
Solubility-Aware Protein Binding Peptide Design Using AlphaFold



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- A library of highly water-soluble peptide sequences is created, and then docking scores and binding affinities are predicted by protein–peptide docking.
- Design peptide sequences that are likely to bind to target proteins using peptide sequence prediction methods such as AfDesign, and then evaluate water solubility and filter out those that exceed water solubility thresholds.
2. Materials and Methods
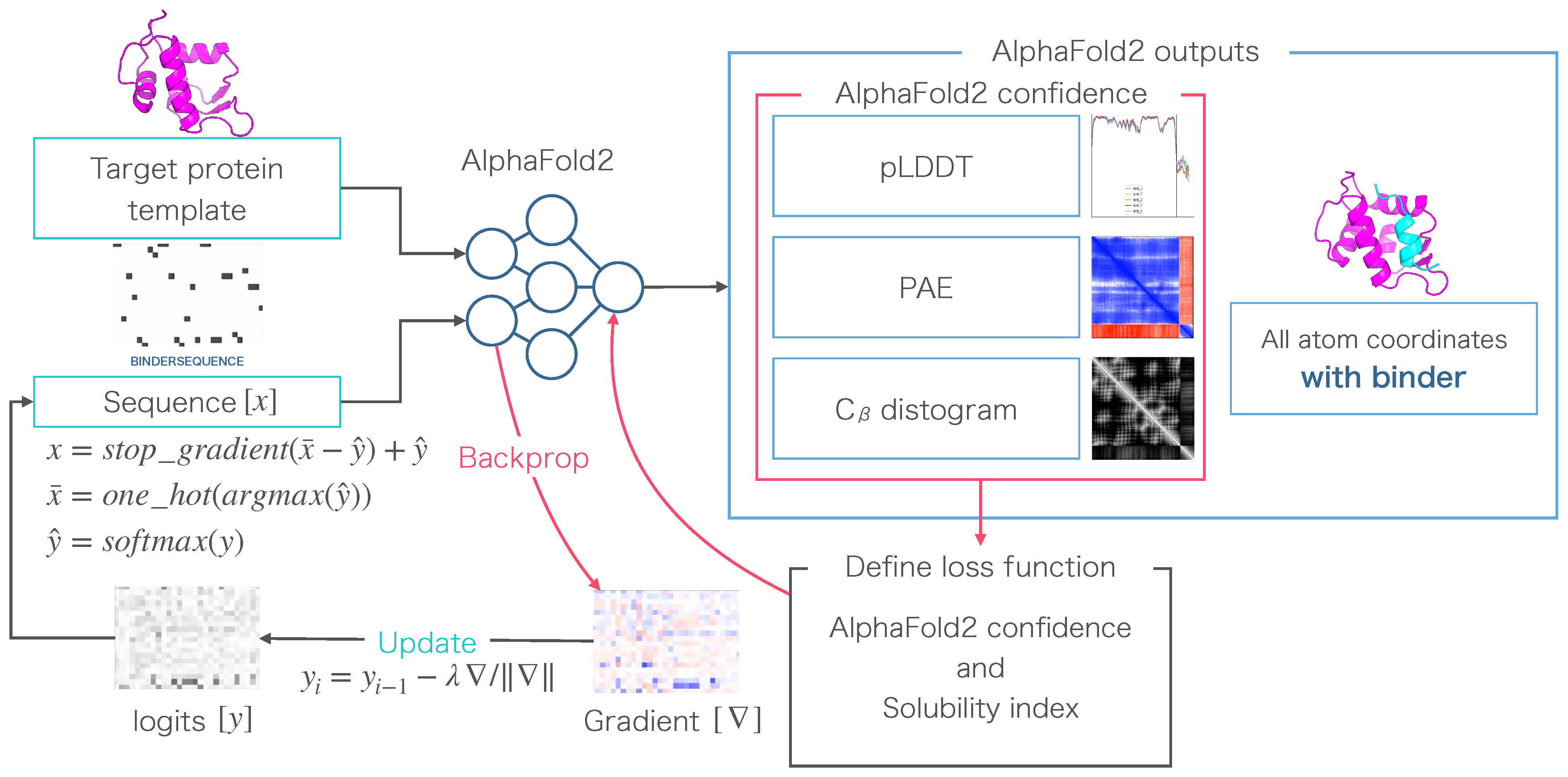
2.1. AfDesign Settings
2.2. Solubility Loss Calculation
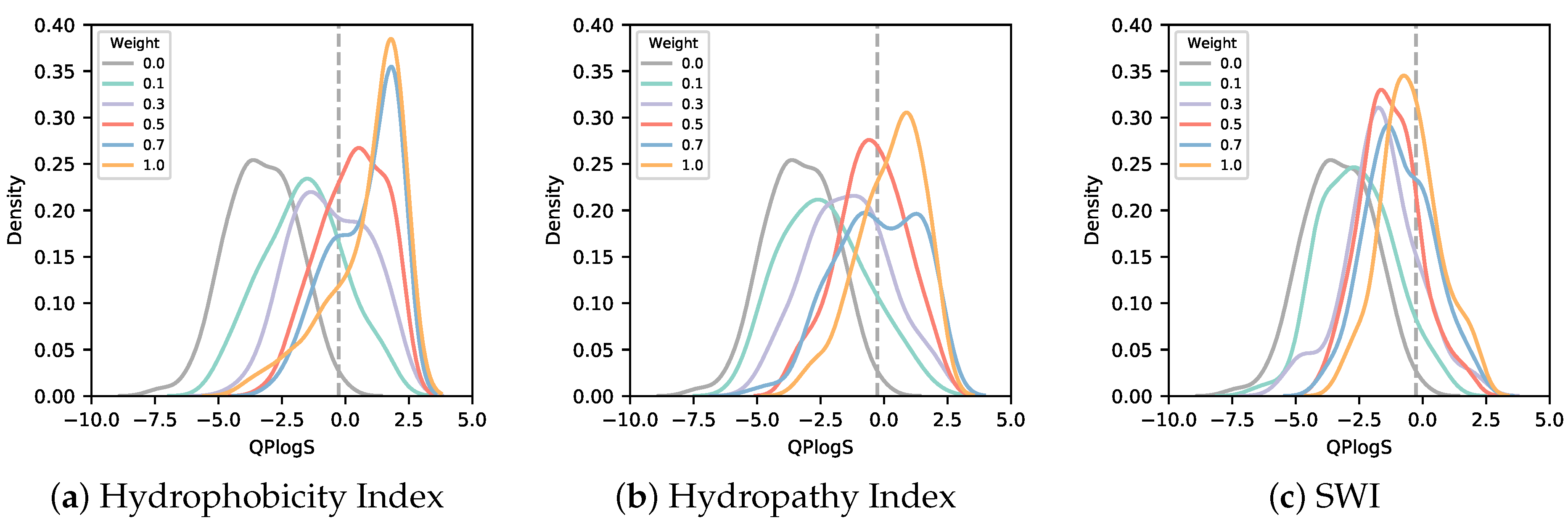
2.3. Calculation of Solubility
2.4. Calculation of Protein–Peptide Binding Affinity
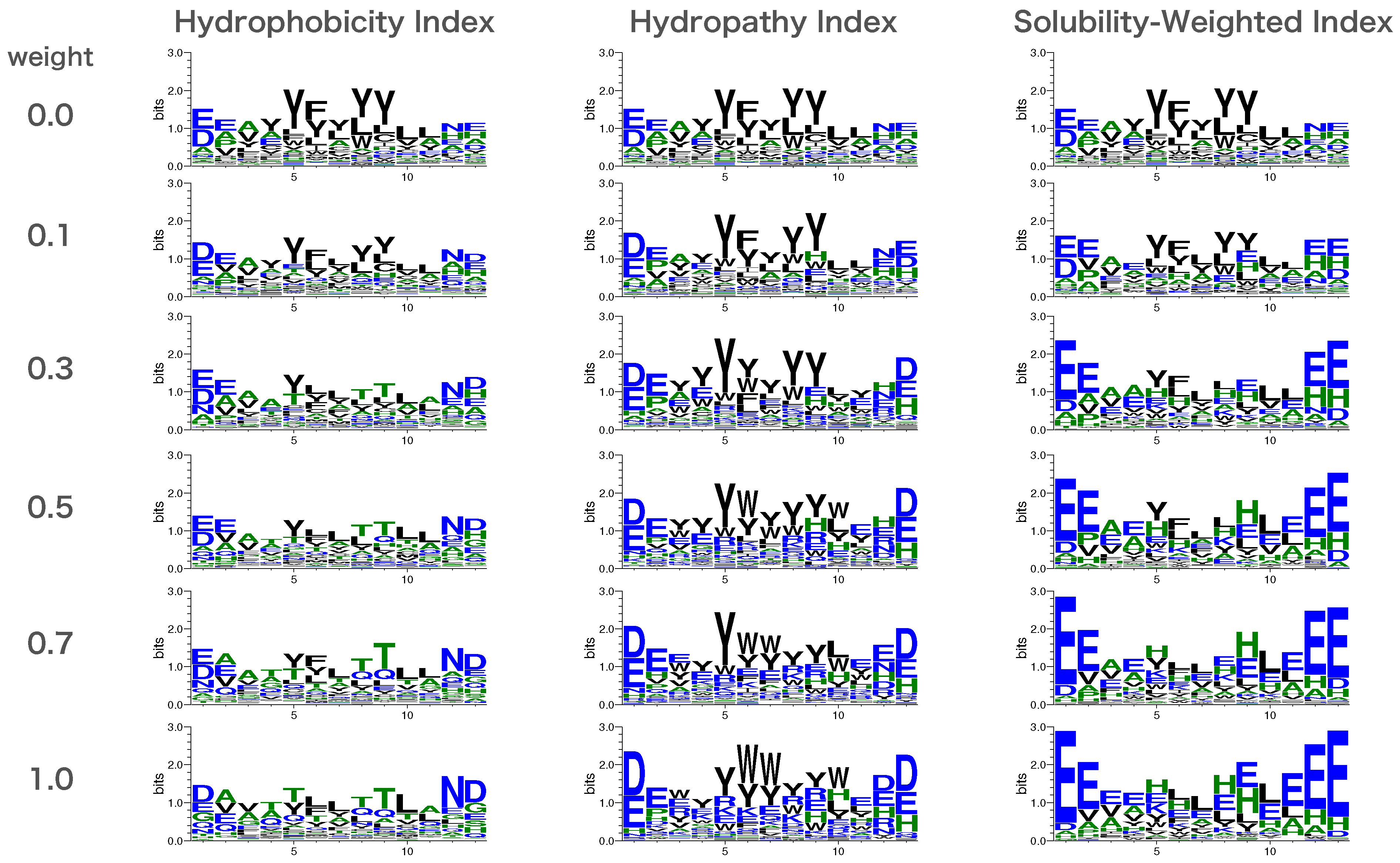
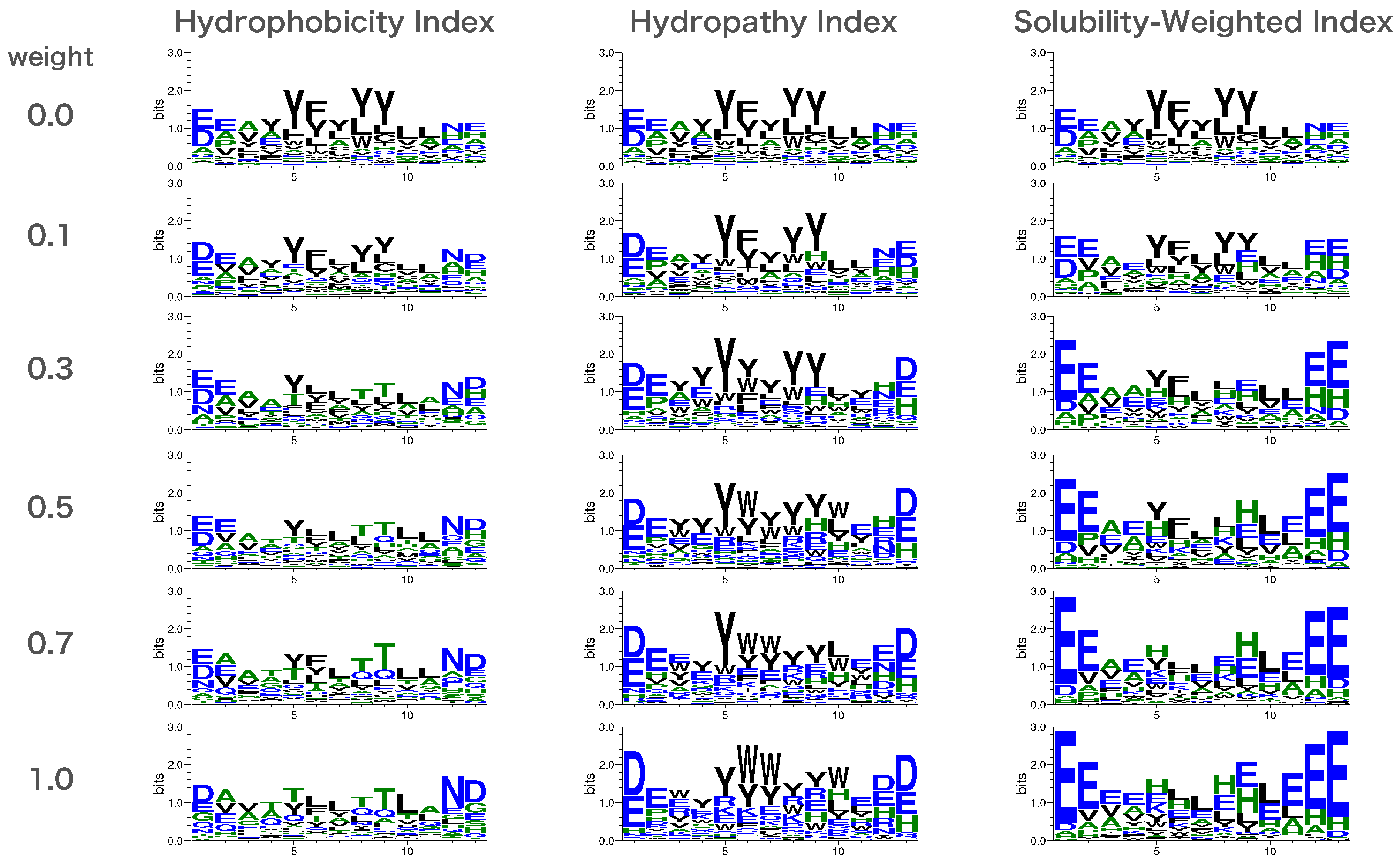
2.5. Creation of Sequence Logos
2.6. Competitive Peptide Binding Predictions Using AlphaFold
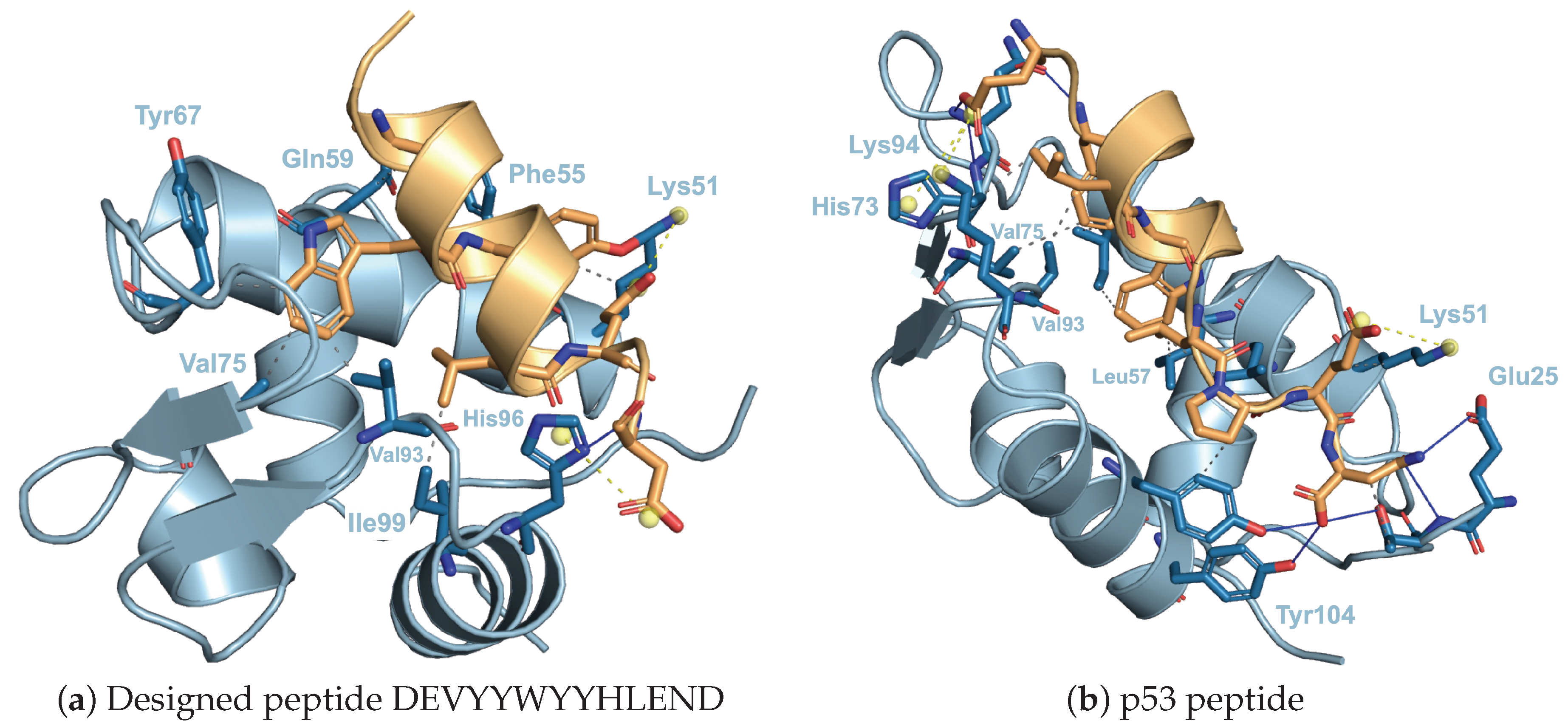
2.7. Interatomic Interactions of MDM2 and Peptide
3. Results
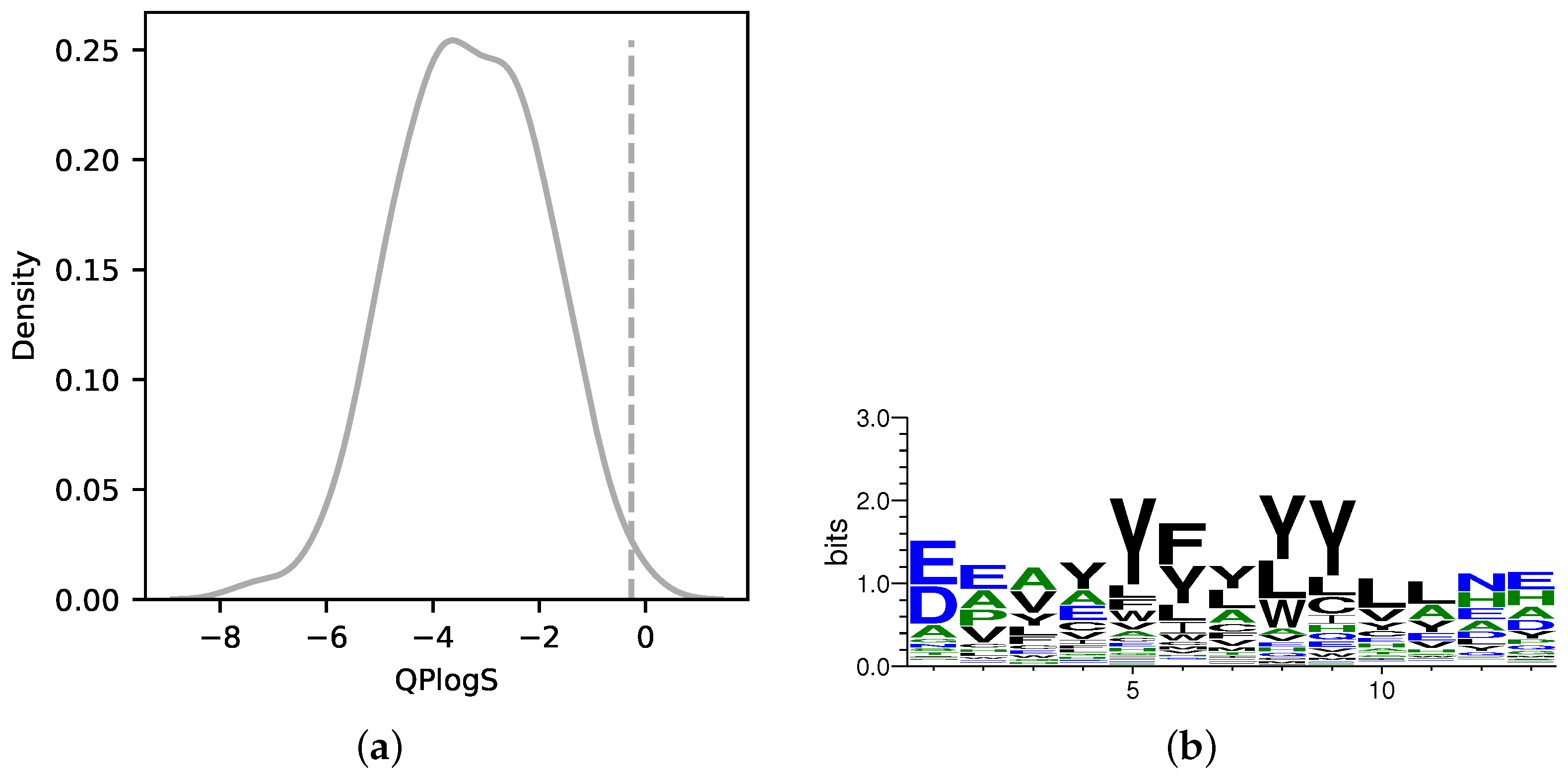
