
Targeting Glioblastoma Stem Cells to Overcome Chemoresistance: An Overview of Current Therapeutic Strategies

 , and
, and
Abstract
1. Introduction
2. Mechanisms of Chemoresistance in GSCs
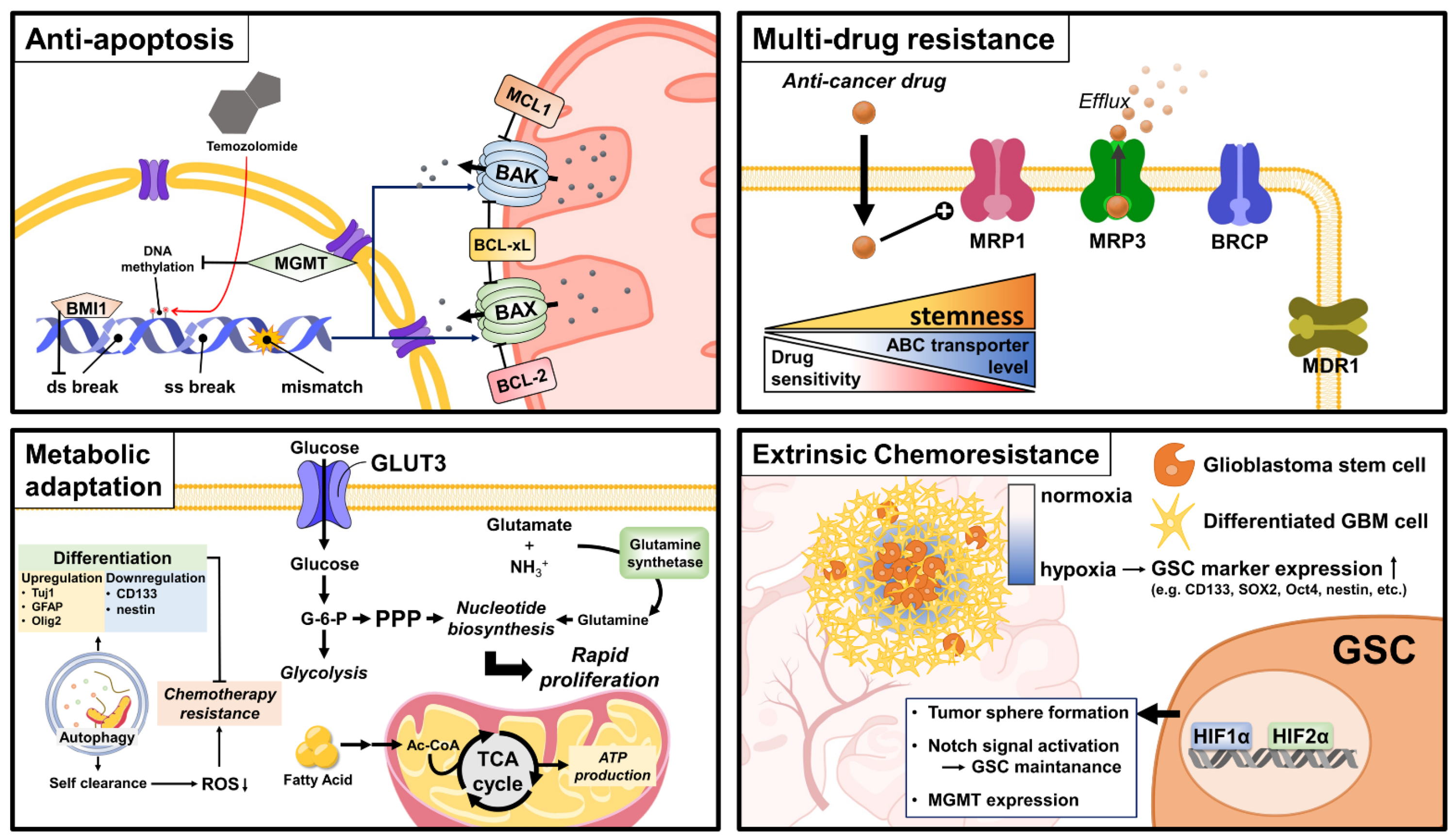
2.1. DNA Repair Systems
2.2. Anti-Apoptosis
2.3. Multidrug Resistance
2.4. Metabolism
2.5. Autophagy
2.6. Extrinsic Chemoresistance
3. Strategies Targeting GSCs
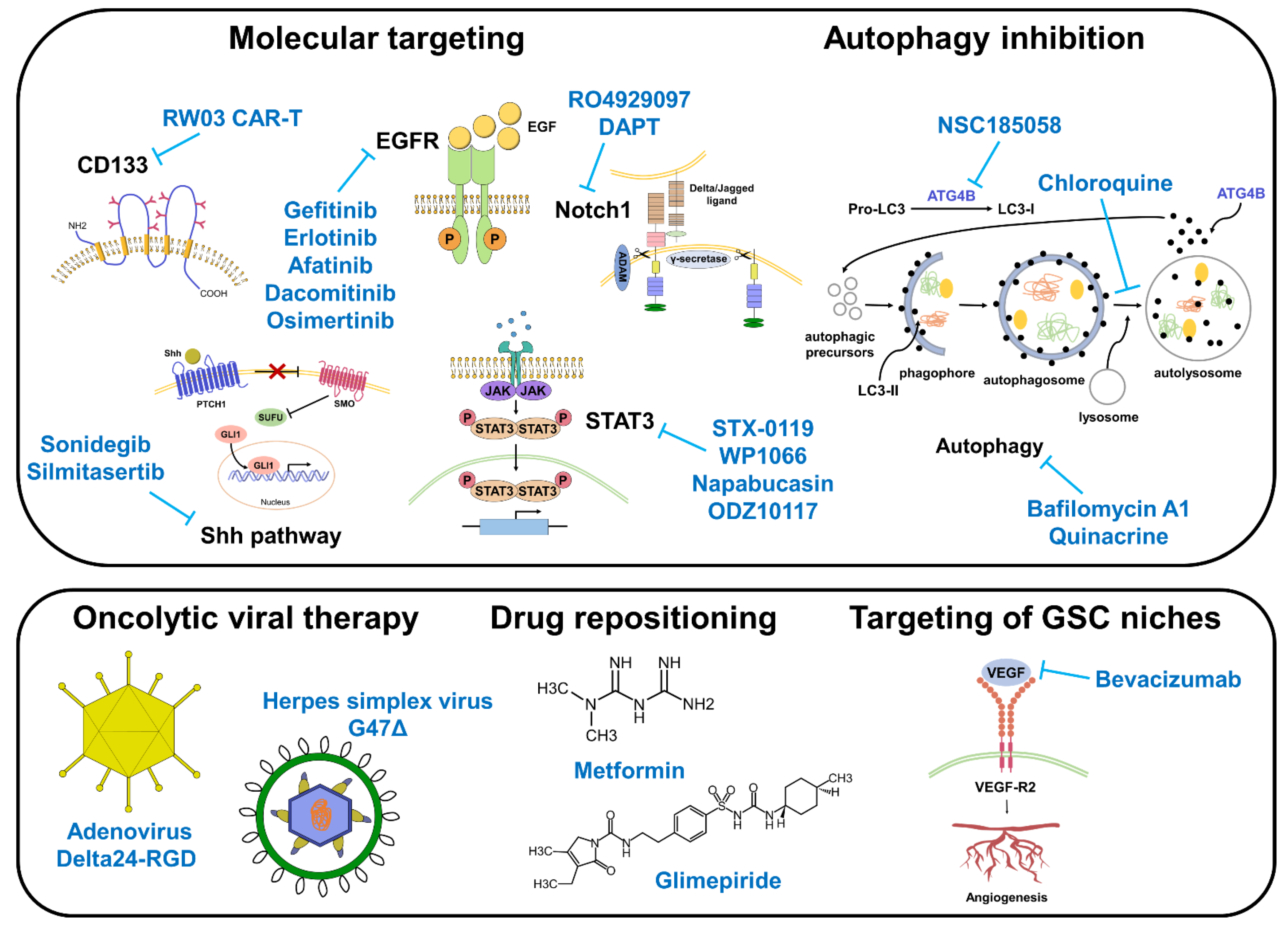
3.1. Targeting GSC Markers and Related Signaling Pathways
3.2. Targeting Autophagy
3.3. Oncolytic Viral Therapy
3.4. Drug Repositioning
3.5. Targeting GSC Niches
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23, iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Blanco, J.; Sanz-Arriazu, L.; Lorenzoni, R.; Blanco-Prieto, M.J. Glioblastoma chemotherapeutic agents used in the clinical setting and in clinical trials: Nanomedicine approaches to improve their efficacy. Int. J. Pharm. 2020, 581, 119283. [Google Scholar] [CrossRef] [PubMed]
- Idoate, M.A.; Díez Valle, R.; Echeveste, J.; Tejada, S. Pathological characterization of the glioblastoma border as shown during surgery using 5-aminolevulinic acid-induced fluorescence. Neuropathology 2011, 31, 575–582. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Wilson, T.A.; Karajannis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [CrossRef]
- Jeon, J.; Lee, S.; Kim, H.; Kang, H.; Youn, H.; Jo, S.; Youn, B.; Kim, H.Y. Revisiting Platinum-Based Anticancer Drugs to Overcome Gliomas. Int. J. Mol. Sci. 2021, 22, 5111. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Nakada, M.; Furuta, T.; Hayashi, Y.; Minamoto, T.; Hamada, J. The strategy for enhancing temozolomide against malignant glioma. Front. Oncol. 2012, 2, 98. [Google Scholar] [CrossRef]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Tirosh, I. The Glioma Stem Cell Model in the Era of Single-Cell Genomics. Cancer Cell 2020, 37, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem. Cell 2019, 24, 25–40. [Google Scholar] [CrossRef]
- Beier, D.; Röhrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008, 68, 5706–5715. [Google Scholar] [CrossRef]
- Gong, X.; Schwartz, P.H.; Linskey, M.E.; Bota, D.A. Neural stem/progenitors and glioma stem-like cells have differential sensitivity to chemotherapy. Neurology 2011, 76, 1126–1134. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’Avella, D.; Basso, G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem. Cells 2010, 28, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Blough, M.D.; Westgate, M.R.; Beauchamp, D.; Kelly, J.J.; Stechishin, O.; Ramirez, A.L.; Weiss, S.; Cairncross, J.G. Sensitivity to temozolomide in brain tumor initiating cells. Neuro-Oncol. 2010, 12, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.; Morey, L. Emerging Roles for Polycomb-Group Proteins in Stem Cells and Cancer. Trends Biochem. Sci. 2019, 44, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Facchino, S.; Chatoo, W.; Balasingam, V.; Ferreira, J.; Bernier, G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J. Neurosci. 2009, 29, 8884–8896. [Google Scholar] [CrossRef] [PubMed]
