
Current Concepts of Vitiligo Immunopathogenesis


{kind=link}
Abstract
1. Introduction
2. Genetic Factors in the Development of Vitiligo
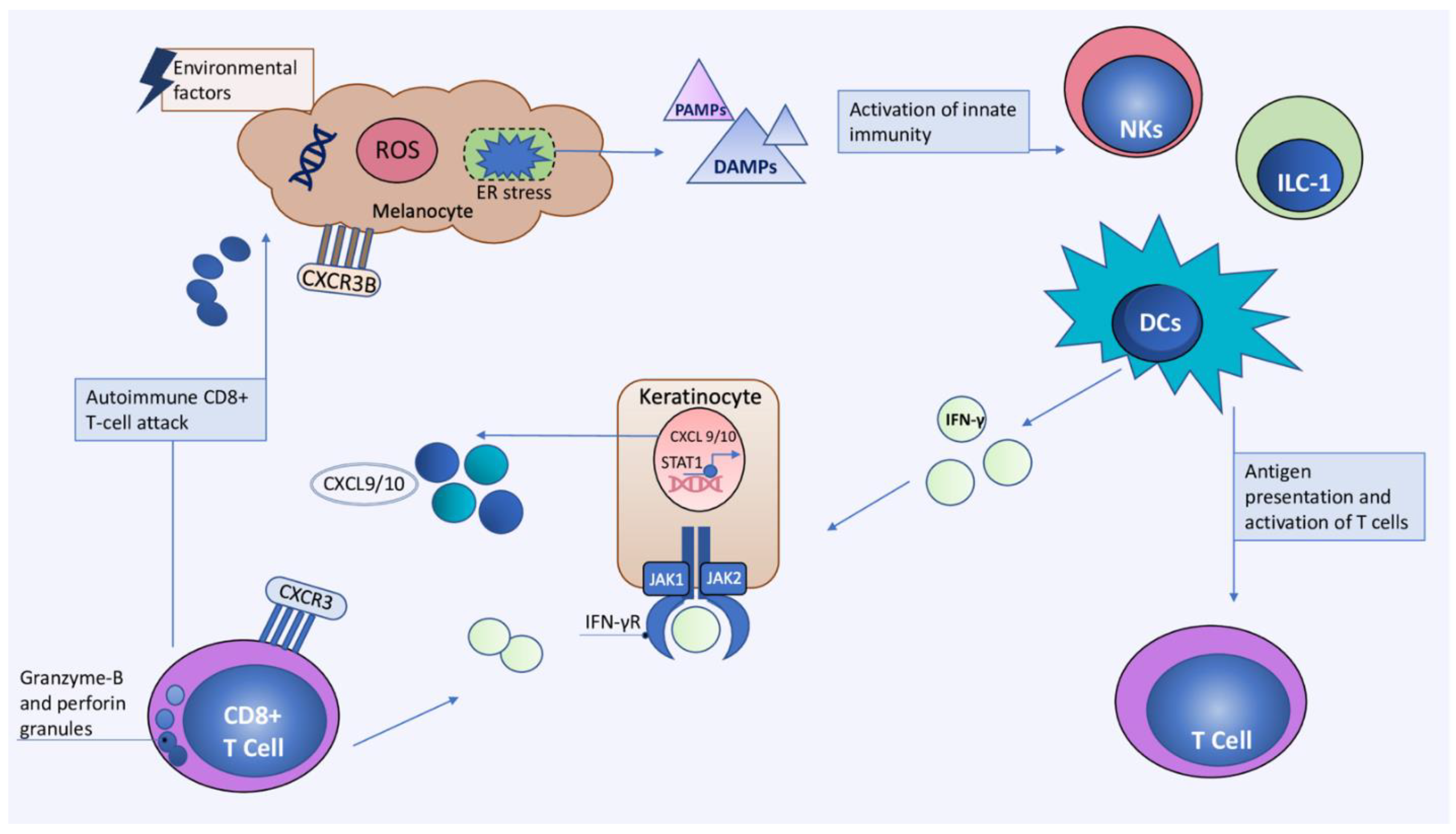
3. Current Concepts in Immunopathogenesis of Vitiligo
3.1. The Role of Oxidative Stress
3.2. The Role of Innate Immunity
3.2.1. The Role of DAMPs and PAMPs
3.2.2. Dendritic Cells (DCs)
3.2.3. Natural Killer (NK) Cells and Innate Lymphoid Cells Type 1 (ILC-1)
3.3. Adaptive Immunity
3.3.1. Cytotoxic CD8+ T cells
3.3.2. Regulatory T Cells
3.3.3. Tissue-Resident Memory T Cells
3.3.4. The Role of B-Lymphocytes
3.4. Main Cytokine Networks
4. Future Perspective of Vitiligo Treatment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alikhan, A.; Felsten, L.M.; Daly, M.; Petronic-Rosic, V. Vitiligo: A comprehensive overview: Part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J. Am. Acad. Dermatol. 2011, 65, 473–491. [Google Scholar] [CrossRef] [PubMed]
- Krüger, C.; Schallreuter, K.U. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int. J. Dermatol. 2012, 51, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, Y.; Shi, M.; Jiang, S.; Cui, S.; Wu, Y.; Gao, X.-H.; Chen, H.-D. The Prevalence of Vitiligo: A Meta-Analysis. PLoS ONE 2016, 11, e0163806. [Google Scholar] [CrossRef]
- Bergqvist, C.; Ezzedine, K. Vitiligo: A Review. Dermatology 2020, 236, 571–592. [Google Scholar] [CrossRef] [PubMed]
- van Geel, N.; Speeckaert, R.; Taieb, A.; Picardo, M.; Böhm, M.; Gawkrodger, D.J.; Schallreuter, K.; Bennett, D.C.; van der Veen, W.; Whitton, M.; et al. Koebner’s phenomenon in vitiligo: European position paper. Pigment Cell Melanoma Res. 2011, 24, 564–573. [Google Scholar] [CrossRef]
- Cohen, B.E.; Mu, E.W.; Orlow, S.J. Comparison of Childhood Vitiligo Presenting with or without Associated Halo Nevi. Pediatr. Dermatol. 2015, 33, 44–48. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, L.-C.; Chen, M.-K.; Liao, Q.-M.; Mao, R.-X.; Han, J.-D. Factors Associated with Development of Vitiligo in Patients with Halo Nevus. Chin. Med. J. 2017, 130, 2703–2708. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Zhu, G.; Zhang, Q.; Wang, G.; Gao, T.; Li, C.; Wang, L.; Jian, Z. A similar local immune and oxidative stress phenotype in vitiligo and halo nevus. J. Dermatol. Sci. 2017, 87, 50–59. [Google Scholar] [CrossRef]
- Jin, Y.; Mailloux, C.M.; Gowan, K.; Riccardi, S.L.; Laberge, G.; Bennett, D.C.; Fain, P.R.; Spritz, R.A. NALP1in Vitiligo-Associated Multiple Autoimmune Disease. N. Engl. J. Med. 2007, 356, 1216–1225. [Google Scholar] [CrossRef]
- Birlea, S.A.; Jin, Y.; Bennett, D.C.; Herbstman, D.M.; Wallace, M.R.; McCormack, W.T.; Kemp, E.H.; Gawkrodger, D.J.; Weetman, A.P.; Picardo, M.; et al. Comprehensive Association Analysis of Candidate Genes for Generalized Vitiligo Supports XBP1, FOXP3, and TSLP. J. Investig. Dermatol. 2011, 131, 371–381. [Google Scholar] [CrossRef]
- Spritz, R.A. Six Decades of Vitiligo Genetics: Genome-Wide Studies Provide Insights into Autoimmune Pathogenesis. J. Investig. Dermatol. 2012, 132, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Spritz, R.A. The genetics of generalized vitiligo: Autoimmune pathways and an inverse relationship with malignant melanoma. Genome Med. 2010, 2, 78. [Google Scholar] [CrossRef] [PubMed]
- Ezzedine, K.; Lim, H.W.; Suzuki, T.; Katayama, I.; Hamzavi, I.; Lan, C.C.E.; Goh, B.K.; Anbar, T.; de Castro, C.S.; Lee, A.Y.; et al. Revised classification/nomenclature of vitiligo and related issues: The Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012, 25, E1–E13. [Google Scholar] [CrossRef]
- Willemsen, M.; Post, N.F.; van Uden, N.O.; Narayan, V.S.; Chielie, S.; Kemp, E.H.; Bekkenk, M.W.; Luiten, R.M. Immunophenotypic Analysis Reveals Differences in Circulating Immune Cells in the Peripheral Blood of Patients with Segmental and Nonsegmental Vitiligo. J. Investig. Dermatol. 2021, 142, 876–883.e3. [Google Scholar] [CrossRef] [PubMed]
- Spritz, R.A.; Santorico, S.A. The Genetic Basis of Vitiligo. J. Investig. Dermatol. 2021, 141, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Spritz, R.A.; Andersen, G.H.L. Genetics of Vitiligo. Dermatol. Clin. 2017, 35, 245–255. [Google Scholar] [CrossRef]
- Majumder, P.P.; Das, S.K.; Li, C.C. A genetical model for vitiligo. Am. J. Hum. Genet. 1988, 43, 119–125. [Google Scholar]
- Nath, S.K.; Majumder, P.P.; Nordlund, J.J. Genetic epidemiology of vitiligo: Multilocus recessivity cross-validated. Am. J. Hum. Genet. 1994, 55, 981–990. [Google Scholar]
- Das, S.K.; Majumder, P.P.; Majumdar, T.K.; Haldar, B. Studies on vitiligo. II. Familial aggregation and genetics. Genet. Epidemiol. 1985, 2, 255–262. [Google Scholar] [CrossRef]
- Birlea, S.A.; Gowan, K.; Fain, P.R.; Spritz, R.A. Genome-Wide Association Study of Generalized Vitiligo in an Isolated European Founder Population Identifies SMOC2, in Close Proximity to IDDM8. J. Investig. Dermatol. 2010, 130, 798–803. [Google Scholar] [CrossRef]
