
Metabolic Profile and Pathological Alterations in the Muscle of Patients with Early-Stage Amyotrophic Lateral Sclerosis

 ,
,  , , , , ,
, , , , ,  , , ,
, , , 
Abstract
1. Introduction
2. Material and Methods
2.1. Subjects’ Recruitment
2.2. Samples Collection
2.3. Metabolomics Analysis
2.4. RNA Extraction and RT-qPCR Analysis
2.5. Mitochondrial Enzymatic Activities
2.6. Electron Microscopy
2.7. Statistical Analysis
2.8. Data Availability
3. Results
3.1. Cohort Description
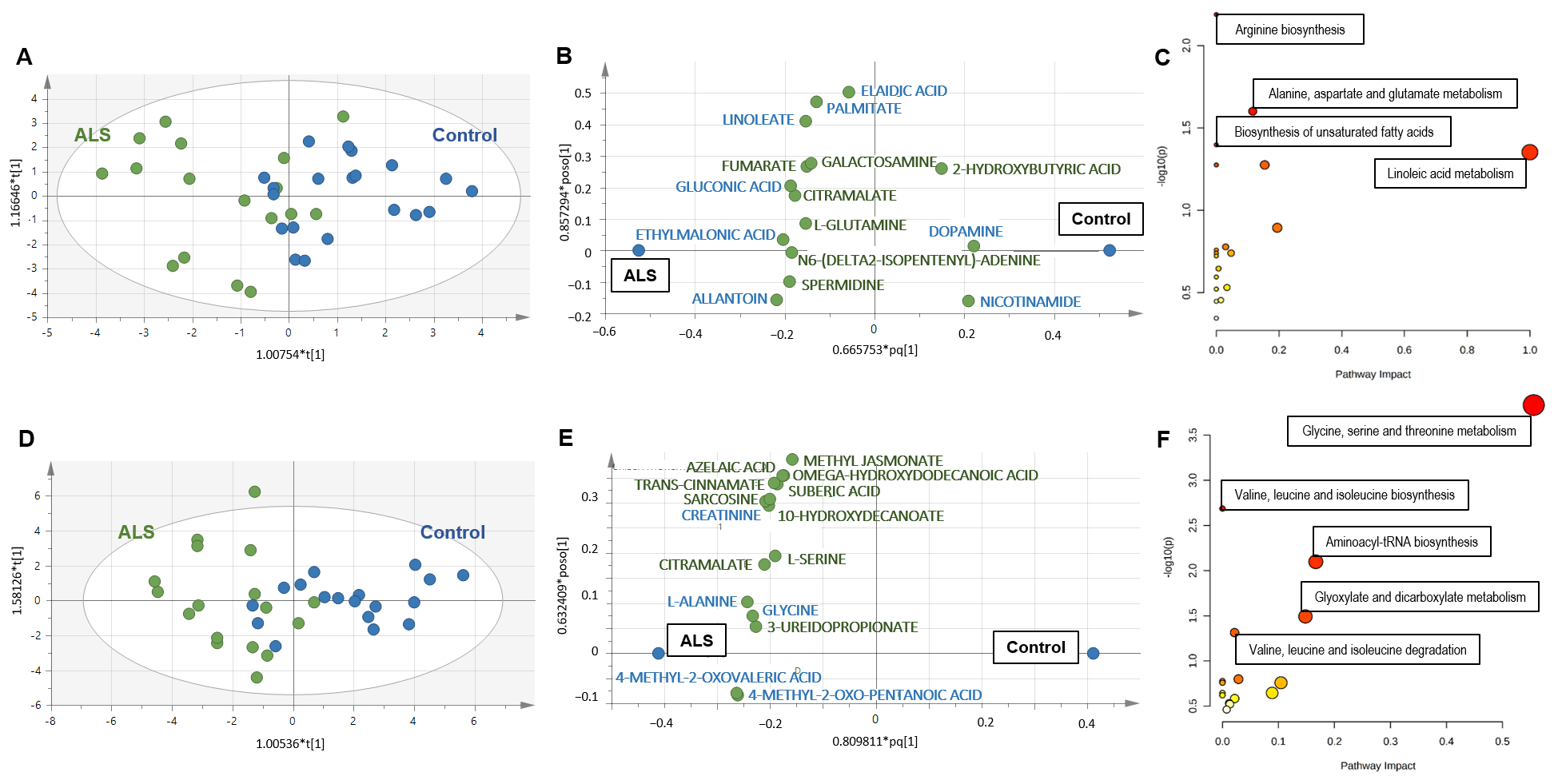
3.2. Metabolomics of Serum
3.2.1. Diagnosis Biomarkers
3.2.2. Biomarkers of Clinical Status
3.2.3. Prognosis Biomarkers
3.3. Metabolomics of Muscle
3.3.1. Diagnosis Biomarkers
3.3.2. Biomarkers of Clinical Status
3.3.3. Prognosis Biomarkers
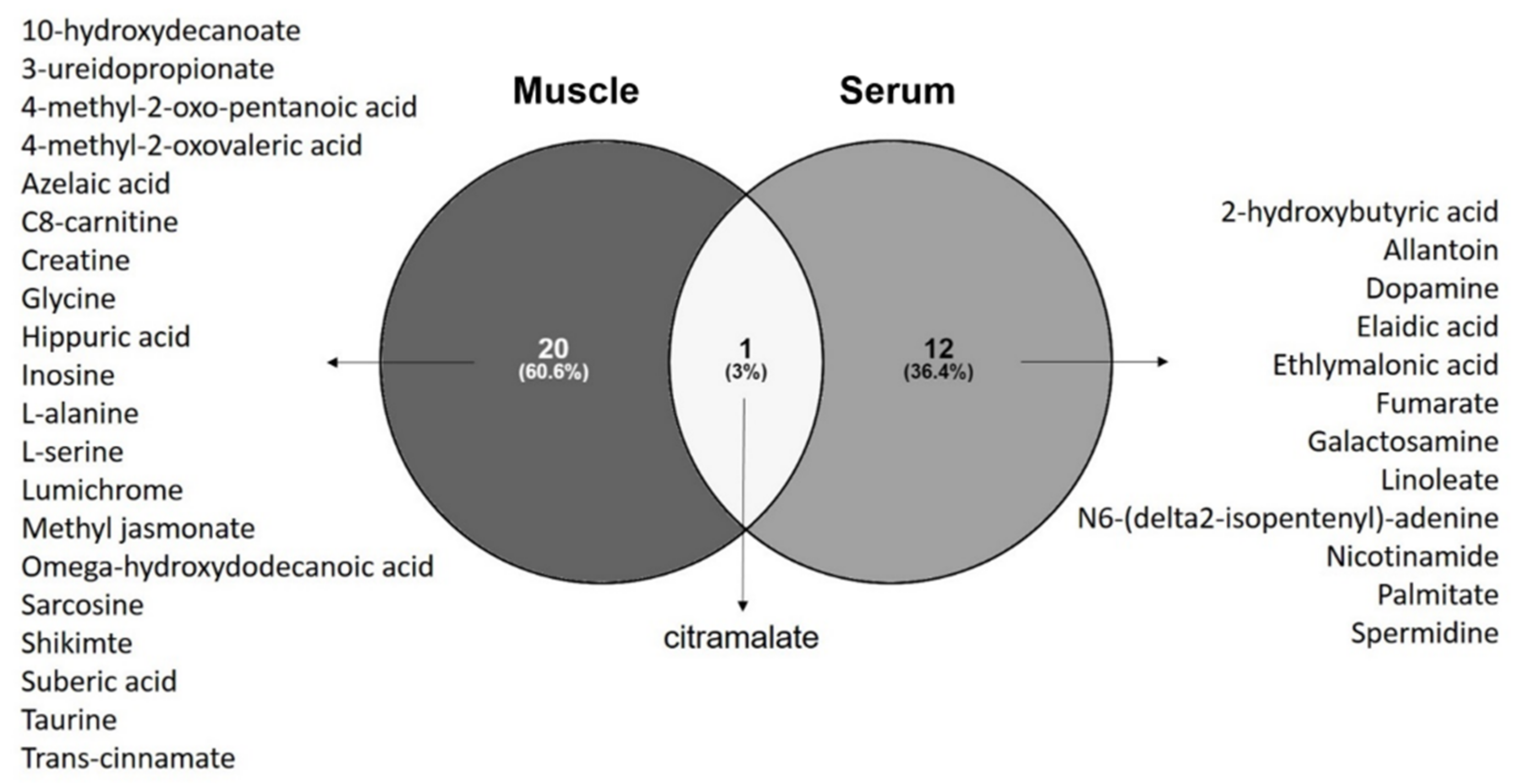
3.4. Common Metabolomics Alterations to Serum and Muscle
3.4.1. Diagnosis Biomarkers
3.4.2. Biomarkers of Clinical Status and for Prognosis
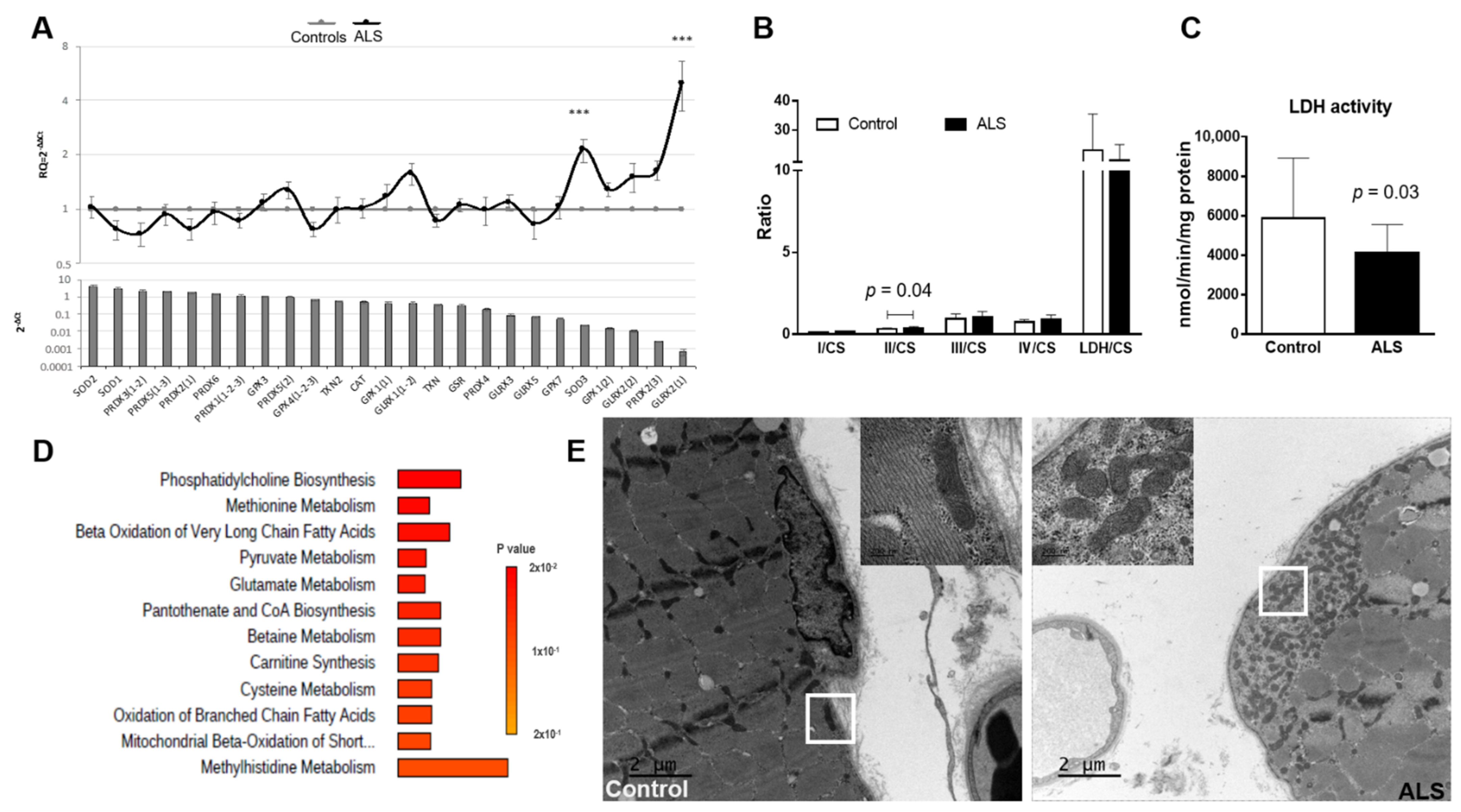
3.5. Antioxidant Genetic Profile of Muscle
3.6. Mitochondrial Enzymatic Activity in Muscle
3.7. Electron Microscopy Analysis of Muscle
4. Discussion
4.1. The Validity of a Panel for Metabolic Biomarkers
4.1.1. High Interest for Diagnosis Biomarkers
4.1.2. Mild Interest for Prognostic Biomarkers
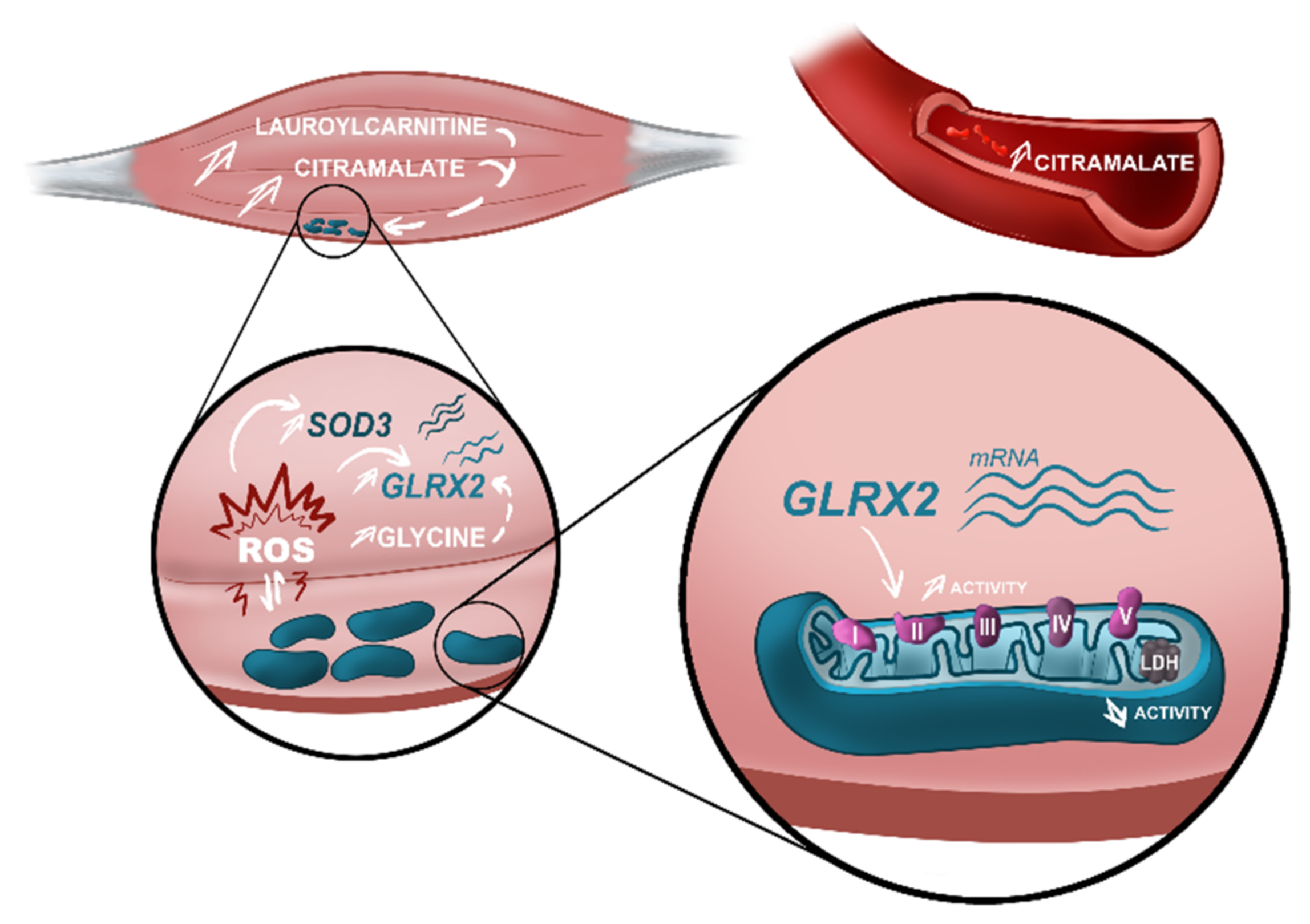
4.2. The Discriminant Ability of Citramalate in Muscle and Serum—New Biomarker for Diagnosis?
4.3. Consistency between Metabolomics Alterations and Pathophysiological Findings
4.3.1. Oxidative Stress Revealed by Superoxide Dismutase 3 (SOD3) and Glutaredoxin-2 (GLRX2(2)) Deregulation Combined with Amino Acids Dysmetabolism
4.3.2. Alterations in Mitochondrial Function and Distribution Reflect in the Intermediary Energetic Metabolism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rocchetti, I.; Taruscio, D.; Pierannunzio, D. Modeling delay to diagnosis for Amiotrophic lateral sclerosis: Under reporting and incidence estimates. BMC Neurol. 2012, 12, 160. [Google Scholar] [CrossRef]
- Shaw, P.J. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1046–1057. [Google Scholar] [CrossRef]
- Blasco, H.; Lanznaster, D.; Veyrat-Durebex, C.; Hergesheimer, R.; Vourch, P.; Maillot, F.; Andres, C.R.; Pradat, P.F.; Corcia, P. Understanding and managing metabolic dysfunction in Amyotrophic Lateral Sclerosis. Expert Rev. Neurother. 2020, 20, 907–919. [Google Scholar] [CrossRef]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in Amyotrophic Lateral Sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef]
- Tracey, T.J.; Kirk, S.E.; Steyn, F.J.; Ngo, S.T. The role of lipids in the central nervous system and their pathological implications in amyotrophic lateral sclerosis. Semin. Cell Dev. Biol. 2021, 112, 69–81. [Google Scholar] [CrossRef]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; De Aguilar, J.-L.G. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- Collis, W.J.; Engel, W.K. Glucose metabolism in five neuromuscular disorders. Neurology 1968, 18, 915–925. [Google Scholar] [CrossRef]
- Koerner, D.R. Abnormal carbohydrate metabolism in amyotrophic lateral sclerosis and Parkinsonism-dementia on Guam. Diabetes 1976, 25, 1055–1065. [Google Scholar] [CrossRef]
- Karpati, G.; Klassen, G.; Tanser, P. The effects of partial chronic denervation on forearm metabolism. Can. J. Neurol. Sci. 1979, 6, 105–112. [Google Scholar] [CrossRef]
- Moxley, R.T.; Griggs, R.C.; Forbes, G.B.; Goldblatt, D.; Donohoe, K. Influence of muscle wasting on oral glucose tolerance testing. Clin. Sci. 1983, 64, 601–609. [Google Scholar] [CrossRef]
- Reyes, E.T.; Perurena, O.H.; Festoff, B.W.; Jorgensen, R.; Moore, W.V. Insulin resistance in amyotrophic lateral sclerosis. J. Neurol. Sci. 1984, 63, 317–324. [Google Scholar] [CrossRef]
- Pradat, P.F.; Bruneteau, G.; Gordon, P.H.; Dupuis, L.; Bonnefont-Rousselot, D.; Simon, D.; Salachas, F.; Corcia, P.; Frochot, V.; Lacorte, J.M.; et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 11, 1–6. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Winkler, K.; Kuznetsov, A.V.; Bartels, C.; Vielhaber, S.; Feistner, H.; Kunz, W.S. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 156, 65–72. [Google Scholar] [CrossRef]