- Facchino, S.; Abdouh, M.; Chatoo, W.; Bernier, G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. 2010, 30, 10096–10111. [Google Scholar] [CrossRef]
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; et al. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol. Cancer Res. 2009, 7, 383–392. [Google Scholar] [CrossRef]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar]
- Hsieh, A.; Ellsworth, R.; Hsieh, D. Hedgehog/GLI1 regulates IGF dependent malignant behaviors in glioma stem cells. J. Cell Physiol. 2011, 226, 1118–1127. [Google Scholar] [CrossRef]
- Fanfone, D.; Idbaih, A.; Mammi, J.; Gabut, M.; Ichim, G. Profiling Anti-Apoptotic BCL-xL Protein Expression in Glioblastoma Tumorspheres. Cancers 2020, 12, 2853. [Google Scholar] [CrossRef]
- Alajez, N.M.; Shi, W.; Hui, A.B.; Yue, S.; Ng, R.; Lo, K.W.; Bastianutto, C.; O’Sullivan, B.; Gullane, P.; Liu, F.F. Targeted depletion of BMI1 sensitizes tumor cells to P53-mediated apoptosis in response to radiation therapy. Cell Death Differ. 2009, 16, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Saraswathy, M.; Gong, S. Different strategies to overcome multidrug resistance in cancer. Biotechnol. Adv. 2013, 31, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Feng, Y.; Kennedy, D. Multidrug-resistant cancer cells and cancer stem cells hijack cellular systems to circumvent systemic therapies, can natural products reverse this? Cell Mol. Life Sci. 2017, 74, 777–801. [Google Scholar] [CrossRef] [PubMed]
- Uribe, D.; Torres, Á.; Rocha, J.D.; Niechi, I.; Oyarzún, C.; Sobrevia, L.; San Martín, R.; Quezada, C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol. Asp. Med. 2017, 55, 140–151. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci 2017, 18, 2362. [Google Scholar] [CrossRef]
- Bi, C.L.; Fang, J.S.; Chen, F.H.; Wang, Y.J.; Wu, J. Chemoresistance of CD133(+) tumor stem cells from human brain glioma. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2007, 32, 568–573. [Google Scholar]
- Zhang, Y.; Wang, S.X.; Ma, J.W.; Li, H.Y.; Ye, J.C.; Xie, S.M.; Du, B.; Zhong, X.Y. EGCG inhibits properties of glioma stem-like cells and synergizes with temozolomide through downregulation of P-glycoprotein inhibition. J. Neurooncol. 2015, 121, 41–52. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Q.; Zhang, H.; Li, G.H.; Feng, S.; Yu, X.Z.; Kong, L.S.; Zhao, L.; Jin, F. Effect of siRNA-Livin on drug resistance to chemotherapy in glioma U251 cells and CD133(+) stem cells. Exp. Med. 2015, 10, 1317–1323. [Google Scholar] [CrossRef][Green Version]
- Jin, Y.; Bin, Z.Q.; Qiang, H.; Liang, C.; Hua, C.; Jun, D.; Dong, W.A.; Qing, L. ABCG2 is related with the grade of glioma and resistance to mitoxantone, a chemotherapeutic drug for glioma. J. Cancer Res. Clin. Oncol. 2009, 135, 1369–1376. [Google Scholar] [CrossRef]
