
Gene Therapy for Acquired and Genetic Cholestasis

 , , and
, , and
Abstract
1. Cholestatic Diseases
1.1. Acquired Cholestasis
1.2. Inherited Cholestasis
2. Current Treatments
2.1. Surgical Procedures: Hurdles and Limitations
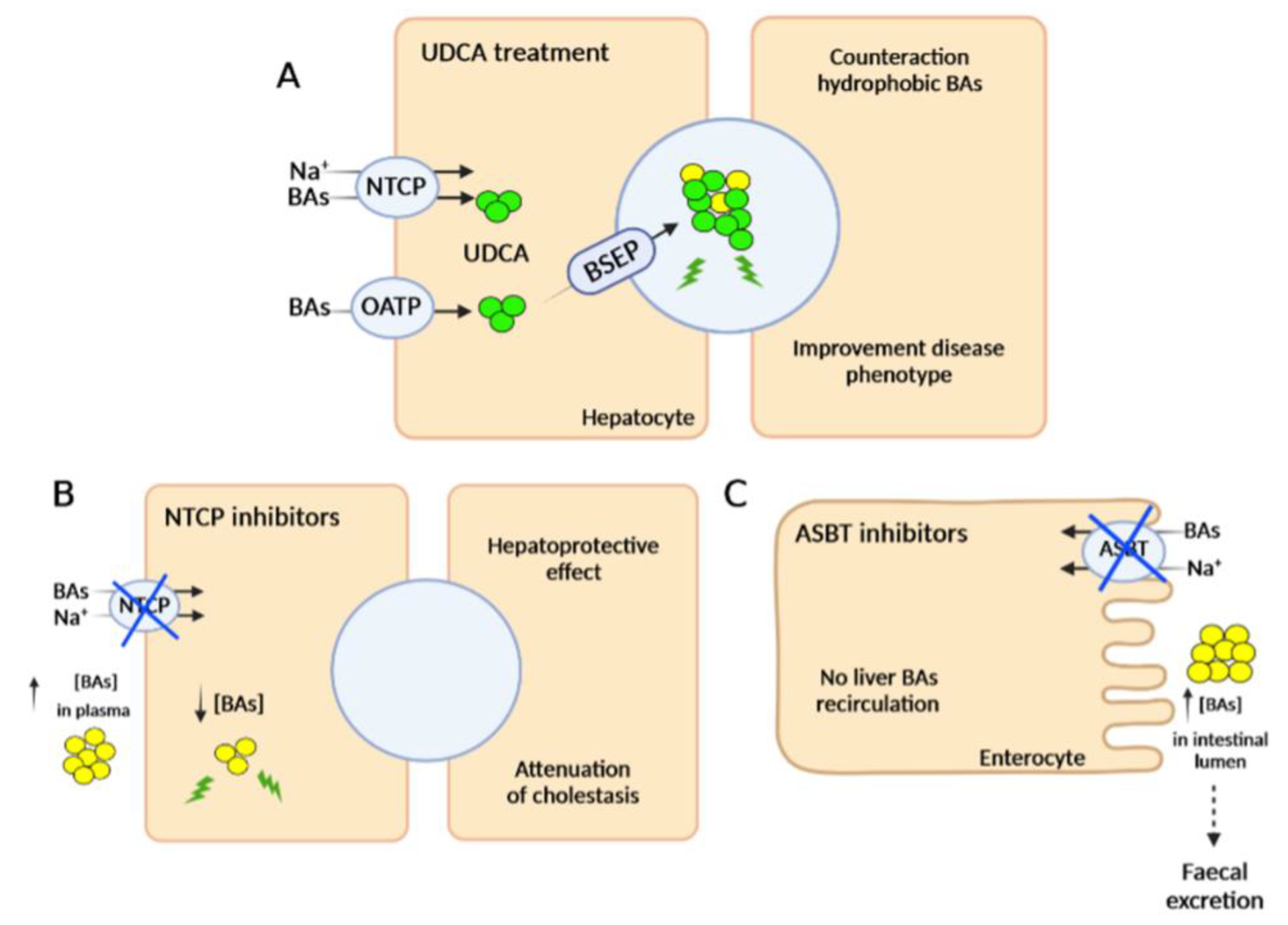
2.2. Pharmacological Therapies
2.2.1. FXR Agonists
2.2.2. Inhibitors of Bile Acid Uptake Transporters
2.2.3. Other Pharmacotherapeutic Agents
3. Gene Therapy
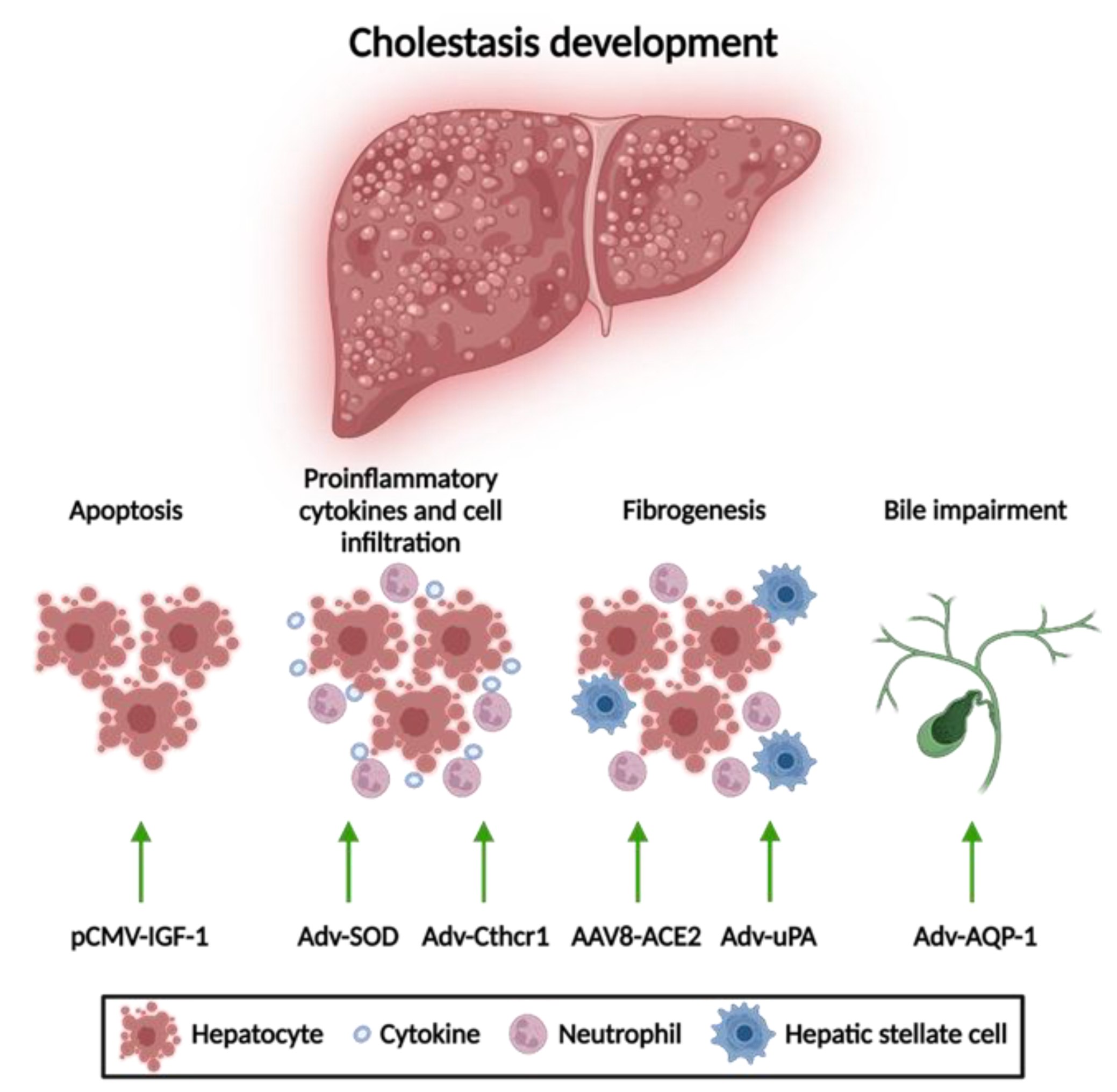
3.1. Gene Therapy for Acquired Cholestasis
3.1.1. Apoptosis Attenuation
3.1.2. Reduction of Mitochondrial Oxidative Stress
3.1.3. Anti-Fibrotic Therapies
3.1.4. Amelioration of Bile Flow
3.2. Gene Therapy for Inherited Cholestasis
3.2.1. Gene Therapy of Genetic Disorders with Associated Cholestasis
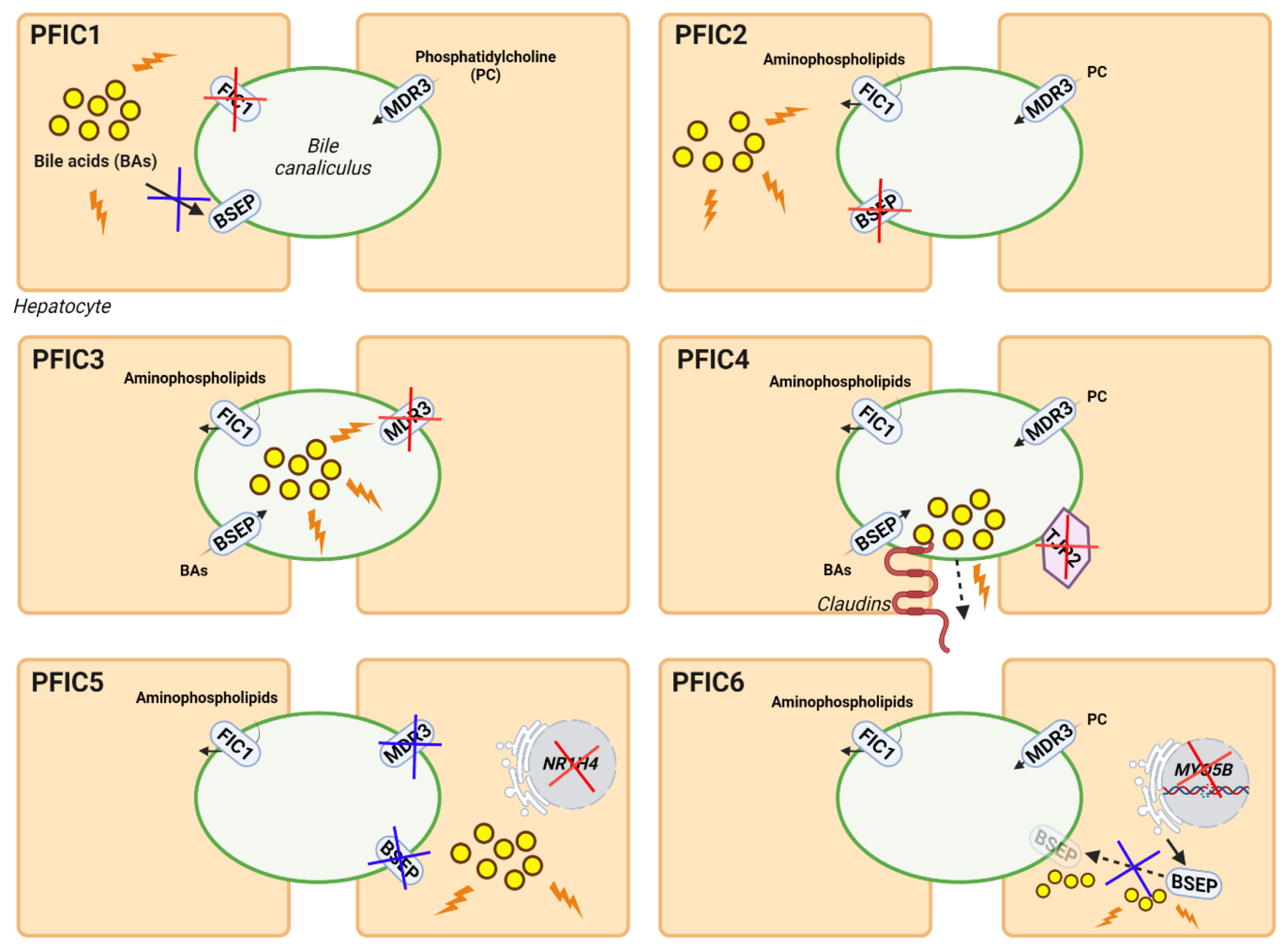
3.2.2. Gene Therapy for PFIC Diseases
Gene Therapy for PFIC3 Based on ABCB4 Supplementation
Gene Therapy for PFIC3 Targeting Mechanisms of Disease
Gene Therapy for Other Types of PFIC
4. Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zollner, G.; Trauner, M. Mechanisms of Cholestasis. Clin. Liver Dis. 2008, 12, 1–26. [Google Scholar] [CrossRef]
- Lee, J.; Boyer, J.L. Molecular Alterations in Hepatocyte Transport Mechanisms in Acquired Cholestatic Liver Disorders. Semin. Liver Dis. 2000, 20, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Yokoda, R.T.; Rodriguez, E.A. Review: Pathogenesis of Cholestatic Liver Diseases. World J. Hepatol. 2020, 12, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.M.; Gershwin, M.E. Primary Biliary Cirrhosis. N. Engl. J. Med. 2005, 353, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary Biliary Cholangitis: Pathogenesis and Therapeutic Opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Beuers, U.; Jones, D.E.J.; Lohse, A.W.; Hudson, M. Primary Sclerosing Cholangitis. Lancet 2018, 391, 2547–2559. [Google Scholar] [CrossRef]
- Reyes, H.; Sjövall, J. Bile Acids and Progesterone Metabolites Intrahepatic Cholestasis of Pregnancy. Ann. Med. 2000, 32, 94–106. [Google Scholar] [CrossRef]
- Pang, S.-Y.; Dai, Y.-M.; Zhang, R.-Z.; Chen, Y.-H.; Peng, X.-F.; Fu, J.; Chen, Z.-R.; Liu, Y.-F.; Yang, L.-Y.; Wen, Z.; et al. Autoimmune Liver Disease-Related Autoantibodies in Patients with Biliary Atresia. World J. Gastroenterol. 2018, 24, 387–396. [Google Scholar] [CrossRef]
- Abbey, P.; Kandasamy, D.; Naranje, P. Neonatal Jaundice. Indian J. Pediatr. 2019, 86, 830–841. [Google Scholar] [CrossRef]
- Visentin, M.; Lenggenhager, D.; Gai, Z.; Kullak-Ublick, G.A. Drug-Induced Bile Duct Injury. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 1498–1506. [Google Scholar] [CrossRef]
- Jacquemin, E. Progressive Familial Intrahepatic Cholestasis. Clin. Res. Hepatol. Gastroenterol. 2012, 36, S26–S35. [Google Scholar] [CrossRef]
- Srivastava, A. Progressive Familial Intrahepatic Cholestasis. J. Clin. Exp. Hepatol. 2014, 4, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Amirneni, S.; Haep, N.; Gad, M.A.; Soto-Gutierrez, A.; Squires, J.E.; Florentino, R.M. Molecular Overview of Progressive Familial Intrahepatic Cholestasis. World J. Gastroenterol. 2020, 26, 7470–7484. [Google Scholar] [CrossRef]
- Imagawa, K.; Hayashi, H.; Sabu, Y.; Tanikawa, K.; Fujishiro, J.; Kajikawa, D.; Wada, H.; Kudo, T.; Kage, M.; Kusuhara, H.; et al. Clinical Phenotype and Molecular Analysis of a Homozygous ABCB11 Mutation Responsible for Progressive Infantile Cholestasis. J. Hum. Genet. 2018, 63, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ospina, N.; Potter, C.J.; Xiao, R.; Manickam, K.; Kim, M.-S.; Kim, K.H.; Shneider, B.L.; Picarsic, J.L.; Jacobson, T.A.; Zhang, J.; et al. Mutations in the Nuclear Bile Acid Receptor FXR Cause Progressive Familial Intrahepatic Cholestasis. Nat. Commun. 2016, 7, 10713. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, E.; Taylor, S.A.; Davit-Spraul, A.; Thébaut, A.; Thomassin, N.; Guettier, C.; Whitington, P.F.; Jacquemin, E. MYO5B Mutations Cause Cholestasis with Normal Serum Gamma-glutamyl Transferase Activity in Children without Microvillous Inclusion Disease. Hepatology 2017, 65, 164–173. [Google Scholar] [CrossRef]
- Luketic, V.A.; Shiffman, M.L. Benign Recurrent Intrahepatic Cholestasis. Clin. Liver Dis. 2004, 8, 133–149. [Google Scholar] [CrossRef]
- Lam, P.; Soroka, C.; Boyer, J. The Bile Salt Export Pump: Clinical and Experimental Aspects of Genetic and Acquired Cholestatic Liver Disease. Semin. Liver Dis. 2010, 30, 125–133. [Google Scholar] [CrossRef]
- Sticova, E.; Jirsa, M. ABCB4 Disease: Many Faces of One Gene Deficiency. Ann. Hepatol. 2020, 19, 126–133. [Google Scholar] [CrossRef]
- Feranchak, A.P.; Sokol, R.J. Cholangiocyte Biology and Cystic Fibrosis Liver Disease. Semin. Liver Dis. 2001, 21, 471–488. [Google Scholar] [CrossRef]
