Abstract
Ischemia-reperfusion (I/R) injury is well-known to be associated with impaired cardiac function, massive arrhythmias, marked alterations in cardiac metabolism and irreversible ultrastructural changes in the heart. Two major mechanisms namely oxidative stress and intracellular Ca2+-overload are considered to explain I/R-induced injury to the heart. However, it is becoming apparent that oxidative stress is the most critical pathogenic factor because it produces myocardial abnormalities directly or indirectly for the occurrence of cardiac damage. Furthermore, I/R injury has been shown to generate oxidative stress by promoting the formation of different reactive oxygen species due to defects in mitochondrial function and depressions in both endogenous antioxidant levels as well as regulatory antioxidative defense systems. It has also been demonstrated to adversely affect a wide variety of metabolic pathways and targets in cardiomyocytes, various resident structures in myocardial interstitium, as well as circulating neutrophils and leukocytes. These I/R-induced alterations in addition to myocardial inflammation may cause cell death, fibrosis, inflammation, Ca2+-handling abnormalities, activation of proteases and phospholipases, as well as subcellular remodeling and depletion of energy stores in the heart. Analysis of results from isolated hearts perfused with or without some antioxidant treatments before subjecting to I/R injury has indicated that cardiac dysfunction is associated with the development of oxidative stress, intracellular Ca2+-overload and protease activation. In addition, changes in the sarcolemma and sarcoplasmic reticulum Ca2+-handling, mitochondrial oxidative phosphorylation as well as myofibrillar Ca2+-ATPase activities in I/R hearts were attenuated by pretreatment with antioxidants. The I/R-induced alterations in cardiac function were simulated upon perfusing the hearts with oxyradical generating system or oxidant. These observations support the view that oxidative stress may be intimately involved in inducing intracellular Ca2+-overload, protease activation, subcellular remodeling, and cardiac dysfunction as a consequence of I/R injury to the heart.
1. Introduction
Although reperfusion of the ischemic myocardium is beneficial for the improvement of cardiac function, delayed reperfusion is known to cause impaired recovery of contractile activity, induce cardiac arrhythmias, enhance metabolic defects, and produce structural damage to cardiomyocytes in the heart [1,2,3,4,5,6,7]. These abnormalities due to reperfusion of the ischemic heart are termed as ischemia-reperfusion (I/R) injury, which is commonly associated with clinical procedures such as angioplasty, thrombolysis, coronary bypass surgery, and cardiac transplantation. Extensive research over the past four decades has shown that two major mechanisms, namely the development of oxidative stress and the occurrence of intracellular Ca2+-overload, as well as myocardial inflammation and alterations in cardiac metabolism, are considered to explain I/R-induced injury to the heart [8,9,10,11,12,13,14,15]. It should also be mentioned that I/R injury not only affects cardiomyocytes and subcellular organelles but is also known to produce dramatic changes in non-cardiomyocyte structures such as vascular smooth muscle, microvasculature, endothelium, fibroblasts, macrophages, mast cells, adrenergic nerve endings, and endogenous renin-angiotensin system, which are present in the myocardial interstitium [5,9,16,17,18]. Furthermore, I/R-induced injury under in vivo conditions is also associated with the activation of neutrophils, leukocytes, platelets, as well as some systemic and central neuro-endocrine systems [7,10,15,19,20,21]. Thus, it can be appreciated that the pathogenesis of I/R-induced injury is of complex nature.
Since oxidative stress, intracellular Ca2+-overload, myocardial inflammation and metabolic defects are inter-related mechanisms and affect each other, it is difficult to identify any one of these to be responsible for the induction of I/R-induced injury. However, the involvement of oxidative stress in inducing a wide variety of cardiac abnormalities directly or indirectly during the development of I/R injury seems most prominent [1,5,10,22,23,24]. This view is based on the fact that oxidative stress has been demonstrated to cause Ca2+-handling abnormalities, apoptosis, necrosis, fibrosis, autophagy, lipid peroxidation, protein oxidation, irreversible cardiomyocyte damage and arrhythmias [5,21,25,26]. In addition, oxidative stress has been shown to produce activation of different proteases, dramatic changes in cardiac gene expression as well as defects in subcellular organelles such as mitochondria, myofibrils, sarcolemma (SL), and sarcoplasmic reticulum (SR) for inducing cardiac dysfunction due to I/R injury in the heart [5,21,27,28,29]. It is also noteworthy that several antioxidants have been reported to exert beneficial effects in attenuating I/R-induced alterations in cardiac function and other myocardial abnormalities [2,30,31,32]. Some of these I/R-induced changes involving oxidative stress, intracellular Ca2+-overload, myocardial inflammation, and cardiac metabolism are depicted schematically in Figure 1. Furthermore, in the present article, we have attempted to summarize the current knowledge regarding the pathophysiology, cardioprotection, and pharmacotherapy of I/R-induced injury to the heart with respect to highlighting its functional significance. Special emphasis has been laid regarding the generation of oxidative stress as well as its implications for inducing molecular and cellular abnormalities during the development of I/R injury to the heart. Particularly, we have outlined the available evidence to show that I/R-induced alterations in the activities of subcellular organelles are not only attenuated by antioxidants but these changes are also simulated upon exposure of the heart to oxidative stress-generating systems.
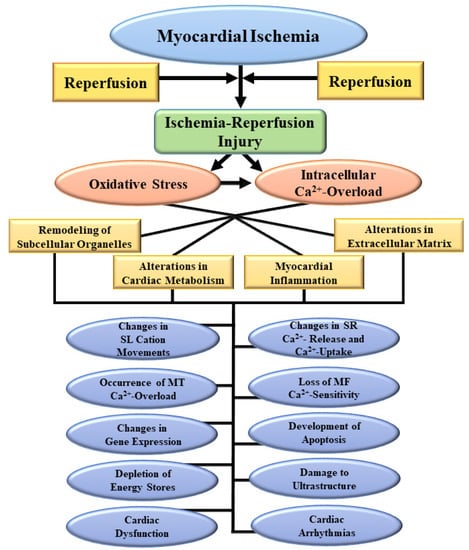
Figure 1.
Some myocardial and subcellular abnormalities due to the development of oxidative stress and intracellular Ca2+-overload as a consequence of ischemia-reperfusion injury. SL, sarcolemma; SR, sarcoplasmic reticulum; MT, mitochondria; MF, myofibrils.
2. I/R-Induced Generation of Oxidative Stress and Its Implication in Heart Disease
Since the status of oxidative stress in the heart is determined by a balance between the formation of reactive oxygen species (ROS) as well as numerous endogenous oxidants and the presence of various antioxidant systems [2,20,21,33,34], it is important to briefly discuss several components of oxidative stress before indicating its involvement in inducing I/R-linked abnormalities. It is pointed out that ROS mainly include superoxide radicals and hydroxyl radicals as well as oxidants such as hydrogen peroxide (H2O2) and hypochlorous acid (HOCl); their concentrations are markedly increased upon induction of I/R injury. On the other hand, the activities of various endogenous enzymes such as superoxide dismutase, catalase, and glutathione peroxidase, which serve as antioxidant defense mechanisms are depressed in I/R-perfused hearts [35,36]. Furthermore, nuclear factor erythroid-2 related factor 2 (Nrf2) and various microRNAs, which regulate different antioxidant systems, were decreased due to I/R injury [21,37]. In addition, several antioxidants such as ascorbic acid, glutathione, ubiquinol 9, and vitamin E were decreased upon subjecting the heart to conditions of oxidative stress [2,38]. These observations are consistent with the view that I/R injury to the heart is associated with the development of oxidative stress both as a consequence of increased formation of ROS as well as depressions in the level of antioxidant defense systems.
It is noteworthy that mitochondria are the major source of ROS because of the impaired electron transport chain and depressed oxidative phosphorylation activity in I/R hearts [11,39,40]. Although several factors are considered to be responsible for I/R-induced production of ROS in mitochondria, uncoupling of mitochondrial proteins as well as metabolic overloading due to increased fatty acid flux and accumulation of succinate have been shown to be involved in proton leakage from mitochondria [41,42]. Furthermore, increased formation of nitric oxide due to elevated levels of nitric oxide synthase in the endothelium has been reported to result in the production of peroxynitrite and subsequent nitrosative stress at initial stages of I/R injury [43]. It is pointed out that ROS production is also increased due to the activation of several cellular and neuronal systems such as various leukocytes (e.g., neutrophils), the sympathetic nervous system, and the renin-angiotensin system during the development of I/R-induced injury to the heart [20,21]. In this regard, myeloperoxidase released from leukocytes has been reported to play an important role through the formation of microbicidal reactive oxidants [44] whereas the activation of monoamine oxidase has been demonstrated to generate H2O2 upon oxidative deamination of catecholamines released from the sympathetic nervous system during the development of I/R-induced injury [45,46]. In addition, angiotensin II formed by the activation of renin-angiotensin system due to I/R injury has been shown to promote the generation of ROS as a consequence of the activation of NADPH oxidase (NOX), present in the plasma membrane (NOX2) and intracellular organelles (NOX4) [21,47]. Thus, a wide variety of enzymes as well as cellular and neuronal systems are involved during the development of oxidative stress.
