
Cell Death-Related Ubiquitin Modifications in Inflammatory Syndromes: From Mice to Men


Abstract
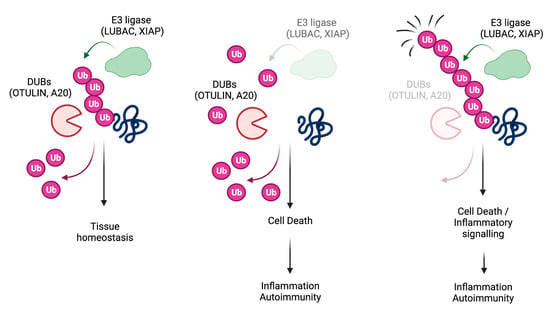
:
1. Introduction
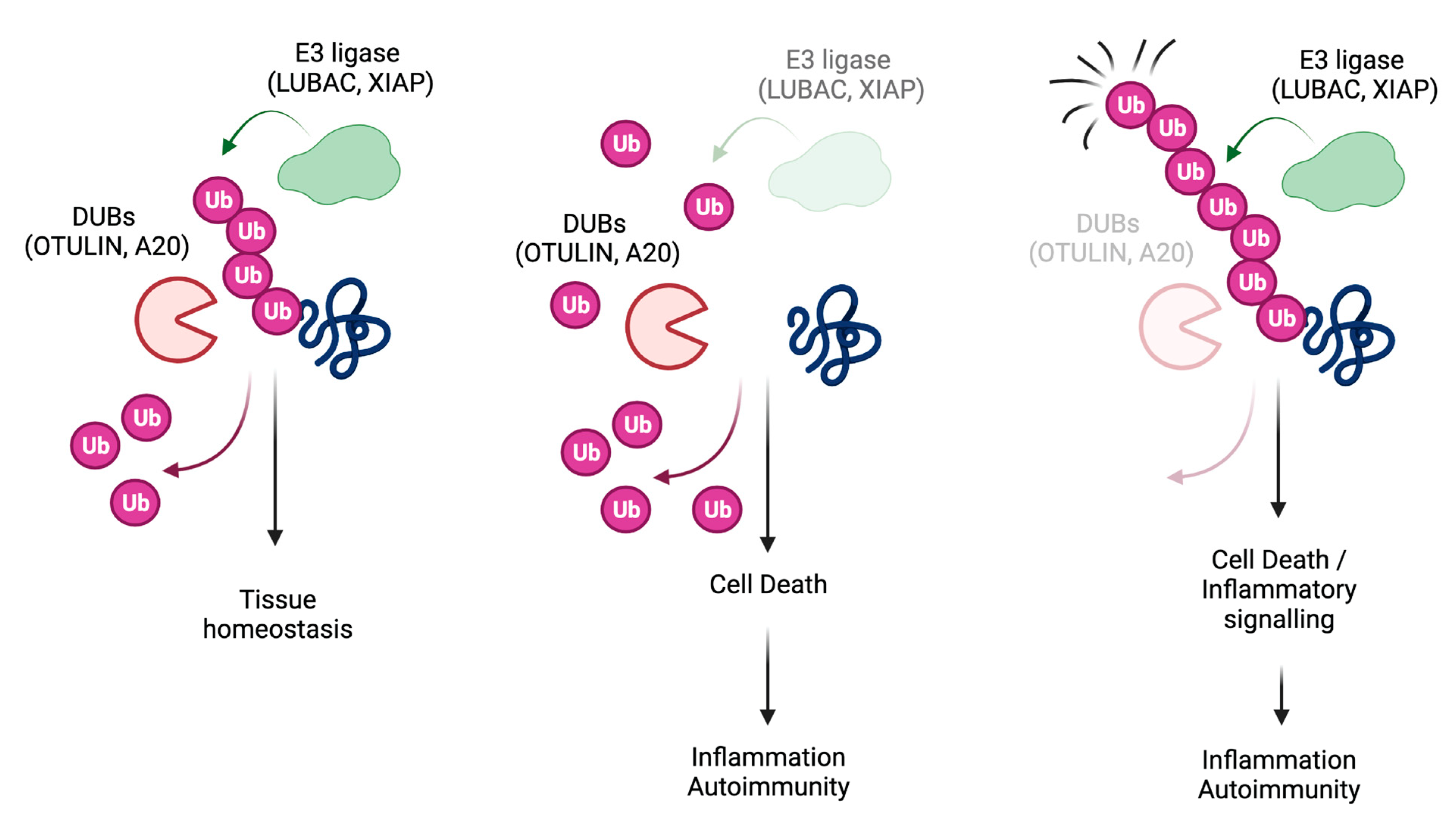
2. TNFR1-Signalling Pathway
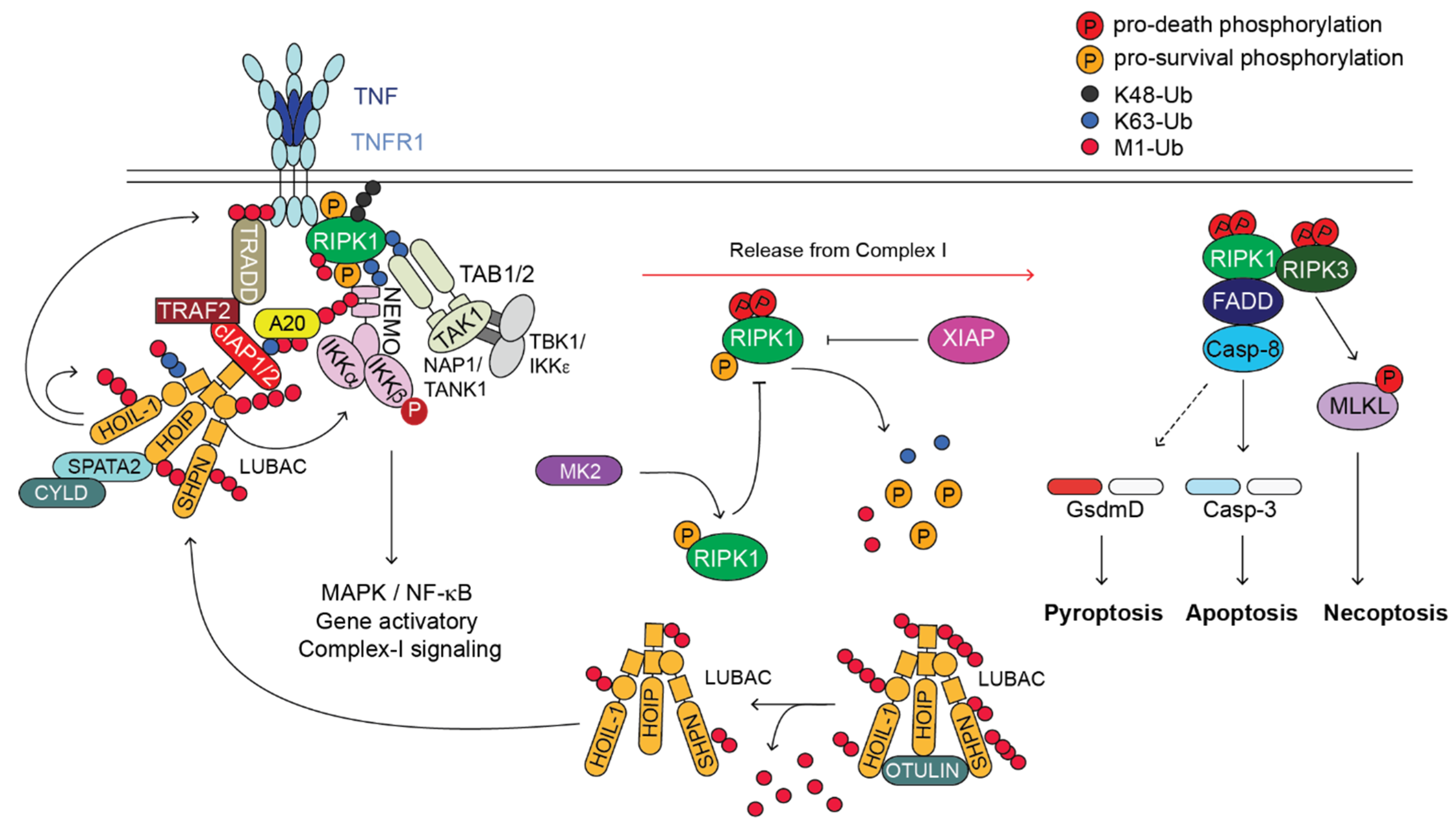
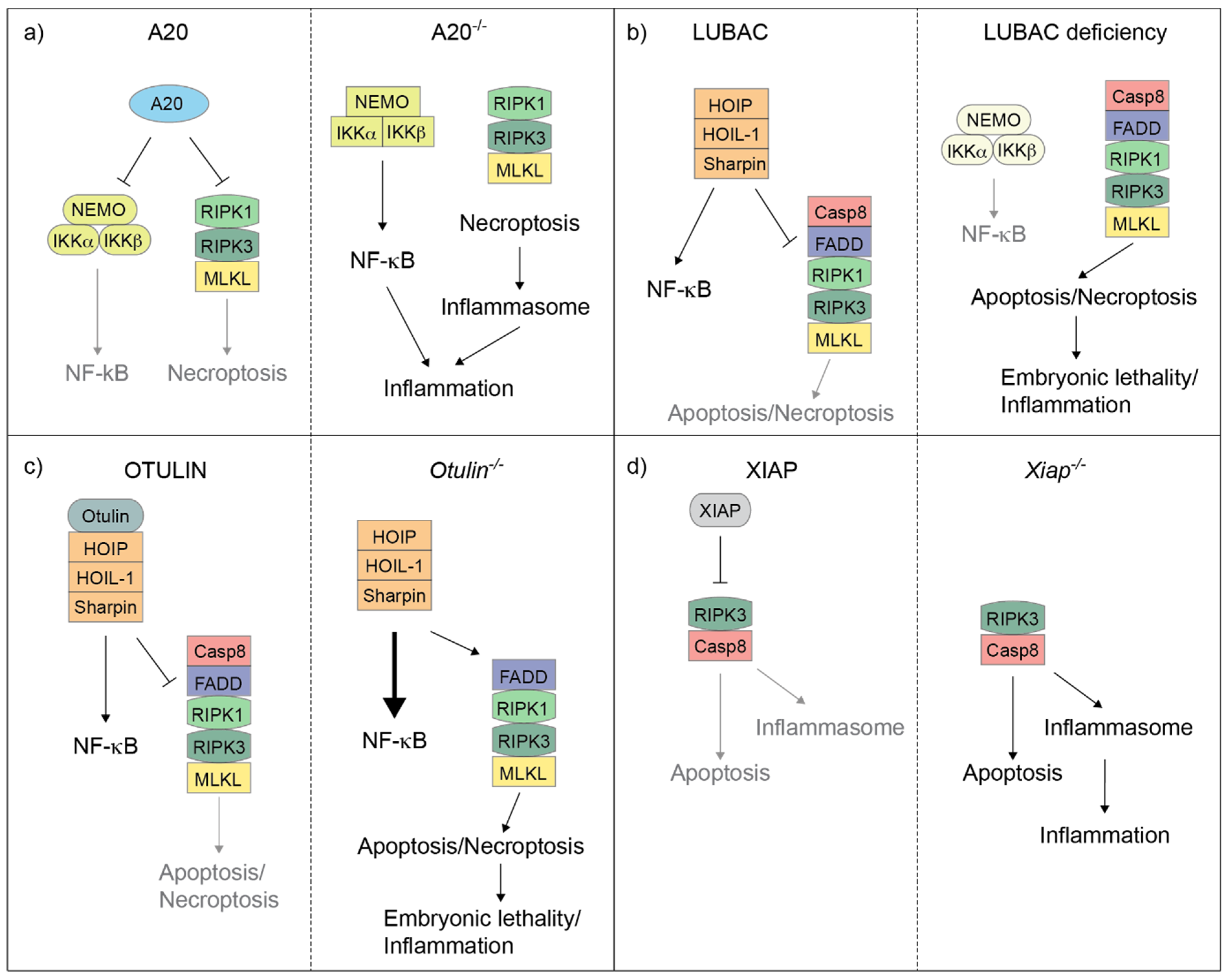
3. LUBAC
4. OTULIN
5. A20
6. XIAP
7. Omics
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Strasser, A.; Vaux, D.L. Cell Death in the Origin and Treatment of Cancer. Mol. Cell 2020, 78, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Legrand, A.J.; Konstantinou, M.; Goode, E.F.; Meier, P. The Diversification of Cell Death and Immunity: Memento Mori. Mol. Cell 2019, 76, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Dixit, V.M. Rescue from a fiery death: A therapeutic endeavor. Science 2019, 366, 688–689. [Google Scholar] [CrossRef]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef] [Green Version]
- Gitlin, A.D.; Heger, K.; Schubert, A.F.; Reja, R.; Yan, D.; Pham, V.C.; Suto, E.; Zhang, J.; Kwon, Y.C.; Freund, E.C.; et al. Integration of innate immune signaling by caspase-8 cleavage of N4BP1. Nature 2020, 587, 275–280. [Google Scholar] [CrossRef]
- Orozco, S.L.; Daniels, B.P.; Yatim, N.; Messmer, M.N.; Quarato, G.; Chen-Harris, H.; Cullen, S.P.; Snyder, A.G.; Ralli-Jain, P.; Frase, S.; et al. RIPK3 Activation Leads to Cytokine Synthesis that Continues after Loss of Cell Membrane Integrity. Cell Rep. 2019, 28, 2275.e5–2287.e5. [Google Scholar] [CrossRef] [Green Version]
- Meier, P.; Banreti, A. Tissue Repair: How to Inflame Your Neighbours. Curr. Biol. 2016, 26, R192–R194. [Google Scholar] [CrossRef] [Green Version]
- Peltzer, N.; Walczak, H. Cell Death and Inflammation—A Vital but Dangerous Liaison. Trends Immunol. 2019, 40, 387–402. [Google Scholar] [CrossRef]
- Annibaldi, A.; Meier, P. Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 2017, 24, 49–65. [Google Scholar] [CrossRef]
- Walczak, H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp.Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Kelliher, M.A.; Grimm, S.; Ishida, Y.; Kuo, F.; Stanger, B.Z.; Leder, P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998, 8, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Rothe, M.; Pan, M.G.; Henzel, W.J.; Ayres, T.M.; Goeddel, D.V. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 1995, 83, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Annibaldi, A.; John, S.W.; Berghe, T.V.; Swatek, K.N.; Ruan, J.; Liccardi, G.; Bianchi, K.; Elliott, P.R.; Choi, S.M.; Coillie, S.V.; et al. Ubiquitin-Mediated Regulation of RIPK1 Kinase Activity Independent of IKK and MK2. Mol. Cell 2018, 69, 566.e5–580.e5. [Google Scholar] [CrossRef]