- Vielhaber, S.; Kunz, D.; Winkler, K.; Wiedemann, F.R.; Kirches, E.; Feistner, H.; Heinze, H.J.; Elger, C.E.; Schubert, W.; Kunz, W.S. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 2000, 123 Pt 7, 1339–1348. [Google Scholar] [CrossRef]
- Dupuis, L.; di Scala, F.; Rene, F.; de Tapia, M.; Oudart, H.; Pradat, P.F.; Meininger, V.; Loeffler, J.P. Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. FASEB J. 2003, 17, 2091–2093. [Google Scholar] [CrossRef]
- Krasnianski, A.; Deschauer, M.; Neudecker, S.; Gellerich, F.N.; Muller, T.; Schoser, B.G.; Krasnianski, M.; Zierz, S. Mitochondrial changes in skeletal muscle in amyotrophic lateral sclerosis and other neurogenic atrophies. Brain 2005, 128, 1870–1876. [Google Scholar] [CrossRef]
- Smittkamp, S.E.; Morris, J.K.; Bomhoff, G.L.; Chertoff, M.E.; Geiger, P.C.; Stanford, J.A. SOD1-G93A mice exhibit muscle-fiber-type-specific decreases in glucose uptake in the absence of whole-body changes in metabolism. Neurodegener. Dis. 2014, 13, 29–37. [Google Scholar] [CrossRef]
- Sassani, M.; Alix, J.J.; McDermott, C.J.; Baster, K.; Hoggard, N.; Wild, J.M.; Mortiboys, H.J.; Shaw, P.J.; Wilkinson, I.D.; Jenkins, T.M. Magnetic resonance spectroscopy reveals mitochondrial dysfunction in amyotrophic lateral sclerosis. Brain A J. Neurol. 2020, 143, 3603–3618. [Google Scholar] [CrossRef]
- Dieme, B.; Mavel, S.; Blasco, H.; Tripi, G.; Bonnet-Brilhault, F.; Malvy, J.; Bocca, C.; Andres, C.R.; Nadal-Desbarats, L.; Emond, P. Metabolomics Study of Urine in Autism Spectrum Disorders Using a Multiplatform Analytical Methodology. J. Proteome Res. 2015, 14, 5273–5282. [Google Scholar] [CrossRef]
- Hounoum, B.M.; Blasco, H.; Nadal-Desbarats, L.; Dieme, B.; Montigny, F.; Andres, C.R.; Emond, P.; Mavel, S. Analytical methodology for metabolomics study of adherent mammalian cells using NMR, GC-MS and LC-HRMS. Anal. Bioanal. Chem. 2015, 407, 8861–8872. [Google Scholar] [CrossRef]
- Bruno, C.; Patin, F.; Bocca, C.; Nadal-Desbarats, L.; Bonnier, F.; Reynier, P.; Emond, P.; Vourc’h, P.; Joseph-Delafont, K.; Corcia, P.; et al. The combination of four analytical methods to explore skeletal muscle metabolomics: Better coverage of metabolic pathways or a marketing argument? J. Pharm. Biomed. Anal. 2018, 148, 273–279. [Google Scholar] [CrossRef]
- Martias, C.; Baroukh, N.; Mavel, S.; Blasco, H.; Lefèvre, A.; Roch, L.; Montigny, F.; Gatien, J.; Schibler, L.; Dufour-Rainfray, D.; et al. Optimization of Sample Preparation for Metabolomics Exploration of Urine, Feces, Blood and Saliva in Humans Using Combined NMR and UHPLC-HRMS Platforms. Molecules 2021, 26, 4111. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.-E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Medja, F.; Allouche, S.; Frachon, P.; Jardel, C.; Malgat, M.; de Camaret, B.M.; Slama, A.; Lunardi, J.; Mazat, J.P.; Lombes, A. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009, 9, 331–339. [Google Scholar] [CrossRef]
- Lanznaster, D.; de Assis, D.R.; Corcia, P.; Pradat, P.F.; Blasco, H. Metabolomics Biomarkers: A Strategy Toward Therapeutics Improvement in ALS. Front. Neurol. 2018, 9, 1126. [Google Scholar] [CrossRef]
- Lanznaster, D.; Veyrat-Durebex, C.; Vourc’h, P.; Andres, C.R.; Blasco, H.; Corcia, P. Metabolomics: A Tool to Understand the Impact of Genetic Mutations in Amyotrophic Lateral Sclerosis. Genes 2020, 11, 537. [Google Scholar] [CrossRef]
- Kumar, A.; Bala, L.; Kalita, J.; Misra, U.K.; Singh, R.L.; Khetrapal, C.L.; Babu, G.N. Metabolomic analysis of serum by (1) H NMR spectroscopy in amyotrophic lateral sclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2010, 411, 563–567. [Google Scholar] [CrossRef]
- Yang, L.; Lv, X.; Du, H.; Wu, D.; Wang, M. Causal effects of serum metabolites on amyotrophic lateral sclerosis: A Mendelian randomization study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 97, 109771. [Google Scholar] [CrossRef]