- Foley, L.M.; Lowe, N.J.; Misheloff, E.; Tiwari, J.L. Association of HLA-DR4 with vitiligo. J. Am. Acad. Dermatol. 1983, 8, 39–40. [Google Scholar] [CrossRef]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Gowan, K.; Riccardi, S.L.; Holland, P.J.; Mailloux, C.M.; Sufit, A.J.D.; Hutton, S.M.; Amadi-Myers, A.; et al. Variant of TYR and Autoimmunity Susceptibility Loci in Generalized Vitiligo. N. Engl. J. Med. 2010, 362, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Hayashi, M.; Jin, Y.; Yorgov, D.; Santorico, S.A.; Holcomb, C.; Rastrou, M.; Erlich, H.; Tengesdal, I.W.; Dagna, L.; et al. MHC class II super-enhancer increases surface expression of HLA-DR and HLA-DQ and affects cytokine production in autoimmune vitiligo. Proc. Natl. Acad. Sci. USA 2016, 113, 1363–1368. [Google Scholar] [CrossRef]
- Fain, P.R.; Babu, S.R.; Bennett, D.C.; Spritz, R.A. HLA class II haplotype DRB1*04-DQB1*0301 contributes to risk of familial generalized vitiligo and arly disease onset. Pigment Cell Res. 2006, 19, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Jin, Y.; Yorgov, D.; Santorico, S.A.; Hagman, J.; Ferrara, T.M.; Jones, K.L.; Cavalli, G.; Dinarello, C.A.; Spritz, R.A. Autoimmune vitiligo is associated with gain-offunction by a transcriptional regulator that elevates expression of HLA-A∗02:01 in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Ren, Y.-Q.; Xiang, L.-H.; Sun, L.-D.; Xu, A.-E.; Gao, X.-H.; Chen, H.-D.; Pu, X.-M.; Wu, R.-N.; Liang, C.-Z.; et al. Genome-wide association study for vitiligo identifies susceptibility loci at 6q27 and the MHC. Nat. Genet. 2010, 42, 614–618. [Google Scholar] [CrossRef]
- Zhu, K.; Lv, Y.-M.; Yin, X.-Y.; Wang, Z.-X.; Sun, L.-D.; He, S.-M.; Cheng, H.; Hu, D.-Y.; Zhang, Z.; Li, Y.; et al. Psoriasis Regression Analysis of MHC Loci Identifies Shared Genetic Variants with Vitiligo. PLoS ONE 2011, 6, e23089. [Google Scholar] [CrossRef]
- Liu, J.; Tang, H.; Zuo, X.; Liang, B.; Wang, P.; Sun, L.; Yang, S.; Zhang, X. A single nucleotide polymorphism rs9468925 of MHC region is associated with clinical features of generalized vitiligo in Chinese Han population. J. Eur. Acad. Dermatol. Venereol. 2011, 26, 1137–1141. [Google Scholar] [CrossRef]
- Birlea, S.A.; Laberge, G.S.; Procopciuc, L.M.; Fain, P.R.; Spritz, R.A. CTLA4and generalized vitiligo: Two genetic association studies and a meta-analysis of published data. Pigment Cell Melanoma Res. 2009, 22, 230–234. [Google Scholar] [CrossRef]
- Kemp, E.H.; Ajjan, R.A.; Waterman, E.A.; Gawkrodger, D.J.; Cork, M.J.; Watson, P.F.; Weetman, A.P. Analysis of a microsatellite polymorphism of the cytotoxic T-lymphocyte antigen-4 gene in patients with vitiligo. Br. J. Dermatol. 1999, 140, 73–78. [Google Scholar] [CrossRef]
- Laberge, G.S.; Birlea, S.A.; Fain, P.R.; Spritz, R.A. The PTPN22-1858C>T (R620W) functional polymorphism is associated with generalized vitiligo in the Romanian population. Pigment Cell Melanoma Res. 2008, 21, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Canton, I.; Akhtar, S.; Gavalas, N.G.; Gawkrodger, D.J.; Blomhoff, A.; Watson, P.F.; Weetman, A.P.; Kemp, E.H. A single-nucleotide polymorphism in the gene encoding lymphoid protein tyrosine phosphatase (PTPN22) confers susceptibility to generalised vitiligo. Genes Immun. 2005, 6, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Ferrara, T.M.; Ben, S.; Riccardi, S.L.; Cole, J.; Gowan, K.; Holland, P.J.; Bennett, D.; et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 2012, 44, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Peach, R.J.; Bajorath, J.; Naemura, J.; Leytze, G.; Greene, J.; Aruffo, A.; Linsley, P.S. Both Extracellular Immunoglobin-like Domains of CD80 Contain Residues Critical for Binding T Cell Surface Receptors CTLA-4 and CD28. J. Biol. Chem. 1995, 270, 21181–21187. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.; Aung, S.; Redmond, W.; Sherman, L.A. Phenotypic and Functional Analysis of Cd8+ T Cells Undergoing Peripheral Deletion in Response to Cross-Presentation of Self-Antigen. J. Exp. Med. 2001, 194, 707–718. [Google Scholar] [CrossRef]
- Basak, P.; Adiloglu, A.; Koc, I.; Taş, T.; Akkaya, V. Evaluation of activatory and inhibitory natural killer cell receptors in non-segmental vitiligo: A flow cytometric study. J. Eur. Acad. Dermatol. Venereol. 2008, 22, 970–976. [Google Scholar] [CrossRef]
- Seya, T.; Matsumoto, M.; Ebihara, T.; Oshiumi, H. Functional evolution of the TICAM-1 pathway for extrinsic RNA sensing. Immunol. Rev. 2008, 227, 44–53. [Google Scholar] [CrossRef]
- Kumeta, H.; Sakakibara, H.; Enokizono, Y.; Ogura, K.; Horiuchi, M.; Matsumoto, M.; Seya, T.; Inagaki, F. The N-terminal domain of TIR domain-containing adaptor molecule-1, TICAM-1. J. Biomol. NMR 2014, 58, 227–230. [Google Scholar] [CrossRef]
- Tang, X.-F.; Zhang, Z.; Hu, D.-Y.; Xu, A.-E.; Zhou, H.-S.; Sun, L.-D.; Gao, M.; Gao, T.-W.; Gao, X.-H.; Chen, H.-D.; et al. Association Analyses Identify Three Susceptibility Loci for Vitiligo in the Chinese Han Population. J. Investig. Dermatol. 2013, 133, 403–410. [Google Scholar] [CrossRef]