3.1. Binder Design Targeting PPI Using AfDesign
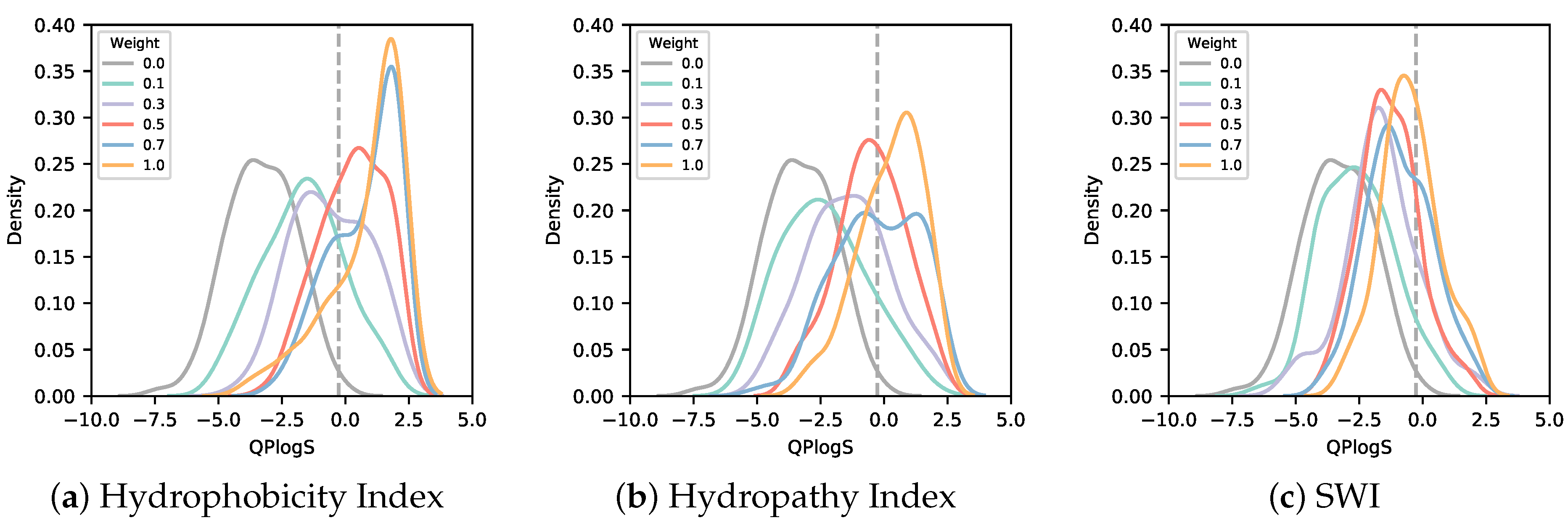
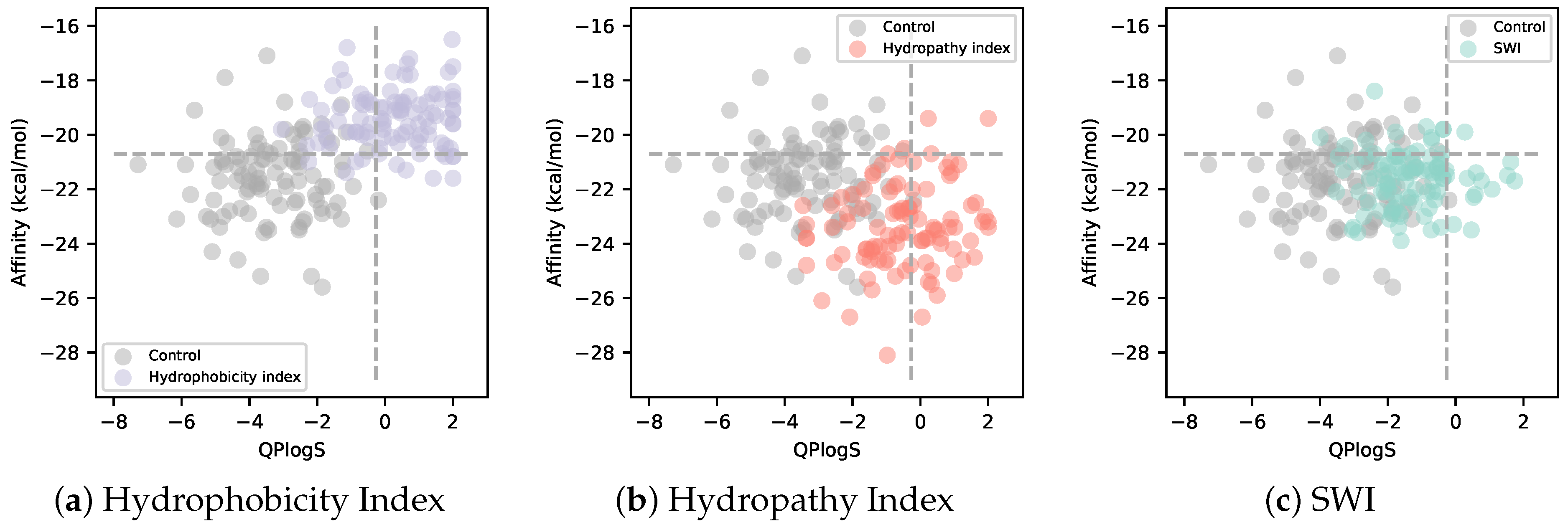
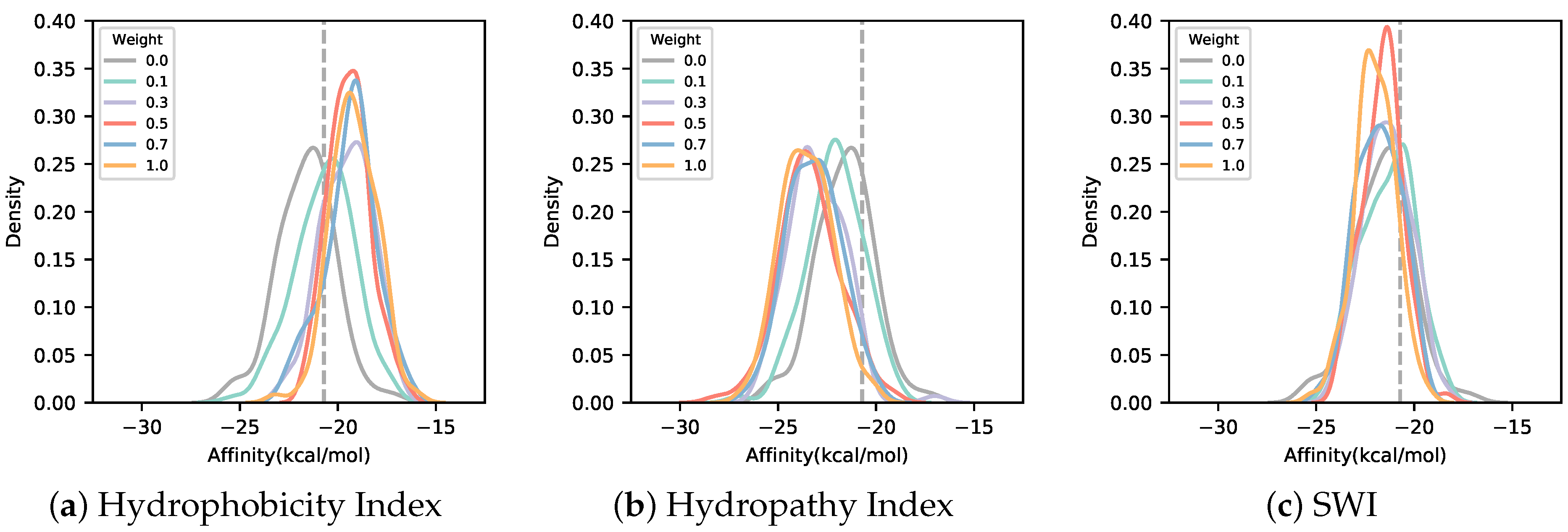
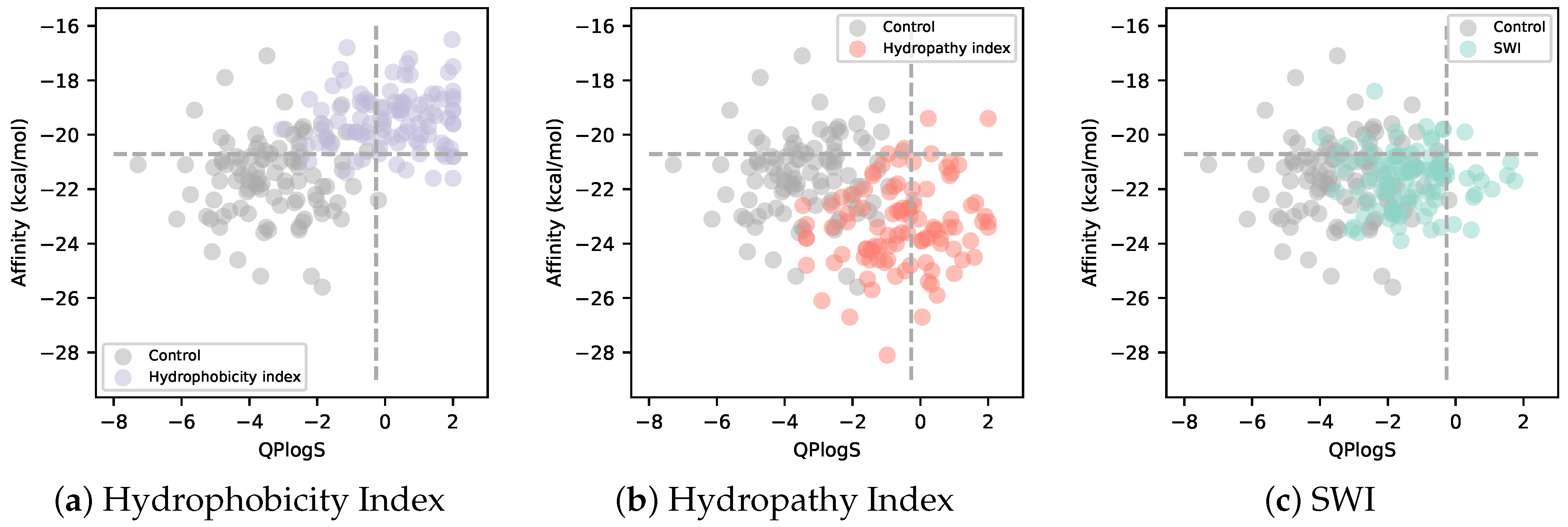
3.2. Solubility-Aware Binder Design Using AfDesign
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonetta, L. Interactome under Construction. Nature 2010, 468, 851–852. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.-J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Toogood, P.L. Inhibition of Protein-Protein Association by Small Molecules: Approaches and Progress. J. Med. Chem. 2002, 45, 1543–1558. [Google Scholar] [CrossRef]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein–protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef]
- Dev, K.K. Making protein interactions druggable: Targeting PDZ domains. Nat. Rev. Drug Discov. 2004, 3, 1047–1056. [Google Scholar] [CrossRef]