- Patin, F.; Corcia, P.; Vourc’h, P.; Nadal-Desbarats, L.; Baranek, T.; Goossens, J.F.; Marouillat, S.; Dessein, A.F.; Descat, A.; Madji Hounoum, B.; et al. Omics to Explore Amyotrophic Lateral Sclerosis Evolution: The Central Role of Arginine and Proline Metabolism. Mol. Neurobiol. 2017, 54, 5361–5374. [Google Scholar] [CrossRef] [PubMed]
- Bereman, M.S.; Kirkwood, K.I.; Sabaretnam, T.; Furlong, S.; Rowe, D.B.; Guillemin, G.J.; Mellinger, A.L.; Muddiman, D.C. Metabolite Profiling Reveals Predictive Biomarkers and the Absence of beta-Methyl Amino-l-alanine in Plasma from Individuals Diagnosed with Amyotrophic Lateral Sclerosis. J. Proteome Res. 2020, 19, 3276–3285. [Google Scholar] [CrossRef]
- Goutman, S.A.; Boss, J.; Guo, K.; Alakwaa, F.M.; Patterson, A.; Kim, S.; Savelieff, M.G.; Hur, J.; Feldman, E.L. Untargeted metabolomics yields insight into ALS disease mechanisms. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Chen, Q.; Zhou, Q.; Zhang, R.; Jin, J.; Hu, F.; Liu, X.; Qin, X.; Kang, L.; Zhao, S.; et al. Characteristics of serum metabolites in sporadic amyotrophic lateral sclerosis patients based on gas chromatography-mass spectrometry. Sci. Rep. 2021, 11, 20786. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Beltran, S.; Bakkouche, S.E.; Couratier, P. Therapeutic news in ALS. Rev. Neurol. 2021, 177, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Mezzapesa, D.M.; D’Errico, E.; Tortelli, R.; Distaso, E.; Cortese, R.; Tursi, M.; Federico, F.; Zoccolella, S.; Logroscino, G.; Dicuonzo, F.; et al. Cortical thinning and clinical heterogeneity in amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e80748. [Google Scholar] [CrossRef] [PubMed]
- Germain, A.; Ruppert, D.; Levine, S.M.; Hanson, M.R. Metabolic profiling of a myalgic encephalomyelitis/chronic fatigue syndrome discovery cohort reveals disturbances in fatty acid and lipid metabolism. Mol. Biosyst. 2017, 13, 371–379. [Google Scholar] [CrossRef]
- Sampey, B.P.; Freemerman, A.J.; Zhang, J.; Kuan, P.F.; Galanko, J.A.; O’Connell, T.M.; Ilkayeva, O.R.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; et al. Metabolomic profiling reveals mitochondrial-derived lipid biomarkers that drive obesity-associated inflammation. PLoS ONE 2012, 7, e38812. [Google Scholar] [CrossRef]
- Zhao, S.; Feng, X.F.; Huang, T.; Luo, H.H.; Chen, J.X.; Zeng, J.; Gu, M.; Li, J.; Sun, X.Y.; Sun, D.; et al. The Association Between Acylcarnitine Metabolites and Cardiovascular Disease in Chinese Patients with Type 2 Diabetes Mellitus. Front. Endocrinol 2020, 11, 212. [Google Scholar] [CrossRef]
- Lim, M.A.; Bence, K.K.; Sandesara, I.; Andreux, P.; Auwerx, J.; Ishibashi, J.; Seale, P.; Kalb, R.G. Genetically altering organismal metabolism by leptin-deficiency benefits a mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 4995–5008. [Google Scholar] [CrossRef]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.W.; Will, A.D.; Peterson, G.W.; Sanchez, A.; Gillan, W.W.; Tan, S.A. Elevated plasma glucagon in amyotrophic lateral sclerosis. Neurology 1992, 42, 1532–1534. [Google Scholar] [CrossRef] [PubMed]
- Lehericey, G.; Le Forestier, N.; Dupuis, L.; Gonzalez-Bermejo, J.; Meininger, V.; Pradat, P.F. Nutritional management in amyotrophic lateral sclerosis: A medical and ethical stake. Presse Med. 2012, 41, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lu, C.J.; Chen, R.C.; Hou, W.H.; Li, C.Y. Risk of Amyotrophic Lateral Sclerosis in Patients with Diabetes: A Nationwide Population-Based Cohort Study. J. Epidemiol. 2015, 25, 445–451. [Google Scholar] [CrossRef]
- Browne, S.E.; Yang, L.; DiMauro, J.P.; Fuller, S.W.; Licata, S.C.; Beal, M.F. Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol. Dis. 2006, 22, 599–610. [Google Scholar] [CrossRef]
- Patin, F.; Baranek, T.; Vourc’h, P.; Nadal-Desbarats, L.; Goossens, J.F.; Marouillat, S.; Dessein, A.F.; Descat, A.; Hounoum, B.M.; Bruno, C.; et al. Combined Metabolomics and Transcriptomics Approaches to Assess the IL-6 Blockade as a Therapeutic of ALS: Deleterious Alteration of Lipid Metabolism. Neurother. J. Am. Soc. Exp. Neurother. 2016, 13, 905–917. [Google Scholar] [CrossRef]