- Pan, F.; Yu, H.; Dang, E.V.; Barbi, J.; Pan, X.; Grosso, J.F.; Jinasena, D.; Sharma, S.M.; McCadden, E.M.; Getnet, D.; et al. Eos Mediates Foxp3-Dependent Gene Silencing in CD4 + Regulatory T Cells. Science 2009, 325, 1142–1146. [Google Scholar] [CrossRef]
- Petukhova, L.; Duvic, M.; Hordinsky, M.; Norris, D.; Price, V.; Shimomura, Y.; Kim, H.; Singh, P.; Lee, A.; Chen, W.V.; et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature 2010, 466, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Hakonarson, H.; Qu, H.-Q.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Casalunovo, T.; Taback, S.P.; Frackelton, E.C.; et al. A Novel Susceptibility Locus for Type 1 Diabetes on Chr12q13 Identified by a Genome-Wide Association Study. Diabetes 2008, 57, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wicker, L.S.; Santamaria, P. IL-2 and its high-affinity receptor: Genetic control of immunoregulation and autoimmunity. Semin. Immunol. 2009, 21, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Sturm, R.A. Multiple genes and locus interactions in susceptibility to vitiligo. J. Investig. Dermatol. 2010, 130, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Faraj, S.; Kemp, E.H.; Gawkrodger, D.J. Patho-immunological mechanisms of vitiligo: The role of the innate and adaptive immunities and environmental stress factors. Clin. Exp. Immunol. 2021, 207, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, N.; Gavalas, N.G.; Weetman, A.P.; Kemp, E.H. Autoimmunity as an aetiological factor in vitiligo. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 865–876. [Google Scholar] [CrossRef]
- Shen, C.; Gao, J.; Sheng, Y.; Dou, J.; Zhou, F.; Zheng, X.; Ko, R.; Tang, X.; Zhu, C.; Yin, X.; et al. Genetic Susceptibility to Vitiligo: GWAS Approaches for Identifying Vitiligo Susceptibility Genes and Loci. Front. Genet. 2016, 7, 3. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Shewmaker, F.; McPhie, P.; Monterroso, B.; Thurber, K.; Wickner, R.B. The repeat domain of the melanosome fibril protein Pmel17 forms the amyloid core promoting melanin synthesis. Proc. Natl. Acad. Sci. USA 2009, 106, 13731–13736. [Google Scholar] [CrossRef]
- Dessinioti, C.; Antoniou, C.; Katsambas, A.; Stratigos, A.J. Melanocortin 1 Receptor Variants: Functional Role and Pigmentary Associations. Photochem. Photobiol. 2011, 87, 978–987. [Google Scholar] [CrossRef]
- Széll, M.; Baltás, E.; Bodai, L.; Bata-Csörgo, Z.; Nagy, N.; Dallos, A.; Pourfarzi, R.; Simics, E.; Kondorosi, I.; Szalai, Z.; et al. The Arg160Trp allele of melanocortin-1 receptor gene might protect against vitiligo. Photochem. Photobiol. 2008, 84, 565–571. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Kanneganti, T.-D. Caspase-7: A protease involved in apoptosis and inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Trapani, J.A.; Sutton, V.R. Granzyme B: Pro-apoptotic, antiviral and antitumor functions. Curr. Opin. Immunol. 2003, 15, 533–543. [Google Scholar] [CrossRef]
- Devadas, S.; Das, J.; Liu, C.; Zhang, L.; Roberts, A.I.; Pan, Z.; Moore, P.A.; Das, G.; Shi, Y. Granzyme B Is Critical for T Cell Receptor-Induced Cell Death of Type 2 Helper T Cells. Immunity 2006, 25, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Kucuksezer, U.C.; Duymaz-Tozkir, J.; Gül, A.; Saruhan-Direskeneli, G. No association of granzyme B gene polymorphism with Behçet’s disease. Clin. Exp. Rheumatol. 2009, 27, S102. [Google Scholar] [PubMed]
- Donn, R.; Ellison, S.; Lamb, R.; Day, T.; Baildam, E.; Ramanan, A.V. Genetic loci contributing to hemophagocytic lymphohistiocytosis do not confer susceptibility to systemic-onset juvenile idiopathic arthritis. Arthritis Care Res. 2008, 58, 869–874. [Google Scholar] [CrossRef]
- Seneschal, J.; Boniface, K.; D’Arino, A.; Picardo, M. An update on Vitiligo pathogenesis. Pigment Cell Melanoma Res. 2021, 34, 236–243. [Google Scholar] [CrossRef]
- Boniface, K.; Seneschal, J.; Picardo, M.; Taïeb, A. Vitiligo: Focus on Clinical Aspects, Immunopathogenesis, and Therapy. Clin. Rev. Allergy Immunol. 2018, 54, 52–67. [Google Scholar] [CrossRef]
- Strassner, J.; Harris, J.E. Understanding mechanisms of autoimmunity through translational research in vitiligo. Curr. Opin. Immunol. 2016, 43, 81–88. [Google Scholar] [CrossRef]
- Migayron, L.; Boniface, K.; Seneschal, J. Vitiligo, From Physiopathology to Emerging Treatments: A Review. Dermatol. Ther. 2020, 10, 1185–1198. [Google Scholar] [CrossRef]
- Xuan, Y.; Yang, Y.; Xiang, L.; Zhang, C. The Role of Oxidative Stress in the Pathogenesis of Vitiligo: A Culprit for Melanocyte Death. Oxidative Med. Cell. Longev. 2022, 2022, 8498472. [Google Scholar] [CrossRef]