- Martín, V.; Sanchez-Sanchez, A.M.; Herrera, F.; Gomez-Manzano, C.; Fueyo, J.; Alvarez-Vega, M.A.; Antolín, I.; Rodriguez, C. Melatonin-induced methylation of the ABCG2/BCRP promoter as a novel mechanism to overcome multidrug resistance in brain tumour stem cells. Br. J. Cancer 2013, 108, 2005–2012. [Google Scholar] [CrossRef]
- Hosokawa, Y.; Takahashi, H.; Inoue, A.; Kawabe, Y.; Funahashi, Y.; Kameda, K.; Sugimoto, K.; Yano, H.; Harada, H.; Kohno, S.; et al. Oct-3/4 modulates the drug-resistant phenotype of glioblastoma cells through expression of ATP binding cassette transporter G2. Biochim. Et Biophys. Acta 2015, 1850, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial⁻Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef]
- Son, B.; Lee, S.; Kim, H.; Kang, H.; Jeon, J.; Jo, S.; Seong, K.M.; Lee, S.J.; Youn, H.; Youn, B. Decreased FBP1 expression rewires metabolic processes affecting aggressiveness of glioblastoma. Oncogene 2020, 39, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lee, S.; Kim, K.; Jeon, J.; Kang, S.G.; Youn, H.; Kim, H.Y.; Youn, B. Downregulated CLIP3 induces radioresistance by enhancing stemness and glycolytic flux in glioblastoma. J. Exp. Clin. Cancer Res. 2021, 40, 282. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef]
- Ye, F.; Zhang, Y.; Liu, Y.; Yamada, K.; Tso, J.L.; Menjivar, J.C.; Tian, J.Y.; Yong, W.H.; Schaue, D.; Mischel, P.S.; et al. Protective properties of radio-chemoresistant glioblastoma stem cell clones are associated with metabolic adaptation to reduced glucose dependence. PLoS ONE 2013, 8, e80397. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- Yang, K.; Niu, L.; Bai, Y.; Le, W. Glioblastoma: Targeting the autophagy in tumorigenesis. Brain Res. Bull. 2019, 153, 334–340. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Das, B.C.; Ray, S.K. Targeting autophagy for combating chemoresistance and radioresistance in glioblastoma. Apoptosis 2018, 23, 563–575. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.; Kim, H.; Lee, H.; Seong, K.M.; Youn, H.; Youn, B. Autophagic Organelles in DNA Damage Response. Front. Cell Dev. Biol. 2021, 9, 668735. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef]
- Dolma, S.; Selvadurai, H.J.; Lan, X.; Lee, L.; Kushida, M.; Voisin, V.; Whetstone, H.; So, M.; Aviv, T.; Park, N.; et al. Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell 2016, 29, 859–873. [Google Scholar] [CrossRef]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef]
- Tao, Z.; Li, T.; Ma, H.; Yang, Y.; Zhang, C.; Hai, L.; Liu, P.; Yuan, F.; Li, J.; Yi, L.; et al. Autophagy suppresses self-renewal ability and tumorigenicity of glioma-initiating cells and promotes Notch1 degradation. Cell Death Dis. 2018, 9, 1063. [Google Scholar] [CrossRef]