- Tefera, T.W.; Borges, K. Neuronal glucose metabolism is impaired while astrocytic TCA cycling is unaffected at symptomatic stages in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Cereb. Blood Flow Metab. 2019, 39, 1710–1724. [Google Scholar] [CrossRef]
- Lazarus, R.; Sparrow, D.; Weiss, S.T. Baseline ventilatory function predicts the development of higher levels of fasting insulin and fasting insulin resistance index: The Normative Aging Study. Eur. Respir. J. 1998, 12, 641–645. [Google Scholar] [CrossRef]
- Milla, C.E.; Warwick, W.J.; Moran, A. Trends in pulmonary function in patients with cystic fibrosis correlate with the degree of glucose intolerance at baseline. Am. J. Respir. Crit. Care Med. 2000, 162, 891–895. [Google Scholar] [CrossRef]
- Hickson, D.A.; Burchfiel, C.M.; Liu, J.; Petrini, M.F.; Harrison, K.; White, W.B.; Sarpong, D.F. Diabetes, impaired glucose tolerance, and metabolic biomarkers in individuals with normal glucose tolerance are inversely associated with lung function: The Jackson Heart Study. Lung 2011, 189, 311–321. [Google Scholar] [CrossRef][Green Version]
- Nakayama, Y.; Shimizu, T.; Matsuda, C.; Haraguchi, M.; Hayashi, K.; Mochizuki, Y.; Nagao, M.; Kawata, A.; Isozaki, E. Non-motor manifestations in ALS patients with tracheostomy and invasive ventilation. Muscle Nerve 2018, 57, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Fallat, R.J.; Jewitt, B.; Bass, M.; Kamm, B.; Norris, F.H., Jr. Spirometry in amyotrophic lateral sclerosis. Arch. Neurol. 1979, 36, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Verma, R.; Garg, R.K.; Singh, M.K.; Shukla, R.; Verma, S.K. Assessment of respiratory functions by spirometry and phrenic nerve studies in patients of amyotrophic lateral sclerosis. J. Neurol. Sci. 2011, 306, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Daghlas, S.A.; Govindarajan, R.; Pooled Resource Open-Access, A.L.S.C.T.C. Relative effects of forced vital capacity and ALSFRS-R on survival in ALS. Muscle Nerve 2021, 64, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Paprotny, L.; Celejewska, A.; Frajberg, M.; Wianowska, D. Development and validation of GC-MS/MS method useful in diagnosing intestinal dysbiosis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1130–1131, 121822. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; De Marchi, F.; Niccolai, E.; Mandrioli, J.; Amedei, A. Gastrointestinal Status and Microbiota Shaping in Amyotrophic Lateral Sclerosis: A New Frontier for Targeting? In Amyotrophic Lateral Sclerosis; Araki, T., Ed.; EXON: Brisbane, Australia, 2021. [Google Scholar] [CrossRef]
- Saha, S.; Singh, S.; Prasad, S.; Mittal, A.; Sharma, A.K.; Chakrabarti, S. Gut Microbiota and Alzheimer’s Disease: Experimental Evidence and Clinical Reality. Curr. Alzheimer Res. 2021, 18, 787–801. [Google Scholar] [CrossRef]
- Yan, Z.; Yang, F.; Cao, J.; Ding, W.; Yan, S.; Shi, W.; Wen, S.; Yao, L. Alterations of gut microbiota and metabolome with Parkinson’s disease. Microb. Pathog. 2021, 160, 105187. [Google Scholar] [CrossRef]
- Wright, M.L.; Fournier, C.; Houser, M.C.; Tansey, M.; Glass, J.; Hertzberg, V.S. Potential Role of the Gut Microbiome in ALS: A Systematic Review. Biol Res. Nurs 2018, 20, 513–521. [Google Scholar] [CrossRef]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef]
- Zeng, Q.; Shen, J.; Chen, K.; Zhou, J.; Liao, Q.; Lu, K.; Yuan, J.; Bi, F. The alteration of gut microbiome and metabolism in amyotrophic lateral sclerosis patients. Sci. Rep. 2020, 10, 12998. [Google Scholar] [CrossRef]
- Chaleckis, R.; Ebe, M.; Pluskal, T.; Murakami, I.; Kondoh, H.; Yanagida, M. Unexpected similarities between the Schizosaccharomyces and human blood metabolomes, and novel human metabolites. Mol. Biosyst. 2014, 10, 2538–2551. [Google Scholar] [CrossRef] [PubMed]