- Wang, Y.; Li, S.; Li, C. Perspectives of New Advances in the Pathogenesis of Vitiligo: From Oxidative Stress to Autoimmunity. Med. Sci. Monit. 2019, 25, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Speeckaert, R.; Dugardin, J.; Lambert, J.; Lapeere, H.; Verhaeghe, E.; Speeckaert, M.; Van Geel, N. Critical appraisal of the oxidative stress pathway in vitiligo: A systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Mosenson, J.A.; Flood, K.; Klarquist, J.; Eby, J.M.; Koshoffer, A.; Boissy, R.E.; Overbeck, A.; Tung, R.C.; Le Poole, I.C. Preferential secretion of inducible HSP70 by vitiligo melanocytes under stress. Pigment Cell Melanoma Res. 2013, 27, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.E.; Harris, T.H.; Weninger, W.; Wherry, E.J.; Hunter, C.A.; Turka, L.A. A Mouse Model of Vitiligo with Focused Epidermal Depigmentation Requires IFN-γ for Autoreactive CD8+ T-Cell Accumulation in the Skin. J. Investig. Dermatol. 2012, 132, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wang, Q.Q.; Wu, J.Q.; Jiang, M.; Chen, L.; Zhang, C.F.; Xiang, L.H. Increased expression of CXCR3 and its ligands in patients with vitiligo and CXCL10 as a potential clinical marker for vitiligo. Br. J. Dermatol. 2016, 174, 1318–1326. [Google Scholar] [CrossRef]
- Passeron, T.; Malmqvst, V.E.; Bzioueche, H.; Marchetti, S.; Rocchi, S.; Tulic, M.K. Increased Activation of Innate Immunity and Pro-Apoptotic CXCR3B in Normal-Appearing Skin on the Lesional Site of Patients with Segmental Vitiligo. J. Investig. Dermatol. 2021, 142, 480–483.e2. [Google Scholar] [CrossRef]
- Tulic, M.K.; Cavazza, E.; Cheli, Y.; Jacquel, A.; Luci, C.; Cardot-Leccia, N.; Hadhiri-Bzioueche, H.; Abbe, P.; Gesson, M.; Sormani, L.; et al. Innate lymphocyte-induced CXCR3B-mediated melanocyte apoptosis is a potential initiator of T-cell autoreactivity in vitiligo. Nat. Commun. 2019, 10, 2178. [Google Scholar] [CrossRef]
- Hassan, A.S.; Kohil, M.M.; Sayed, S.S.E.; Mahmoud, S.B. Immunohistochemical study of perforin and apoptosis stimulation fragment ligand (FasL)in active vitiligo. Arch. Dermatol. Res. 2021, 313, 453–460. [Google Scholar] [CrossRef]
- Rashighi, M.; Harris, J.E. Vitiligo Pathogenesis and Emerging Treatments. Dermatol. Clin. 2017, 35, 257–265. [Google Scholar] [CrossRef]
- Xie, H.; Zhou, F.; Liu, L.; Zhu, G.; Li, Q.; Li, C.; Gao, T. Vitiligo: How do oxidative stress-induced autoantigens trigger autoimmunity? J. Dermatol. Sci. 2016, 81, 3–9. [Google Scholar] [CrossRef]
- He, Y.; Li, S.; Zhang, W.; Dai, W.; Cui, T.; Wang, G.; Gao, T.; Li, C. Dysregulated autophagy increased melanocyte sensitivity to H2O2-induced oxidative stress in vitiligo. Sci. Rep. 2017, 7, 42394. [Google Scholar] [CrossRef] [PubMed]
- Schallreuter, K.U.; Moore, J.; Wood, J.M.; Beazley, W.D.; Gaze, D.C.; Tobin, D.; Marshall, H.S.; Panske, A.; Panzig, E.; Hibberts, N.A. In Vivo and In Vitro Evidence for Hydrogen Peroxide (H2O2) Accumulation in the Epidermis of Patients with Vitiligo and its Successful Removal by a UVB-Activated Pseudocatalase. J. Investig. Dermatol. Symp. Proc. 1999, 4, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Jadeja, S.D.; Mayatra, J.M.; Vaishnav, J.; Shukla, N.; Begum, R. A Concise Review on the Role of Endoplasmic Reticulum Stress in the Development of Autoimmunity in Vitiligo Pathogenesis. Front. Immunol. 2021, 11, 624566. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Yang, L.; Wu, H.; Li, W.; Ma, X.; Katayama, I.; Zhang, H. New insight into the role of exosomes in vitiligo. Autoimmun. Rev. 2020, 19, 102664. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, R.; Guo, X.; Zou, Y.; Chen, S.; Zhou, K.; Chen, Y.; Li, Y.; Gao, S.; Wu, Y. Identification of TYR, TYRP1, DCT and LARP7 as related biomarkers and immune infiltration characteristics of vitiligo via comprehensive strategies. Bioengineered 2021, 12, 2214–2227. [Google Scholar] [CrossRef]
- Jimbow, K.; Chen, H.; Park, J.S.; Thomas, P.D. Increased sensitivity of melanocytes to oxidative stress and abnormal expression of tyrosinase-related protein in vitiligo. Br. J. Dermatol. 2001, 144, 55–65. [Google Scholar] [CrossRef]
- Manga, P.; Choudhury, N. The unfolded protein and integrated stress response in melanoma and vitiligo. Pigment Cell Melanoma Res. 2020, 34, 204–211. [Google Scholar] [CrossRef]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2017, 19, 109–120. [Google Scholar] [CrossRef]
- Shrestha, N.; Reinert, R.B.; Qi, L. Endoplasmic Reticulum Protein Quality Control in β Cells. Semin. Cell Dev. Biol. 2020, 103, 59–67. [Google Scholar] [CrossRef]
- Todd, D.J.; Lee, A.H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Toosi, S.; Orlow, S.; Manga, P. Vitiligo-Inducing Phenols Activate the Unfolded Protein Response in Melanocytes Resulting in Upregulation of IL6 and IL8. J. Investig. Dermatol. 2012, 132, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Jadeja, S.D.; Vaishnav, J.; Mansuri, M.S.; Shah, C.; Mayatra, J.M.; Shah, A.; Begum, R. Investigation of the Role of Interleukin 6 in Vitiligo Pathogenesis. Immunol. Investig. 2020, 51, 120–137. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef] [PubMed]
- Mou, K.; Liu, W.; Miao, Y.; Cao, F.; Li, P. HMGB1 deficiency reduces H2O2-induced oxidative damage in human melanocytes via the Nrf2 pathway. J. Cell Mol. Med. 2018, 22, 6148–6156. [Google Scholar] [CrossRef]
- Arowojolu, O.A.; Orlow, S.; Elbuluk, N.; Manga, P. The nuclear factor (erythroid-derived 2)-like 2 (NRF2) antioxidant response promotes melanocyte viability and reduces toxicity of the vitiligo-inducing phenol monobenzone. Exp. Dermatol. 2017, 26, 637–644. [Google Scholar] [CrossRef]