Generation of oxidative stress and nitrosative stress due to I/R injury are not only known to cause cardiac dysfunction, these pathological factors have also been shown to be involved in the formation of pro-inflammatory agents as well as disruption of different signal transduction pathways [20,21,48,49]. Particularly, these pathogenic factors are known to induce myocardial cell damage, apoptosis, necrosis, and fibrosis as well as different defects in cardiac gene expression and myocardial metabolism in hearts subjected to I/R injury. There also occurs the activation and infiltration of polymorphonuclear leukocytes which will promote the development of myocardial infarct and disorganization of several adhesion molecules. Oxidative stress for a prolonged period also induces defects in the endothelial function for the formation of nitric oxide (a vasodilator molecule) and subsequently reduce or block the blood flow to the myocardium. Prolonged oxidative stress in the heart will produce Ca2+-handling abnormalities in cardiomyocytes and defects in the SL, SR, mitochondria, and myofibrils mainly as a consequence of the activation of proteases and phospholipases [5,12,20,21,26,50]. A schematic representation of some of the events involved in the generation of oxidative stress in I/R heart and its implications in remodeling of subcellular organelles and cardiac dysfunction are depicted in Figure 2. Depression in SL Na+-K+ ATPase will produce marked changes in the concentration of Na+ and K+ in cardiomyocytes which may explain the development of cardiac arrhythmias associated with I/R injury. Furthermore, alterations in SL Na+-Ca2+ exchange, Ca2+-channels and Ca2+-gating mechanisms as well as SR Ca2+-release and Ca2+-pump ATPase due to oxidative stress may account for Ca2+-handling abnormalities in cardiomyocytes, and changes in contraction and relaxation processes in I/R hearts. Oxidative stress is also considered to promote the occurrence of mitochondrial Ca2+-overload and induce defects in the process of ATP production and release different cytotoxic agents including cytochrome C for the development of I/R-induced injury to the heart. Some events associated with I/R-induced development and consequence of intracellular Ca2+-transport systems indirectly through oxidative stress are shown in Figure 3. In addition to inducing intracellular Ca2+-overload, oxidative stress has been demonstrated to produce varying degrees of defects in myofibrillar proteins for the loss of Ca2+-sensitivity in myofibrils and subsequent depression in cardiac dysfunction due to I/R injury. Thus, oxidative stress can be viewed as the most prominent pathophysiologic factor for the induction of I/R injury to the heart.
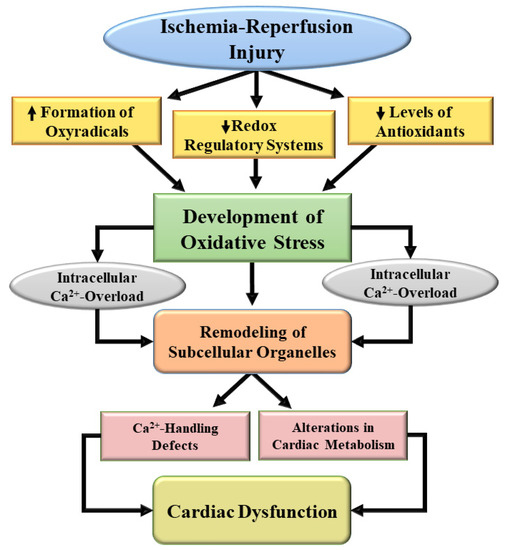
Figure 2.
A schematic representation of mechanisms for the development of oxidative stress and subsequent subcellular defects leading to cardiac dysfunction due to ischemia-reperfusion. ↑, increase; ↓, decrease.
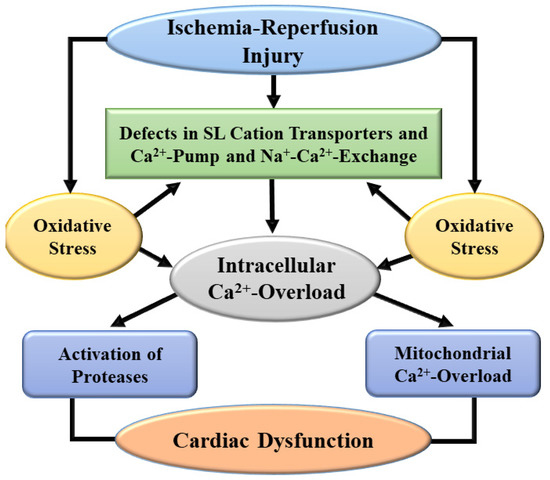
Figure 3.
A schematic representation of mechanisms for the development of intracellular Ca2+-overload and subsequent subcellular defects leading to cardiac dysfunction due to ischemia-reperfusion injury. SL, sarcolemma.
3. Pathophysiological Aspects of I/R-Induced Injury
The pathophysiology of I/R injury to the heart is of complex nature because it involves the effects of reperfusion which are superimposed upon those of myocardial ischemia [30,51,52,53,54,55,56]. The ischemic insult by occluding the coronary arteries is associated with the lack of oxygen/hypoxia, inability of mitochondria to oxidize substrate, depression of oxidative phosphorylation, and accumulation of hydrogen in cardiomyocytes. The initial events due to myocardial ischemia result in the stimulation of SL Na+-H+ exchange and SL Na+-Ca2+ exchange systems as well as elevation in the intracellular concentration of Ca2+, depression in energy production, occurrence of some ultrastructural damage, and cessation of contractile activity. These varying degrees of alterations in the heart are dependent upon the duration of myocardial ischemia at early stages. However, if the reperfusion is carried out after a certain period of myocardial ischemia, the changes in cardiac metabolism, Ca2+-handling ultrastructure, and contractile function become irreversible and are commonly called as lethal reperfusion injury of the heart. It should be emphasized that I/R injury is not only limited to inducing marked abnormalities in cardiomyocytes but other structures and cells in myocardial interstitium including endothelium and smooth muscle cells of coronary vessels are adversely affected. In addition, I/R injury to the heart is known to produce dramatic changes in macrophages, leukocytes, and platelets, as well as the sympathetic nervous system and the renin-angiotensin system for promoting inflammation and oxidative stress. Thus, I/R injury is considered to result in cellular death (formation of infarct) of the ischemic myocardium by a wide variety of pathological mechanisms.
In addition to oxidative stress and intracellular Ca2+-overload, myocardial I/R injury has been shown to release various inflammatory cytokines such as tumor-necrosis factor α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) [57,58,59,60,61]. All these mechanisms are interlinked and are considered to be closely involved in causing I/R-induced myocardial cell damage, apoptosis, necrosis, and fibrosis as well as calcium overload, cardiac arrhythmias, and heart dysfunction. Different regulatory noncoding RNAs including long noncoding RNAs (lncRNA) and microRNAs (miRNAs) have also been reported to play a critical role in the initiation and progression of I/R-induced injury through the expression of target genes for oxidative stress and inflammation [62,63,64]. Thioredoxin-interacting protein has been demonstrated not only to sensitize cardiomyocytes to oxidative stress-induced apoptosis but has also been implicated in the regulation of inflammatory response and glucose homeostasis during the development of I/R injury to the heart [65]. Recently, different arachidonic acid metabolic pathways such as cyclooxygenase pathway, lipoxygenase pathway, and cytochrome P450 monooxygenase pathway have been suggested to be involved in the development of I/R injury [66].
It is noteworthy that I/R injury to the heart not only produces abnormalities in cardiomyocytes for the induction of contractile dysfunction but has also been shown to cause defect in the endothelium for inducing no-flow phenomenon in coronary circulation [67,68,69]. Such a defect in endothelial dysfunction is elicited by oxidative stress and inflammation upon infiltration of leukocytes as well as activation of fibroblasts due to I/R injury [68,69]. Furthermore, upregulation of platelet surface receptors and release of immunomodulatory mediators have been shown to be involved in the modification of endothelial function during the development of myocardial I/R injury [70]. It should be noted that changes in mitochondrial function due to I/R-induced injury not only participate in generating oxidative stress but these organelles are also adversely affected by I/R injury to the heart [71,72]. Particularly there occurs mitochondrial Ca2+-overload, which will further depress the energy-producing ability and impair the recovery of cardiac function as a consequence of I/R injury. A combination of both Ca2+-overload and oxidative stress is considered to open mitochondrial permeability transition pores involving the participation of both PKC-δ and PKC-ε, and thus releases different proteins for the activation of apoptosis due to I/R injury [73,74]. In addition, I/R-induced injury to the heart has been shown to produce endoplasmic reticulum stress leading to the accumulation of unfolded proteins, and cause Ca2+-handling abnormalities due to a marked release of Ca2+ as a consequence of SR ryanodine receptor oxidation [75,76]. However, the individual contribution of oxidative stress, inflammation and intracellular Ca2+-overload in the genesis of these different myocardial alterations during the development of I/R injury remains to be investigated.