- Dynek, J.N.; Goncharov, T.; Dueber, E.C.; Fedorova, A.V.; Izrael-Tomasevic, A.; Phu, L.; Helgason, E.; Fairbrother, W.J.; Deshayes, K.; Kirkpatrick, D.S.; et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010, 29, 4198–4209. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol.Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Ikeda, F.; Deribe, Y.L.; Skanland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.L.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rikard, J.A.; Anderton, H.; Wong, W.W.-L.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; de Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ori, D.; Kato, H.; Sanjo, H.; Tartey, S.; Mino, T.; Akira, S.; Takeuchi, O. Essential roles of K63-linked polyubiquitin-binding proteins TAB2 and TAB3 in B cell activation via MAPKs. J. Immunol. 2013, 190, 4037–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaco, I.; Annibaldi, A.; Lalaoui, N.; Wilson, R.; Tenev, T.; Laurien, L.; Kim, C.; Jamal, K.; Wicky Hohn, S.; Liccardi, G.; et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol. Cell 2017, 66, 698–710. [Google Scholar] [CrossRef]
- Lafont, E.; Draber, P.; Rieser, E.; Reichert, M.; Kupka, S.; de Miguel, D.; Draberova, H.; von Massenhausen, A.; Bhamra, A.; Henderson, S.; et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 2018, 12, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Kupka, S.; de Miguel, D.; Draber, P.; Martino, L.; Surinova, S.; Rittinger, K.; Walczak, H. SPATA2-Mediated Binding of CYLD to HOIP Enables CYLD Recruitment to Signaling Complexes. Cell Rep. 2016, 16, 2271–2280. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.R.; Leske, D.; Wagstaff, J.; Schlicher, L.; Berridge, G.; Maslen, S.; Timmermann, F.; Ma, B.; Fischer, R.; Freund, S.M.V.; et al. Regulation of CYLD activity and specificity by phosphorylation and ubiquitin-binding CAP-Gly domains. Cell Rep. 2021, 37, 109777. [Google Scholar] [CrossRef]
- Ting, A.T.; Bertrand, M.J. More to Life than NF-kappaB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, e10888–e10897. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. Embo. J. 2019, 38, e101638. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, N.; Darding, M.; Walczak, H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016, 11, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the immune system. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef]
- Yabal, M.; Muller, N.; Adler, H.; Knies, N.; Gross, C.J.; Damgaard, R.B.; Kanegane, H.; Ringelhan, M.; Kaufmann, T.; Heikenwalder, M.; et al. XIAP Restricts TNF- and RIP3-Dependent Cell Death and Inflammasome Activation. Cell Rep. 2014, 28, 1796–1808. [Google Scholar] [CrossRef] [Green Version]
- Peltzer, N.; Darding, M.; Montinaro, A.; Draber, P.; Draberova, H.; Kupka, S.; Rieser, E.; Fisher, A.; Hutchinson, C.; Taraborrelli, L.; et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 2018, 557, 112–117. [Google Scholar] [CrossRef]
- Seymour, R.E.; Hasham, M.G.; Cox, G.A.; Shultz, L.D.; Hogenesch, H.; Roopenian, D.C.; Sundberg, J.P. Spontaneous mutations in the mouse Sharpin gene result in multiorgan inflammation, immune system dysregulation and dermatitis. Genes Immun. 2007, 8, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Rickard, J.A.; Anderton, H.; Etemadi, N.; Nachbur, U.; Darding, M.; Peltzer, N.; Lalaoui, N.; Lawlor, K.E.; Vanyai, H. