
TGF-β as a Key Modulator of Astrocyte Reactivity: Disease Relevance and Therapeutic Implications


{kind=link}
{kind=link}
Abstract
1. Introduction
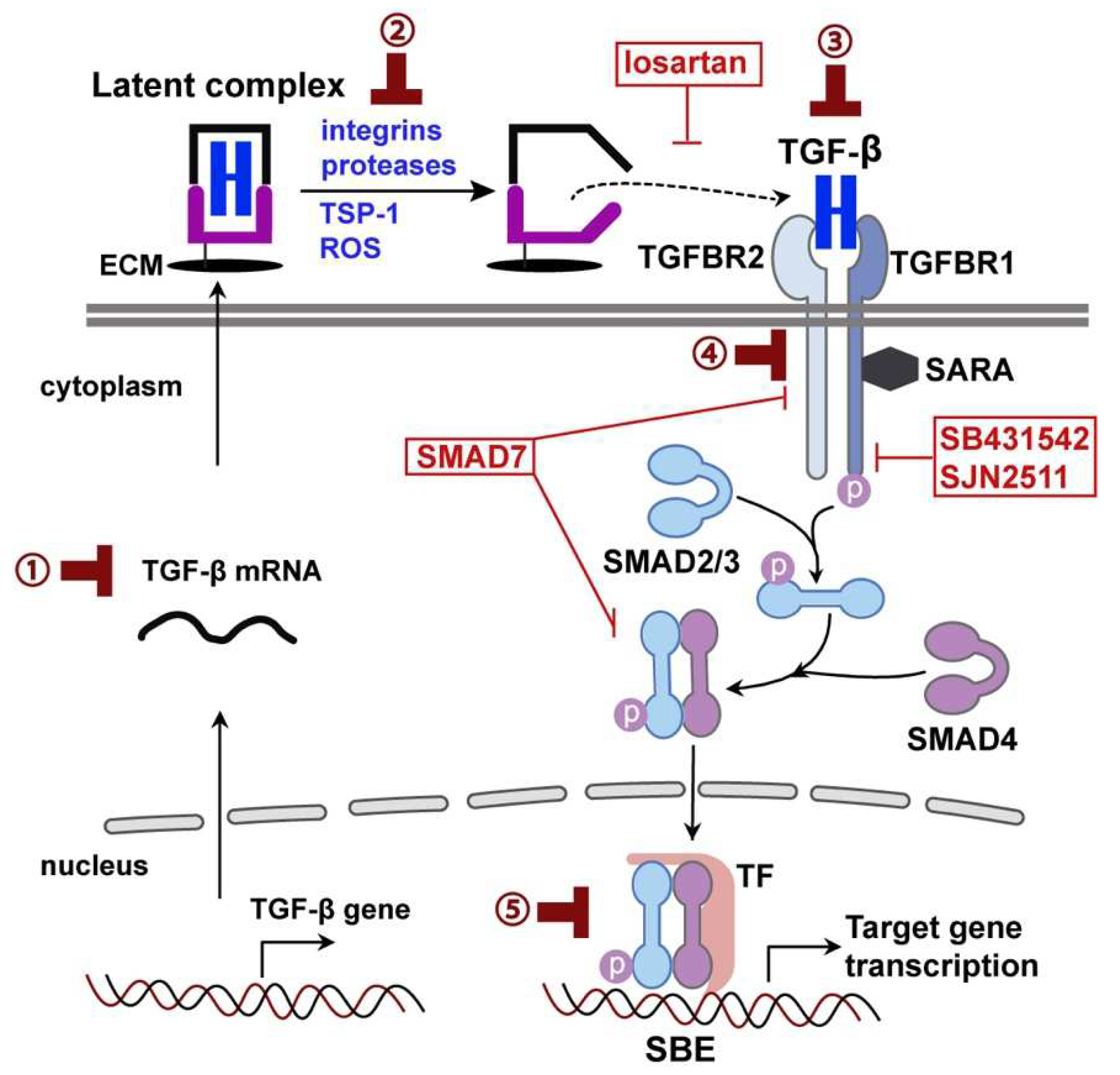
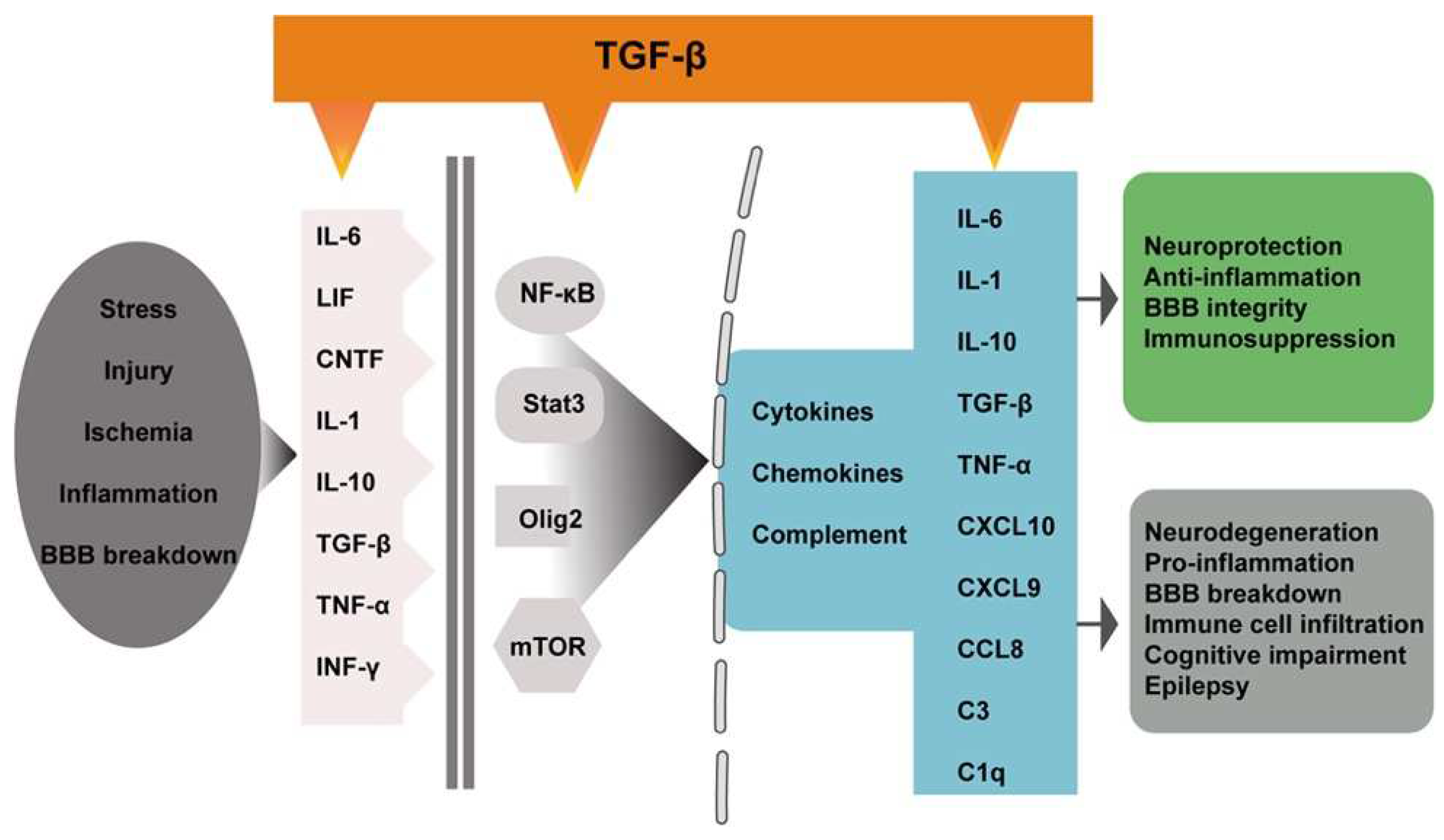
2. TGF-β Signaling in the Brain
3. Astrocytes Reactivity
4. TGF-β Regulation of Astrocyte Reactivity
5. Astrocytic TGF-β Signaling in CNS Diseases
5.1. Traumatic Brain Injury
5.2. Stroke
5.3. Aging
5.4. Alzheimer’s Disease
5.5. Parkinson’s Disease
5.6. Amyotrophic Lateral Sclerosis
5.7. Multiple Sclerosis
5.8. Huntington’s Disease
5.9. Epilepsy
6. Opportunities and Challenges of Targeting TGF-β Signaling in the Brain
7. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| APP | amyloid precursor protein |
| AQP4 | aquaporin 4 |
| α-syn | α-synuclein |
| BBB | blood–brain barrier |
| BMP | bone morphogenetic proteins |
| C3 | third complement component |
| C9ORF72 | Chromosome 9 Open Reading Frame 72 |
| CBD CBF | corticobasal degeneration cerebral blood flow |
| CCL | chemokine (C-C motif) ligand |
| CNS | central nervous system |
| CTGF | connective tissue growth factors |
| CXCL | chemokine (C-X-C motif) ligand |
| DA | dopaminergic |
| DAT DLB | dopamine transporter dementia with Lewy bodies |
| EAE | autoimmune encephalomyelitis |
| ECM | extracellular matrix |
| EEG | electroencephalogram |
| GABA | γ-aminobutiryc acid |
| GDF | growth differentiation factors |
| GFAP | glial fibrillary acidic protein |
| HD HTT ICV | Huntington’s disease huntingtin intracerebroventricular |
| IFN | interferon |
| IL | interleukin |
| iPSC | induced pluripotent stem cell |
| LPS mHTT | Lipopolysaccharide mutant huntingtin |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MS | multiple sclerosis |
| MSA mTOR | multiple system atrophy mechanistic/mammalian target of rapamycin |
| NFκB | nuclear factor kappa B |
| PAI-1 | plasminogen activator inhibitor-1 |
| PD | Parkinson’s disease |
| PSP ROS | progressive supranuclear palsy reactive oxygen species |
| SARA | Smad anchor for receptor activation |
| SBE | Smad binding element |
| scRNA-seq | single-cell RNA sequencing |
| SMAD | Sma- and Mad-related protein |
| SNpc | substantia nigra pars compacta |
| snRNA-seq | single-nucleus RNA sequencing |
| SOD1 | superoxide dismutase type 1 |
| STAT3 | signal transducer and activator of transcription 3 |
| TBI | traumatic brain injury |
| TGF-β | transforming growth factor-β |
| TGFBR1 | transforming growth factor-β receptor 1 |
| TGFBR2 | transforming growth factor-β receptor 2 |
| TLR | toll-like receptor |
| TLE | temporal lobe epilepsy |
| TNF | tumor necrosis factor |
| TSP-1 | thrombospondin-1 |
References
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Vahsen, B.F.; Gray, E.; Thompson, A.G.; Ansorge, O.; Anthony, D.C.; Cowley, S.A.; Talbot, K.; Turner, M.R. Non-neuronal cells in amyotrophic lateral sclerosis—from pathogenesis to biomarkers. Nat. Rev. Neurol. 2021, 17, 333–348. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V.; Rodriguez-Arellano, J.J.; Zorec, R. Astroglia in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 273–324. [Google Scholar] [CrossRef]
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGFbeta superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Schlecht, A.; Vallon, M.; Wagner, N.; Ergun, S.; Braunger, B.M. TGFbeta-Neurotrophin Interactions in Heart, Retina, and Brain. Biomolecules 2021, 11, 1360. [Google Scholar] [CrossRef]