- Mitchell, E.; Gilbert, M.; Loomes, K.M. Alagille Syndrome. Clin. Liver Dis. 2018, 22, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhao, J.; Peng, X.-M.; Qian, Y.-Y.; Zhao, X.-M.; Zhou, W.-H.; Wang, J.-S.; Wu, B.-B.; Wang, H.-J. Cholestasis as a Dominating Symptom of Patients with CYP27A1 Mutations: An Analysis of 17 Chinese Infants. J. Clin. Lipidol. 2021, 15, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M. Liver Transplantation for Inborn Errors of Liver Metabolism. J. Inherit. Metab. Dis. 2006, 29, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F. PBC-Transplantation and Disease Recurrence. Best Pract. Res. Clin. Gastroenterol. 2018, 34–35, 107–111. [Google Scholar] [CrossRef]
- Jadlowiec, C.C. Liver Transplantation: Current Status and Challenges. World J. Gastroenterol. 2016, 22, 4438. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, L.-Y.; Zhu, Z.-J.; Wei, L.; Qu, W.; Zeng, Z.-G. Liver Transplantation for Progressive Familial Intrahepatic Cholestasis. Ann. Transplant. 2018, 23, 666–673. [Google Scholar] [CrossRef]
- Kubitz, R.; Dröge, C.; Kluge, S.; Stross, C.; Walter, N.; Keitel, V.; Häussinger, D.; Stindt, J. Autoimmune BSEP Disease: Disease Recurrence After Liver Transplantation for Progressive Familial Intrahepatic Cholestasis. Clin. Rev. Allergy Immunol. 2015, 48, 273–284. [Google Scholar] [CrossRef]
- Stindt, J.; Kluge, S.; Dröge, C.; Keitel, V.; Stross, C.; Baumann, U.; Brinkert, F.; Dhawan, A.; Engelmann, G.; Ganschow, R.; et al. Bile Salt Export Pump-Reactive Antibodies Form a Polyclonal, Multi-Inhibitory Response in Antibody-Induced Bile Salt Export Pump Deficiency. Hepatology 2016, 63, 524–537. [Google Scholar] [CrossRef]
- Bull, L.N.; Pawlikowska, L.; Strautnieks, S.; Jankowska, I.; Czubkowski, P.; Dodge, J.L.; Emerick, K.; Wanty, C.; Wali, S.; Blanchard, S.; et al. Outcomes of Surgical Management of Familial Intrahepatic Cholestasis 1 and Bile Salt Export Protein Deficiencies. Hepatol. Commun. 2018, 2, 515–528. [Google Scholar] [CrossRef]
- Bjørnland, K.; Hukkinen, M.; Gatzinsky, V.; Arnell, H.; Pakarinen, M.P.; Almaas, R.; Svensson, J.F. Partial Biliary Diversion May Promote Long-Term Relief of Pruritus and Native Liver Survival in Children with Cholestatic Liver Diseases. Eur. J. Pediatr. Surg. 2021, 31, 341–346. [Google Scholar] [CrossRef]
- Cielecka-Kuszyk, J.; Lipiński, P.; Szymańska, S.; Ismail, H.; Jankowska, I. Long-Term Follow-up in Children with Progressive Familial Intrahepatic Cholestasis Type 2 after Partial External Biliary Diversion with Focus on Histopathological Features. Pol. J. Pathol. 2019, 70, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, C.; Bhardwaj, T.; Bass, L.M.; Superina, R.A. Outcomes Following Partial External Biliary Diversion in Patients with Progressive Familial Intrahepatic Cholestasis. J. Pediatr. Surg. 2017, 52, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Lal, B.B. Recent Updates on Progressive Familial Intrahepatic Cholestasis Types 1, 2 and 3: Outcome and Therapeutic Strategies. World J. Hepatol. 2022, 14, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Abbas, N.; Quraishi, M.N.; Trivedi, P. Emerging Drugs for the Treatment of Primary Sclerosing Cholangitis. Curr. Opin. Pharmacol. 2022, 62, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, D.; Arab, J.P.; Arrese, M. UDCA, NorUDCA, and TUDCA in Liver Diseases: A Review of Their Mechanisms of Action and Clinical Applications. In Bile Acids and Their Receptors; Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2019; pp. 237–264. [Google Scholar]
- Manna, L.B.; Papacleovoulou, G.; Flaviani, F.; Pataia, V.; Qadri, A.; Abu-Hayyeh, S.; McIlvride, S.; Jansen, E.; Dixon, P.; Chambers, J.; et al. Ursodeoxycholic Acid Improves Feto-Placental and Offspring Metabolic Outcomes in Hypercholanemic Pregnancy. Sci. Rep. 2020, 10, 10361. [Google Scholar] [CrossRef]
- Carey, E.J.; Ali, A.H.; Lindor, K.D. Primary Biliary Cirrhosis. Lancet 2015, 386, 1565–1575. [Google Scholar] [CrossRef]
- Gordo-Gilart, R.; Andueza, S.; Hierro, L.; Martínez-Fernández, P.; D’Agostino, D.; Jara, P.; Alvarez, L. Functional Analysis of ABCB4 Mutations Relates Clinical Outcomes of Progressive Familial Intrahepatic Cholestasis Type 3 to the Degree of MDR3 Floppase Activity. Gut 2015, 64, 147–155. [Google Scholar] [CrossRef]
- Ghonem, N.S.; Assis, D.N.; Boyer, J.L. Fibrates and Cholestasis. Hepatology 2015, 62, 635–643. [Google Scholar] [CrossRef]
- Stapelbroek, J.M.; van Erpecum, K.J.; Klomp, L.W.J.; Houwen, R.H.J. Liver Disease Associated with Canalicular Transport Defects: Current and Future Therapies. J. Hepatol. 2010, 52, 258–271. [Google Scholar] [CrossRef]
- Fickert, P.; Hirschfield, G.M.; Denk, G.; Marschall, H.-U.; Altorjay, I.; Färkkilä, M.; Schramm, C.; Spengler, U.; Chapman, R.; Bergquist, A.; et al. NorUrsodeoxycholic Acid Improves Cholestasis in Primary Sclerosing Cholangitis. J. Hepatol. 2017, 67, 549–558. [Google Scholar] [CrossRef]