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife 2014, 3, e03464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Redouane, Y.; Lopez-Mosqueda, J.; Shiraishi, R.; Romanowska, M.; Lutzmayer, S.; Kuiper, J.; Martinez, C.; Dikic, I.; Pasparakis, M.; et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife 2014, 3, 03422. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, N.; Rieser, E.; Taraborrelli, L.; Draber, P.; Darding, M.; Pernaute, B.; Shimizu, Y.; Sarr, A.; Draberova, H.; Montinaro, A.; et al. HOIP Deficiency Causes Embryonic Lethality by Aberrant TNFR1-Mediated Endothelial Cell Death. Cell Rep. 2014, 9, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Taraborrelli, L.; Peltzer, N.; Montinaro, A.; Kupka, S.; Rieser, E.; Hartwig, T.; Sarr, A.; Darding, M.; Draber, P.; Haas, T.L.; et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 2018, 9, 3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Tu, H.; Liu, G.; Zheng, G.; Wang, M.; Li, L.; Zhao, X.; Lin, X. RNF31 Regulates Skin Homeostasis by Protecting Epidermal Keratinocytes from Cell Death. J. Immunol. 2018, 200, 4117–4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Peltzer, N.; Sevko, A.; Lafont, E.; Sarr, A.; Draberova, H.; Walczak, H. The linear ubiquitin chain assembly complex acts as a liver tumor suppressor and inhibits hepatocyte apoptosis and hepatitis. Hepatology 2017, 24, 1963–1978. [Google Scholar] [CrossRef]
- Teh, C.E.; Lalaoui, N.; Jain, R.; Policheni, A.N.; Heinlein, M.; Alvarez-Diaz, S.; Sheridan, J.; Rieser, E.; Deuser, S.; Darding, M.; et al. Linear ubiquitin chain assembly complex coordinates late thymic T-cell differentiation and regulatory T-cell homeostasis. Nat. Commun. 2016, 7, 13353. [Google Scholar] [CrossRef]
- Sasaki, Y.; Sano, S.; Nakahara, M.; Murata, S.; Kometani, K.; Aiba, Y.; Sakamoto, S.; Watanabe, Y.; Tanaka, K.; Kurosaki, T.; et al. Defective immune responses in mice lacking LUBAC-mediated linear ubiquitination in B cells. Embo J. 2013, 32, 2463–2476. [Google Scholar] [CrossRef] [Green Version]
- Annibaldi, A.; Meier, P. Ripk1 and haematopoiesis: A case for LUBAC and Ripk3. Cell Death Differ. 2018, 25, 1361. [Google Scholar] [CrossRef]
- Stieglitz, B.; Morris-Davies, A.C.; Koliopoulos, M.G.; Christodoulou, E.; Rittinger, K. LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep. 2012, 13, 840–846. [Google Scholar] [CrossRef]
- Fuseya, Y.; Fujita, H.; Kim, M.; Ohtake, F.; Nishide, A.; Sasaki, K.; Saeki, Y.; Tanaka, K.; Takahashi, R.; Iwai, K. The HOIL-1L ligase modulates immune signalling and cell death via monoubiquitination of LUBAC. Nat. Cell Biol. 2020, 22, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Kelsall, I.R.; McCrory, E.H.; Xu, Y.; Scudamore, C.L.; Nanda, S.K.; Mancebo-Gamella, P.; Wood, N.T.; Knebel, A.; Matthews, S.J.; Cohen, P. HOIL-1-catalysed ubiquitylation of unbranched glucosaccharides and its activation by ubiquitin oligomers. bioRxiv 2021. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israël, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Kim, C.; Bradfield, J.; Guo, Y.; Toskala, E.; Otieno, F.G.; Hou, C.; Thomas, K.; Cardinale, C.; Lyon, G.J.; et al. Whole-genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome. Med. 2013, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Krenn, M.; Salzer, E.; Simonitsch-Klupp, I.; Rath, J.; Wagner, M.; Haack, T.B.; Strom, T.M.; Schänzer, A.; Kilimann, M.W.; Schmidt, R.L.J.; et al. Mutations outside the N-terminal part of RBCK1 may cause polyglucosan body myopathy with immunological dysfunction: Expanding the genotype-phenotype spectrum. J. Neurol. 2018, 265, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Boisson, B.; Laplantine, E.; Dobbs, K.; Cobat, A.; Tarantino, N.; Hazen, M.; Lidov, H.G.W.; Hopkins, G.; Du, L.; Belkadi, A.; et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 2015, 212, 939–951. [Google Scholar] [CrossRef] [Green Version]
- Oda, H.; Beck, D.B.; Kuehn, H.S.; Moura, N.S.; Hoffmann, P.; Ibarra, M.; Stoddard, J.; Tsai, W.L.; Gutierrez-Cruz, G.; Gadina, M.; et al. Second Case of HOIP Deficiency Expands Clinical Features and Defines Inflammatory Transcriptome Regulated by LUBAC. Front. Immunol. 2019, 10, 479. [Google Scholar] [CrossRef] [Green Version]
- Asanomi, Y.; Shigemizu, D.; Miyashita, A.; Mitsumori, R.; Mori, T.; Hara, N.; Ito, K.; Niida, S.; Ikeuchi, T.; Ozaki, K. A rare functional variant of SHARPIN attenuates the inflammatory response and associates with increased risk of late-onset Alzheimer’s disease. Mol. Med. 2019, 25, 20. [Google Scholar] [CrossRef] [Green Version]
- Asanomi, Y.; Shigemizu, D.; Akiyama, S.; Miyashita, A.; Mitsumori, R.; Hara, N.; Ikeuchi, T.; Niida, S.; Ozaki, K. A functional variant of SHARPIN confers increased risk of late-onset Alzheimer’s disease. J. Hum. Genet. 2022, 67, 203–208. [Google Scholar] [CrossRef]
- Rivkin, E.; Almeida, S.M.; Ceccarelli, D.F.; Juang, Y.-C.; MacLean, T.A.; Srikumar, T.; Huang, H.; Dunham, W.H.; Fukumura, R.; Xie, G.; et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature 2013, 498, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.; McKenzie, A.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinngrebe, J.; Moepps, B.; Monecke, T.; Gierschik, P.; Schlichtig, F.; Barth, T.F.E.; Strauß, G.; Boldrin, E.; Posovszky, C.; Schulz, A.; et al. Compound heterozygous variants in OTULIN are associated with fulminant atypical late-onset ORAS. EMBO Mol. Med. 2022, 14, e14901. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yu, X.; Demirkaya, E.; Deuitch, N.; Stone, D.; Tsai, W.L.; Kuehn, H.S.; Wang, H.; Yang, D.; Park, Y.H.; et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA 2016, 113, 10127–10132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heger, K.; Wickliffe, K.E.; Ndoja, A.; Zhang, J.; Murthy, A.; Dugger, D.L.; Maltzman, A.; de Sousa e Melo, F.; Hung, J.; Zeng, Y.; et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 2018, 559, 120–124. [Google Scholar] [CrossRef]
- Hoste, E.; Lecomte, K.; Annusver, K.; Vandamme, N.; Roels, J.; Maschalidi, S.; Verboom, L.; Vikkula, H.-K.; Sze, M.; Van Hove, L.; et al. OTULIN maintains skin homeostasis by controlling keratinocyte death and stem cell identity. Nat. Commun. 2021, 12, 5913. [Google Scholar] [CrossRef]