- Meyers, E.A.; Kessler, J.A. TGF-beta Family Signaling in Neural and Neuronal Differentiation, Development, and Function. Cold Spring Harb. Perspect. Biol. 2017, 9, 2244. [Google Scholar] [CrossRef]
- Kandasamy, M.; Anusuyadevi, M.; Aigner, K.M.; Unger, M.S.; Kniewallner, K.M.; de Sousa, D.M.B.; Altendorfer, B.; Mrowetz, H.; Bogdahn, U.; Aigner, L. TGF-beta Signaling: A Therapeutic Target to Reinstate Regenerative Plasticity in Vascular Dementia? Aging Dis. 2020, 11, 828–850. [Google Scholar] [CrossRef]
- Diniz, L.P.; Matias, I.; Siqueira, M.; Stipursky, J.; Gomes, F.C.A. Astrocytes and the TGF-beta1 Pathway in the Healthy and Diseased Brain: A Double-Edged Sword. Mol. NeuroBiol. 2019, 56, 4653–4679. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Koyama, Y. Signaling molecules regulating phenotypic conversions of astrocytes and glial scar formation in damaged nerve tissues. Neurochem. Int. 2014, 78, 35–42. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef]
- Luo, J.; Wyss-Coray, T. Bioluminescence analysis of Smad-dependent TGF-beta signaling in live mice. Methods Mol. Biol. 2009, 574, 193–202. [Google Scholar] [CrossRef]
- Luo, J.; Lin, A.H.; Masliah, E.; Wyss-Coray, T. Bioluminescence imaging of Smad signaling in living mice shows correlation with excitotoxic neurodegeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 18326–18331. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ho, P.P.; Buckwalter, M.S.; Hsu, T.; Lee, L.Y.; Zhang, H.; Kim, D.K.; Kim, S.J.; Gambhir, S.S.; Steinman, L.; et al. Glia-dependent TGF-beta signaling, acting independently of the TH17 pathway, is critical for initiation of murine autoimmune encephalomyelitis. J. Clin. Investig. 2007, 117, 3306–3315. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.H.; Luo, J.; Mondshein, L.H.; ten Dijke, P.; Vivien, D.; Contag, C.H.; Wyss-Coray, T. Global analysis of Smad2/3-dependent TGF-beta signaling in living mice reveals prominent tissue-specific responses to injury. J. Immunol. 2005, 175, 547–554. [Google Scholar] [CrossRef]
- Pandya, V.A.; Patani, R. Region-specific vulnerability in neurodegeneration: Lessons from normal ageing. Ageing Res. Rev. 2021, 67, 101311. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.; Bryant, S.; Luo, J.; Kiesler, P.; Koontz, S.; Warren, J.; Malech, H.; Kang, E.; Dveksler, G. Recombinant Pregnancy-Specific Glycoprotein 1 Has a Protective Role in a Murine Model of Acute Graft-versus-Host Disease. Biol. Blood Marrow Transplant. 2019, 25, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Vest, R.T.; Chou, C.C.; Zhang, H.; Haney, M.S.; Li, L.; Laqtom, N.N.; Chang, B.; Shuken, S.; Nguyen, A.; Yerra, L.; et al. Small molecule C381 targets the lysosome to reduce inflammation and ameliorate disease in models of neurodegeneration. Proc. Natl. Acad. Sci. USA 2022, 119, e2121609119. [Google Scholar] [CrossRef] [PubMed]
- Sousa Vde, O.; Romao, L.; Neto, V.M.; Gomes, F.C. Glial fibrillary acidic protein gene promoter is differently modulated by transforming growth factor-beta 1 in astrocytes from distinct brain regions. Eur. J. Neurosci. 2004, 19, 1721–1730. [Google Scholar] [CrossRef]