- Mali, Y.; Zisapels, N. Gain of interaction of ALS-linked G93A superoxide dismutase with cytosolic malate dehydrogenase. Neurobiol. Dis. 2008, 32, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Mali, Y.; Zisapel, N. A novel decoy that interrupts G93A-superoxide dismutase gain of interaction with malate dehydrogenase improves survival in an amyotrophic lateral sclerosis cell model. J. Med. Chem. 2009, 52, 5442–5448. [Google Scholar] [CrossRef]
- Mali, Y.; Zisapel, N. VEGF up-regulation by G93A superoxide dismutase and the role of malate-aspartate shuttle inhibition. Neurobiol. Dis. 2010, 37, 673–681. [Google Scholar] [CrossRef]
- Ling, J.; Soll, D. Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proc. Natl. Acad. Sci. USA 2010, 107, 4028–4033. [Google Scholar] [CrossRef] [PubMed]
- Ham, D.J.; Gleeson, B.G.; Chee, A.; Baum, D.M.; Caldow, M.K.; Lynch, G.S.; Koopman, R. L-Citrulline Protects Skeletal Muscle Cells from Cachectic Stimuli through an iNOS-Dependent Mechanism. PLoS ONE 2015, 10, e0141572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Schmitt-Ulms, G.; Sato, C.; Xi, Z.; Zhang, Y.; Zhou, Y.; St George-Hyslop, P.; Rogaeva, E. Drug Repositioning for Alzheimer’s Disease Based on Systematic ‘omics’ Data Mining. PLoS ONE 2016, 11, e0168812. [Google Scholar] [CrossRef]
- Miller, O.G.; Mieyal, J.J. Critical Roles of Glutaredoxin in Brain Cells-Implications for Parkinson’s Disease. Antioxid. Redox Signal 2019, 30, 1352–1368. [Google Scholar] [CrossRef] [PubMed]
- Seco-Cervera, M.; Gonzalez-Cabo, P.; Pallardo, F.V.; Roma-Mateo, C.; Garcia-Gimenez, J.L. Thioredoxin and Glutaredoxin Systems as Potential Targets for the Development of New Treatments in Friedreich’s Ataxia. Antioxidants 2020, 9, 1257. [Google Scholar] [CrossRef]
- Jia, J.; Zeng, X.; Xu, G.; Wang, Z. The Potential Roles of Redox Enzymes in Alzheimer’s Disease: Focus on Thioredoxin. ASN Neuro 2021, 13, 1759091421994351. [Google Scholar] [CrossRef]
- Carroll, M.C.; Outten, C.E.; Proescher, J.B.; Rosenfeld, L.; Watson, W.H.; Whitson, L.J.; Hart, P.J.; Jensen, L.T.; Cizewski, C.V. The effects of glutaredoxin and copper activation pathways on the disulfide and stability of Cu, Zn superoxide dismutase. J. Biol. Chem. 2006, 281, 28648–28656. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Zaldiernas, C.; Lu, J.; Zheng, Y.; Yang, H.; Blasi, J.; Solsona, C.; Holmgren, A. Cellular Redox Systems Impact the Aggregation of Cu, Zn Superoxide Dismutase Linked to Familial Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2016, 291, 17197–17208. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Fiorenzo, P.; Nencini, M.; Cozzolino, M.; Pesaresi, M.G.; Valle, C.; Sepe, S.; Moreno, S.; Carri, M.T. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 2010, 19, 4529–4542. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Kramer, P.A.; Duan, J.; Gaffrey, M.J.; Shukla, A.K.; Wang, L.; Bammler, T.K.; Qian, W.J.; Marcinek, D.J. Fatiguing contractions increase protein S-glutathionylation occupancy in mouse skeletal muscle. Redox Biol. 2018, 17, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Chen, C.L.; Pfeiffer, D.R.; Zweier, J.L. Mitochondrial complex II in the post-ischemic heart: Oxidative injury and the role of protein S-glutathionylation. J. Biol. Chem. 2007, 282, 32640–32654. [Google Scholar] [CrossRef] [PubMed]
- Ost, M.; Keipert, S.; van Schothorst, E.M.; Donner, V.; van der Stelt, I.; Kipp, A.P.; Petzke, K.J.; Jove, M.; Pamplona, R.; Portero-Otin, M.; et al. Muscle mitohormesis promotes cellular survival via serine/glycine pathway flux. FASEB J. 2015, 29, 1314–1328. [Google Scholar] [CrossRef]
- Nikkanen, J.; Forsstrom, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamaki, J.; Roivainen, A.; Marjamaki, P.; et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab. 2016, 23, 635–648. [Google Scholar] [CrossRef]
- Koopman, R.; Caldow, M.K.; Ham, D.J.; Lynch, G.S. Glycine metabolism in skeletal muscle: Implications for metabolic homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 237–242. [Google Scholar] [CrossRef]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 2008, 108, 227–238. [Google Scholar] [CrossRef]