4. Cardioprotection in Hearts Subjected to I/R Injury
In order to attenuate the adverse effects of oxidative/nitrosative stress, inflammation and intracellular Ca2+-overload in I/R hearts, different redox-based strategies involving endogenous components have been attempted to prevent I/R-induced myocardial cell damage and cardiac dysfunction [77,78,79]. Particularly, interventions such as ischemic preconditioning, ischemic postconditioning, and remote conditioning have been demonstrated to exert cardioprotective actions in improving cardiac performance as well as limiting infarct size and preventing adverse cardiac remodeling due to I/R injury [80,81,82]. It is pointed out that there are several other conditions such as hibernation of myocardium and early stages of myocardial ischemia but their cardioprotective effects against I/R injury have not been examined in details. Although the exact mechanisms for the beneficial effects of these cardioprotective interventions are not fully understood, reductions in the formation of ROS or reactive nitrogen species, levels of lipid peroxidation products and oxidized DNA/RNA bases content as well as activators of redox-based signaling and mitochondrial modulators have been implicated. Modulation of endogenous reducing mechanisms such as thioredoxin and glutathione systems, which are known to scavenge oxyradicals and reduce oxidized proteins through thiol disulfide exchange reactions, is also considered to be involved in cardioprotection [83]. Heme oxygenase-1 protein, which degrades the oxidant heme and generates the antioxidant bilirubin and anti-inflammatory carbon monoxide, has been reported to participate in the intrinsic defense mechanisms for protecting I/R injury [84]. Furthermore, peroxisome proliferator-activated receptor γ (PPARγ), which regulates the gene expression of enzymes involved in glucose and lipid metabolism, was observed as an excellent target for cardioprotection against I/R injury because of its ability to attenuate oxidative stress and inhibit inflammatory response [85]. It is also noteworthy that different gaseous molecules such as nitric oxide, hydrogen sulfide, and hydrogen have been shown to prevent I/R injury to the heart because of their antioxidative, anti-inflammatory, antiproteolytic, and antiapoptotic activities [86,87,88].
Several experimental studies have indicated that cardioprotection by ischemic preconditioning is associated with increases in some enzymes as well as translational and transcriptional factors, which are involved in the regulation of innate detoxifying and antioxidant systems in I/R hearts [89,90,91,92,93]. In this regard, it is pointed out that the elevated level of O-linked β-N-acetylglucosamine (O-GlcNAc), which modifies numerous biological processes post-translationally, has been shown to be involved in reduction of intracellular Ca2+-overload, attenuation of mitochondrial permeability transition pore opening, suppression of endoplasmic reticulum stress, and modification of inflammatory response [89]. The ischemic preconditioning-induced activation of hypoxia-inducible factor-1α (HIF-1α), an oxygen sensitive transcription factor, has been reported to improve mitochondrial function, decrease oxidative stress, interact with non-coding RNAs, and activate cardioprotective signaling pathway as well as downstream protective genes [90]. The activation of mitochondrial aldehyde dehydrogenase 2, which detoxify reactive aldehydes, has also been shown to play a central role in cardioprotection because it inhibits opening of the mitochondrial permeability transition pores, attenuates autophagy, and prevents I/R-induced arrhythmias [91,92]. Furthermore, increased levels of redox-sensitive microRNAs, which regulate some components of the cellular antioxidants, interact with proteasomes and modify DNA repair system, have been implicated in cardioprotection of I/R hearts [93]. The activation of Nrf2, a transcription factor that controls the expression of various antioxidant genes, has also been demonstrated to play a pivotal role in enhancing endogenous antioxidant defenses in hearts subjected to I/R injury or myocardial infarction [94,95,96]. It should be mentioned that the induction of stem cells in I/R hearts, where these can differentiate into target tissues and produce trophic paracrine signaling to suppress injury, has been claimed to be of potential therapeutic value [97]. However, it needs to be emphasized that in spite of the wide variety strategies, which have been identified to explain the mechanisms of cardioprotection, a great deal of future research work needs to be carried out to make any meaningful conclusion.
5. Pharmacotherapy of I/R Injury to the Heart
In view of the complex pathophysiology of I/R injury involving oxidative stress, inflammation, intracellular Ca2+-overload and metabolic defects, various types of pharmacological agents, acting on diverse molecular targets, have been shown to exert beneficial effects in different experimental models of I/R injury [2,4,5,14,20,26,31]. Several antioxidants, Ca2+-antagonists, β-adrenoceptor blockers, angiotensin II antagonists, and metabolic modulators have been reported to improve cardiac function, prevent arrhythmias, and attenuate cellular damage in hearts subjected to I/R injury [20,29,30,98,99]. Some phosphodiesterase inhibitors such as pentoxifylline have been shown to prevent I/R-induced cardiac dysfunction by reducing the activation of NF-κB and TNF-α content [100], whereas several TNF-α inhibitors including etanercept exert therapeutic effects by reducing myocardial inflammation and oxidative stress [25,101]. Both leupeptin and compound MDL28170 (inhibitors of matrix metalloproteinases) were observed to prevent I/R injury by depressing the activation of proteolytic enzymes in the heart [26,102,103]. Furthermore, various aldosterone receptor antagonists and sodium-glucose cotransporter 2 (SGLT2) inhibitors have been observed to attenuate I/R injury as well as infarct size due to myocardial infarction by multiple mechanisms including inflammation and oxidative stress [104,105]. Because of the multifactorial basis of I/R injury, a wide variety of drugs such as cyclosporin, colchicine, tocilizumab, glucagon-like peptide 1 antagonist and modulators of different protein kinases (acting at different target sites) have been demonstrated to limit myocardial infarct size as well as prevent cardiac arrhythmias, cellular necrosis, apoptosis, and metabolic defects [99,106,107,108]. In addition, these agents have been shown to promote endothelial and vascular functions, enhance flow, and improve cardiac function.
Since the antioxidant reserve is depressed in hearts exposed to I/R injury for a prolonged period, it is considered appropriate to enhance the endogenous antioxidant systems (either by inducing increases in their activities or by supplementation with exogenous antioxidant) if the adverse effects of I/R injury have to be reversed [29,30,109,110]. In this regard, exercise-induced increases in endogenous antioxidants and nutritional supplements with polyphenolic compounds from foods such as grapes, cocoa, and soy have been reported to limit I/R-induced myocardial cell damage [110]. While several synthetic antioxidants, N-acetylcysteine and N-mercaptopropionylglycine, have been shown to attenuate I/R injury in animal experimentation, clinical trials have not provided indisputable evidence for favorable action by using these antioxidants in humans [31,32,111]. Untargeted applications of insufficient doses and/or delayed administration following I/R injury may explain the ineffectiveness of these antioxidants in clinical studies [34,112]. On the other hand, a systematic approach for the use of antioxidant vitamins was proposed to offer a novel opportunity to ameliorate the lethal I/R injury [108]. In fact, different studies have revealed that dietary antioxidant vitamins such as vitamin A, C, E, and β-carotene are effective in preventing major cardiovascular events associated with I/R injury [24,113,114]. Some other antioxidants including lazaroid U83836E and liproxstatin-1 have been reported to exert protective effects against I/R injury by targeting protein kinase C and ferroptosis, respectively [115,116]. A non-enzymatic antioxidant selenium, which is an essential component of oxyradical scavengers such as glutathione peroxidase and thioredoxin reductase, has been demonstrated to attenuate I/R injury by regulating the gene expression of these selenoenzymes [117,118].