- de Sampaio e Spohr, T.C.; Martinez, R.; da Silva, E.F.; Neto, V.M.; Gomes, F.C. Neuro-glia interaction effects on GFAP gene: A novel role for transforming growth factor-beta1. Eur. J. Neurosci. 2002, 16, 2059–2069. [Google Scholar] [CrossRef]
- Buckwalter, M.S.; Yamane, M.; Coleman, B.S.; Ormerod, B.K.; Chin, J.T.; Palmer, T.; Wyss-Coray, T. Chronically increased transforming growth factor-beta1 strongly inhibits hippocampal neurogenesis in aged mice. Am. J. Pathol. 2006, 169, 154–164. [Google Scholar] [CrossRef]
- O’Neil, S.M.; Hans, E.E.; Jiang, S.; Wangler, L.M.; Godbout, J.P. Astrocyte immunosenescence and deficits in interleukin 10 signaling in the aged brain disrupt the regulation of microglia following innate immune activation. Glia 2022, 70, 913–934. [Google Scholar] [CrossRef]
- Baxter, P.S.; Dando, O.; Emelianova, K.; He, X.; McKay, S.; Hardingham, G.E.; Qiu, J. Microglial identity and inflammatory responses are controlled by the combined effects of neurons and astrocytes. Cell Rep. 2021, 34, 108882. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- McConnell, H.L.; Li, Z.; Woltjer, R.L.; Mishra, A. Astrocyte dysfunction and neurovascular impairment in neurological disorders: Correlation or causation? Neurochem. Int. 2019, 128, 70–84. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Reid, J.K.; Kuipers, H.F. She Doesn’t Even Go Here: The Role of Inflammatory Astrocytes in CNS Disorders. Front. Cell. Neurosci. 2021, 15, 704884. [Google Scholar] [CrossRef]
- Leng, K.; Rooney, B.; Kim, H.; Xia, W.; Koontz, M.; Krawczyk, M.; Zhang, Y.; Ullian, E.M.; Fancy, S.P.J.; Schrag, M.S.; et al. CRISPRi screens in human astrocytes elucidate regulators of distinct inflammatory reactive states. bioRxiv 2021. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Adler, D.I.; Couthouis, J.; Liddelow, S.A.; Gitler, A.D.; Barres, B.A. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat. Commun. 2020, 11, 3753. [Google Scholar] [CrossRef]
- Hasel, P.; Rose, I.V.L.; Sadick, J.S.; Kim, R.D.; Liddelow, S.A. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat. Neurosci. 2021. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Munch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Pekna, M.; Pekny, M. The Complement System: A Powerful Modulator and Effector of Astrocyte Function in the Healthy and Diseased Central Nervous System. Cells 2021, 10, 1812. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Lennon, V.A.; Liu, Y.U.; Bosco, D.B.; Li, Y.; Yi, M.H.; Zhu, J.; Wei, S.; Wu, L.J. Astrocyte-microglia interaction drives evolving neuromyelitis optica lesion. J. Clin. Investig. 2020, 130, 4025–4038. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef]
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Kohl, J.; Zheng, H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Guttikonda, S.R.; Sikkema, L.; Tchieu, J.; Saurat, N.; Walsh, R.M.; Harschnitz, O.; Ciceri, G.; Sneeboer, M.; Mazutis, L.; Setty, M.; et al. Fully defined human pluripotent stem cell-derived microglia and tri-culture system model C3 production in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 343–354. [Google Scholar] [CrossRef]
- Taylor, X.; Cisternas, P.; Jury, N.; Martinez, P.; Huang, X.; You, Y.; Redding-Ochoa, J.; Vidal, R.; Zhang, J.; Troncoso, J.; et al. Activated endothelial cells induce a distinct type of astrocytic reactivity. Commun. Biol. 2022, 5, 282. [Google Scholar] [CrossRef]