- Ullah, R.; Jo, M.H.; Riaz, M.; Alam, S.I.; Saeed, K.; Ali, W.; Rehman, I.U.; Ikram, M.; Kim, M.O. Glycine, the smallest amino acid, confers neuroprotection against D-galactose-induced neurodegeneration and memory impairment by regulating c-Jun N-terminal kinase in the mouse brain. J. Neuroinflamm. 2020, 17, 303. [Google Scholar] [CrossRef] [PubMed]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; de Leon, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: Prevention by mitochondrial-targeted antioxidants. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef] [PubMed]
- Calio, M.L.; Henriques, E.; Siena, A.; Bertoncini, C.R.A.; Gil-Mohapel, J.; Rosenstock, T.R. Mitochondrial Dysfunction, Neurogenesis, and Epigenetics: Putative Implications for Amyotrophic Lateral Sclerosis Neurodegeneration and Treatment. Front. Neurosci. 2020, 14, 679. [Google Scholar] [CrossRef] [PubMed]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef]
- Cassina, P.; Miquel, E.; Martinez-Palma, L.; Cassina, A. Glial Metabolic Reprogramming in Amyotrophic Lateral Sclerosis. Neuroimmunomodulation 2021, 28, 204–212. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; Lopez-Blanch, R.; Jihad-Jebbar, A.; Valles, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci 2021, 22, 6352. [Google Scholar] [CrossRef]
- Zhang, S.; Zeng, X.; Ren, M.; Mao, X.; Qiao, S. Novel metabolic and physiological functions of branched chain amino acids: A review. J. Anim. Sci. Biotechnol. 2017, 8, 10. [Google Scholar] [CrossRef]
- Testa, D.; Caraceni, T.; Fetoni, V. Branched-chain amino acids in the treatment of amyotrophic lateral sclerosis. J. Neurol. 1989, 236, 445–447. [Google Scholar] [CrossRef]
- Group, T.I.A.S. Branched-chain amino acids and amyotrophic lateral sclerosis: A treatment failure? The Italian ALS Study Group. Neurology 1993, 43, 2466–2470. [Google Scholar] [CrossRef]
- Tandan, R.; Bromberg, M.B.; Forshew, D.; Fries, T.J.; Badger, G.J.; Carpenter, J.; Krusinski, P.B.; Betts, E.F.; Arciero, K.; Nau, K. A controlled trial of amino acid therapy in amyotrophic lateral sclerosis: I. Clinical, functional, and maximum isometric torque data. Neurology 1996, 47, 1220–1226. [Google Scholar] [CrossRef]
- Gannon, N.P.; Schnuck, J.K.; Vaughan, R.A. BCAA Metabolism and Insulin Sensitivity-Dysregulated by Metabolic Status? Mol. Nutr. Food Res. 2018, 62, e1700756. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; De Aguilar, J.L.G.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.J.; Lacomblez, L.; Loeffler, J.P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, M.K.; Lee, E.; Bradburn, M.; McDermott, C.J.; Shaw, P.J. Effect of lipid profile on prognosis in the patients with amyotrophic lateral sclerosis: Insights from the olesoxime clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Fergani, A.; Oudart, H.; De Aguilar, J.L.G.; Fricker, B.; Rene, F.; Hocquette, J.F.; Meininger, V.; Dupuis, L.; Loeffler, J.P. Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J. Lipid Res. 2007, 48, 1571–1580. [Google Scholar] [CrossRef]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.L.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.P.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Osmundsen, H.; Hovik, R. Beta-oxidation of polyunsaturated fatty acids. Biochem. Soc. Trans. 1988, 16, 420–422. [Google Scholar] [CrossRef]
- Chegary, M.; Brinke, H.; Ruiter, J.P.; Wijburg, F.A.; Stoll, M.S.; Minkler, P.E.; van Weeghel, M.; Schulz, H.; Hoppel, C.L.; Wanders, R.J.; et al. Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim. Biophys. Acta 2009, 1791, 806–815. [Google Scholar] [CrossRef]
- Grimaldi, P.A. Fatty acid regulation of gene expression. Curr. Opin. Clin. Nutr Metab. Care 2001, 4, 433–437. [Google Scholar] [CrossRef]
- Brand, A.; Gil, S.; Seger, R.; Yavin, E. Lipid constituents in oligodendroglial cells alter susceptibility to H2O2-induced apoptotic cell death via ERK activation. J. Neurochem. 2001, 76, 910–918. [Google Scholar] [CrossRef]
- Nowak, J.Z. Oxidative stress, polyunsaturated fatty acids-derived oxidation products and bisretinoids as potential inducers of CNS diseases: Focus on age-related macular degeneration. Pharmacol. Rep. 2013, 65, 288–304. [Google Scholar] [CrossRef]