- van den Boorn, J.G.; Picavet, D.I.; van Swieten, P.F.; van Veen, H.A.; Konijnenberg, D.; van Veelen, P.A.; van Capel, T.; Jong, E.C.D.; Reits, E.A.; Drijfhout, J.W.; et al. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J. Investig. Dermatol. 2011, 131, 1240–1251. [Google Scholar] [CrossRef]
- Jian, Z.; Tang, L.; Yi, X.; Liu, B.; Zhang, Q.; Zhu, G.; Wang, G.; Gao, T.; Li, C. Aspirin induces Nrf2-mediated transcriptional activation of haem oxygenase-1 in protection of human melanocytes from H2O2-induced oxidative stress. J. Cell. Mol. Med. 2016, 20, 1307–1318. [Google Scholar] [CrossRef]
- Chang, Y.; Li, S.; Guo, W.; Yang, Y.; Zhang, W.; Zhang, Q.; He, Y.; Yi, X.; Cui, T.; An, Y.; et al. Simvastatin Protects Human Melanocytes from H2O2-Induced Oxidative Stress by Activating Nrf2. J. Investig. Dermatol. 2017, 137, 1286–1296. [Google Scholar] [CrossRef]
- Zhou, T.; Li, D.; Deng, Y. Update on the role of noncoding RNAs in vitiligo. Chin. Med. J. 2021, 135, 793–795. [Google Scholar] [CrossRef]
- Sun, X.; Wang, T.; Huang, B.; Ruan, G.; Xu, A. ΜicroRNA-421 participates in vitiligo development through regulating human melanocyte survival by targeting receptor-interacting serine/threonine kinase 1. Mol. Med. Rep. 2020, 21, 858–866. [Google Scholar] [PubMed]
- Mansuri, M.S.; Singh, M.; Begum, R. miRNA signatures and transcriptional regulation of their target genes in vitiligo. J. Dermatol. Sci. 2016, 84, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Mansuri, M.S.; Singh, M.; Dwivedi, M.; Laddha, N.C.; Marfatia, Y.S.; Begum, R. MicroRNA profiling reveals differentially expressed microRNA signatures from the skin of patients with nonsegmental vitiligo. Br. J. Dermatol. 2014, 171, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhang, W.; Guo, S.; Jian, Z.; Li, S.; Li, K.; Ge, R.; Dai, W.; Wang, G.; Gao, T.; et al. Oxidative stress-induced overexpression of miR-25: The mechanism underlying the degeneration of melanocytes in vitiligo. Cell Death Differ. 2015, 23, 496–508. [Google Scholar] [CrossRef]
- Sahoo, A.; Lee, B.; Boniface, K.; Seneschal, J.; Sahoo, S.K.; Seki, T.; Wang, C.; Das, S.; Han, X.; Steppie, M.; et al. MicroRNA-211 Regulates Oxidative Phosphorylation and Energy Metabolism in Human Vitiligo. J. Investig. Dermatol. 2017, 137, 1965–1974. [Google Scholar] [CrossRef]
- Delmas, V.; Larue, L. Molecular and cellular basis of depigmentation in vitiligo patients. Exp. Dermatol. 2018, 28, 662–666. [Google Scholar] [CrossRef]
- Boukhedouni, N.; Martins, C.; Darrigade, A.S.; Drullion, C.; Rambert, J.; Barrault, C.; Garnier, J.; Jacquemin, C.; Thiolat, D.; Lucchese, F.; et al. Type-1 cytokines regulate MMP-9 production and E-cadherin disruption to promote melanocyte loss in vitiligo. JCI Insight. 2020, 5, e133772. [Google Scholar]
- Bordignon, M.; Castellani, C.; Fedrigo, M.; Thiene, G.; Peserico, A.; Alaibac, M.; Angelini, A. Role of alpha5beta1 integrin and MIA (melanoma inhibitory activity) in the pathogenesis of vitiligo. J. Dermatol. Sci. 2013, 71, 142–145. [Google Scholar] [CrossRef]
- Bordignon, M.; Luisetto, R.; Valente, M.L.; Fedrigo, M.; Castellani, C.; Angelini, A.; Alaibac, M. Melanoma Inhibitory Activity (MIA) Is Able to Induce Vitiligo-Like Depigmentation in an in vivo Mouse Model by Direct Injection in the Tail. Front. Med. 2020, 7, 430. [Google Scholar] [CrossRef]
- Boniface, K.; Passeron, T.; Seneschal, J.; Tulic, M.K. Targeting Innate Immunity to Combat Cutaneous Stress: The Vitiligo Perspective. Front. Immunol. 2021, 12, 613056. [Google Scholar] [CrossRef]
- Yu, R.; Broady, R.; Huang, Y.; Wang, Y.; Yu, J.; Gao, M.; Levings, M.; Wei, S.; Zhang, S.; Xu, A.; et al. Transcriptome Analysis Reveals Markers of Aberrantly Activated Innate Immunity in Vitiligo Lesional and Non-Lesional Skin. PLoS ONE 2012, 7, e51040. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Boniface, K.; Vergier, B.; Mossalayi, D.; Taieb, A.; Ezzedine, K.; Seneschal, J. Type I interferon signature in the initiation of the immune response in vitiligo. Pigment Cell Melanoma Res. 2014, 27, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Gholijani, N.; Yazdani, M.-R.; Dastgheib, L. Predominant role of innate pro-inflammatory cytokines in vitiligo disease. Arch. Dermatol. Res. 2019, 312, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Andersen, G.; Yorgov, D.; Ferrara, T.M.; Ben, S.; Brownson, K.M.; Holland, P.J.; Birlea, S.A.; Siebert, J.; Hartmann, A.; et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat. Genet. 2016, 48, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Yi, X.; Chen, J.; Kang, P.; Chen, X.; Chen, J.; Cui, T.; Chang, Y.; Ye, Z.; Ni, Q.; et al. Intracellular virus sensor MDA5 exacerbates vitiligo by inducing the secretion of chemokines in keratinocytes under virus invasion. Cell Death Dis. 2020, 11, 453. [Google Scholar] [CrossRef] [PubMed]
- Frisoli, M.L.; Essien, K.; Harris, J.E. Vitiligo: Mechanisms of Pathogenesis and Treatment. Annu. Rev. Immunol. 2020, 38, 621–648. [Google Scholar] [CrossRef]