- Price, B.R.; Johnson, L.A.; Norris, C.M. Reactive astrocytes: The nexus of pathological and clinical hallmarks of Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101335. [Google Scholar] [CrossRef]
- Xue, X.; Zhang, W.; Zhu, J.; Chen, X.; Zhou, S.; Xu, Z.; Hu, G.; Su, C. Aquaporin-4 deficiency reduces TGF-beta1 in mouse midbrains and exacerbates pathology in experimental Parkinson’s disease. J. Cell Mol. Med. 2019, 23, 2568–2582. [Google Scholar] [CrossRef]
- Nataf, S.; Barritault, M.; Pays, L. A Unique TGFB1-Driven Genomic Program Links Astrocytosis, Low-Grade Inflammation and Partial Demyelination in Spinal Cord Periplaques from Progressive Multiple Sclerosis Patients. Int. J. Mol. Sci. 2017, 18, 2097. [Google Scholar] [CrossRef]
- Nataf, S.; Guillen, M.; Pays, L. TGFB1-Mediated Gliosis in Multiple Sclerosis Spinal Cords Is Favored by the Regionalized Expression of HOXA5 and the Age-Dependent Decline in Androgen Receptor Ligands. Int. J. Mol. Sci. 2019, 20, 5934. [Google Scholar] [CrossRef]
- Yu, P.; Wang, H.; Katagiri, Y.; Geller, H.M. An in vitro model of reactive astrogliosis and its effect on neuronal growth. Methods Mol. Biol. 2012, 814, 327–340. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, Y.; Xie, F.; Zhong, Y.; Wang, Y.; Xu, M.; Feng, J.; Charish, J.; Monnier, P.P.; Qin, X. RGMa mediates reactive astrogliosis and glial scar formation through TGFbeta1/Smad2/3 signaling after stroke. Cell Death Differ. 2018, 25, 1503–1516. [Google Scholar] [CrossRef]
- Lu, M.; Yan, X.F.; Si, Y.; Chen, X.Z. CTGF Triggers Rat Astrocyte Activation and Astrocyte-Mediated Inflammatory Response in Culture Conditions. Inflammation 2019, 42, 1693–1704. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Lin, C.; Sanan, D.A.; Mucke, L.; Masliah, E. Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am. J. Pathol. 2000, 156, 139–150. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Masliah, E.; Mallory, M.; McConlogue, L.; Johnson-Wood, K.; Lin, C.; Mucke, L. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature 1997, 389, 603–606. [Google Scholar] [CrossRef]
- Gaertner, R.F.; Wyss-Coray, T.; Von Euw, D.; Lesne, S.; Vivien, D.; Lacombe, P. Reduced brain tissue perfusion in TGF-beta 1 transgenic mice showing Alzheimer’s disease-like cerebrovascular abnormalities. NeuroBiol. Dis. 2005, 19, 38–46. [Google Scholar] [CrossRef]
- Lesne, S.; Docagne, F.; Gabriel, C.; Liot, G.; Lahiri, D.K.; Buee, L.; Plawinski, L.; Delacourte, A.; MacKenzie, E.T.; Buisson, A.; et al. Transforming growth factor-beta 1 potentiates amyloid-beta generation in astrocytes and in transgenic mice. J. Biol. Chem. 2003, 278, 18408–18418. [Google Scholar] [CrossRef]
- McCarty, J.H. alphavbeta8 integrin adhesion and signaling pathways in development, physiology and disease. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Cambier, S.; Gline, S.; Mu, D.; Collins, R.; Araya, J.; Dolganov, G.; Einheber, S.; Boudreau, N.; Nishimura, S.L. Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: An angiogenic control switch. Am. J. Pathol. 2005, 166, 1883–1894. [Google Scholar] [CrossRef]
