 ZINC150340408 | 8 –8.7 | 8 –7.3 | 6 –7.6 | 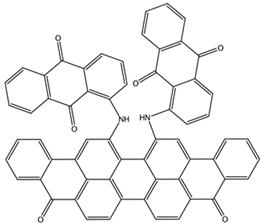 |
| NCI688806 ZINC5543121 ZINC5543124 ZINC107044834 ZINC107044838 ZINC107044843 ZINC107044846 | 9 –8.7 | 6 –7.6 | 8 –7.4 |  |
| NCI58783 Na atom changed to hydrogen | 10 –8.6 | 9 –6.5 | 10 –6.4 | 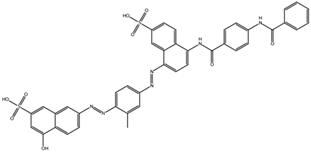 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adzhubei, A.A.; Tolstova, A.P.; Strelkova, M.A.; Mitkevich, V.A.; Petrushanko, I.Y.; Makarov, A.A. Interaction Interface of Aβ42 with Human Na,K-ATPase Studied by MD and ITC and Inhibitor Screening by MD. Biomedicines 2022, 10, 1663. https://doi.org/10.3390/biomedicines10071663
Adzhubei AA, Tolstova AP, Strelkova MA, Mitkevich VA, Petrushanko IY, Makarov AA. Interaction Interface of Aβ42 with Human Na,K-ATPase Studied by MD and ITC and Inhibitor Screening by MD. Biomedicines. 2022; 10(7):1663. https://doi.org/10.3390/biomedicines10071663
Chicago/Turabian StyleAdzhubei, Alexei A., Anna P. Tolstova, Maria A. Strelkova, Vladimir A. Mitkevich, Irina Yu. Petrushanko, and Alexander A. Makarov. 2022. "Interaction Interface of Aβ42 with Human Na,K-ATPase Studied by MD and ITC and Inhibitor Screening by MD" Biomedicines 10, no. 7: 1663. https://doi.org/10.3390/biomedicines10071663
APA StyleAdzhubei, A. A., Tolstova, A. P., Strelkova, M. A., Mitkevich, V. A., Petrushanko, I. Y., & Makarov, A. A. (2022). Interaction Interface of Aβ42 with Human Na,K-ATPase Studied by MD and ITC and Inhibitor Screening by MD. Biomedicines, 10(7), 1663. https://doi.org/10.3390/biomedicines10071663