- Chazouillères, O. 24-Norursodeoxycholic Acid in Patients with Primary Sclerosing Cholangitis: A New “Urso Saga” on the Horizon? J. Hepatol. 2017, 67, 446–447. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zeng, M.; Han, Y.; Yan, H.; Tang, H.; Sheng, J.; Hu, H.; Cheng, L.; Xie, Q.; Zhu, Y.; et al. A Multicenter, Randomized, Double-Blind Trial Comparing the Efficacy and Safety of TUDCA and UDCA in Chinese Patients with Primary Biliary Cholangitis. Medicine 2016, 95, e5391. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Nevens, F.; Shiffman, M.L.; Drenth, J.P.H.; Bowlus, C.L.; Vargas, V.; Andreone, P.; Hirschfield, G.M.; Pencek, R.; Malecha, E.S.; et al. Long-Term Efficacy and Safety of Obeticholic Acid for Patients with Primary Biliary Cholangitis: 3-Year Results of an International Open-Label Extension Study. Lancet Gastroenterol. Hepatol. 2019, 4, 445–453. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. A Randomized, Placebo-Controlled, Phase II Study of Obeticholic Acid for Primary Sclerosing Cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, D.; de Vincentis, A.; Malinverno, F.; Viganò, M.; Alvaro, D.; Pompili, M.; Picciotto, A.; Palitti, V.P.; Russello, M.; Storato, S.; et al. Real-World Experience with Obeticholic Acid in Patients with Primary Biliary Cholangitis. JHEP Rep. 2021, 3, 100248. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, Q.; Zeng, S.; Lou, Y.; Li, X.; Hu, M.; Lu, L.; Liu, Z. Disordered Farnesoid X Receptor Signaling Is Associated with Liver Carcinogenesis in Abcb11 -deficient Mice. J. Pathol. 2021, 255, 412–424. [Google Scholar] [CrossRef]
- Fiorucci, S.; Di Giorgio, C.; Distrutti, E. Obeticholic Acid: An Update of Its Pharmacological Activities in Liver Disorders. In Bile Acids and Their Receptors; Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2019; pp. 283–295. [Google Scholar]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, X.; Wei, H.; Dai, S.; Qu, L.; Chen, X.; Guo, M.; Chen, Y. Structural Insight into the Molecular Mechanism of Cilofexor Binding to the Farnesoid X Receptor. Biochem. Biophys. Res. Commun. 2022, 595, 1–6. [Google Scholar] [CrossRef]
- Massafra, V.; Pellicciari, R.; Gioiello, A.; van Mil, S.W.C. Progress and Challenges of Selective Farnesoid X Receptor Modulation. Pharmacol. Ther. 2018, 191, 162–177. [Google Scholar] [CrossRef]
- van de Wiel, S.M.W.; Bijsmans, I.T.G.W.; Van Mil, S.W.C.; van de Graaf, S.F.J. Identification of FDA-Approved Drugs Targeting the Farnesoid X Receptor. Sci. Rep. 2019, 9, 2193. [Google Scholar] [CrossRef]
- Baghdasaryan, A.; Chiba, P.; Trauner, M. Clinical Application of Transcriptional Activators of Bile Salt Transporters. Mol. Asp. Med. 2014, 37, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, J.J.; Brouwer, K.L.R.; Malinen, M.M. Novel Insights into the Organic Solute Transporter Alpha/Beta, OSTα/β: From the Bench to the Bedside. Pharmacol. Ther. 2020, 211, 107542. [Google Scholar] [CrossRef]
- Slijepcevic, D.; van de Graaf, S.F.J. Bile Acid Uptake Transporters as Targets for Therapy. Dig. Dis. 2017, 35, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Slijepcevic, D.; Roscam Abbing, R.L.P.; Fuchs, C.D.; Haazen, L.C.M.; Beuers, U.; Trauner, M.; Oude Elferink, R.P.J.; van de Graaf, S.F.J. Na+-Taurocholate Cotransporting Polypeptide Inhibition Has Hepatoprotective Effects in Cholestasis in Mice. Hepatology 2018, 68, 1057–1069. [Google Scholar] [CrossRef]
- Kamath, B.M.; Stein, P.; Houwen, R.H.J.; Verkade, H.J. Potential of Ileal Bile Acid Transporter Inhibition as a Therapeutic Target in Alagille Syndrome and Progressive Familial Intrahepatic Cholestasis. Liver Int. 2020, 40, 1812–1822. [Google Scholar] [CrossRef] [PubMed]
- Karpen, S.J.; Kelly, D.; Mack, C.; Stein, P. Ileal Bile Acid Transporter Inhibition as an Anticholestatic Therapeutic Target in Biliary Atresia and Other Cholestatic Disorders. Hepatol. Int. 2020, 14, 677–689. [Google Scholar] [CrossRef]
- Deeks, E.D. Odevixibat: First Approval. Drugs 2021, 81, 1781–1786. [Google Scholar] [CrossRef]
- Mayo, M.J.; Pockros, P.J.; Jones, D.; Bowlus, C.L.; Levy, C.; Patanwala, I.; Bacon, B.; Luketic, V.; Vuppalanchi, R.; Medendorp, S.; et al. A Randomized, Controlled, Phase 2 Study of Maralixibat in the Treatment of Itching Associated with Primary Biliary Cholangitis. Hepatol. Commun. 2019, 3, 365–381. [Google Scholar] [CrossRef]
- Shirley, M. Maralixibat: First Approval. Drugs 2022, 82, 71–76. [Google Scholar] [CrossRef]
- Gonzales, E.; Hardikar, W.; Stormon, M.; Baker, A.; Hierro, L.; Gliwicz, D.; Lacaille, F.; Lachaux, A.; Sturm, E.; Setchell, K.D.R.; et al. Efficacy and Safety of Maralixibat Treatment in Patients with Alagille Syndrome and Cholestatic Pruritus (ICONIC): A Randomised Phase 2 Study. Lancet 2021, 398, 1581–1592. [Google Scholar] [CrossRef]