By virtue of their ability to function as the major source for generating oxidative stress during the development of I/R injury, mitochondria are regarded as a main target of several pharmacological agents for improving cardiac function [27,119,120,121,122,123]. Cardioprotection by various therapeutic interventions is achieved by attenuating alterations in different mitochondrial events such as oxidative phosphorylation, mitochondrial membrane potential, Ca2+-overload, permeability transition pore formation, leakage of different apoptotic and necrotic factors, mitochondrial cardiolipin content, as well as NAPDH oxidase 2 activity. Alteration of I/R-induced changes in the heart and the left ventricular function by gypenoside, a herbal medicine, was associated with preservation of mitochondrial enzymatic activities of complex 1, II, and IV in the respiratory chain as well as the activity of citrate synthase for energy generation [124]. The beneficial effects of cyclosporine A in I/R injury were found to be due to its action on desensitizing the mitochondrial permeability transition pore opening in the myocardium [125,126]. Likewise, the antioxidant activity of melatonin in reducing the adverse effects of I/R injury was also shown to be related to its inhibitory action on the mitochondrial permeability transition pore opening as well as up-regulation of cytochrome c oxidase activity [127,128]. In addition, nicorandil, a mitochondrial ATP-sensitive potassium channel opener, has been demonstrated to prevent I/R-induced injury to the myocardium, alleviate cardiomyocyte necrosis, attenuate endothelial dysfunction, and improve blood flow as well as cardiac function [129]. These experimental observations indicate that several cardiac alterations induced by I/R injury are prevented by treatment with a wide variety of pharmacological agents including antioxidants; however, extensive research work needs to be carried out to establish if these interventions are able to reverse the I/R-induced myocardial abnormalities. It is desirable to include all doses and administration routes of these drugs for indicating their cardioprotective effects against I/R injury, but these issues require detailed description and thus are not considered within the scope of this article.
6. Evidence for the Role of Oxidative Stress in I/R-Induced Cardiac Dysfunction and Subcellular Defects
Although several experimental studies have revealed that oxidative stress generated during the development of I/R injury is associated with cardiac dysfunction, occurrence of intracellular Ca2+-overload, metabolic abnormalities, and subcellular defects for Ca2+-handling, the cause-and-effect relationships among these alterations are not fully understood. Accordingly, we have analyzed some of the existing information to provide evidence for the role of oxidative stress in inducing cardiac dysfunction and subcellular defects as a consequence of I/R injury upon perfusing the isolated hearts in the absence and presence of an oxyradical scavenging mixture or antioxidants [130,131,132,133,134,135,136,137,138]. Furthermore, data were also analyzed to examine if I/R-induced adverse effects in the heart are simulated upon perfusing with an oxyradical generating systems or H2O2, a well-known oxidant. For the purpose of inducing I/R injury, isolated perfused rat hearts were subjected to 30 min of global ischemia followed by different periods of reperfusion whereas the effects of oxidative stress were examined by perfusing the hearts with an oxyradical generating mixture (xanthine plus xanthine oxidase) or H2O2 for 30 min. The data in Table 1 show that various parameters of the left ventricular (LV) function such as developed pressure, end-diastolic pressure, +dP/dt and -dP/dt were markedly depressed by I/R injury whereas the LV end-diastolic pressure, as well as H2O2, malondialdehyde and total Ca2+ content were increased. These alterations in I/R-induced cardiac function, oxidative stress parameters, and Ca2+ content were greatly attenuated by the presence of superoxide dismutase plus catalase in the perfusion medium (Table 1). The data in Table 2 indicate that depressions in both LV-developed pressure and SL Na+-K+ ATPase activity by I/R injury were associated with increases in the activity of both calpain and matrix metalloproteinase enzymes; these effects of I/R on cardiac function, Na+-K+ ATPase and proteolytic enzyme activities were markedly attenuated by the presence of antioxidants such as N-acetylcysteine and mercaptopropionylglycine. Furthermore, the adverse effects of I/R injury on all these parameters were simulated upon perfusing the heart with xanthine plus xanthine oxidase mixture or H2O2 (Table 2). It may also be noted that the activities of SL Na+-Ca2+ exchange and ATP-dependent Ca2+-uptake as well as SL Ca2+-stimulated ATPase were depressed upon subjecting the heart to I/R injury and these alterations were prevented by the presence of superoxide dismutase plus catalase mixture (Table 3). It may also be seen from Table 3 that SL Na+-Ca2+ exchange and Ca2+-pump activities were depressed upon perfusing the heart with xanthine plus xanthine oxidase and these changes were prevented by the presence of superoxide dismutase plus catalase mixture in the perfusion medium.

Table 1.
Influence of ischemia-reperfusion (I/R) with or without oxyradical scavenger mixture (SOD plus CAT) on cardiac function and myocardial markers for oxidative stress as well as Ca2+-content in isolated perfused hearts.
Table 2.
Influence of ischemia-reperfusion (I/R) with or without some antioxidants as well as perfusion with xanthine plus xanthine oxidase (X + XO) or H2O2 on cardiac function, sarcolemmal Na+-K+ ATPase activity and protease activities in isolated perfused hearts.
Table 3.
Influence of ischemia-reperfusion (I/R) with or without oxyradical scavenger (SOD plus CAT) as well as perfusion with xanthine plus xanthine oxidase (X + XO) or H2O2 on sarcolemmal Na+-Ca2+ exchange, Ca2+-uptake and Ca2+-stimulated ATPase activities in isolated perfused hearts.
The data in Table 4 show that depression of the LV-developed pressure was also associated with decreases in different SR activities such as Ca2+-uptake, Ca2+-pump ATPase, Ca2+-release, and ryanodine binding upon subjecting the heart to I/R injury or perfusion with xanthine with xanthine plus xanthine oxidase as well as H2O2. It may also be seen from Table 4 that I/R-induced depressions in SR Ca2+-pump and Ca2+-release activities were prevented by the presence of superoxide dismutase plus catalase in the perfusion medium. Furthermore, data in Table 5 indicate that depressions in both LV-developed pressure and LV end-diastolic pressure by I/R-injury and perfusion with xanthine plus xanthine oxidase or H2O2 were associated with depressed mitochondrial state 3 respiration and oxidative phosphorylation. These adverse effects of I/R on cardiac function and mitochondrial function were markedly attenuated by the presence of superoxide dismutase plus catalase mixture in the perfusion medium (Table 5). Subjecting the hearts to I/R injury as well as perfusion with xanthine plus xanthine oxidase or H2O2 also showed depression in LV-developed pressure and myofibrillar Ca2+-stimulated ATPase activity (Table 6). These effects of I/R injury were prevented by the presence of an oxyradical scavenger (superoxide dismutase plus catalase mixture) as well as by an antioxidant (N-acetylcysteine). Although I/R injury did not affect myofibrillar Mg2+-ATPase, the activity of this enzyme was increased upon perfusion with xanthine plus xanthine oxidase as well as H2O2 (Table 6); the exact reason for the activation of myofibrillar Mg2+-ATPase by oxyradical generating system or oxidant is not clear at present. Nonetheless, the overall information described here indicates that there is a linear association between the depression in cardiac performance and changes in subcellular functions related to cation homeostasis, Ca2+-handling, energy production and generation of contractile activity during the development of I/R injury. It is noteworthy that I/R-induced alterations in cardiac function and subcellular activities were not only attenuated by oxyradical scavengers or antioxidants but these changes were also simulated upon perfusing the heart with oxyradical generating system or oxidant. These observations provide a compelling evidence that oxidative stress plays a critical role in the pathophysiology of I/R injury.
Table 4.
Influence of ischemia-reperfusion (I/R) with or without oxyradical scavenger (SOD plus CAT) as well as perfusion with xanthine plus xanthine oxidase (X + XO) or H2O2 on cardiac function and sarcoplasmic reticular Ca2+-uptake and Ca2+-release activities in isolated perfused hearts.
Table 5.
Influence of ischemia-reperfusion (I/R) in the absence or presence of oxyradical scavenger mixture (SOD plus CAT) as well as perfusion with oxyradical generating mixture (X plus XO) or H2O2 on cardiac function and mitochondrial function in isolated perfused hearts.
Table 6.
Influence of ischemia-reperfusion (I/R) with or without oxyradical scavenger and antioxidant as well as perfusion with xanthine plus xanthine oxidase (X + XO) or H2O2 on cardiac functions and myofibrillar ATPase activities in isolated perfused hearts.
7. Concluding Remarks
From the foregoing discussion, it is evident that there occurs a lack of oxygen, accumulation of intracellular H+ due to the inability of mitochondria to oxidize substrates, increase in the concentration of intracellular Ca2+ due to the activation of SL Na+-H+ exchange and Na+-Ca2+ exchange systems, as well as loss of contractile activity in the ischemic heart. All these alterations are reversible if the reperfusion is carried out during early periods of the ischemic insult but delayed reperfusion has been shown to produce irreversible changes in inflammation and depletion of energy stores in the myocardium. Mitochondrial defects in respiratory chain as well as changes in different enzymes such as NADPH oxidase, nitric oxide synthase, and monoamine oxidase are the major sources of oxyradicals and oxidants in I/R hearts. Excessive formation of reactive oxygen/nitrogen species and depression in the different antioxidant defense systems result in oxidative/nitrosative stress during the development of I/R injury. Furthermore, the occurrence of intracellular Ca2+-overload appears to be due to increased membrane permeability as well as changes in SL and SR Ca2+-handling systems. On the other hand, I/R-induced myocardial inflammation is a consequence of the release of inflammatory cytokines including TNF-α from macrophages in the cardiac interstitium as well as peripheral neutrophils which enter the injured myocardium. It is difficult to clearly indicate the cause-and-effect of oxidative stress, myocardial inflammation, or intracellular Ca2+-overload with I/R injury because adverse effects of these pathological factors are inter-related. In this regard, it is pointed out that both oxidative stress and myocardial inflammation are known to cause subcellular Ca2+-handling abnormalities, mitochondrial Ca2+-overload, and depression in energy production. Ca2+-abnormalities in SL and SR membranes as well as loss of myofibrillar Ca2+-sensitivity have also been shown to occur due to the activation of different proteases and modification of cardiac gene expression by both intracellular Ca2+-overload and oxidative stress. Furthermore, opening of mitochondrial permeability transition pore and leakage of various mitochondrial cytotoxic components as well as different apoptotic and necrotic factors in the cytoplasm have been reported to occur by oxidative stress in combination with mitochondrial Ca2+-overload. Taken together, these observations and other information in the literature suggest that oxidative stress plays a pivotal role in the development of I/R-induced cardiac dysfunction and myocardial cell damage. Some salient events involving oxidative stress, intracellular Ca2+-overload, as well as SL, SR, myofibrillar, and mitochondrial defects for the occurrence of cardiac dysfunction due to I/R-induced injury are depicted in Figure 4.