- Levandowski, C.B.; Mailloux, C.M.; Ferrara, T.M.; Gowan, K.; Ben, S.; Jin, Y.; McFann, K.K.; Holland, P.J.; Fain, P.R.; Dinarello, C.A.; et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1β processing via the NLRP1 inflammasome. Proc. Natl. Acad. Sci. USA 2013, 110, 2952–2956. [Google Scholar] [CrossRef]
- Traks, T.; Keermann, M.; Karelson, M.; Rätsep, R.; Reimann, E.; Silm, H.; Vasar, E.; Kõks, S.; Kingo, K. Polymorphisms in Toll-like receptor genes are associated with vitiligo. Front. Genet. 2015, 6, 278. [Google Scholar] [CrossRef]
- Doss, R.W.; El-Rifaie, A.-A.A.; Abdel-Wahab, A.M.; Gohary, Y.M.; Rashed, L.A. Heat shock protein-70 expression in vitiligo and its relation to the disease activity. Indian J. Dermatol. 2016, 61, 408–412. [Google Scholar] [CrossRef]
- Henning, S.W.; Fernandez, M.F.; Mahon, J.P.; Duff, R.; Azarafrooz, F.; Guevara-Patiño, J.A.; Rademaker, A.W.; Salzman, A.L.; Le Poole, I.C. HSP70iQ435A-Encoding DNA Repigments Vitiligo Lesions in Sinclair Swine. J. Investig. Dermatol. 2018, 138, 2531–2539. [Google Scholar] [CrossRef]
- Cui, T.; Zhang, W.; Li, S.; Chen, X.; Chang, Y.; Yi, X.; Kang, P.; Yang, Y.; Chen, J.; Liu, L.; et al. Oxidative Stress–Induced HMGB1 Release from Melanocytes: A Paracrine Mechanism Underlying the Cutaneous Inflammation in Vitiligo. J. Investig. Dermatol. 2019, 139, 2174–2184.e4. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, E.; Seo, J.; Oh, S.H. Impact of high-mobility group box 1 on melanocytic survival and its involvement in the pathogenesis of vitiligo. Br. J. Dermatol. 2017, 176, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.; Jin, L.; Yi, X.; Dang, E.; Yang, Y.; Li, C.; Gao, T. Oxidative Stress–Induced Calreticulin Expression and Translocation: New Insights into the Destruction of Melanocytes. J. Investig. Dermatol. 2014, 134, 183–191. [Google Scholar] [CrossRef]
- Dwivedi, M.; Laddha, N.C.; Begum, R. Viral causes of Vitiligo: A New Perspective for Vitiligo Pathogenesis. Virol. Immunol. J. 2018, 2, 000181. [Google Scholar]
- Ganju, P.; Nagpal, S.; Mohammed, M.H.; Kumar, P.N.; Pandey, R.; Natarajan, V.T.; Mande, S.S.; Gokhale, R.S. Microbial community profiling shows dysbiosis in the lesional skin of Vitiligo subjects. Sci. Rep. 2016, 6, 18761. [Google Scholar] [CrossRef] [PubMed]
- Dellacecca, E.R.; Cosgrove, C.; Mukhatayev, Z.; Akhtar, S.; Engelhard, V.H.; Rademaker, A.W.; Knight, K.L.; Le Poole, I.C. Antibiotics Drive Microbial Imbalance and Vitiligo Development in Mice. J. Investig. Dermatol. 2020, 140, 676–687.e6. [Google Scholar] [CrossRef]
- Bzioueche, H.; Simonyté Sjödin, K.; West, C.E.; Khemis, A.; Rocchi, S.; Passeron, T.; Tulic, M.K. Analysis of Matched Skin and Gut Microbiome of Patients with Vitiligo Reveals Deep Skin Dysbiosis: Link with Mitochondrial and Immune Changes. J. Investig. Dermatol. 2021, 141, 2280–2290. [Google Scholar] [CrossRef]
- Conrad, C.; Gilliet, M. Type I IFNs at the Interface between Cutaneous Immunity and Epidermal Remodeling. J. Investig. Dermatol. 2012, 132, 1759–1762. [Google Scholar] [CrossRef]
- Jacquemin, C.; Rambert, J.; Guillet, S.; Thiolat, D.; Boukhedouni, N.; Doutre, M.; Darrigade, A.; Ezzedine, K.; Blanco, P.; Taieb, A.; et al. Heat shock protein 70 potentiates interferon alpha production by plasmacytoid dendritic cells: Relevance for cutaneous lupus and vitiligo pathogenesis. Br. J. Dermatol. 2017, 177, 1367–1375. [Google Scholar] [CrossRef]
- Singh, A.; Das, D.; Kurra, S.; Arava, S.; Gupta, S.; Sharma, A. Dendritic cells and their associated pro-inflammatory cytokines augment to the inflammatory milieu in vitiligo skin. Cytokine 2021, 148, 155598. [Google Scholar] [CrossRef]
- Srivastava, N.; Bishnoi, A.; Parsad, D.; Kumaran, M.S.; Vinay, K.; Gupta, S. Dendritic cells sub-sets are associated with inflammatory cytokine production in progressive vitiligo disease. Obs. Study 2021, 313, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, N.; Skroza, N.; Tolino, E.; Mambrin, A.; Anzalone, A.; Balduzzi, V.; Colapietra, D.; Marchesiello, A.; Michelini, S.; Proietti, I.; et al. IL-17 and its role in inflammatory, autoimmune, and oncological skin diseases: State of art. Int. J. Dermatol. 2020, 59, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Halder, R.M.; Walters, C.S.; Johnson, B.A.; Chakrabarti, S.G.; Kenney, J.A. Aberrations in T lymphocytes and natural killer cells in vitiligo: A flow cytometric study. J. Am. Acad. Dermatol. 1986, 14, 733–737. [Google Scholar] [CrossRef]
- Regazzetti, C.; Joly, F.; Marty, C.; Rivier, M.; Mehul, B.; Reiniche, P.; Mounier, C.; Rival, Y.; Piwnica, D.; Cavalié, M.; et al. Transcriptional Analysis of Vitiligo Skin Reveals the Alteration of WNT Pathway: A Promising Target for Repigmenting Vitiligo Patients. J. Investig. Dermatol. 2015, 135, 3105–3114. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Luo, Y.; Chang, C.; Wu, H.; Ding, Y.; Xiao, R. The Emerging Epigenetic Role of CD8+T Cells in Autoimmune Diseases: A Systematic Review. Front. Immunol. 2019, 10, 856. [Google Scholar] [CrossRef]