- Mazzetti, M.; Marconi, G.; Mancinelli, M.; Benedetti, A.; Marzioni, M.; Maroni, L. The Management of Cholestatic Liver Diseases: Current Therapies and Emerging New Possibilities. J. Clin. Med. 2021, 10, 1763. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.R.; Soroka, C.J.; Hagey, L.R.; Boyer, J.L. Sirtuin 1 Activation Alleviates Cholestatic Liver Injury in a Cholic Acid-Fed Mouse Model of Cholestasis. Hepatology 2016, 64, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Claveria-Cabello, A.; Colyn, L.; Uriarte, I.; Latasa, M.U.; Arechederra, M.; Herranz, J.M.; Alvarez, L.; Urman, J.M.; Martinez-Chantar, M.L.; Banales, J.M.; et al. Dual Pharmacological Targeting of HDACs and PDE5 Inhibits Liver Disease Progression in a Mouse Model of Biliary Inflammation and Fibrosis. Cancers 2020, 12, 3748. [Google Scholar] [CrossRef]
- Mareux, E.; Lapalus, M.; Amzal, R.; Almes, M.; Aït-Slimane, T.; Delaunay, J.; Adnot, P.; Collado-Hilly, M.; Davit-Spraul, A.; Falguières, T.; et al. Functional Rescue of an ABCB11 Mutant by Ivacaftor: A New Targeted Pharmacotherapy Approach in Bile Salt Export Pump Deficiency. Liver Int. 2020, 40, 1917–1925. [Google Scholar] [CrossRef]
- Cançado, G.G.L.; Couto, C.A.; Guedes, L.V.; Braga, M.H.; Terrabuio, D.R.B.; Cançado, E.L.R.; Ferraz, M.L.G.; Villela-Nogueira, C.A.; Nardelli, M.J.; Faria, L.C.; et al. Fibrates for the Treatment of Primary Biliary Cholangitis Unresponsive to Ursodeoxycholic Acid: An Exploratory Study. Front. Pharmacol. 2022, 12, 818089. [Google Scholar] [CrossRef]
- Joutsiniemi, T.; Timonen, S.; Leino, R.; Palo, P.; Ekblad, U. Ursodeoxycholic Acid in the Treatment of Intrahepatic Cholestasis of Pregnancy: A Randomized Controlled Trial. Arch. Gynecol. Obstet. 2014, 289, 541–547. [Google Scholar] [CrossRef]
- Hopf, C.; Grieshaber, R.; Hartmann, H.; Hinrichsen, H.; Eisold, M.; Cordes, H.-J.; Greinwald, R.; Rust, C. Therapeutic Equivalence of Ursodeoxycholic Acid Tablets and Ursodeoxycholic Acid Capsules for the Treatment of Primary Biliary Cirrhosis. Clin. Pharmacol. Drug Dev. 2013, 2, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Poupon, R.E.; Eschwège, E.; Poupon, R.; Poupon, R.; Poupon, R.E.; Eschwège, E.; Attali, P.; Capron, J.P.; Erlinger, S.; Beaugrand, M.; et al. Ursodeoxycholic Acid for the Treatment of Primary Biliary Cirrhosis. J. Hepatol. 1990, 11, 16–21. [Google Scholar] [CrossRef]
- Jacquemin, E.; Hermans, D.; Myara, A.; Habes, D.; Debray, D.; Hadchouel, M.; Sokal, E.M.; Bernard, O. Ursodeoxycholic Acid Therapy in Pediatric Patients with Progressive Familial Intrahepatic Cholestasis. Hepatology 1997, 25, 519–523. [Google Scholar] [CrossRef]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.H.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Luketic, V.; Chapman, R.; Hirschfield, G.M.; Poupon, R.; Schramm, C.; Vincent, C.; Rust, C.; Parés, A.; Mason, A.; et al. A Randomized Trial of Obeticholic Acid Monotherapy in Patients with Primary Biliary Cholangitis. Hepatology 2018, 67, 1890–1902. [Google Scholar] [CrossRef]
- Schramm, C.; Hirschfield, G.; Mason, A.L.; Wedemeyer, H.; Klickstein, L.; Neelakantham, S.; Koo, P.; Sanni, J.; Badman, M.; Jones, D. Early Assessment of Safety and Efficacy of Tropifexor, a Potent Non Bile-Acid FXR Agonist, in Patients with Primary Biliary Cholangitis: An Interim Analysis of an Ongoing Phase 2 Study. J. Hepatol. 2018, 68, S103. [Google Scholar] [CrossRef]
- Slavetinsky, C.; Sturm, E. Odevixibat and Partial External Biliary Diversion Showed Equal Improvement of Cholestasis in a Patient with Progressive Familial Intrahepatic Cholestasis. BMJ Case Rep. 2020, 13, e234185. [Google Scholar] [CrossRef]
- Baumann, U.; Sturm, E.; Lacaille, F.; Gonzalès, E.; Arnell, H.; Fischler, B.; Jørgensen, M.H.; Thompson, R.J.; Mattsson, J.P.; Ekelund, M.; et al. Effects of Odevixibat on Pruritus and Bile Acids in Children with Cholestatic Liver Disease: Phase 2 Study. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101751. [Google Scholar] [CrossRef]
- Hegade, V.S.; Kendrick, S.F.W.; Dobbins, R.L.; Miller, S.R.; Thompson, D.; Richards, D.; Storey, J.; Dukes, G.E.; Corrigan, M.; Oude Elferink, R.P.J.; et al. Effect of Ileal Bile Acid Transporter Inhibitor GSK2330672 on Pruritus in Primary Biliary Cholangitis: A Double-Blind, Randomised, Placebo-Controlled, Crossover, Phase 2a Study. Lancet 2017, 389, 1114–1123. [Google Scholar] [CrossRef]
- Cholangitis, H. GLIMMER Trial-A Randomized, Double-Blind, Placebo-Controlled Study of Linerixibat, an Inhibitor of the Ileal Bile Acid Transporter, in the Treatment of Cholestatic Pruritus in Primary Biliary Cholangitis. Gastroenterol. Hepatol. 2021, 17, 11–12. [Google Scholar]