- Zhuang, W.; Long, L.; Zheng, B.; Ji, W.; Yang, N.; Zhang, Q.; Liang, Z. Curcumin promotes differentiation of glioma-initiating cells by inducing autophagy. Cancer Sci. 2012, 103, 684–690. [Google Scholar] [CrossRef]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.Z.; Long, L.M.; Ji, W.J.; Liang, Z.Q. Rapamycin induces differentiation of glioma stem/progenitor cells by activating autophagy. Chin. J. Cancer 2011, 30, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells--much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvironment: Shaping the phenotype of cancer stem-like cells. Neuro-Oncology 2017, 19, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef]
- Persano, L.; Pistollato, F.; Rampazzo, E.; Della Puppa, A.; Abbadi, S.; Frasson, C.; Volpin, F.; Indraccolo, S.; Scienza, R.; Basso, G. BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1α stability and MGMT expression. Cell Death Dis. 2012, 3, e412. [Google Scholar] [CrossRef]
- Yin, J.; Ge, X.; Shi, Z.; Yu, C.; Lu, C.; Wei, Y.; Zeng, A.; Wang, X.; Yan, W.; Zhang, J.; et al. Extracellular vesicles derived from hypoxic glioma stem-like cells confer temozolomide resistance on glioblastoma by delivering miR-30b-3p. Theranostics 2021, 11, 1763–1779. [Google Scholar] [CrossRef]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e6. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib–A phase II trial. Mol. Cancer 2011, 10, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Giglio, P.; Hu, J.; Groves, M.; Merrell, R.; Conrad, C.; Phuphanich, S.; Puduvalli, V.K.; Loghin, M.; Paleologos, N.; et al. A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J. Neurooncol. 2016, 126, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro-Oncology 2013, 15, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y.; et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro-Oncology 2015, 17, 430–439. [Google Scholar] [CrossRef]
- Cardona, A.F.; Jaramillo-Velásquez, D.; Ruiz-Patiño, A.; Polo, C.; Jiménez, E.; Hakim, F.; Gómez, D.; Ramón, J.F.; Cifuentes, H.; Mejía, J.A.; et al. Efficacy of osimertinib plus bevacizumab in glioblastoma patients with simultaneous EGFR amplification and EGFRvIII mutation. J. Neurooncol. 2021, 154, 353–364. [Google Scholar] [CrossRef]
- Gilbert, C.A.; Daou, M.C.; Moser, R.P.; Ross, A.H. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res. 2010, 70, 6870–6879. [Google Scholar] [CrossRef]
- Hung, H.C.; Liu, C.C.; Chuang, J.Y.; Su, C.L.; Gean, P.W. Inhibition of Sonic Hedgehog Signaling Suppresses Glioma Stem-Like Cells Likely Through Inducing Autophagic Cell Death. Front. Oncol. 2020, 10, 1233. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Li, W.; Hang, C.; Dai, Y. Inhibition of Casein Kinase II by CX-4945, But Not Yes-associated protein (YAP) by Verteporfin, Enhances the Antitumor Efficacy of Temozolomide in Glioblastoma. Transl. Oncol. 2020, 13, 70–78. [Google Scholar] [CrossRef]