- Cacabelos, D.; Ayala, V.; Granado-Serrano, A.B.; Jove, M.; Torres, P.; Boada, J.; Cabre, R.; Ramirez-Nunez, O.; Gonzalo, H.; Soler-Cantero, A.; et al. Interplay between TDP-43 and docosahexaenoic acid-related processes in amyotrophic lateral sclerosis. Neurobiol. Dis. 2016, 88, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Zoll, J.; Ribera, F.; Tranchant, C.; Warter, J.M.; Lonsdorfer, J.; Lampert, E. Mitochondrial respiratory chain function in skeletal muscle of ALS patients. Ann. Neurol. 2002, 52, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Vielhaber, S.; Winkler, K.; Kirches, E.; Kunz, D.; Buchner, M.; Feistner, H.; Elger, C.E.; Ludolph, A.C.; Riepe, M.W.; Kunz, W.S. Visualization of defective mitochondrial function in skeletal muscle fibers of patients with sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 1999, 169, 133–139. [Google Scholar] [CrossRef]
- Soraru, G.; Vergani, L.; Fedrizzi, L.; D’Ascenzo, C.; Polo, A.; Bernazzi, B.; Angelini, C. Activities of mitochondrial complexes correlate with nNOS amount in muscle from ALS patients. Neuropathol. Appl. Neurobiol. 2007, 33, 204–211. [Google Scholar] [CrossRef]
- Al-Sarraj, S.; King, A.; Cleveland, M.; Pradat, P.F.; Corse, A.; Rothstein, J.D.; Leigh, P.N.; Abila, B.; Bates, S.; Wurthner, J.; et al. Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. Acta Neuropathol. Commun. 2014, 2, 165. [Google Scholar] [CrossRef]
- Lynch, E.; Semrad, T.; Belsito, V.S.; FitzGibbons, C.; Reilly, M.; Hayakawa, K.; Suzuki, M. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis. Model. Mech. 2019, 12, dmm039552. [Google Scholar] [CrossRef]
- Badu-Mensah, A.; Guo, X.; McAleer, C.W.; Rumsey, J.W.; Hickman, J.J. Functional skeletal muscle model derived from SOD1-mutant ALS patient iPSCs recapitulates hallmarks of disease progression. Sci. Rep. 2020, 10, 14302. [Google Scholar] [CrossRef]
- Mandler, R.N.; Baca, J.M. Muscle lactate dehydrogenase activity is decreased in murine motor neuron disease. Brain Res. 1992, 576, 337–338. [Google Scholar] [CrossRef]
- Cederblad, G.; Bylund, A.C.; Holm, J.; Schersten, T. Carnitine concentration in relation to enzyme activities and substrate utilization in human skeletal muscles. Scand. J. Clin. Lab. Investig. 1976, 36, 547–552. [Google Scholar] [CrossRef]
- Joardar, A.; Manzo, E.; Zarnescu, D.C. Metabolic Dysregulation in Amyotrophic Lateral Sclerosis: Challenges and Opportunities. Curr. Genet. Med. Rep. 2017, 5, 108–114. [Google Scholar] [CrossRef]
- Afifi, A.K.; Aleu, F.P.; Goodgold, J.; MacKay, B. Ultrastructure of atrophic muscle in amyotrophic lateral sclerosis. Neurology 1966, 16, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Inoue, K.; Munakata, S.; Nagao, K.; Shimizu, N. Muscle pathology in the early stage of Guillain-Barre syndrome. Eur. Neurol. 1998, 39, 141–147. [Google Scholar] [CrossRef]
- Chung, M.J.; Suh, Y.L. Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct. Pathol. 2002, 26, 3–7. [Google Scholar] [CrossRef] [PubMed]
- De Rose, F.; Marotta, R.; Talani, G.; Catelani, T.; Solari, P.; Poddighe, S.; Borghero, G.; Marrosu, F.; Sanna, E.; Kasture, S.; et al. Differential effects of phytotherapic preparations in the hSOD1 Drosophila melanogaster model of ALS. Sci. Rep. 2017, 7, 41059. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Lee, S.H.; Yang, E.J. Bojungikgi-tang Improves Muscle and Spinal Cord Function in an Amyotrophic Lateral Sclerosis Model. Mol. Neurobiol. 2019, 56, 2394–2407. [Google Scholar] [CrossRef]
- Higgins, C.M.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef]
- Abbasabadi, A.O.; Javanian, A.; Nikkhah, M.; Meratan, A.A.; Ghiasi, P.; Nemat-Gorgani, M. Disruption of mitochondrial membrane integrity induced by amyloid aggregates arising from variants of SOD1. Int. J. Biol. Macromol. 2013, 61, 212–217. [Google Scholar] [CrossRef]