- Sanyal, A.J.; Ling, L.; Beuers, U.; DePaoli, A.M.; Lieu, H.D.; Harrison, S.A.; Hirschfield, G.M. Potent Suppression of Hydrophobic Bile Acids by Aldafermin, an FGF19 Analogue, across Metabolic and Cholestatic Liver Diseases. JHEP Rep. 2021, 3, 100255. [Google Scholar] [CrossRef] [PubMed]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef]
- Schattenberg, J.M.; Pares, A.; Kowdley, K.V.; Heneghan, M.A.; Caldwell, S.; Pratt, D.; Bonder, A.; Hirschfield, G.M.; Levy, C.; Vierling, J.; et al. A Randomized Placebo-Controlled Trial of Elafibranor in Patients with Primary Biliary Cholangitis and Incomplete Response to UDCA. J. Hepatol. 2021, 74, 1344–1354. [Google Scholar] [CrossRef]
- Kremer, A.E.; Mayo, M.J.; Hirschfield, G.; Levy, C.; Bowlus, C.L.; Jones, D.E.; Steinberg, A.; McWherter, C.A.; Choi, Y. Seladelpar Improved Measures of Pruritus, Sleep, and Fatigue and Decreased Serum Bile Acids in Patients with Primary Biliary Cholangitis. Liver Int. 2022, 42, 112–123. [Google Scholar] [CrossRef]
- High, K.A.; Roncarolo, M.G. Gene Therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Szabó, G.T.; Mahiny, A.J.; Vlatkovic, I. COVID-19 mRNA vaccines: Platforms and current developments. Mol. Ther. 2022, 30, 1850–1868. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene Therapy Comes of Age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Waddington, S.N.; Alexander, I.E.; Gissen, P. Gene Therapy for Monogenic Liver Diseases: Clinical Successes, Current Challenges and Future Prospects. J. Inherit. Metab. Dis. 2017, 40, 497–517. [Google Scholar] [CrossRef]
- van der Laan, L.J.; Wang, Y.; Tilanus, H.W.; Janssen, H.L.; Pan, Q. AAV-Mediated Gene Therapy for Liver Diseases: The Prime Candidate for Clinical Application? Expert Opin. Biol. Ther. 2011, 11, 315–327. [Google Scholar] [CrossRef]
- Maestro, S.; Weber, N.D.; Zabaleta, N.; Aldabe, R.; Gonzalez-Aseguinolaza, G. Novel Vectors and Approaches for Gene Therapy in Liver Diseases. JHEP Rep. 2021, 3, 100300. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and Safety of Voretigene Neparvovec (AAV2-HRPE65v2) in Patients with RPE65 -Mediated Inherited Retinal Dystrophy: A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Mariotti, V.; Strazzabosco, M.; Fabris, L.; Calvisi, D.F. Animal Models of Biliary Injury and Altered Bile Acid Metabolism. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 1254–1261. [Google Scholar] [CrossRef]
- Fernández-Ramos, D.; Fernández-Tussy, P.; Lopitz-Otsoa, F.; Gutiérrez-de-Juan, V.; Navasa, N.; Barbier-Torres, L.; Zubiete-Franco, I.; Simón, J.; Fernández, A.F.; Arbelaiz, A.; et al. MiR-873-5p Acts as an Epigenetic Regulator in Early Stages of Liver Fibrosis and Cirrhosis. Cell Death Dis. 2018, 9, 958. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.P.-C.; Lee, L.-M.; Lin, T.-J.; Sheen-Chen, S.-M.; Lin, J.-W.; Chiu, W.-T.; Wang, C.-C.; Hung, K.-S. Gene Transfer of IGF1 Attenuates Hepatocellular Apoptosis After Bile Duct Ligation. J. Surg. Res. 2011, 167, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Froh, M.; Wheeler, M.; Smutney, O.; Lehmann, T.; Thurman, R. Viral Gene Delivery of Superoxide Dismutase Attenuates Experimental Cholestasis-Induced Liver Fibrosis in the Rat. Gene Ther. 2002, 9, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Miao, Q.; Zhong, W.; Zhang, H.; Wang, Q.; Peng, Y.; Chen, X.; Guo, C.; Shen, L.; Yang, F.; et al. Treatment of Cholestatic Fibrosis by Altering Gene Expression of Cthrc1: Implications for Autoimmune and Non-Autoimmune Liver Disease. J. Autoimmun. 2015, 63, 76–87. [Google Scholar] [CrossRef]
- Miranda-Díaz, A.; Rincón, A.R.; Salgado, S.; Vera-Cruz, J.; Gálvez, J.; Islas, M.C.; Berumen, J.; Aguilar Cordova, E.; Armendáriz-Borunda, J. Improved Effects of Viral Gene Delivery of Human UPA plus Biliodigestive Anastomosis Induce Recovery from Experimental Biliary Cirrhosis. Mol. Ther. 2004, 9, 30–37. [Google Scholar] [CrossRef]
- Salgado, S.; Garcia, J.; Vera, J.; Siller, F.; Bueno, M.; Miranda, A.; Segura, A.; Grijalva, G.; Segura, J.; Orozco, H.; et al. Liver Cirrhosis Is Reverted by Urokinase-Type Plasminogen Activator Gene Therapy. Mol. Ther. 2000, 2, 545–551. [Google Scholar] [CrossRef]
- Mak, K.Y.; Chin, R.; Cunningham, S.C.; Habib, M.R.; Torresi, J.; Sharland, A.F.; Alexander, I.E.; Angus, P.W.; Herath, C.B. ACE2 Therapy Using Adeno-Associated Viral Vector Inhibits Liver Fibrosis in Mice. Mol. Ther. 2015, 23, 1434–1443. [Google Scholar] [CrossRef]
- Yang, T.; Poenisch, M.; Khanal, R.; Hu, Q.; Dai, Z.; Li, R.; Song, G.; Yuan, Q.; Yao, Q.; Shen, X.; et al. Therapeutic HNF4A MRNA Attenuates Liver Fibrosis in a Preclinical Model. J. Hepatol. 2021, 75, 1420–1433. [Google Scholar] [CrossRef]
- Marrone, J.; Lehmann, G.L.; Soria, L.R.; Pellegrino, J.M.; Molinas, S.; Marinelli, R.A. Adenoviral Transfer of Human Aquaporin -1 Gene to Rat Liver Improves Bile Flow in Estrogen-Induced Cholestasis. Gene Ther. 2014, 21, 1058–1064. [Google Scholar] [CrossRef]