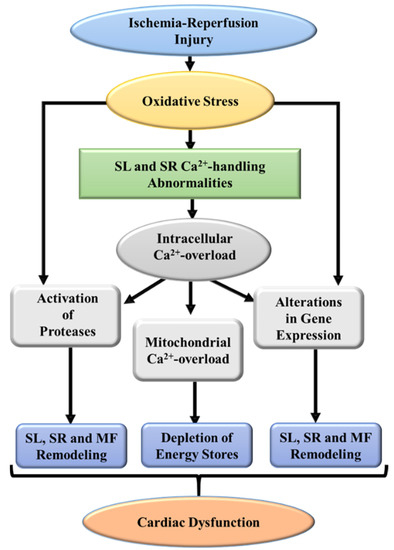
Figure 4.
A schematic representation of some major events indicating defects in subcellular organelles leading to cardiac dysfunction due to ischemia-reperfusion injury. SL, sarcolemma; SR, sarcoplasmic reticulum; MF, myofibrils.
Several pathophysiological studies in different experimental models of I/R injury have identified various major targets in the myocardium for the development of cardioprotective strategies. Some of these entities include metabolic defects, Ca2+-handling abnormalities, lipid peroxidation, protease activation, signal transduction for apoptosis and necrosis, transcriptional factors for maintaining redox homeostasis, and mitochondrial K+-ATP channels. Although numerous pharmacological agents, including Ca2+-antagonists, β-adrenoceptor blockers, angiotensin II antagonists, metabolic modulators, cyclosporin A, and nicorandil have shown to exert beneficial effects in attenuating I/R injury in animal studies, their results in human clinical trials are not conclusive. On the other hand, strategies such as ischemic preconditioning and ischemic postconditioning, which enhance antioxidant levels, have shown a great promise for cardioprotection in both animals and clinical investigations. Likewise, antioxidant vitamins (vitamin A, C, E, and β-carotene), unlike synthetic antioxidants, have been demonstrated to improve cardiac function and reduce myocardial damage. Such observations support the concept that oxidative stress may be intimately involved in the pathophysiology of I/R-induced injury to the heart. This view is further attested by the observations that I/R-induced alterations in cardiac contractile activity, oxidative stress markers, Ca2+-handling by SL and SR as well as myofibrillar proteins and mitochondrial function were attenuated by oxyradical scavenging mixture. Furthermore, all these I/R-induced adverse effects in the heart were simulated upon perfusion with an oxyradical generating system or an oxidant. Thus, there is great challenge for directing the future research activities for developing appropriate antioxidant interventions for the prevention and/or therapy of I/R injury to the heart.
Funding
Some of the research work quoted in this article was supported by the Slovak Research and Development Agency and the Ministry of Education, Science, Research and Sport of the Slovak Republic (Grant numbers: APVV-15-0607, APVV-20-0242, VEGA 1/0016/20 and VEGA 2/0104/20).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The infrastructure support for this project was provided by St. Boniface Hospital Albrechtsen Research Centre. We wish to thank Andrea Opsima for typing this manuscript.
Conflicts of Interest
The authors declare no conflict of interest. It may be mentioned that all authors have contributed in the preparation and editing this article and have approved its submission for its publication.
References
- Hoffman, J.W.; Gilbert, T.B.; Poston, R.S.; Silldorff, E.P. Myocardial reperfusion injury: Etiology, mechanisms, and therapies. J. Extra Corpor. Technol. 2004, 36, 391–411. [Google Scholar] [PubMed]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef]
- Neuzil, J.; Rayner, B.S.; Lowe, H.C.; Witting, P.K. Oxidative stress in myocardial ischemia reperfusion injury: A renewed focus on a long-standing area of heart research. Redox Rep. 2005, 10, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Milei, J.; Grana, D.R.; Forcada, P.; Ambrosio, G. Mitochondrial oxidative and structural damage in ischemia-reperfusion in human myocardium. Current knowledge and future directions. Front. Biosci. 2007, 12, 1124–1130. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Saini, H.K.; Tappia, P.S.; Sethi, R.; Mengi, S.A.; Gupta, S.K. Potential role and mechanisms of subcellular remodeling in cardiac dysfunction due to ischemic heart disease. J. Cardiovasc. Med. 2007, 8, 238–250. [Google Scholar] [CrossRef]
- Rodrigo, R.; Libuy, M.; Feliu, F.; Hasson, D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Dis. Markers 2013, 35, 773–790. [Google Scholar] [CrossRef]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/reperfusion injury following acute myocardial infarction: A critical issue for clinicians and forensic pathologists. Mediators Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef]
- Zhou, T.; Chuang, C.C.; Zuo, L. Molecular characterization of reactive oxygen species in myocardial ischemia-reperfusion injury. Biomed. Res. Int. 2015, 2015, 864946. [Google Scholar] [CrossRef]
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: Revisited. Oxidative Med. Cell. Longev. 2016, 2016, 165450. [Google Scholar] [CrossRef]
- Xiang, M.; Lu, Y.; Xin, L.; Gao, J.; Shang, C.; Jiang, Z.; Lin, H.; Fang, X.; Qu, Y.; Wang, Y.; et al. Role of oxidative stress in reperfusion following myocardial ischemia and its treatments. Oxidative Med. Cell. Longev. 2021, 2021, 6614009. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.L.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Machackova, J.; Dhalla, N.S. Role of reactive oxygen species in ischemic preconditioning of subcellular organelles in the heart. Antioxid. Redox Signal. 2004, 6, 393–404. [Google Scholar] [CrossRef]
- Steffens, S.; Montecucco, F.; Mach, F. The inflammatory response as a target to reduce myocardial ischemia and reperfusion injury. Thromb. Haemost. 2009, 102, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Monassier, J.P. Reperfusion injury in acute myocardial infarction. From bench to cath lab. Part I: Basic considerations. Arch. Cardiovasc. Dis. 2008, 101, 491–500. [Google Scholar] [CrossRef]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative stress as a mechanism for functional alterations in cadiac hypertrophy and heart failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef]
- Bhullar, S.K.; Shah, A.K.; Dhalla, N.S. Role of angiotensin II in the development of subcellular remodeling in heart failure. Explor. Med. 2021, 2, 352–371. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Bhullar, S.K.; Shah, A.K. Future scope and challenges for congestive heart failure: Moving towards development of pharmacotherapy. Can. J. Physiol. Pharmacol. 2022. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Shah, A.K.; Nusier, M. Mechanisms of cardiac dysfunction in heart failure due to myocardial infarction. J. Integr. Cardiol. Open Access 2019, 2, 3–7. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Bartekova, M.; Adameova, A. Involvement of oxidative stress in the development of subcellular defects and heart disease. Biomedicines 2022, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Xu, Y.J.; Zhang, M.; Liu, P.P.; Kirshenbaum, L.A.; Dhalla, N.S. Role of tumor necrosis factor-alpha and other cytokines in ischemia-reperfusion-induced injury in the heart. Exp. Clin. Cardiol. 2005, 10, 213–222. [Google Scholar] [PubMed]
- Ambrosio, G.; Zweier, J.L.; Flaherty, J.T. The relationship between oxygen radical generation and impairment of myocardial energy metabolism following post-ischemic reperfusion. J. Mol. Cell. Cardiol. 1991, 23, 1359–1374. [Google Scholar] [CrossRef]
- Sinning, C.; Westermann, D.; Clemmensen, P. Oxidative stress in ischemia and reperfusion: Current concepts, novel ideas and future perspectives. Biomark. Med. 2017, 11, 11031–11040. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Matsui, Y.; Sadoshima, J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid. Redox Signal. 2007, 9, 1373–1381. [Google Scholar] [CrossRef]
- Muller, A.L.; Hryshko, L.V.; Dhalla, N.S. Extracellular and intracellular proteases in cardiac dysfunction due to ischemia-reperfusion injury. Int. J. Cardiol. 2013, 164, 39–47. [Google Scholar] [CrossRef]