- Oka, S.; Hirai, J.; Yasukawa, T.; Nakahara, Y.; Inoue, Y.H. A correlation of reactive oxygen species accumulation by depletion of superoxide dismutases with age-dependent impairment in the nervous system and muscles of Drosophila adults. Biogerontology 2015, 16, 485–501. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef]
- Debska-Vielhaber, G.; Miller, I.; Peeva, V.; Zuschratter, W.; Walczak, J.; Schreiber, S.; Petri, S.; Machts, J.; Vogt, S.; Szczepanowska, J.; et al. Impairment of mitochondrial oxidative phosphorylation in skin fibroblasts of SALS and FALS patients is rescued by in vitro treatment with ROS scavengers. Exp. Neurol. 2021, 339, 113620. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Controls | ALS | p |
|---|---|---|---|
| Age | 56.9 ± 19.2 | 65.9 ± 9.9 | 0.0793 |
| Sex (men) | 8/20 (40%) | 8/17 (47%) | 0.746 * |
| Weight (kg) | 69.4 ± 13.6 | 64.2 ± 15.8 | 0.299 |
| BMI (kg/m2) | 24.9 ± 3.8 | 23.5 ± 4.0 | 0.3024 |
| Age at onset (years) | - | 65.0 ± 9.5 | |
| Disease duration (from onset; months) | - | 11.1 ± 6.8 | |
| Diagnosis delay (from onset; months) | - | 9.3 ± 4.7 | |
| Spinal onset | - | 64.7% | |
| FVC at diagnosis (%) | - | 98.0 ± 7.1 | |
| ALSFRS-r at diagnosis | 34.7 ± 7.2 | ||
| Parameters of disease progression | |||
| FVC variation (%) | −19.66 ± 10.6 | ||
| Weight variation (%) | −4.65 ± 9.0 | ||
| ALSFRS-r variation (%) | −23.43 ± 17.9 | ||
| Muscle | |||
| Metabolites | p Value | Metabolites | p Value |
| Diagnosis | Prognosis—ALSFRS-r | ||
| C8-carnitine | 0.075 | 4-guanidinobutanoate | 0.0202 |
| Lumichrome | 0.09 | Glucuronolactone | 0.0299 |
| L-alanine | 0.007 | Sn-glycero-3-phosphocholine | 0.0482 |
| 3-ureidopropionate | 0.009 | ||
| Glycine | 0.009 | Prognosis—weight variation | |
| Citramalate | 0.04 | 4-hydroxy-L-proline | 0.0105 |
| 4-methyl-2-oxo-pentanoic acid | 0.04 | ||
| Taurine | 0.05 | ||
| 4-methyl-2-oxovaleric acid | 0.05 | ||
| Hippuric acid | 0.06 | ||
| 10-hydroxydecanoate | 0.06 | ||
| Shikimate | 0.07 | ||
| Inosine | 0.07 | ||
| Suberic acid | 0.09 | ||
| Trans-cinnamate | 0.09 | ||
| Serum | |||
| Metabolites | p Value | Metabolites | p Value |
| Prognosis—weight variation | Prognosis—ALSFRS-r | ||
| 5,6-dihydro-uracil | 0.0023 | 10-hydroxydecanoate | 0.043 |
| Deoxycarnitine | 0.0032 | Elaidic acid | 0.0456 |
| Trans-aconitate | 0.0057 | ||
| L-glutamic acid | 0.0071 | ||
| Linoleate | 0.0116 | ||
| L-tryptophan | 0.013 | ||
| Malate | 0.0159 | ||
| Isocitric acid | 0.0192 | ||
| Succinate | 0.0258 | ||
| O-acetyl-carnitine | 0.0273 | ||
| Adipic acid | 0.0406 | ||
| Nicotinate | 0.0414 | ||
| Lactate | 0.0448 | ||
| Cytidine | 0.0459 | ||
| Norleucine | 0.0466 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanznaster, D.; Bruno, C.; Bourgeais, J.; Emond, P.; Zemmoura, I.; Lefèvre, A.; Reynier, P.; Eymieux, S.; Blanchard, E.; Vourc'h, P.; et al. Metabolic Profile and Pathological Alterations in the Muscle of Patients with Early-Stage Amyotrophic Lateral Sclerosis. Biomedicines 2022, 10, 1307. https://doi.org/10.3390/biomedicines10061307
Lanznaster D, Bruno C, Bourgeais J, Emond P, Zemmoura I, Lefèvre A, Reynier P, Eymieux S, Blanchard E, Vourc'h P, et al. Metabolic Profile and Pathological Alterations in the Muscle of Patients with Early-Stage Amyotrophic Lateral Sclerosis. Biomedicines. 2022; 10(6):1307. https://doi.org/10.3390/biomedicines10061307
Chicago/Turabian StyleLanznaster, Débora, Clément Bruno, Jérôme Bourgeais, Patrick Emond, Ilyess Zemmoura, Antoine Lefèvre, Pascal Reynier, Sébastien Eymieux, Emmanuelle Blanchard, Patrick Vourc'h, and et al. 2022. "Metabolic Profile and Pathological Alterations in the Muscle of Patients with Early-Stage Amyotrophic Lateral Sclerosis" Biomedicines 10, no. 6: 1307. https://doi.org/10.3390/biomedicines10061307
APA StyleLanznaster, D., Bruno, C., Bourgeais, J., Emond, P., Zemmoura, I., Lefèvre, A., Reynier, P., Eymieux, S., Blanchard, E., Vourc'h, P., Andres, C. R., Bakkouche, S. E., Herault, O., Favard, L., Corcia, P., & Blasco, H. (2022). Metabolic Profile and Pathological Alterations in the Muscle of Patients with Early-Stage Amyotrophic Lateral Sclerosis. Biomedicines, 10(6), 1307. https://doi.org/10.3390/biomedicines10061307