- Hirota, S.; Liu, Q.; Lee, H.S.; Hossain, M.G.; Lacy-Hulbert, A.; McCarty, J.H. The astrocyte-expressed integrin alphavbeta8 governs blood vessel sprouting in the developing retina. Development 2011, 138, 5157–5166. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Ding, Z.; Ho, P.P.; Luo, J.; Agrawal, A.N.; Srinagesh, H.; Axtell, R.; Zhang, H.; Platten, M.; Wyss-Coray, T.; et al. Angiotensin II sustains brain inflammation in mice via TGF-beta. J. Clin. Investig. 2010, 120, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Senatorov, V.V., Jr.; Morrissey, C.S.; Lippmann, K.; Vazquez, O.; Milikovsky, D.Z.; Gu, F.; Parada, I.; Prince, D.A.; Becker, A.J.; et al. TGFbeta signaling is associated with changes in inflammatory gene expression and perineuronal net degradation around inhibitory neurons following various neurological insults. Sci. Rep. 2017, 7, 7711. [Google Scholar] [CrossRef] [PubMed]
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J. Neurosci. 2010, 30, 5843–5854. [Google Scholar] [CrossRef] [PubMed]
- Senatorov, V.V., Jr.; Friedman, A.R.; Milikovsky, D.Z.; Ofer, J.; Saar-Ashkenazy, R.; Charbash, A.; Jahan, N.; Chin, G.; Mihaly, E.; Lin, J.M.; et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFbeta signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Sakamoto, T.; Yamanaka, I.; Sassa, Y.; Kubota, T.; Ueno, H.; Ohnishi, Y.; Ishibashi, T. Photocoagulation-induced retinal gliosis is inhibited by systemically expressed soluble TGF-beta receptor type II via adenovirus mediated gene transfer. Lab. Investig. 2002, 82, 863–870. [Google Scholar] [CrossRef]
- Levy, N.; Milikovsky, D.Z.; Baranauskas, G.; Vinogradov, E.; David, Y.; Ketzef, M.; Abutbul, S.; Weissberg, I.; Kamintsky, L.; Fleidervish, I.; et al. Differential TGF-beta Signaling in Glial Subsets Underlies IL-6-Mediated Epileptogenesis in Mice. J. Immunol. 2015, 195, 1713–1722. [Google Scholar] [CrossRef]
- Cacheaux, L.P.; Ivens, S.; David, Y.; Lakhter, A.J.; Bar-Klein, G.; Shapira, M.; Heinemann, U.; Friedman, A.; Kaufer, D. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J. Neurosci. 2009, 29, 8927–8935. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Dhandapani, K.M.; Hadman, M.; De Sevilla, L.; Wade, M.F.; Mahesh, V.B.; Brann, D.W. Astrocyte protection of neurons: Role of transforming growth factor-beta signaling via a c-Jun-AP-1 protective pathway. J. Biol. Chem. 2003, 278, 43329–43339. [Google Scholar] [CrossRef]
- Yu, Z.; Yu, P.; Chen, H.; Geller, H.M. Targeted inhibition of KCa3.1 attenuates TGF-beta-induced reactive astrogliosis through the Smad2/3 signaling pathway. J. Neurochem. 2014, 130, 41–49. [Google Scholar] [CrossRef]
- Tatomir, A.; Beltrand, A.; Nguyen, V.; Boodhoo, D.; Mekala, A.; Cudrici, C.; Badea, T.C.; Muresanu, D.F.; Rus, V.; Rus, H. RGC-32 Regulates Generation of Reactive Astrocytes in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Guo, X.; Kimura, A.; Namekata, K.; Harada, C.; Arai, N.; Takeda, K.; Ichijo, H.; Harada, T. ASK1 signaling regulates phase-specific glial interactions during neuroinflammation. Proc. Natl. Acad. Sci. USA 2022, 119. [Google Scholar] [CrossRef]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef]
- Rojas, A.; Padidam, M.; Cress, D.; Grady, W.M. TGF-beta receptor levels regulate the specificity of signaling pathway activation and biological effects of TGF-beta. Biochim. Biophys. Acta 2009, 1793, 1165–1173. [Google Scholar] [CrossRef]
- Langley, B.; D’Annibale, M.A.; Suh, K.; Ayoub, I.; Tolhurst, A.; Bastan, B.; Yang, L.; Ko, B.; Fisher, M.; Cho, S.; et al. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J. Neurosci. 2008, 28, 163–176. [Google Scholar] [CrossRef]