- Marrone, J.; Soria, L.R.; Danielli, M.; Lehmann, G.L.; Larocca, M.C.; Marinelli, R.A. Hepatic Gene Transfer of Human Aquaporin-1 Improves Bile Salt Secretory Failure in Rats with Estrogen-induced Cholestasis. Hepatology 2016, 64, 535–548. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.D.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Lumbreras, S.; Ricobaraza, A.; Baila-Rueda, L.; Gonzalez-Aparicio, M.; Mora-Jimenez, L.; Uriarte, I.; Bunuales, M.; Avila, M.A.; Monte, M.J.; Marin, J.J.G.; et al. Gene Supplementation of CYP27A1 in the Liver Restores Bile Acid Metabolism in a Mouse Model of Cerebrotendinous Xanthomatosis. Mol. Ther.-Methods Clin. Dev. 2021, 22, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Collaud, F.; Bortolussi, G.; Guianvarc’h, L.; Aronson, S.J.; Bordet, T.; Veron, P.; Charles, S.; Vidal, P.; Sola, M.S.; Rundwasser, S.; et al. Preclinical Development of an AAV8-HUGT1A1 Vector for the Treatment of Crigler-Najjar Syndrome. Mol. Ther.-Methods Clin. Dev. 2019, 12, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Ronzitti, G.; Bortolussi, G.; van Dijk, R.; Collaud, F.; Charles, S.; Leborgne, C.; Vidal, P.; Martin, S.; Gjata, B.; Sola, M.S.; et al. A Translationally Optimized AAV-UGT1A1 Vector Drives Safe and Long-Lasting Correction of Crigler-Najjar Syndrome. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16049. [Google Scholar] [CrossRef]
- Ginocchio, V.M.; Ferla, R.; Auricchio, A.; Brunetti-Pierri, N. Current Status on Clinical Development of Adeno-Associated Virus-Mediated Liver-Directed Gene Therapy for Inborn Errors of Metabolism. Hum. Gene Ther. 2019, 30, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.N.; Thompson, R.J. Progressive Familial Intrahepatic Cholestasis. Clin. Liver Dis. 2018, 22, 657–669. [Google Scholar] [CrossRef]
- Bosma, P.J.; Wits, M.; Oude-Elferink, R.P.J. Gene Therapy for Progressive Familial Intrahepatic Cholestasis: Current Progress and Future Prospects. Int. J. Mol. Sci. 2020, 22, 273. [Google Scholar] [CrossRef]
- de Vree, J.M.L.; Ottenhoff, R.; Bosma, P.J.; Smith, A.J.; Aten, J.; Oude Elferink, R.P.J. Correction of Liver Disease by Hepatocyte Transplantation in a Mouse Model of Progressive Familial Intrahepatic Cholestasis. Gastroenterology 2000, 119, 1720–1730. [Google Scholar] [CrossRef]
- Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Yoshida, S.; Nasser, I.; Ke, Q.; Kang, P.M.; Popov, Y. A New Mdr2−/− Mouse Model of Sclerosing Cholangitis with Rapid Fibrosis Progression, Early-Onset Portal Hypertension, and Liver Cancer. Am. J. Pathol. 2015, 185, 325–334. [Google Scholar] [CrossRef]
- Aronson, S.J.; Bakker, R.S.; Shi, X.; Duijst, S.; Bloemendaal, L.T.; de Waart, D.R.; Verheij, J.; Ronzitti, G.; Elferink, R.P.O.; Beuers, U.; et al. Liver-Directed Gene Therapy Results in Long-Term Correction of Progressive Familial Intrahepatic Cholestasis Type 3 in Mice. J. Hepatol. 2019, 71, 153–162. [Google Scholar] [CrossRef]
- Weber, N.D.; Odriozola, L.; Martínez-García, J.; Ferrer, V.; Douar, A.; Bénichou, B.; González-Aseguinolaza, G.; Smerdou, C. Gene Therapy for Progressive Familial Intrahepatic Cholestasis Type 3 in a Clinically Relevant Mouse Model. Nat. Commun. 2019, 10, 5694. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Cao, J.; Huang, P.; An, P.; Badlani, D.; Vaid, K.A.; Zhao, S.; Wang, D.Q.-H.; Zhuo, J.; Yin, L.; et al. Synthetic Human ABCB4 MRNA Therapy Rescues Severe Liver Disease Phenotype in a BALB/c.Abcb4 Mouse Model of PFIC3. J. Hepatol. 2021, 74, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.D.; Martínez-García, J.; González-Aseguinolaza, G. Comment on “Synthetic Human ABCB4 MRNA Therapy Rescues Severe Liver Disease Phenotype in a BALB/c.Abcb4 Mouse Model of PFIC3”. J. Hepatol. 2022, 76, 749–751. [Google Scholar] [CrossRef] [PubMed]
- Siew, S.M.; Cunningham, S.C.; Zhu, E.; Tay, S.S.; Venuti, E.; Bolitho, C.; Alexander, I.E. Prevention of Cholestatic Liver Disease and Reduced Tumorigenicity in a Murine Model of PFIC Type 3 Using Hybrid AAV-PiggyBac Gene Therapy. Hepatology 2019, 70, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- Rajapaksha, I.G.; Angus, P.W.; Herath, C.B. Current Therapies and Novel Approaches for Biliary Diseases. World J. Gastrointest. Pathophysiol. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Li, J.; Zhu, X.; Zhang, M.; Zhang, Y.; Ye, S.; Leng, Y.; Yang, T.; Kong, L.; Zhang, H. Limb Expression 1-like (LIX1L) Protein Promotes Cholestatic Liver Injury by Regulating Bile Acid Metabolism. J. Hepatol. 2021, 75, 400–413. [Google Scholar] [CrossRef]
- Ceci, L.; Francis, H.; Zhou, T.; Giang, T.; Yang, Z.; Meng, F.; Wu, N.; Kennedy, L.; Kyritsi, K.; Meadows, V.; et al. Knockout of the Tachykinin Receptor 1 in the Mdr2−/− (Abcb4−/−) Mouse Model of Primary Sclerosing Cholangitis Reduces Biliary Damage and Liver Fibrosis. Am. J. Pathol. 2020, 190, 2251–2266. [Google Scholar] [CrossRef]