- Ashizawa, T.; Miyata, H.; Iizuka, A.; Komiyama, M.; Oshita, C.; Kume, A.; Nogami, M.; Yagoto, M.; Ito, I.; Oishi, T.; et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int. J. Oncol. 2013, 43, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Lee, H.; Park, C.G.; Jeong, A.J.; Lee, S.H.; Noh, K.H.; Park, J.B.; Lee, C.G.; Paek, S.H.; Kim, H.; et al. STAT3 Inhibitor ODZ10117 Suppresses Glioblastoma Malignancy and Prolongs Survival in a Glioblastoma Xenograft Model. Cells 2020, 9, 722. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855.e8. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Briceño, E.; López-González, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; Decarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef]
- Piper, K.; DePledge, L.; Karsy, M.; Cobbs, C. Glioma Stem Cells as Immunotherapeutic Targets: Advancements and Challenges. Front. Oncol. 2021, 11, 615704. [Google Scholar] [CrossRef]
- Wang, C.H.; Chiou, S.H.; Chou, C.P.; Chen, Y.C.; Huang, Y.J.; Peng, C.A. Photothermolysis of glioblastoma stem-like cells targeted by carbon nanotubes conjugated with CD133 monoclonal antibody. Nanomedicine 2011, 7, 69–79. [Google Scholar] [CrossRef]
- Aldape, K.D.; Ballman, K.; Furth, A.; Buckner, J.C.; Giannini, C.; Burger, P.C.; Scheithauer, B.W.; Jenkins, R.B.; James, C.D. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 2004, 63, 700–707. [Google Scholar] [CrossRef]
- Emlet, D.R.; Gupta, P.; Holgado-Madruga, M.; Del Vecchio, C.A.; Mitra, S.S.; Han, S.Y.; Li, G.; Jensen, K.C.; Vogel, H.; Xu, L.W.; et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 2014, 74, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Fernandez-Bruno, M.; Bracht, J.W.P.; Rosell, R. EGFR first- and second-generation TKIs-there is still place for them in EGFR-mutant NSCLC patients. Transl. Cancer Res. 2019, 8, S23–S47. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Du, X.; Yang, B.; An, Q.; Assaraf, Y.G.; Cao, X.; Xia, J. Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovation 2021, 2, 100103. [Google Scholar] [CrossRef] [PubMed]
- Colclough, N.; Chen, K.; Johnström, P.; Strittmatter, N.; Yan, Y.; Wrigley, G.L.; Schou, M.; Goodwin, R.; Varnäs, K.; Adua, S.J.; et al. Preclinical Comparison of the Blood-brain barrier Permeability of Osimertinib with Other EGFR TKIs. Clin. Cancer Res. 2021, 27, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Peled, N.; Kian, W.; Inbar, E.; Goldstein, I.M.; Zemel, M.; Rotem, O.; Rozenblum, A.B.; Nechushtan, H.; Dudnik, E.; Levin, D.; et al. Osimertinib in advanced EGFR-mutant lung adenocarcinoma with asymptomatic brain metastases: An open-label, 3-arm, phase II pilot study. Neurooncol. Adv. 2022, 4, vdab188. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Cho, H.J.; Choi, Y.D.; Oh, I.J.; Kim, Y.C. A Phase II Trial of Osimertinib in the Second-Line Treatment of Non-small Cell Lung Cancer with the EGFR T790M Mutation, Detected from Circulating Tumor DNA: LiquidLung-O-Cohort 2. Cancer Res. Treat. 2019, 51, 777–787. [Google Scholar] [CrossRef]
- Liu, X.; Chen, X.; Shi, L.; Shan, Q.; Cao, Q.; Yue, C.; Li, H.; Li, S.; Wang, J.; Gao, S.; et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 219. [Google Scholar] [CrossRef]