- Jin, L.; Wang, W.; Fang, G. Targeting Protein-Protein Interaction by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein–protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity While Sparing Platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Shin, W.-H.; Kumazawa, K.; Imai, K.; Hirokawa, T.; Kihara, D. Current challenges and opportunities in designing protein–protein interaction targeted drugs. Adv. Appl. Bioinform. Chem. 2020, 13, 11–25. [Google Scholar]
- Truong, J.; George, A.; Holien, J.K. Analysis of Physicochemical Properties of Protein–Protein Interaction Modulators Suggests Stronger Alignment with the “Rule of Five”. RSC Med. Chem. 2021, 12, 1731–1749. [Google Scholar] [CrossRef]
- Kosugi, T.; Ohue, M. Quantitative Estimate Index for Early-Stage Screening of Compounds Targeting Protein-Protein Interactions. Int. J. Mol. Sci. 2021, 22, 10925. [Google Scholar] [CrossRef]
- Wodak, S.J.; Janin, J. Structural Basis of Macromolecular Recognition. Adv. Protein Chem. 2002, 61, 9–73. [Google Scholar]
- Conte, L.L.; Chothia, C.; Janin, J. The Atomic Structure of Protein-Protein Recognition Sites11Edited by A. R. Fersht. J. Mol. Biol. 1999, 285, 2177–2198. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.C.; Gestwicki, J.E. Features of Protein–Protein Interactions That Translate into Potent Inhibitors: Topology, Surface Area and Affinity. Expert Rev. Mol. Med. 2012, 14, e16. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.A.; McClendon, C.L. Reaching for High-Hanging Fruit in Drug Discovery at Protein–Protein Interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Cabri, W.; Cantelmi, P.; Corbisiero, D.; Fantoni, T.; Ferrazzano, L.; Martelli, G.; Mattellone, A.; Tolomelli, A. Therapeutic Peptides Targeting PPI in Clinical Development: Overview, Mechanism of Action and Perspectives. Front. Mol. Biosci. 2021, 8, 565. [Google Scholar] [CrossRef]
- Wu, C.-H.; Liu, I.-J.; Lu, R.-M.; Wu, H.-C. Advancement and Applications of Peptide Phage Display Technology in Biomedical Science. J. Biomed. Sci. 2016, 23, 8. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, N.; Miyamoto-Sato, E.; Husimi, Y.; Yanagawa, H. In Vitro Virus: Bonding of MRNA Bearing Puromycin at the 3′-Terminal End to the