- Tusell, J.M.; Saura, J.; Serratosa, J. Absence of the cell cycle inhibitor p21Cip1 reduces LPS-induced NO release and activation of the transcription factor NF-kappaB in mixed glial cultures. Glia 2005, 49, 52–58. [Google Scholar] [CrossRef]
- Docagne, F.; Nicole, O.; Gabriel, C.; Fernandez-Monreal, M.; Lesne, S.; Ali, C.; Plawinski, L.; Carmeliet, P.; MacKenzie, E.T.; Buisson, A.; et al. Smad3-dependent induction of plasminogen activator inhibitor-1 in astrocytes mediates neuroprotective activity of transforming growth factor-beta 1 against NMDA-induced necrosis. Mol. Cell Neurosci. 2002, 21, 634–644. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhao, L.; Ma, N.; Ye, Z.; Jiang, Z.; Chu, C. Up-regulated Complement 3 Production by Toll-like receptor 9/ Transforming Growth Factor-Beta 1/Complement 3 Pathway in Whole Blood Cells of Lupus Thrombocytopenia. Arch. Rheumatol. 2017, 32, 275–283. [Google Scholar] [CrossRef]
- Li, Y.; Song, D.; Song, Y.; Zhao, L.; Wolkow, N.; Tobias, J.W.; Song, W.; Dunaief, J.L. Iron-induced Local Complement Component 3 (C3) Up-regulation via Non-canonical Transforming Growth Factor (TGF)-beta Signaling in the Retinal Pigment Epithelium. J. Biol. Chem. 2015, 290, 11918–11934. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Mickler, E.A.; Cummings, O.W.; Sandusky, G.E.; Weber, D.J.; Gracon, A.; Woodruff, T.; Wilkes, D.S.; Vittal, R. Crosstalk between TGF-beta1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J. 2014, 28, 4223–4234. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Ko, J.S.; Lee, A.R.; Shin, B.S.; Choi, S.J.; Lee, J.J.; Kim, H.S.; Lee, S.M. Downregulation of constitutive and cytokine-induced complement 3 expression by morphine in rat astrocytes. Curr. Ther. Res. Clin. Exp. 2011, 72, 23–35. [Google Scholar] [CrossRef] [PubMed]
- da Cunha, A.; Jefferson, J.A.; Jackson, R.W.; Vitkovic, L. Glial cell-specific mechanisms of TGF-beta 1 induction by IL-1 in cerebral cortex. J. NeuroImmunol. 1993, 42, 71–85. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Weise, S.C.; Villarreal, A.; Heidrich, S.; Dehghanian, F.; Schachtrup, C.; Nestel, S.; Schwarz, J.; Thedieck, K.; Vogel, T. TGFβ-Signaling and FOXG1-Expression Are a Hallmark of Astrocyte Lineage Diversity in the Murine Ventral and Dorsal Forebrain. Front. Cell. Neurosci. 2018, 12, 448. [Google Scholar] [CrossRef]
- John Lin, C.C.; Yu, K.; Hatcher, A.; Huang, T.W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017, 20, 396–405. [Google Scholar] [CrossRef]
- Buosi, A.S.; Matias, I.; Araujo, A.P.B.; Batista, C.; Gomes, F.C.A. Heterogeneity in Synaptogenic Profile of Astrocytes from Different Brain Regions. Mol. NeuroBiol. 2018, 55, 751–762. [Google Scholar] [CrossRef]
- Bradley, R.A.; Shireman, J.; McFalls, C.; Choi, J.; Canfield, S.G.; Dong, Y.; Liu, K.; Lisota, B.; Jones, J.R.; Petersen, A.; et al. Regionally specified human pluripotent stem cell-derived astrocytes exhibit different molecular signatures and functional properties. Development 2019, 146, dev170910. [Google Scholar] [CrossRef]
- Ohlig, S.; Clavreul, S.; Thorwirth, M.; Simon-Ebert, T.; Bocchi, R.; Ulbricht, S.; Kannayian, N.; Rossner, M.; Sirko, S.; Smialowski, P.; et al. Molecular diversity of diencephalic astrocytes reveals adult astrogenesis regulated by Smad4. EMBO J. 2021, 40, e107532. [Google Scholar] [CrossRef]