- Marin, W.; Marin, D.; Ao, X.; Liu, Y. Mitochondria as a therapeutic target for cardiac ischemia-reperfusion injury (review). Int. J. Mol. Med. 2021, 47, 485–499. [Google Scholar] [CrossRef]
- Ostadal, P.; Elmoselhi, A.B.; Zdobnicka, I.; Lukas, A.; Elimban, V.; Dhalla, N.S. Role of oxidative stress in ischemia-reperfusion-induced changes in Na+, K+-ATPase isoform expression in rat heart. Antioxid. Redox Signal. 2004, 6, 914–923. [Google Scholar] [CrossRef]
- Netticadan, T.; Temsah, R.; Osada, M.; Dhalla, N.S. Status of Ca2+/calmodulin protein kinase phosphorylation of cardiac SR proteins in ischemia-reperfusion. Am. J. Physiol. 1999, 277, C384–C391. [Google Scholar] [CrossRef]
- Marczin, N.; El-Habashi, N.; Hoare, G.S.; Bundy, R.E.; Yacoub, M. Antioxidants in myocardial ischemia-reperfusion injury: Therapeutic potential and basic mechanisms. Arch. Biochem. Biophys. 2003, 420, 222–236. [Google Scholar] [CrossRef]
- Bartekova, M.; Barancik, M.; Ferenczyova, K.; Dhalla, N.S. Beneficial effects of N-acetylcysteine and N–N-mercaptopropionylglycine on Ischemia reperfusion injury in the heart. Curr. Med. Chem. 2018, 25, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Bartekova, M.; Adameova, A.; Gorbe, A.; Ferenczyova, K.; Pechanova, O.; Lazou, A.; Dhalla, N.S.; Ferdinandy, P.; Giricz, Z. Natural and synthetic antioxidants targeting cardiac oxidative stress and redox signaling in cardiometabolic diseases. Free Radic. Biol. Med. 2021, 169, 446–477. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Kleikers, P.W.M.; Wingler, K.; Hermans, J.J.R.; Diebold, I.; Altenhofer, S.; Radermacher, K.A.; Janssen, B.; Gorlach, A.; Schmidt, H.H.H.W. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef]
- Ferrari, R.; Ceconi, C.; Curello, S.; Guarnieri, C.; Caldarera, C.M.; Albertini, A.; Visioli, O. Oxygen-mediated myocardial damage during ischemia and reperfusion: Role of the cellular defenses against oxygen toxicity. J. Mol. Cell. Cardiol. 1985, 17, 937–945. [Google Scholar] [CrossRef]
- Arduini, A.; Mezzetti, A.; Porreca, E.; Lapenna, D.; DeJulia, J.; Marzio, L.; Polidoro, G.; Cuccurullo, F. Effect of ischemia and reperfusion on antioxidant enzymes and mitochondrial inner membrane proteins in perfused rat heart. Biochim. Biophys. Acta 1988, 970, 113–121. [Google Scholar] [CrossRef]
- Padmavathi, G.; Ramkumar, K.M. MicroRNA mediated regulation of the major redox homeostasis switch, Nrf2, and its impact on oxidative stress-induced ischemic/reperfusion injury. Arch. Biochem. Biophys. 2021, 698, 108725. [Google Scholar] [CrossRef]
- Haramaki, N.; Stewart, D.B.; Aggarwal, S.; Ikeda, H.; Reznick, A.Z.; Packer, L. Networking antioxidants in the isolated rat heart are selectively depleted by ischemia-reperfusion. Free. Radic. Biol. Med. 1998, 25, 329–339. [Google Scholar] [CrossRef]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef]
- Murphy, M.P. Understanding and preventing mitochondrial oxidative damage. Biochem. Soc. Trans. 2016, 44, 1219–1226. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy substrate metabolism and mitochondrial oxidative stress in cardiac ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef]
- Liu, P.; Hock, C.E.; Nagele, R.; Wong, P.Y.K. Formation of nitric oxide, superoxide, and peroxynitrite in myocardial ischemia-reperfusion injury in rats. Am. J. Physiol. 1997, 272, H2327–H2336. [Google Scholar] [CrossRef] [PubMed]
- Anatoliotakis, N.; Defteros, S.; Bouras, G.; Giannopoulos, G.; Tsounis, D.; Angelidis, C.; Kaoukis, A.; Stefanadis, C. Myeloperoxidase: Expressing inflammation and oxidative stress in cardiovascular disease. Curr. Top. Med. Chem. 2013, 13, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Kaluderic, N.; Carpi, A.; Menabo, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidase (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Santin, Y.; Parini, A. Monoamine oxidase-A, serotonin and norepinephrine: Synergistic players in cardiac physiology and pathology. J. Neural Transm. 2018, 125, 1627–1634. [Google Scholar] [CrossRef]
- Harrison, D.G.; Cai, H.; Landmesser, U.; Griendling, K.K. Interactions of angiotensin II with NADPH oxidase, oxidant stress and cardiovascular stress. J. Renin Angiotensin Aldosterone Syst. 2003, 4, 51–61. [Google Scholar] [CrossRef]
- Kaminski, K.A.; Bonda, T.A.; Korecki, J.; Musial, W.J. Oxidative stress and neutrophil activation—The two keystones of ischemia/reperfusion injury. Int. J. Cardiol. 2002, 86, 41–59. [Google Scholar] [CrossRef]
- Li, B.; Tian, J.; Sun, Y.; Xu, T.R.; Chi, R.F.; Zhang, X.L.; Hu, X.L.; Zhang, Y.A.; Qin, F.Z.; Zhang, W.F. Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction. Biochim. Biophys. Acta 2015, 1852, 805–815. [Google Scholar] [CrossRef]
- Luo, J.; Xuan, Y.T.; Gu, Y.; Prabhu, S.D. Prolonged oxidative stress inverts the cardiac force-frequency relation: Role of altered calcium handling and myofilament calcium responsiveness. J. Mol. Cell. Cardiol. 2006, 40, 64–75. [Google Scholar] [CrossRef]
- Sharma, G.P.; Varley, K.G.; Kim, S.W.; Barwinsky, J.; Cohen, M.; Dhalla, N.S. Alterations in energy metabolism and ultrastructure upon reperfusion of the ischemic myocardium after coronary occlusion. Am. J. Cardiol. 1975, 36, 234–243. [Google Scholar] [CrossRef]
- Nayler, W.G.; Panagiotopoulos, S.; Elz, J.S.; Daly, M.J. Calcium-mediated damage during post-ischemic reperfusion. J. Mol. Cell. Cardiol. 1988, 20 Suppl 2, 41–54. [Google Scholar] [CrossRef]
- Murphy, J.G.; Smith, T.W.; Marsh, J.D. Mechanisms of reoxygenation-induced calcium overload in cultured chick embryo heart cells. Am. J. Physiol. 1988, 254, H1133–H1141. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Reimer, K.A. The cell biology of acute myocardial ischemia. Annu. Rev. Med. 1991, 42, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Marban, E. Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 1999, 79, 609–634. [Google Scholar] [CrossRef] [PubMed]
- Piper, M.H.; Meuter, K.; Schafer, C. Cellular mechanisms of ischemia-reperfusion injury. Ann. Thorac. Surg. 2003, 75, S644–S648. [Google Scholar] [CrossRef]
- Bhosale, G.; Sharpe, J.A.; Sundier, S.; Duchen, M. Calcium signaling as mediator of cell energy demand and a trigger to cell death. Ann. N. Y. Acad. Sci. 2015, 1350, 107–116. [Google Scholar] [CrossRef]
- Badalzadeh, R.; Mokhtari, B.; Yavari, R. Contribution of apoptosis in myocardial reperfusion injury and loss of cardioprotection in diabetes mellitus. J. Physiol. Sci. 2015, 65, 201–215. [Google Scholar] [CrossRef]
- Ferrari, R.; Guardigli, G.; Mele, D.; Percoco, G.F.; Ceconi, C.; Curello, S. Oxidative stress during myocardial ischemia and heart failure. Curr. Pharm. Des. 2004, 10, 1699–1711. [Google Scholar] [CrossRef]
- Neri, M.; Fineschi, V.; Di Paolo, M.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Cerretani, D. Cardiac oxidative stress and inflammation cytokines response after myocardial infarction. Curr. Vasc. Pharmacol. 2015, 13, 26–36. [Google Scholar] [CrossRef]
- Li, X.; Zhang, F.; Zhou, H.; Hu, Y.; Guo, D.; Fang, X.; Chen, Y. Interplay of TNF-α, soluble TNF receptors and oxidative stress in coronary chronic total occlusion of the oldest patients with coronary heart disease. Cytokine 2020, 125, 154836. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Qu, Y.; Chen, H.; Qian, J. Insight into long noncoding RNA-miRNA-mRNA axes in myocardial ischemia-reperfusion injury: The implications for mechanism and therapy. Epigenomics 2019, 11, 1733–1748. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sun, W.; Guo, Z.; Liu, B.; Yu, H.; Zhang, J. Long noncoding RNAs in myocardial ischemia-reperfusion inury. Oxidative Med. Cell. Longev. 2021, 2021, 8889123. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, Z.; Fan, Z.; Yang, Y.; Lu, C. Involvement of non-coding RNAs in the pathogenesis of myocardial ischemia/reperfusion injury (review). Intl. J. Mol. Med. 2021, 47, 42. [Google Scholar] [CrossRef]