- Lili, Y.; Yi, W.; Ji, Y.; Yue, S.; Weimin, S.; Ming, L. Global Activation of CD8+ Cytotoxic T Lymphocytes Correlates with an Impairment in Regulatory T Cells in Patients with Generalized Vitiligo. PLoS ONE 2012, 7, e37513. [Google Scholar] [CrossRef]
- Yang, L.; Wei, Y.; Sun, Y.; Shi, W.; Yang, J.; Zhu, L.; Li, M. Interferon-gamma Inhibits Melanogenesis and Induces Apoptosis in Melanocytes: A Pivotal Role of CD8+ Cytotoxic T Lymphocytes in Vitiligo. Acta Derm. Venereol. 2015, 95, 664–670. [Google Scholar] [CrossRef]
- Wańkowicz-Kalińska, A.; van den Wijngaard, R.M.J.G.J.; Tigges, B.J.; Westerhof, W.; Ogg, G.S.; Cerundolo, V.; Storkus, W.; Das, P.K. Immunopolarization of CD4+ and CD8+ T Cells to Type-1–Like is Associated with Melanocyte Loss in Human Vitiligo. Lab. Investig. 2003, 83, 683–695. [Google Scholar] [CrossRef]
- Richmond, J.M.; Bangari, D.S.; Essien, K.I.; Currimbhoy, S.D.; Groom, J.R.; Pandya, A.G.; Youd, M.E.; Luster, A.D.; Harris, J.E. Keratinocyte-Derived Chemokines Orchestrate T-Cell Positioning in the Epidermis during Vitiligo and May Serve as Biomarkers of Disease. J. Investig. Dermatol. 2017, 137, 350–358. [Google Scholar] [CrossRef]
- Bergqvist, C.; Ezzedine, K. Vitiligo: A focus on pathogenesis and its therapeutic implications. J. Dermatol. 2021, 48, 252–270. [Google Scholar] [CrossRef] [PubMed]
- Benzekri, L.; Gauthier, Y. Clinical markers of vitiligo activity. J. Am. Acad. Dermatol. 2017, 76, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.; Powrie, F. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2001, 2, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Giri, P.S.; Dwivedi, M.; Begum, R. Decreased suppression of CD8 + and CD4 + T cells by peripheral regulatory T cells in generalized vitiligo due to reduced NFATC1 and FOXP3 proteins. Exp. Dermatol. 2020, 29, 759–775. [Google Scholar] [CrossRef]
- Giri, P.S.; Dwivedi, M.; Laddha, N.C.; Begum, R.; Bharti, A.H. Altered expression of nuclear factor of activated T cells, forkhead box P3, and immune-suppressive genes in regulatory T cells of generalized vitiligo patients. Pigment Cell Melanoma Res. 2020, 33, 566–578. [Google Scholar] [CrossRef]
- ben Ahmed, M.; Zaraa, I.; Rekik, R.; Elbeldi-Ferchiou, A.; Kourda, N.; Belhadj Hmida, N.; Abdeladhim, M.; Karoui, O.; ben Osman, A.; Mokni, M.; et al. Functional defects of peripheral regulatory T lymphocytes in patients with progressive vitiligo. Pigment Cell Melanoma Res. 2012, 25, 9–109. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, B.-X.; Shen, N.; Dong, X.-J.; Zhang, C.; Mao-Qiang, M.; Zhu, J.; Li, Y.-Z.; Man, M.-Q.; Tu, C.-X. Regulatory T cells from active non-segmental vitiligo exhibit lower suppressive ability on CD8+CLA+ T cells. Eur. J. Dermatol. 2014, 24, 676–682. [Google Scholar] [CrossRef]
- Klarquist, J.; Denman, C.J.; Hernandez, C.; Wainwright, D.; Strickland, F.M.; Overbeck, A.; Mehrotra, S.; Nishimura, M.I.; Le Poole, I.C. Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res. 2010, 23, 276–286. [Google Scholar] [CrossRef]
- le Poole, I.C.; Mehrotra, S. Replenishing Regulatory T Cells to Halt Depigmentation in Vitiligo. J. Investig. Dermatol. Symp. Proc. 2017, 18, S38–S45. [Google Scholar] [CrossRef]
- Eby, J.M.; Kang, H.-K.; Tully, S.T.; Bindeman, W.E.; Peiffer, D.S.; Chatterjee, S.; Mehrotra, S.; Le Poole, I.C. CCL22 to Activate Treg Migration and Suppress Depigmentation in Vitiligo. J. Investig. Dermatol. 2015, 135, 1574–1580. [Google Scholar] [CrossRef]
- Dwivedi, M.; Kemp, E.H.; Laddha, N.C.; Mansuri, M.S.; Weetman, A.P.; Begum, R. Regulatory T cells in vitiligo: Implications for pathogenesis and therapeutics. Autoimmun. Rev. 2015, 14, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A. Resident memory T cells in human health and disease. Sci. Transl. Med. 2015, 7, 269rv1. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Gebhardt, T.; Heath, W.; Carbone, F.R. Cutting Edge: Local Recall Responses by Memory T Cells Newly Recruited to Peripheral Nonlymphoid Tissues. J. Immunol. 2008, 181, 5837–5841. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.W.; Kupper, T.S. T cells and the skin: From protective immunity to inflammatory skin disorders. Nat. Rev. Immunol. 2019, 19, 490–502. [Google Scholar] [CrossRef]
- Cheuk, S.; Schlums, H.; Sérézal, I.G.; Martini, E.; Chiang, S.C.; Marquardt, N.; Gibbs, A.; Detlofsson, E.; Introini, A.; Forkel, M.; et al. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017, 46, 287–300. [Google Scholar] [CrossRef]
- Richmond, J.M.; Strassner, J.P.; Zapata, L., Jr.; Garg, M.; Riding, R.L.; Refat, M.A.; Fan, X.; Azzolino, V.; Tovar-Garza, A.; Tsurushita, N.; et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci. Transl. Med. 2018, 10, eaam7710. [Google Scholar] [CrossRef]
- Boniface, K.; Jacquemin, C.; Darrigade, A.-S.; Dessarthe, B.; Martins, C.; Boukhedouni, N.; Vernisse, C.; Grasseau, A.; Thiolat, D.; Rambert, J.; et al. Vitiligo Skin Is Imprinted with Resident Memory CD8 T Cells Expressing CXCR3. J. Investig. Dermatol. 2017, 138, 355–364. [Google Scholar] [CrossRef]