- Schünke, H.; Göbel, U.; Dikic, I.; Pasparakis, M. OTULIN inhibits RIPK1-mediated keratinocyte necroptosis to prevent skin inflammation in mice. Nat. Commun. 2021, 12, 5912. [Google Scholar] [CrossRef]
- Damgaard, R.B.; Jolin, H.E.; Allison, M.E.D.; Davies, S.E.; Titheradge, H.L.; McKenzie, A.N.J.; Komander, D. OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death Differ. 2020, 27, 1457–1474. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef]
- Graham, R.R.; Cotsapas, C.; Davies, L.; Hackett, R.; Lessard, C.J.; Leon, J.M.; Burtt, N.P.; Guiducci, C.; Parkin, M.; Gates, C.; et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 1059–1061. [Google Scholar] [CrossRef]
- Plenge, R.M.; Cotsapas, C.; Davies, L.; Price, A.L.; Bakker, P.I.W.D.; Maller, J.; Pe’Er, I.; Burtt, N.P.; Blumenstiel, B.; DeFelice, M.; et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat. Genet. 2007, 39, 1477–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, W.; Wellcome Trust Case Control Consortium; Barton, A.; Walker, N.; Eyre, S.; Hinks, A.; Bowes, J.; Donn, R.; Symmons, D.; Hider, S.; et al. Rheumatoid arthritis association at 6q23. Nat. Genet. 2007, 39, 1431–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musone, S.L.; Taylor, K.E.; Lu, T.T.; Nititham, J.; Ferreira, R.C.; Ortmann, W.; Shifrin, N.; Petri, M.A.; Kamboh, M.I.; Manzi, S.; et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 1062–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.; et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Fung, E.Y.; Smyth, D.J.; Howson, J.M.; Cooper, J.D.; Walker, N.M.; Stevens, H.; Wicker, L.S.; Todd, J.A. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun. 2009, 10, 188–191. [Google Scholar] [CrossRef]
- Polykratis, A.; Martens, A.; Eren, R.O.; Shirasaki, Y.; Yamagishi, M.; Yamaguchi, Y.; Uemura, S.; Miura, M.; Holzmann, B.; Kollias, G.; et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 2019, 21, 731–742. [Google Scholar] [CrossRef]
- Wahida, A.; Müller, M.; Hiergeist, A.; Popper, B.; Steiger, K.; Branca, C.; Tschurtschenthaler, M.; Engleitner, T.; Donakonda, S.; De Coninck, J.; et al. XIAP restrains TNF-driven intestinal inflammation and dysbiosis by promoting innate immune responses of Paneth and dendritic cells. Sci. Immunol. 2021, 6, eabf7235. [Google Scholar] [CrossRef]
- Strigli, A.; Gopalakrishnan, S.; Zeissig, Y.; Basic, M.; Wang, J.; Schwerd, T.; Doms, S.; Peuker, K.; Hartwig, J.; Harder, J.; et al. Deficiency in X-linked inhibitor of apoptosis protein promotes susceptibility to microbial triggers of intestinal inflammation. Sci. Immunol. 2021, 6, eabf7473. [Google Scholar] [CrossRef]
- Rigaud, S.; Fondanèche, M.-C.; Lambert, N.C.; Pasquier, B.; Mateo, V.; Soulas-Sprauel, P.; Galicier, L.; Le Deist, F.; Rieux-Laucat, F.; Revy, P.; et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 2006, 444, 110–114. [Google Scholar] [CrossRef]
- Crowley, E.; Warner, N.; Pan, J.; Khalouei, S.; Elkadri, A.; Fiedler, K.; Foong, J.; Turinsky, A.L.; Bronte-Tinkew, D.; Zhang, S.; et al. Prevalence and Clinical Features of Inflammatory Bowel Diseases Associated With Monogenic Variants, Identified by Whole-Exome Sequencing in 1000 Children at a Single Center. Gastroenterology 2020, 158, 2208–2220. [Google Scholar] [CrossRef]