C-Terminal End of Its Encoded Protein on the Ribosome in Vitro. FEBS Lett. 1997, 414, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.W.; Szostak, J.W. RNA-Peptide Fusions for the in Vitro Selection of Peptides And Proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar] [CrossRef] [Green Version]
- Das, P.; Sercu, T.; Wadhawan, K.; Padhi, I.; Gehrmann, S.; Cipcigan, F.; Chenthamarakshan, V.; Strobelt, H.; dos Santos, C.; Chen, P.-Y.; et al. Accelerated Antimicrobial Discovery via Deep Generative Models and Molecular Dynamics Simulations. Nat. Biomed. Eng. 2021, 5, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, A.; Zhang, A.; Reymond, J.-L. Populating Chemical Space with Peptides Using a Genetic Algorithm. J. Chem. Inf. Model. 2020, 60, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, A.; Silva, D.-A.; Rocklin, G.J.; Hicks, D.R.; Vergara, R.; Murapa, P.; Bernard, S.M.; Zhang, L.; Lam, K.-H.; Yao, G.; et al. Massively Parallel de Novo Protein Design for Targeted Therapeutics. Nature 2017, 550, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Strauch, E.-M.; Bernard, S.M.; La, D.; Bohn, A.J.; Lee, P.S.; Anderson, C.E.; Nieusma, T.; Holstein, C.A.; Garcia, N.K.; Hooper, K.A.; et al. Computational Design of Trimeric Influenza-Neutralizing Proteins Targeting the Hemagglutinin Receptor Binding Site. Nat. Biotechnol. 2017, 35, 667–671. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.-A.; Yu, S.; Ulge, U.Y.; Spangler, J.B.; Jude, K.M.; Labão-Almeida, C.; Ali, L.R.; Quijano-Rubio, A.; Ruterbusch, M.; Leung, I.; et al. De Novo Design of Potent and Selective Mimics of IL-2 and IL-15. Nature 2019, 565, 186–191. [Google Scholar] [CrossRef]
- Baran, D.; Pszolla, M.G.; Lapidoth, G.D.; Norn, C.; Dym, O.; Unger, T.; Albeck, S.; Tyka, M.D.; Fleishman, S.J. Principles for Computational Design of Binding Antibodies. Proc. Natl. Acad. Sci. USA 2017, 114, 10900–10905. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Gulsevin, A.; Meiler, J. Benchmarking Peptide Structure Prediction with AlphaFold2. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tsaban, T.; Varga, J.K.; Avraham, O.; Ben-Aharon, Z.; Khramushin, A.; Schueler-Furman, O. Harnessing Protein Folding Neural Networks for Peptide–Protein Docking. Nat. Commun. 2022, 13, 176. [Google Scholar] [CrossRef] [PubMed]
- Anishchenko, I.; Pellock, S.J.; Chidyausiku, T.M.; Ramelot, T.A.; Ovchinnikov, S.; Hao, J.; Bafna, K.; Norn, C.; Kang, A.; Bera, A.K.; et al. De Novo Protein Design by Deep Network Hallucination. Nature 2021, 600, 547–552. [Google Scholar] [CrossRef]
- Cao, L.; Coventry, B.; Goreshnik, I.; Huang, B.; Park, J.S.; Jude, K.M.; Marković, I.; Kadam, R.U.; Verschueren, K.H.G.; Verstraete, K.; et al. Design of Protein Binding Proteins from Target Structure Alone. Nature 2022, 605, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Jendrusch, M.; Korbel, J.O.; Sadiq, S.K. AlphaDesign: A de Novo Protein Design Framework Based on AlphaFold. bioRxiv 2021. [Google Scholar] [CrossRef]
- GitHub, AfDesign (v1.0.2); GitHub: San Francisco, CA, USA, 2022. Available online: https://github.com/sokrypton/ColabDesign/tree/main/af(accessed on 14 March 2022).