- Frączek, A.; Owczarczyk-Saczonek, A.; Placek, W. The Role of TRM Cells in the Pathogenesis of Vitiligo—A Review of the Current State-Of-The-Art. Int. J. Mol. Sci. 2020, 21, 3552. [Google Scholar] [CrossRef]
- Boniface, K.; Seneschal, J. Vitiligo as a skin memory disease: The need for early intervention with immunomodulating agents and a maintenance therapy to target resident memory T cells. Exp. Dermatol. 2019, 28, 656–661. [Google Scholar] [CrossRef]
- Jacquemin, C.; Martins, C.; Lucchese, F.; Thiolat, D.; Taieb, A.; Seneschal, J.; Boniface, K. NKG2D Defines a Subset of Skin Effector Memory CD8 T Cells with Proinflammatory Functions in Vitiligo. J. Investig. Dermatol. 2019, 140, 1143–1153.e5. [Google Scholar] [CrossRef]
- Martins, C.; Darrigade, A.; Jacquemin, C.; Barnetche, T.; Taieb, A.; Ezzedine, K.; Boniface, K.; Seneschal, J. Phenotype and function of circulating memory T cells in human vitiligo. Br. J. Dermatol. 2020, 183, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, S.; Hu, W.; Wang, M.; Abudoureyimu, D.; Luo, D.; Li, T.; Long, L.; Zeng, H.; Cheng, C.; et al. Comprehensive Analysis of Cell Population Dynamics and Related Core Genes During Vitiligo Development. Front. Genet. 2021, 12, 627092. [Google Scholar] [CrossRef] [PubMed]
- Kemp, E.H.; Gavalas, N.G.; Gawkrodger, D.J.; Weetman, A.P. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun. Rev. 2007, 6, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Raam, L.; Kaleviste, E.; Šunina, M.; Vaher, H.; Saare, M.; Prans, E.; Pihlap, M.; Abram, K.; Karelson, M.; Peterson, P.; et al. Lymphoid Stress Surveillance Response Contributes to Vitiligo Pathogenesis. Front. Immunol. 2018, 9, 2707. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, S.; Lei, J.; Hu, W.; Chen, R.; Lin, F.; Xu, A.-E. Role of chemokines and the corresponding receptors in vitiligo: A pilot study. J. Dermatol. 2018, 45, 31–38. [Google Scholar] [CrossRef]
- Toussirot, É.; Aubin, F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: An analytical and comprehensive overview. RMD Open 2016, 2, e000239. [Google Scholar] [CrossRef]
- Chen, X.; Guo, W.; Chang, Y.; Chen, J.; Kang, P.; Yi, X.; Cui, T.; Guo, S.; Xiao, Q.; Jian, Z.; et al. Oxidative stress-induced IL-15 trans-presentation in keratinocytes contributes to CD8+ T cells activation via JAK-STAT pathway in vitiligo. Free Radic. Biol. Med. 2019, 139, 80–91. [Google Scholar] [CrossRef]
- Singh, R.K.; Lee, K.M.; Vujkovic-Cvijin, I.; Ucmak, D.; Farahnik, B.; Abrouk, M.; Nakamura, M.; Zhu, T.H.; Bhutani, T.; Wei, M.; et al. The role of IL-17 in vitiligo: A review. Autoimmun. Rev. 2016, 15, 397–404. [Google Scholar] [CrossRef]
- Zhou, J.; An, X.; Dong, J.; Wang, Y.; Zhong, H.; Duan, L.; Ling, J.; Ping, F.; Shang, J. IL-17 induces cellular stress microenvironment of melanocytes to promote autophagic cell apoptosis in vitiligo. FASEB J. 2018, 32, 4899–4916. [Google Scholar] [CrossRef]
- Sushama, S.; Dixit, N.; Gautam, R.K.; Arora, P.; Khurana, A.; Anubhuti, A. Cytokine profile (IL-2, IL-6, IL-17, IL-22, and TNF-α) in vitiligo—New insight into pathogenesis of disease. J. Cosmet. Dermatol. 2019, 18, 337–341. [Google Scholar] [CrossRef]
- Speeckaert, R.; Mylle, S.; Van Geel, N. IL-17A is not a treatment target in progressive vitiligo. Pigment Cell Melanoma Res. 2019, 32, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Richmond, J.M.; Masterjohn, E.; Chu, R.; Tedstone, J.; Youd, M.E.; Harris, J.E. CXCR3 Depleting Antibodies Prevent and Reverse Vitiligo in Mice. J. Investig. Dermatol. 2017, 137, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Mobasher, P.; Guerra, R.; Li, S.J.; Frangos, J.; Ganesan, A.; Huang, V. Open-label pilot study of tofacitinib 2% for the treatment of refractory vitiligo. Br. J. Dermatol. 2020, 182, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Rosmarin, D.; Pandya, A.G.; Lebwohl, M.; Grimes, P.; Hamzavi, I.; Gottlieb, A.B.; Butler, K.; Kuo, F.; Sun, K.; Ji, T.; et al. Ruxolitinib cream for treatment of vitiligo: A randomised, controlled, phase 2 trial. Lancet 2020, 396, 110–120. [Google Scholar] [CrossRef]
- McKesey, J.; Pandya, A.G. A pilot study of 2% tofacitinib cream with narrowband ultraviolet B for the treatment of facial vitiligo. J. Am. Acad. Dermatol. 2019, 81, 646–648. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hlača, N.; Žagar, T.; Kaštelan, M.; Brajac, I.; Prpić-Massari, L. Current Concepts of Vitiligo Immunopathogenesis. Biomedicines 2022, 10, 1639. https://doi.org/10.3390/biomedicines10071639
Hlača N, Žagar T, Kaštelan M, Brajac I, Prpić-Massari L. Current Concepts of Vitiligo Immunopathogenesis. Biomedicines. 2022; 10(7):1639. https://doi.org/10.3390/biomedicines10071639
Chicago/Turabian StyleHlača, Nika, Tina Žagar, Marija Kaštelan, Ines Brajac, and Larisa Prpić-Massari. 2022. "Current Concepts of Vitiligo Immunopathogenesis" Biomedicines 10, no. 7: 1639. https://doi.org/10.3390/biomedicines10071639
APA StyleHlača, N., Žagar, T., Kaštelan, M., Brajac, I., & Prpić-Massari, L. (2022). Current Concepts of Vitiligo Immunopathogenesis. Biomedicines, 10(7), 1639. https://doi.org/10.3390/biomedicines10071639