- Hrdinka, M.; Gyrd-Hansen, M. The Met1-Linked Ubiquitin Machinery: Emerging Themes of (De)regulation. Mol. Cell 2017, 68, 265–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, V.M.; Green, S.; Sarma, V.; Holzman, L.B.; Wolf, F.W.; O’Rourke, K.; Ward, P.A.; Prochownik, E.V.; Marks, R.M. Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J. Biol. Chem. 1990, 265, 2973–2978. [Google Scholar] [CrossRef]
- Opipari, A.W., Jr.; Hu, H.M.; Yabkowitz, R.; Dixit, V.M. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J. Biol. Chem. 1992, 267, 12424–12427. [Google Scholar] [CrossRef]
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000, 289, 2350–2354. [Google Scholar] [CrossRef] [PubMed]
- De Dainichi, A.T.; Rathinam, C.V.; Ghosh, S. The deubiquitinase activity of A20 is dispensable for NF-kappaB signaling. EMBO Rep. 2014, 15, 775–783. [Google Scholar]
- Lu, T.T.; Onizawa, M.; Hammer, G.; Turer, E.E.; Yin, Q.; Damko, E.; Agelidis, A.; Shifrin, N.; Advincula, R.; Barrera, J.; et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity 2013, 38, 896–905. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; Newton, K.; Seshasayee, D.; Kusam, S.; Lam, C.; Zhang, J.; Popovych, N.; Helgason, E.; Schoeffler, A.; Jeet, S.; et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 2015, 528, 370–375. [Google Scholar] [CrossRef]
- Skaug, B.; Chen, J.; Du, F.; He, J.; Ma, A.; Chen, Z.J. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol. Cell 2011, 44, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, F.; Nishimasu, H.; Ishitani, R.; Goto, E.; Noguchi, T.; Mio, K.; Kamei, K.; Ma, A.; Iwai, K.; Nureki, O. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-κB regulation. Embo J. 2012, 31, 3856–3870. [Google Scholar] [CrossRef]
- Martens, A.; van Loo, G. A20 at the Crossroads of Cell Death, Inflammation, and Autoimmunity. Cold Spring Harb. Perspect. Biol. 2020, 12, a036418. [Google Scholar] [CrossRef]
- Walle, L.V.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.-D.; Van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, A.; Malynn, B.A. A20: Linking a complex regulator of ubiquitylation to immunity and human disease. Nat. Rev. Immunol. 2012, 12, 774–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Shiozaki, E.; Srinivasula, S.M.; Wu, Q.; Datta, P.; Alnemri, E.S.; Shi, Y. Structural basis of caspase-7 inhibition by XIAP. Cell 2001, 104, 769–780. [Google Scholar] [CrossRef]
- Shiozaki, E.N.; Chai, J.; Rigotti, D.J.; Riedl, S.J.; Li, P.; Srinivasula, S.M.; Alnemri, E.S.; Fairman, R.; Shi, Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003, 11, 519–527. [Google Scholar] [CrossRef]
- Fesik, S.W.; Shi, Y. Structural biology. Controlling the caspases. Science 2001, 294, 1477–1478. [Google Scholar] [CrossRef]
- Scott, F.L.; Denault, J.B.; Riedl, S.J.; Shin, H.; Renatus, M.; Salvesen, G.S. XIAP inhibits caspase-3 and -7 using two binding sites: Evolutionarily conserved mechanism of IAPs. Embo J. 2005, 24, 645–655. [Google Scholar] [CrossRef]