- Moulson, A.J.; Squair, J.W.; Franklin, R.J.M.; Tetzlaff, W.; Assinck, P. Diversity of Reactive Astrogliosis in CNS Pathology: Heterogeneity or Plasticity? Front. Cell Neurosci. 2021, 15, 703810. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Buki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Scholten, J.; Vasterling, J.J.; Grimes, J.B. Traumatic brain injury clinical practice guidelines and best practices from the VA state of the art conference. Brain Inj. 2017, 31, 1246–1251. [Google Scholar] [CrossRef]
- McCrory, P.; Meeuwisse, W.; Dvorak, J.; Aubry, M.; Bailes, J.; Broglio, S.; Cantu, R.C.; Cassidy, D.; Echemendia, R.J.; Castellani, R.J.; et al. Consensus statement on concussion in sport-the 5(th) international conference on concussion in sport held in Berlin, October 2016. Br. J. Sports Med. 2017, 51, 838–847. [Google Scholar] [CrossRef]
- Fehily, B.; Fitzgerald, M. Repeated Mild Traumatic Brain Injury: Potential Mechanisms of Damage. Cell Transplant. 2017, 26, 1131–1155. [Google Scholar] [CrossRef]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef]
- Ma, X.; Aravind, A.; Pfister, B.J.; Chandra, N.; Haorah, J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol. NeuroBiol. 2019, 56, 5332–5345. [Google Scholar] [CrossRef]
- Petersen, A.; Soderstrom, M.; Saha, B.; Sharma, P. Animal models of traumatic brain injury: A review of pathophysiology to biomarkers and treatments. Exp. Brain Res. 2021, 239, 2939–2950. [Google Scholar] [CrossRef]
- Williams, A.J.; Hartings, J.A.; Lu, X.C.; Rolli, M.L.; Dave, J.R.; Tortella, F.C. Characterization of a new rat model of penetrating ballistic brain injury. J. Neurotrauma 2005, 22, 313–331. [Google Scholar] [CrossRef]
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 2020, 8, 389. [Google Scholar] [CrossRef]
- McGinn, M.J.; Povlishock, J.T. Pathophysiology of Traumatic Brain Injury. Neurosurg. Clin. N. Am. 2016, 27, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Wu, H. Astrocytes in the Traumatic Brain Injury: The Good and the Bad. Exp. Neurol. 2022, 348, 113943. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Rosa, J.M.; Farre-Alins, V.; Ortega, M.C.; Navarrete, M.; Lopez-Rodriguez, A.B.; Palomino-Antolin, A.; Fernandez-Lopez, E.; Vila-Del Sol, V.; Decouty, C.; Narros-Fernandez, P.; et al. TLR4 pathway impairs synaptic number and cerebrovascular functions through astrocyte activation following traumatic brain injury. Br. J. Pharmacol. 2021, 178, 3395–3413. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.; Apgar, C.; Shapiro, L.A. Astrocyte Hypertrophy Contributes to Aberrant Neurogenesis after Traumatic Brain Injury. Neural. Plast. 2016, 2016, 1347987. [Google Scholar] [CrossRef]
- Zhu, L.; Ramboz, S.; Hewitt, D.; Boring, L.; Grass, D.S.; Purchio, A.F. Non-invasive imaging of GFAP expression after neuronal damage in mice. Neurosci. Lett. 2004, 367, 210–212. [Google Scholar] [CrossRef]
- Li, L.; Yerra, L.; Chang, B.; Mathur, V.; Nguyen, A.; Luo, J. Acute and late administration of colony stimulating factor 1 attenuates chronic cognitive impairment following mild traumatic brain injury in mice. Brain Behav. Immun. 2021, 94, 274–288. [Google Scholar] [CrossRef]