- Makhlin, I.; Salinas, R.D.; Zhang, D.; Jacob, F.; Ming, G.L.; Song, H.; Saxena, D.; Dorsey, J.F.; Nasrallah, M.P.; Morrissette, J.J.; et al. Clinical activity of the EGFR tyrosine kinase inhibitor osimertinib in EGFR-mutant glioblastoma. CNS Oncol. 2019, 8, Cns43. [Google Scholar] [CrossRef]
- Olsauskas-Kuprys, R.; Zlobin, A.; Osipo, C. Gamma secretase inhibitors of Notch signaling. OncoTargets Ther. 2013, 6, 943–955. [Google Scholar] [CrossRef]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H.; et al. A high Notch pathway activation predicts response to γ secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem. Cells 2014, 32, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Aoki, K.; Hirai, N.; Fujita, S.; Iwama, J.; Hiramoto, Y.; Ishii, M.; Sato, K.; Nakayama, H.; Harashina, J.; et al. Effect of Notch expression in glioma stem cells on therapeutic response to chemo-radiotherapy in recurrent glioblastoma. Brain Tumor. Pathol. 2015, 32, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Pan, E.; Supko, J.G.; Kaley, T.J.; Butowski, N.A.; Cloughesy, T.; Jung, J.; Desideri, S.; Grossman, S.; Ye, X.; Park, D.M. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J. Neurooncol. 2016, 130, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef]
- Torrisi, F.; Alberghina, C.; Lo Furno, D.; Zappalà, A.; Valable, S.; Li Volti, G.; Tibullo, D.; Vicario, N.; Parenti, R. Connexin 43 and Sonic Hedgehog Pathway Interplay in Glioblastoma Cell Proliferation and Migration. Biology 2021, 10, 767. [Google Scholar] [CrossRef]
- Fu, J.; Rodova, M.; Nanta, R.; Meeker, D.; Van Veldhuizen, P.J.; Srivastava, R.K.; Shankar, S. NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro. Oncol. 2013, 15, 691–706. [Google Scholar] [CrossRef]
- Nitta, R.T.; Gholamin, S.; Feroze, A.H.; Agarwal, M.; Cheshier, S.H.; Mitra, S.S.; Li, G. Casein kinase 2α regulates glioblastoma brain tumor-initiating cell growth through the β-catenin pathway. Oncogene 2015, 34, 3688–3699. [Google Scholar] [CrossRef]
- Nitta, R.T.; Bolin, S.; Luo, E.; Solow-Codero, D.E.; Samghabadi, P.; Purzner, T.; Aujla, P.S.; Nwagbo, G.; Cho, Y.J.; Li, G. Casein kinase 2 inhibition sensitizes medulloblastoma to temozolomide. Oncogene 2019, 38, 6867–6879. [Google Scholar] [CrossRef]
- Stechishin, O.D.; Luchman, H.A.; Ruan, Y.; Blough, M.D.; Nguyen, S.A.; Kelly, J.J.; Cairncross, J.G.; Weiss, S. On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro. Oncol. 2013, 15, 198–207. [Google Scholar] [CrossRef]
- Shi, Y.; Guryanova, O.A.; Zhou, W.; Liu, C.; Huang, Z.; Fang, X.; Wang, X.; Chen, C.; Wu, Q.; He, Z.; et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 2018, 10, eaah6816. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Xue, Y.; Yan, X.; Li, J.; Wu, Y.; Guo, R.; Zhang, J.; Zhang, L.; Li, Y.; Liu, Y.; et al. Autophagy inhibitor chloroquine induces apoptosis of cholangiocarcinoma cells via endoplasmic reticulum stress. Oncol. Lett. 2018, 16, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef]