- Damgaard, R.B.; Nachbur, U.; Yabal, M.; Wong, W.W.-L.; Fiil, B.K.; Kastirr, M.; Rieser, E.; Rickard, J.A.; Bankovacki, A.; Peschel, C.; et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. 2012, 46, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, K.E.; Feltham, R.; Yabal, M.; Conos, S.A.; Chen, K.W.; Ziehe, S.; Graß, C.; Zhan, Y.; Nguyen, T.A.; Hall, C.; et al. XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1beta Activation and Cell Death as a Consequence of TLR-MyD88-Induced cIAP1-TRAF2 Degradation. Cell Rep. 2017, 20, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Mudde, A.C.A.; Booth, C.; Marsh, R.A. Evolution of Our Understanding of XIAP Deficiency. Front. Pediatr. 2021, 9, 660520. [Google Scholar] [CrossRef]
- Filipovich, A.H.; Zhang, K.; Snow, A.L.; Marsh, R.A. X-linked lymphoproliferative syndromes: Brothers or distant cousins? Blood 2010, 116, 3398–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, R.A.; Madden, L.; Kitchen, B.J.; Mody, R.; McClimon, B.; Jordan, M.B.; Bleesing, J.J.; Zhang, K.; Filipovich, A.H. XIAP deficiency: A unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood 2010, 116, 1079–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeissig, Y.; Petersen, B.-S.; Milutinovic, S.; Bosse, E.; Mayr, G.; Peuker, K.; Hartwig, J.; Keller, A.; Kohl, M.; Laass, M.W.; et al. XIAP variants in male Crohn’s disease. Gut 2015, 64, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.J.; Bushell, M.; Jeffrey, I.W.; Pain, V.M.; Morley, S.J. Translation initiation factor modifications and the regulation of protein synthesis in apoptotic cells. Cell Death Differ. 2000, 7, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Tanzer, M.C.; Frauenstein, A.; Stafford, C.A.; Phulphagar, K.; Mann, M.; Meissner, F. Quantitative and Dynamic Catalogs of Proteins Released during Apoptotic and Necroptotic Cell Death. Cell Rep. 2020, 30, 1260.e5–1270.e5. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Mouse | Human | |||
|---|---|---|---|---|
| Gene | Tissue | Phenotype | Mutation | Phenotype |
| Hoip/ Hoil-1 | Full body deletion | -Embryonic lethality -Excessive endothelial cell death [39,43] | Deletion/ nonsense/ missense | -Autoinflammation (e.g., abdominal pain) -Immunodeficiency (recurrent bacterial infections) -Amylopectin-like deposit in the muscle -Cardiomyopathy -Progressive muscular weakness -Immune cells hyperresponsive to IL-1β stimulation -Delayed NF-kB activation in non-immune cells -Lymphopenia [53,54,55,56] |
| Skin-specific deletion | -Excessive keratinocyte death -Severe skin inflammation [44,45] | |||
| T cell-specific deletion | -Severe T cell depletion -Delayed NF-κB activation in TNF- and TCR-induced-signalling pathways [47] | |||
| Hoip | Liver-specific deletion | -Hepatocyte death-driven inflammation -Hepatocellular carcinoma [46] | ||
| B cell-specific deletion | -Impaired CD40 signalling and antibody production [48] | |||
| Hoil-1 | Catalytic inactive (Full body) | -Increased NF-κB activation -Protection from cell death -Glycogen deposition [51] | ||