- Giang, S.; Gordon, R.L.; Haas, K.B. A Diagnostic Quagmire: PFIC5 Presenting as a Rare Cause of Neonatal Cholestasis. ACG Case Rep. J. 2021, 8, e00558. [Google Scholar] [CrossRef]
- Sambrotta, M.; Strautnieks, S.; Papouli, E.; Rushton, P.; Clark, B.E.; Parry, D.A.; Logan, C.V.; Newbury, L.J.; Kamath, B.M.; Ling, S.; et al. Mutations in TJP2 Cause Progressive Cholestatic Liver Disease. Nat. Genet. 2014, 46, 326–328. [Google Scholar] [CrossRef]
- Overeem, A.W.; Li, Q.; Qiu, Y.; Cartón-García, F.; Leng, C.; Klappe, K.; Dronkers, J.; Hsiao, N.; Wang, J.; Arango, D.; et al. A Molecular Mechanism Underlying Genotype-Specific Intrahepatic Cholestasis Resulting From MYO5B Mutations. Hepatology 2020, 72, 213–229. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Patterson, A.D.; Wang, Y.; Krausz, K.W.; Neale, G.; Thomas, S.; Nachagari, D.; Vogel, P.; Vore, M.; et al. Abcb11 Deficiency Induces Cholestasis Coupled to Impaired β-Fatty Acid Oxidation in Mice. J. Biol. Chem. 2012, 287, 24784–24794. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Busuttil, R.W.; Lipshutz, G.S. RH10 Provides Superior Transgene Expression in Mice When Compared with Natural AAV Serotypes for Neonatal Gene Therapy. J. Gene Med. 2010, 12, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic MRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed]
- Rivière, C.; Danos, O.; Douar, A.M. Long-Term Expression and Repeated Administration of AAV Type 1, 2 and 5 Vectors in Skeletal Muscle of Immunocompetent Adult Mice. Gene Ther. 2006, 13, 1300–1308. [Google Scholar] [CrossRef]
- Ros-Gañán, I.; Hommel, M.; Trigueros-Motos, L.; Tamarit, B.; Rodríguez-García, E.; Salas, D.; Pérez, G.; Douar, A.; Combal, J.P.; Benichou, B.; et al. Optimising the IgG-degrading Enzyme Treatment Regimen for Enhanced Adeno-associated Virus Transduction in the Presence of Neutralising Antibodies. Clin. Transl. Immunol. 2022, 11, e1375. [Google Scholar] [CrossRef]
- Meliani, A.; Boisgerault, F.; Hardet, R.; Marmier, S.; Collaud, F.; Ronzitti, G.; Leborgne, C.; Verdera, H.C.; Sola, M.S.; Charles, S.; et al. Antigen-Selective Modulation of AAV Immunogenicity with Tolerogenic Rapamycin Nanoparticles Enables Successful Vector Re-Administration. Nat. Commun. 2018, 9, 4098. [Google Scholar] [CrossRef]
- Buscara, L.; Gross, D.-A.; Daniele, N. Of RAAV and Men: From Genetic Neuromuscular Disorder Efficacy and Toxicity Preclinical Studies to Clinical Trials and Back. J. Pers. Med. 2020, 10, 258. [Google Scholar] [CrossRef]
- Chand, D.; Mohr, F.; McMillan, H.; Tukov, F.F.; Montgomery, K.; Kleyn, A.; Sun, R.; Tauscher-Wisniewski, S.; Kaufmann, P.; Kullak-Ublick, G. Hepatotoxicity Following Administration of Onasemnogene Abeparvovec (AVXS-101) for the Treatment of Spinal Muscular Atrophy. J. Hepatol. 2021, 74, 560–566. [Google Scholar] [CrossRef]
- Suzuki, M.; Singh, R.N.; Crystal, R.G. Regulatable Promoters for Use in Gene Therapy Applications: Modification of the 5′-Flanking Region of the CFTR Gene with Multiple CAMP Response Elements to Support Basal, Low-Level Gene Expression That Can Be Upregulated by Exogenous Agents That Raise Int. Hum. Gene Ther. 1996, 7, 1883–1893. [Google Scholar] [CrossRef]
- Martínez-García, J.; Molina, M.; Odriozola, L.; Molina, A.; González-Aseguinolaza, G.; Weber, N.D.; Smerdou, C. A Minimal Bile Salt Excretory Pump Promoter Allows Bile Acid-Driven Physiological Regulation of Transgene Expression from a Gene Therapy Vector. Cell Biosci. 2022, 12. [Google Scholar] [CrossRef]
- Chen, S.-J.; Johnston, J.; Sandhu, A.; Bish, L.T.; Hovhannisyan, R.; Jno-Charles, O.; Sweeney, H.L.; Wilson, J.M. Enhancing the Utility of Adeno-Associated Virus Gene Transfer through Inducible Tissue-Specific Expression. Hum. Gene Ther. Methods 2013, 24, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.G.; Romero, Z.; Muñoz, P.; Cobo, M.; Benabdellah, K.; Martin, F. Physiological and Tissue-Specific Vectors for Treatment of Inherited Diseases. Gene Ther. 2011, 18, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Konkle, B.A.; Walsh, C.E.; Escobar, M.A.; Josephson, N.C.; Young, G.; von Drygalski, A.; McPhee, S.W.J.; Samulski, R.J.; Bilic, I.; de la Rosa, M.; et al. BAX 335 Hemophilia B Gene Therapy Clinical Trial Results: Potential Impact of CpG Sequences on Gene Expression. Blood 2021, 137, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.F. Codon Modification and PAMPs in Clinical AAV Vectors: The Tortoise or the Hare? Mol. Ther. 2020, 28, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In Vivo Genome Editing via CRISPR/Cas9 Mediated Homology-Independent Targeted Integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug Name | Indication | Current Status | Clinical Trial | Sponsor [Reference] | ||