- Wang, B.F.; Yoshioka, J. The emerging role of thioredoxin-interacting protein in myocardial ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 219–229. [Google Scholar] [CrossRef]
- Zhang, C.; He, M.; Ni, L.; He, K.; Su, K.; Deng, Y.; Li, Y.; Xia, H. The role of arachidonic acid metabolism in myocardial ischemia-reperfusion injury. Cell Biochem. Biophys. 2020, 78, 255–265. [Google Scholar] [CrossRef]
- Laude, K.; Richard, V.; Thuillez, C. Coronary endothelial cells: A target of ischemia reperfusion and its treatment? Arch. Mal. Coeur. Vaiss. 2004, 97, 250–254. [Google Scholar]
- Rohrbach, S.; Troidl, C.; Hamm, C.; Schulz, R. Ischemia and reperfusion related myocardial inflammation: A network of cells and mediators targeting the cardiomyocyte. IUBMB Life 2015, 67, 110–119. [Google Scholar] [CrossRef][Green Version]
- Gunata, M.; Parlakpinar, H. A review of myocardial ischemia/reperfusion injury: Pathophysiology, experimental models, biomarkers, genetics and pharmacological treatment. Cell. Biochem. Func. 2021, 39, 190–217. [Google Scholar] [CrossRef]
- Schanze, N.; Bode, C.; Duerschmied, D. Platelet contributions to myocardial ischemia/reperfusion injury. Front. Immunol. 2019, 10, 1260. [Google Scholar] [CrossRef]
- Sadek, H.A.; Nulton-Persson, A.C.; Szweda, P.A.; Szweda, L.I. Cardiac ischemia/reperfusion, aging, and redox-dependent alterations in mitochondrial function. Arch. Biochem. Biophys. 2003, 420, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Vercesi, A.E.; Kowaltowski, A.J.; Oliveira, H.C.F.; Castilho, R.F. Mitchondrial Ca2+ transport, permeability transition and oxidative stress in cell death: Implications in cardiotoxicity, neurodegeneration and dyslipidemias. Front. Biosci. 2006, 11, 2554–2564. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. Mitochondria and reperfusion injury of the heart—A holey death but not beyond salvation. J. Bioenerg, Biomembr. 2009, 41, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shi, D.; Guo, M. The roles of PKC-δ and PKC-ε in myocardial ischemia/reperfusion injury. Pharmacol. Res. 2021, 170, 105716. [Google Scholar] [CrossRef]
- Ruan, Y.; Zeng, J.; Jin, Q.; Chu, M.; Ji, K.; Wang, Z.; Li, L. Endoplasmic reticulum stress serves an important role in cardiac ischemia/reperfusion injury (review). Exp. Ther. Med. 2020, 20, 268. [Google Scholar] [CrossRef]
- Zima, A.V.; Mazurek, S.R. Functional impact of ryanodine receptor oxidation on intracellular calcium regulation in the heart. Rev. Physiol. Biochem. Pharmacol. 2016, 171, 39–62. [Google Scholar] [CrossRef]
- Frohlich, G.M.; Meier, P.; White, S.K.; Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury: Looking beyond primary PCI. Eur. Heart J. 2013, 34, 1714–1722. [Google Scholar] [CrossRef]
- Pagliaro, P.; Penna, C. Redox signalling and cardioprotection: Translatability and mechanism. Br. J. Pharmacol. 2015, 172, 1974–1995. [Google Scholar] [CrossRef]
- Daiber, A.; Andreadou, I.; Oelze, M.; Davidson, S.M.; Hausenloy, D.J. Discovery of new therapeutic redox targets for cardioprotection against ischemia/reperfusion injury and heart failure. Free Radic. Biol. Med. 2021, 163, 325–343. [Google Scholar] [CrossRef]
- Tappia, P.S.; Shah, A.K.; Ramjiawan, B.; Dhalla, N.S. Modification of ischemia/reperfusion-induced alterations in subcellular organelles by ischemic preconditioning. Int. J. Mol. Sci. 2022, 23, 3425. [Google Scholar] [CrossRef]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1723–H1741. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidties, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, N.; Oka, S.; Sadoshima, J. Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic. Biol. Med. 2017, 109, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.L.; Ho, Y.C.; Yet, S.F. A central role of heme oxygenase-1 in cardiovascular protection. Antioxid. Redox Signal. 2011, 15, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.B.; Chen, X.; Zhou, X.Y.; Wang, X.B. The role of peroxisome proliferator-activated receptor γ in mediating cardioprotection against ischemia/reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 46–56. [Google Scholar] [CrossRef]
- Wang, W.L.; Ge, T.Y.; Chen, X.; Mao, Y.; Zhu, Y.Z. Advances in the protective mechanism of NO, H2S, and H2 in myocardial ischemic injury. Front. Cardiovasc. Med. 2020, 7, 588206. [Google Scholar] [CrossRef] [PubMed]
- Dongo, E.; Hornyak, I.; Benko, Z.S.; Kiss, L. The cardioprotective potential of hydrogen sulfide in myocardial ischemia/reperfusion injury (review). Acta Physiol. Hung 2011, 98, 369–381. [Google Scholar] [CrossRef]
- Chohan, P.K.; Singh, R.B.; Dhalla, N.S.; Netticadan, T. L-arginine administration recovers sarcoplasmic reticulum function in ischemic reperfused hearts by preventing calpain activation. Cardiovasc. Res. 2006, 69, 152–163. [Google Scholar] [CrossRef]
- Jensen, R.V.; Andreadou, I.; Hausenloy, D.J.; Botker, H.E. The role of O-GlcNAcylation for protection against ischemia-reperfusion injury. Int. J. Mol. Sci. 2019, 20, 404. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, P.; Zhong, J.; Cheng, Y.; Chen, H.; He, Y.; Chen, C. HIF-1α in myocardial ischemia-reperfusion injury (review). Mol. Med. Rep. 2021, 23, 352. [Google Scholar] [CrossRef]
- Ding, J.; Yang, Z.; Ma, H.; Zhang, H. Mitochondrial aldehyde dehydrogenase in myocardial ischemic and ischemia-reperfusion injury. Adv. Exp. Med. Biol. 2019, 1193, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Zhang, H.; Shengshou, H. Mitochondrial aldehyde dehydrogenase 2 activation and cardioprotection. J. Mol. Cell. Cardiol. 2013, 55, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, T.; Gomes, A.V. MicroRNAs in the regulation of cellular redox status and its implications in myocardial ischemia-reperfusion injury. Redox Biol. 2020, 36, 101607. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Sun, W.; Zhang, Z.; Zheng, Y. The role of Nrf2-mediated pathway in cardiac remodeling and heart failure. Oxidative Med. Cell. Longev. 2014, 2014, 260429. [Google Scholar] [CrossRef]
- Bocci, V.; Valachi, G. Nrf2 activation as target to implement therapeutic treatments. Front. Chem. 2015, 3, 4. [Google Scholar] [CrossRef]
- Barzegar, M.; Kaur, G.; Wang, Y.; Boyer, C.J.; Alexander, J.S. Potential therapeutic roles of stem cells in ischemia-reperfusion injury. Stem Cells Res. 2019, 37, 101421. [Google Scholar] [CrossRef]
- Jeroudi, M.O.; Hartley, C.J.; Bolli, R. Myocardial reperfusion injury: Role of oxygen radicals and potential therapy with antioxidants. Am. J. Cardiol 1994, 73, 2B–7B. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Rodriguez-Sinovas, A.; Ruiz-Meena, M.; Inserte, J. Protection against myocardial ischemia-reperfusion injury in clinical practice. Rev. Esp. Cardiol. 2014, 67, 394–404. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Y.J.; Saini, H.K.; Turan, B.; Liu, P.P.; Dhalla, N.S. Pentoxifylline attenuates cardiac dysfunction and reduces TNF-α level in ischemic-reperfused heart. Am. J. Physiol. Heart. Circ. Physiol. 2005, 289, H832–H839. [Google Scholar] [CrossRef]