- Fosgerau, K.; Hoffmann, T. Peptide Therapeutics: Current Status and Future Directions. Drug Discov. 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, D.; Chatterjee, D.K. Enhancement of Soluble Protein Expression through the Use of Fusion Tags. Curr. Opin. Biotechnol. 2006, 17, 353–358. [Google Scholar] [CrossRef]
- Wu, S.-J.; Luo, J.; O’Neil, K.T.; Kang, J.; Lacy, E.R.; Canziani, G.; Baker, A.; Huang, M.; Tang, Q.M.; Raju, T.S.; et al. Structure-Based Engineering of a Monoclonal Antibody for Improved Solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Mant, C.T.; Kovacs, J.M.; Kim, H.-M.; Pollock, D.D.; Hodges, R.S. Intrinsic Amino Acid Side-Chain Hydrophilicity/Hydrophobicity Coefficients Determined by Reversed-Phase High-Performance Liquid Chromatography of Model Peptides: Comparison with Other Hydrophilicity/Hydrophobicity Scales. Pept. Sci. 2009, 92, 573–595. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, B.K.; Gardner, P.P.; Lim, C.S. Solubility-Weighted Index: Fast and Accurate Prediction of Protein Solubility. Bioinformatics 2020, 36, 4691–4698. [Google Scholar] [CrossRef]
- Argos, P.; Rao, J.K.M.; Hargrave, P.A. Structural Prediction of Membrane-Bound Proteins. Eur. J. Biochem. 1982, 128, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sanner, M.F. AutoDock CrankPep: Combining Folding and Docking to Predict Protein–Peptide Complexes. Bioinformatics 2019, 35, 5121–5127. [Google Scholar] [CrossRef] [PubMed]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and Glutamine: Using Hydrogen Atom Contacts in the Choice of Side-Chain Amide Orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Perez, A. AlphaFold Encodes the Principles to Identify High Affinity Peptide Binders. bioRxiv 2022. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the Scope of the Protein-Ligand Interaction Profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Norn, C.; Wicky, B.I.M.; Juergens, D.; Liu, S.; Kim, D.; Tischer, D.; Koepnick, B.; Anishchenko, I.; Foldit Players; Baker, D.; et al. Protein Sequence Design by Conformational Landscape Optimization. Proc. Natl. Acad. Sci. USA 2021, 118, e2017228118. [Google Scholar] [CrossRef]
- Weng, G.; Gao, J.; Wang, Z.; Wang, E.; Hu, X.; Yao, X.; Cao, D.; Hou, T. Comprehensive Evaluation of Fourteen Docking Programs on Protein–Peptide Complexes. J. Chem. Theory Comput. 2020, 16, 3959–3969. [Google Scholar] [CrossRef]
- Baek, M. Twitter Post: Adding a Big Enough Number for Residue_Index Feature Is Enough to Model Hetero-Complex Using AlphaFold (green&cyan: Crystal Structure/Magenta: Predicted Model w/residue_index Modification). Available online: https://twitter.com/minkbaek/status/1417538291709071362 (accessed on 20 July 2021).
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.-H.; Christoffer, C.W.; Kihara, D. In Silico Structure-Based Approaches to Discover Protein-Protein Interaction-Targeting Drugs. Methods 2017, 131, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.; Li, Z.; Kasprzak, A.; Li, B.; Sebti, S.; Guida, W.; Schönbrunn, E.; Chen, J. Structure-Based Design of High Affinity Peptides Inhibiting the Interaction of P53 with MDM2 and MDMX. J. Biol. Chem. 2010, 285, 2174–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosugi, T.; Ohue, M. Solubility-Aware Protein Binding Peptide Design Using AlphaFold. Biomedicines 2022, 10, 1626. https://doi.org/10.3390/biomedicines10071626
Kosugi T, Ohue M. Solubility-Aware Protein Binding Peptide Design Using AlphaFold. Biomedicines. 2022; 10(7):1626. https://doi.org/10.3390/biomedicines10071626
Chicago/Turabian StyleKosugi, Takatsugu, and Masahito Ohue. 2022. "Solubility-Aware Protein Binding Peptide Design Using AlphaFold" Biomedicines 10, no. 7: 1626. https://doi.org/10.3390/biomedicines10071626
APA StyleKosugi, T., & Ohue, M. (2022). Solubility-Aware Protein Binding Peptide Design Using AlphaFold. Biomedicines, 10(7), 1626. https://doi.org/10.3390/biomedicines10071626