|---|---|---|---|---|---|---|
| FXR agonists | Bile acids | UDCA (Actigall/Ursodiol/Ursofalk) | ICP | Phase III Phase IV | NCT01576458 NCT01510860 | Turku University Hospital [68] Pharma GmbH [69] |
| PBC | Approved | Sanofi-Synthelabo [70] | ||||
| PFIC3 | [71] | |||||
| Nor-UDCA | PSC | Phase II | NCT01755507 | Pharma GmbH [41] | ||
| TUDCA (Taurolite) | PBC | Phase III | NCT01857284 | Beijing Friendship Hospital [43] | ||
| OCA (INT-747/Ocaliva) | PBC | Phase II Phase III | NCT00570765 NCT01473524 | Intercept Pharmaceuticals [44,45,72,73] | ||
| PSC | Phase II | NCT02177136 | ||||
| Non-bile acids | Cilofexor (CILO) | PSC | Phase I/II | NCT02943460 | Gilead Sciences [49] | |
| Tropifexor (LJN452) | PBC | Phase II | NCT02516605 | Novartis Pharmaceuticals [74] | ||
| EDP-305 | PBC | Phase II | NCT03394924 | Enanta Pharmaceuticals | ||
| ASBT inhibitors | Odevixibat (A4250) | ALGS | Phase III | NCT04674761 | Albireo [75,76] | |
| PFIC | Approved | |||||
| Maralixibat (LUM001) | ALGS | Approved | Mirum Pharmaceuticals, Inc. [61] | |||
| PFIC | Phase III | NCT02057718 NCT03905330 | ||||
| Linerixibat (GSK2330672) | PBC | Phase III | NCT02966834 NCT04167358 | GlaxoSmithKline [77,78] | ||
| Volixibat (SHP626) | ICP PBC PSC | Phase II | NCT04718961 NCT05050136 NCT04663308 | Mirum Pharmaceuticals, Inc. | ||
| Other pharmacotherapeutic agents | Aldafermin (NGM282) | PBC | Phase II | NCT02026401 | NGM Biopharmaceuticals, Inc. [79] | |
| PSC | NCT02704364 | |||||
| Bezafibrate | PBC | Phase III | NCT01654731 | Hôpitaux de Paris [80] | ||
| Elafibranor | PBC | Phase II | NCT03124108 | Genfit [81] | ||
| Seladelpar (MBX-8025) | PBC | Phase III | NCT03602560 | CymaBay Therapeutics, Inc. [82] | ||

| Aronson et al. [112] | Weber et al. [113] | Siew et al. [116] | Wei et al. [114] | |
|---|---|---|---|---|
| Strain Background | C57BL/6 Abcb4-/- | FVB Abcb4-/- | FVB Abcb4-/- | BALB/c Abcb4-/- |
| Phenotype | Mild (requiring cholate-enriched diet) | Severe (similar to patients) | Severe (similar to patients) | More severe |
| Vector | AAV8 | AAV8 | Hybrid AAV-piggyBac transposon | LNP |
| Dose | 5 × 1013 vg/kg | 1 × 1014 vg/kg | ~2 × 1014 vg/kg | 1.0 mg/kg |
| Age of treatment | 10-week-old | 2- or 5-week-old | Newborn | 4-week-old |
| Outcomes | Increased biliary PC and cholesterol content. Rescue of serum ALT, ALP and bilirubin levels. Prevention of liver fibrosis. | Increased biliary PC. Rescue of serum transaminases, ALP and BA levels. Improvement of the degree of hepatosplenomegaly. Prevention and reversal of liver fibrosis. | Increased biliary PC. Decreased hepatomegaly and serum parameters (ALT, ALP, BAs). Reduced liver fibrosis and liver tumor incidence. | Increased biliary PC (10–25% WT) and %BW. Decreased hepatomegaly and serum parameters (ALT, ALP, BAs). Normalization of liver fibrosis and portal hypertension. |
| Advantages | Long-term correction. No risk of mutagenesis. | Granted orphan drug designation. Long-term prevention and correction at early and late stages of PFIC3, respectively. No risk of mutagenesis. | Long-term correction. Preventing genome loss by hepatocellular proliferation during liver growth. | No risk of mutagenesis. Less immune responses. |
| Disadvantages | Need for challenge with BA-enriched dietary supplementation (model). Need to evaluate efficacy in younger mice more representative of the age of patients. Risks of using a high viral dose. | Loss of long-term therapeutic effect in half of the females treated with a single dose. Need to address the immune response based on anti-AAV neutralizing antibody for repeated administrations of the vector. Risks of using a high viral dose. | Risk of mutagenesis. Transposase overexpression Lack of serotype that efficiently transduces human hepatocytes. | Less durable expression. Requires repeated parenteral dosing. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-García, J.; Molina, A.; González-Aseguinolaza, G.; Weber, N.D.; Smerdou, C. Gene Therapy for Acquired and Genetic Cholestasis. Biomedicines 2022, 10, 1238. https://doi.org/10.3390/biomedicines10061238
Martínez-García J, Molina A, González-Aseguinolaza G, Weber ND, Smerdou C. Gene Therapy for Acquired and Genetic Cholestasis. Biomedicines. 2022; 10(6):1238. https://doi.org/10.3390/biomedicines10061238
Chicago/Turabian StyleMartínez-García, Javier, Angie Molina, Gloria González-Aseguinolaza, Nicholas D. Weber, and Cristian Smerdou. 2022. "Gene Therapy for Acquired and Genetic Cholestasis" Biomedicines 10, no. 6: 1238. https://doi.org/10.3390/biomedicines10061238
APA StyleMartínez-García, J., Molina, A., González-Aseguinolaza, G., Weber, N. D., & Smerdou, C. (2022). Gene Therapy for Acquired and Genetic Cholestasis. Biomedicines, 10(6), 1238. https://doi.org/10.3390/biomedicines10061238