- Zhou, X.; Sheng, X.; Chen, M.; Wang, Z.; Yu, L.; Jiang, H. Tumor necrosis factor-α inhibitor: A promising therapeutic approach for attenuating myocardial ischemia-reperfusion by antioxidants stress. Int. J. Cardiol. 2015, 190, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Dhalla, N.S. Ischemia-reperfusion-induced changes in sarcolemmal Na+-K+-ATPase are due to the activation of calpain in the heart. Can. J. Physiol. Pharmacol. 2010, 88, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.L.; Freed, D.; Dhalla, N.S. Activation of proteases and changes in Na+-K+-ATPase subunits in hearts subjected to ischemia-reperfusion. J. Appl. Physiol. 2013, 114, 351–361. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dragasevic, N.; Jakovljevic, V.; Zivkovic, V.; Draginic, N.; Andjic, M.; Bolevich, S.; Jovic, S. The role of aldosterone inhibitors in cardiac ischemia-reperfusion injury. Can. J. Physiol. Pharmacol. 2021, 99, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Bell, R.M.; Botker, H.E.; Zuurbier, C.J. SGLT2 inhibitors reduce infarct size in reperfused ischemic heart and improve cardiac function during ischemic episodes in preclinical models. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165770. [Google Scholar] [CrossRef]
- Rout, A.; Tantry, U.S.; Novakovic, M.; Sukhi, A.; Gurbel, P.A. Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert. Opin. Pharmacother. 2020, 21, 1851–1865. [Google Scholar] [CrossRef]
- Correa, F.; Martinez-Abundis, E.; Hernandez-Resendiz, S.; Garcia, N.; Buelna-Chontal, M.; Arreguin, F.; Zazueta, C. Pharmacological strategies to contend against myocardial reperfusion damage: Diverse chemicals for multiple targets. Curr. Med. Chem. 2010, 17, 2261–2273. [Google Scholar] [CrossRef]
- Kryzwonos-Zawadzka, A.; Franczak, A.; Sawicki, G.; Wozniak, M.; Bil-Lula, I. Multidrug prevention or therapy of ischemia-reperfusion injury of the heart—Mini-review. Environ. Toxicol. Pharmacol. 2017, 55, 55–59. [Google Scholar] [CrossRef]
- Wang, W.; Kang, P.M. Oxidative stress and antioxidant treatments in cardiovascular diseases. Antioxidants 2020, 9, 1292. [Google Scholar] [CrossRef]
- Hamilton, K.L. Antioxidants and cardioprotection. Med. Sci. Sports Exerc. 2007, 39, 1544–1553. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Jaquet, V. Reactive oxygen species in myocardial reperfusion injury: From physiopathology to therapeutic approaches. Curr. Pharm. Biotechnol. 2012, 13, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Libuy, M.; Feliu, F.; Hasson, D. Molecular basis of cardioprotective effect of antioxidant vitamins in myocardial infarction. Biomed. Res. Int. 2013, 2013, 437613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.Y.; Xu, X.; Li, X.C. Cardiovascular diseases: Oxidative damage and antioxidant protection. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3091–3096. [Google Scholar]
- Leopold, J.A. Antioxidants and coronary artery disease: From pathophysiology to preventive therapy. Coron. Artery. Dis. 2015, 26, 176–183. [Google Scholar] [CrossRef]
- Lai, L.N.; Zhang, X.J.; Zhang, X.Y.; Song, L.H.; Guo, C.H.; Lei, J.W.; Song, X.L. Lazaroid U83836E protects the heart against ischemia reperfusion injury via inhibition of oxidative stress and activation of PKC. Mol. Med. Rep. 2016, 13, 3993–4000. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moya-Lillo, J.; Rojas-Sole, C.; Munoz-Salamanca, D.; Panieri, E.; Saso, L.; Rodrigo, R. Targeting ferrptosis against ischemia-reperfusion cardiac injury. Antioxidants 2021, 10, 667. [Google Scholar] [CrossRef] [PubMed]
- Boucher, F.R.; Jouan, M.G.; Moro, C.; Rakotavo, A.N.; Tanguy, S.; de Leiris, J. Does selenium exert cardioprotective effects against oxidative stress in myocardial ischemia? Acta Physiol. Hung. 2008, 95, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Venardos, K.M.; Perkins, A.; Headrick, J.; Kaye, D.M. Myocardial ischemia-reperfusion injury, antioxidant enzymes systems, and selenium: A review. Curr. Med. Chem. 2007, 14, 1539–1549. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Bienengraeber, M.; Stowe, D.F. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2011, 2, 13. [Google Scholar] [CrossRef]
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef]
- Lotz, C.; Herrmann, J.; Notz, Q.; Meybohm, P.; Kehl, F. Mitochondria and pharmacologic cardiac conditioning at the heart of ischemic injury. Int. J. Mol. Sci. 2021, 22, 3224. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: Implications for pharmacological cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1341–H1352. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowaski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NAPDH oxidase 2 (NOX2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes. PLoS ONE 2016, 11, 145750. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guan, Q.; Guo, L.; Zhang, H.; Pang, X.; Cheng, Y.; Zhang, X.; Sun, Y. Gypenosides alleviate myocardial ischemia-reperfusion injury via attenuation of oxidative stress and preservation of mitochondrial function in rat heart. Cell Stress Chaperones 2016, 21, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Perrelli, M.G.; Pagliaro, P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: Therapeutic implications. Antioxid. Redox Signal. 2013, 18, 556–599. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Parodi-Rullan, R.; Khuchua, Z.; Kuznetsov, A.V. Mitochondrial permeability transition in cardiac ischemia-reperfusion: Wether cyclophilin D is a viable target for cardioprotection? Cell. Mol. Life Sci. 2017, 74, 2795–2813. [Google Scholar] [CrossRef]
- Halladin, N.L. Oxidaitve and inflammatory biomarkers of ischemia and reperfusion injuries. Dan. Med. J. 2015, 62, B5054. [Google Scholar]
- Giacomo, C.G.; Antonio, M. Melatonin in cardiac ischemia/reperfusion-induced mitochondrial adaptive changes. Cardiovasc. Hematol. Disord. Drug Targets 2007, 7, 163–169. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, D.; Jiang, Z.; Ling, W.; Qian, G. Protective effect of nicorandil on cardiac microvascular injury: Role of mitochondrial integrity. Oxidative Med. Cell. Longev. 2021, 2021, 4665632. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Golfman, L.; Takeda, S.; Takeda, N.; Nagano, M. Evidence for the role of oxidative stress in acute ischemic heart disease: A brief review. Can. J. Cardiol. 1999, 15, 587–593. [Google Scholar]
- Singh, R.B.; Hryshko, L.; Freed, D.; Dhalla, N.S. Activation of proteolytic enzymes and depression of the sarcolemma Na+-K+-ATPase in ischemia-reperfused heart may be mediated through oxidative stress. Can. J. Physiol. Pharmacol. 2012, 90, 249–260. [Google Scholar] [CrossRef]
- Dixon, I.M.C.; Kaneko, M.; Hata, T.; Panagia, V.; Dhalla, N.S. Alterations in cardiac membrane Ca2+ transport during oxidative stress. Mol. Cell. Biochem. 1990, 99, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Dhalla, N.S. Effect of oxygen free radicals on cardiac contractile activity and sarcolemmal Na+-Ca2+ exchange. J. Cardiovasc. Pharmacol. Ther. 1996, 1, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Dhalla, N.S. Relationship between mechanical dysfunction and depression of sarcolemmal Ca2+-pump activity in hearts perfused with oxygen free radicals. Mol. Cell. Biochem. 1996, 160/161, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Temsah, R.M.; Netticadan, T.; Chapman, D.; Takeda, S.; Mochizuki, S.; Dhalla, N.S. Alterations in sarcoplasmic reticulum function and gene expression in ischemic-reperfused rat heart. Am. J. Physiol. 1999, 277, H584–H594. [Google Scholar] [CrossRef]
- Makazan, Z.; Saini, H.K.; Dhalla, N.S. Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1986–H1994. [Google Scholar] [CrossRef]
- Maddika, S.; Elimban, V.; Chapman, D.; Dhalla, N.S. Role of oxidative stress in ischemia-reperfusion-induced alterations in myofibrillar ATPase activities and gene expression in the heart. Can. J. Physiol. Pharmacol. 2009, 87, 120–129. [Google Scholar] [CrossRef]
- Suzuki, S.; Kaneko, M.; Chapman, D.C.; Dhalla, N.S. Alterations in cardiac contractile proteins due to oxygen free radicals. Biochim. Biophys. Acta. 1991, 1074, 95–100. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).