- Firat, E.; Weyerbrock, A.; Gaedicke, S.; Grosu, A.L.; Niedermann, G. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote γ-irradiation-induced cell death in primary stem-like glioma cells. PLoS ONE 2012, 7, e47357. [Google Scholar] [CrossRef]
- Dey, M.; Ulasov, I.V.; Tyler, M.A.; Sonabend, A.M.; Lesniak, M.S. Cancer stem cells: The final frontier for glioma virotherapy. Stem. Cell Rev. Rep. 2011, 7, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: Role of autophagic cell death. J. Natl. Cancer Instig. 2007, 99, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- van den Bossche, W.B.L.; Kleijn, A.; Teunissen, C.E.; Voerman, J.S.A.; Teodosio, C.; Noske, D.P.; van Dongen, J.J.M.; Dirven, C.M.F.; Lamfers, M.L.M. Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro. Oncol. 2018, 20, 1494–1504. [Google Scholar] [CrossRef]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T., Jr.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.C.; et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef]
- Maraka, S.; Groves, M.D.; Mammoser, A.G.; Melguizo-Gavilanes, I.; Conrad, C.A.; Tremont-Lukats, I.W.; Loghin, M.E.; O’Brien, B.J.; Puduvalli, V.K.; Sulman, E.P.; et al. Phase 1 lead-in to a phase 2 factorial study of temozolomide plus memantine, mefloquine, and metformin as postradiation adjuvant therapy for newly diagnosed glioblastoma. Cancer 2019, 125, 424–433. [Google Scholar] [CrossRef]
- Würth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem. Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Aderetti, D.A.; Hira, V.V.V.; Molenaar, R.J.; van Noorden, C.J.F. The hypoxic peri-arteriolar glioma stem cell niche, an integrated concept of five types of niches in human glioblastoma. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, N.; Chen, T.C.; Hofman, F.M. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett. 2016, 380, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Shiras, A. Cancer stem cell-vascular endothelial cell interactions in glioblastoma. Biochem. Biophys. Res. Commun. 2016, 473, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, M.; Pavlakis, N.; Wheeler, H.; Grant, R.; Simes, J.; Khasraw, M. Anti-angiogenic therapy for high-grade glioma. Cochrane Database Syst. Rev. 2018, 11, Cd008218. [Google Scholar] [CrossRef]
- Sandmann, T.; Bourgon, R.; Garcia, J.; Li, C.; Cloughesy, T.; Chinot, O.L.; Wick, W.; Nishikawa, R.; Mason, W.; Henriksson, R.; et al. Patients With Proneural Glioblastoma May Derive Overall Survival Benefit From the Addition of Bevacizumab to First-Line Radiotherapy and Temozolomide: Retrospective Analysis of the AVAglio Trial. J. Clin. Oncol. 2015, 33, 2735–2744. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, P.; Fu, Z.; Chen, X.; Liu, N.; Lu, A.; Li, R.; Shi, L.; Pu, P.; Kang, C.; et al. Glioma cells enhance endothelial progenitor cell angiogenesis via VEGFR-2, not VEGFR-1. Oncol. Rep. 2008, 20, 1457–1463. [Google Scholar]