| Sharpin | Full body mutation | -Chronic proliferative dermatitis -Liver inflammation -Peyer’s patches loss -Splenomegaly [40] | Missense | Risk factor for LOAD (Late-onset Alzheimer disease) [58,59] |
| Otulin | Full body deletion | -Embryonic lethality -Loss of vascularization [60] | Loss of function | -ORAS (OTULIN-related autoinflammatory syndrome) or Otulipenia -Systemic sterile inflammation (e.g., joint swelling, prolonged fever, diarrhoea, panniculitis) -Developmental delay -Increased linear ubiquitination and NF-κB activation -Infliximab (monoclonal anti-TNF antibody) ameliorates the symptoms [61,62,63] |
| Full body conditional deletion | Decreased survival [61] | |||
| Myeloid-cell specific deletion | -Severe acute systemic inflammation -NF-κB hyperactivation -Excessive cytokine production [61] | |||
| Catalytic inactivation (full body) | -Embryonic lethality -Deregulated endothelial cell death [64] | |||
| Skin-specific deletion | -Deregulated keratinocyte death -Severe skin inflammation [65,66] | |||
| Liver-specific deletion | -TNFR1-driven hepatocyte death -Compensatory proliferation -Hepatocellular carcinoma [67] | |||
| A20 | Full body deletion | -Perinatal lethality [68] -Cell death-dependent multiorgan inflammation (e.g., liver, kidneys, joints) | Nonsense | -Early onset systemic inflammation (e.g., arthritis, oral and genital ulcers, SLE-like disease, central nervous system vasculitis) -Patients’ derived immune cells have elevated cytokine levels, are hyperresponsive to inflammasome activation. Fibroblasts have increased NF-kB activation following TNF stimulation [69,70,71,72,73,74,75] |
| Myeloid cell-specific deletion | -Cell death-dependent joint inflammation [76] | |||
| ZnF7 mutation (full body) | -Cell death-dependent joint inflammation [76] | |||
| Xiap | Full body deletion | -Low grade ileal inflammation, TNFR1- and TNFR2-dependent -Increased sensitivity to pathogenic bacterial strain -Cell death-dependent reduction in Paneth cells and dendritic cells -Cell death is the trigger of ileal inflammation [77,78] | Deletion/ insertion/ nonsense/ missense/ frameshift/ intronic | -Familia hemophagocytic lymphohystiocystiosis (FHLH) or XLP2 -High risk IBD -Excessive activation of macrophages and dendritic cells following EBV infection [79,80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peltzer, N.; Annibaldi, A. Cell Death-Related Ubiquitin Modifications in Inflammatory Syndromes: From Mice to Men. Biomedicines 2022, 10, 1436. https://doi.org/10.3390/biomedicines10061436
Peltzer N, Annibaldi A. Cell Death-Related Ubiquitin Modifications in Inflammatory Syndromes: From Mice to Men. Biomedicines. 2022; 10(6):1436. https://doi.org/10.3390/biomedicines10061436
Chicago/Turabian StylePeltzer, Nieves, and Alessandro Annibaldi. 2022. "Cell Death-Related Ubiquitin Modifications in Inflammatory Syndromes: From Mice to Men" Biomedicines 10, no. 6: 1436. https://doi.org/10.3390/biomedicines10061436
APA StylePeltzer, N., & Annibaldi, A. (2022). Cell Death-Related Ubiquitin Modifications in Inflammatory Syndromes: From Mice to Men. Biomedicines, 10(6), 1436. https://doi.org/10.3390/biomedicines10061436