- Yao, X.; Ping, Y.; Liu, Y.; Chen, K.; Yoshimura, T.; Liu, M.; Gong, W.; Chen, C.; Niu, Q.; Guo, D.; et al. Vascular endothelial growth factor receptor 2 (VEGFR-2) plays a key role in vasculogenic mimicry formation, neovascularization and tumor initiation by Glioma stem-like cells. PLoS ONE 2013, 8, e57188. [Google Scholar] [CrossRef]
- Reardon, D.A.; Turner, S.; Peters, K.B.; Desjardins, A.; Gururangan, S.; Sampson, J.H.; McLendon, R.E.; Herndon, J.E., 2nd; Jones, L.W.; Kirkpatrick, J.P.; et al. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J. Natl. Compr. Cancer Netw. 2011, 9, 414–427. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | Compound | Phase | NCT ID | Disease | Reference |
|---|---|---|---|---|---|
| CD133 | RW03 CAR-T | N/A | N/A | GBM | [71] |
| EGFR | Gefitinib | Phase II | NCT00052208 | Newly diagnosed GBM | [72] |
| NCT00250887 | Recurrent GBM | [73] | |||
| Erlotinib | Phase II | NCT00525525 | Newly diagnosed GBM | [74] | |
| NCT00445588 | Newly diagnosed GBM and Recurrent GBM | [75] | |||
| Afatinib | Phase II | NCT00727506 | Recurrent GBM | [76] | |
| Dacomitinib | Phase II | NCT01520870 | Recurrent GBM | [76] | |
| Osimertinib | N/A | N/A | GBM | [77] | |
| Notch1 | RO4929097 | Phase II | NCT01122901 | Newly diagnosed GBM and Recurrent GBM | N/A |
| DAPT | N/A | N/A | GBM | [78] | |
| Shh pathway | Sonidegib | N/A | N/A | GBM | [79] |
| Silmitasertib | N/A | N/A | GBM | [80] | |
| STAT3 | STX-0119 | N/A | N/A | Recurrent GBM | [81] |
| WP1066 | Phase I | NCT01904123 | Newly diagnosed GBM and Recurrent GBM | N/A | |
| Napabucasin | Phase I/II | NCT02315534 | Newly diagnosed GBM and Recurrent GBM | N/A | |
| ODZ10117 | N/A | N/A | GBM | [82] | |
| Autophagy | NSC185058 | N/A | N/A | GBM | [83] |
| Chloroquine | Phase III | NCT00224978 | Newly diagnosed GBM and Recurrent GBM | [84] | |
| Bafilomycin A1 | N/A | N/A | GBM | [85] | |
| Quinacrine | N/A | N/A | GBM | [86] | |
| VEGF | Bevacizumab | Approved | Recurrent GBM | [87] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.; Lee, H.; Kim, D.; Kim, B.; Kang, J.; Kim, H.Y.; Youn, H.; Youn, B. Targeting Glioblastoma Stem Cells to Overcome Chemoresistance: An Overview of Current Therapeutic Strategies. Biomedicines 2022, 10, 1308. https://doi.org/10.3390/biomedicines10061308
Kang H, Lee H, Kim D, Kim B, Kang J, Kim HY, Youn H, Youn B. Targeting Glioblastoma Stem Cells to Overcome Chemoresistance: An Overview of Current Therapeutic Strategies. Biomedicines. 2022; 10(6):1308. https://doi.org/10.3390/biomedicines10061308
Chicago/Turabian StyleKang, Hyunkoo, Haksoo Lee, Dahye Kim, Byeongsoo Kim, JiHoon Kang, Hae Yu Kim, HyeSook Youn, and BuHyun Youn. 2022. "Targeting Glioblastoma Stem Cells to Overcome Chemoresistance: An Overview of Current Therapeutic Strategies" Biomedicines 10, no. 6: 1308. https://doi.org/10.3390/biomedicines10061308
APA StyleKang, H., Lee, H., Kim, D., Kim, B., Kang, J., Kim, H. Y., Youn, H., & Youn, B. (2022). Targeting Glioblastoma Stem Cells to Overcome Chemoresistance: An Overview of Current Therapeutic Strategies. Biomedicines, 10(6), 1308. https://doi.org/10.3390/biomedicines10061308