- Luo, J.; Nguyen, A.; Villeda, S.; Zhang, H.; Ding, Z.; Lindsey, D.; Bieri, G.; Castellano, J.M.; Beaupre, G.S.; Wyss-Coray, T. Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front. Neurol. 2014, 5, 12. [Google Scholar] [CrossRef]
- Abdelhak, A.; Foschi, M.; Abu-Rumeileh, S.; Yue, J.K.; D’Anna, L.; Huss, A.; Oeckl, P.; Ludolph, A.C.; Kuhle, J.; Petzold, A.; et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat. Rev. Neurol. 2022, 18, 158–172. [Google Scholar] [CrossRef]
- Lenzlinger, P.M.; Morganti-Kossmann, M.C.; Laurer, H.L.; McIntosh, T.K. The duality of the inflammatory response to traumatic brain injury. Mol. NeuroBiol. 2001, 24, 169–181. [Google Scholar] [CrossRef]
- Rimaniol, A.C.; Lekieffre, D.; Serrano, A.; Masson, A.; Benavides, J.; Zavala, F. Biphasic transforming growth factor-beta production flanking the pro-inflammatory cytokine response in cerebral trauma. Neuroreport 1995, 7, 133–136. [Google Scholar] [CrossRef]
- Komuta, Y.; Teng, X.; Yanagisawa, H.; Sango, K.; Kawamura, K.; Kawano, H. Expression of transforming growth factor-beta receptors in meningeal fibroblasts of the injured mouse brain. Cell. Mol. Neurobiol. 2010, 30, 101–111. [Google Scholar] [CrossRef]
- Divolis, G.; Stavropoulos, A.; Manioudaki, M.; Apostolidou, A.; Doulou, A.; Gavriil, A.; Dafnis, I.; Chroni, A.; Mummery, C.; Xilouri, M.; et al. Activation of both transforming growth factor-beta and bone morphogenetic protein signalling pathways upon traumatic brain injury restrains pro-inflammatory and boosts tissue reparatory responses of reactive astrocytes and microglia. Brain Commun. 2019, 1, fcz028. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, B.; Wu, X.; Yang, Z.; Huang, T.; Guo, X.; Guo, D.; Liu, Z.; Song, J. TGFbeta1 alleviates axonal injury by regulating microglia/macrophages alternative activation in traumatic brain injury. Brain Res. Bull. 2020, 161, 21–32. [Google Scholar] [CrossRef]
- Izzy, S.; Liu, Q.; Fang, Z.; Lule, S.; Wu, L.; Chung, J.Y.; Sarro-Schwartz, A.; Brown-Whalen, A.; Perner, C.; Hickman, S.E.; et al. Time-Dependent Changes in Microglia Transcriptional Networks Following Traumatic Brain Injury. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Wang, X.Y.; Ba, Y.C.; Xiong, L.L.; Li, X.L.; Zou, Y.; Zhu, Y.C.; Zhou, X.F.; Wang, T.H.; Wang, F.; Tian, H.L.; et al. Endogenous TGFbeta1 Plays a Crucial Role in Functional Recovery After Traumatic Brain Injury Associated with Smad3 Signal in Rats. Neurochem. Res. 2015, 40, 1671–1680. [Google Scholar] [CrossRef]
- Patel, R.K.; Prasad, N.; Kuwar, R.; Haldar, D.; Abdul-Muneer, P.M. Transforming growth factor-beta 1 signaling regulates neuroinflammation and apoptosis in mild traumatic brain injury. Brain Behav. Immun. 2017, 64, 244–258. [Google Scholar] [CrossRef]
- Xiong, J.; Gao, Y.; Li, X.; Li, K.; Li, Q.; Shen, J.; Han, Z.; Zhang, J. Losartan Treatment Could Improve the Outcome of TBI Mice. Front. Neurol. 2020, 11, 992. [Google Scholar] [CrossRef]
- Li, T.; Zhang, P.; Yuan, B.; Zhao, D.; Chen, Y.; Zhang, X. Thrombin-induced TGF-beta1 pathway: A cause of communicating hydrocephalus post subarachnoid hemorrhage. Int. J. Mol. Med. 2013, 31, 660–666. [Google Scholar] [CrossRef]
- Mhatre, M.; Nguyen, A.; Kashani, S.; Pham, T.; Adesina, A.; Grammas, P. Thrombin, a mediator of neurotoxicity and memory impairment. NeuroBiol. Aging 2004, 25, 783–793. [Google Scholar] [CrossRef]
- Piao, C.S.; Holloway, A.L.; Hong-Routson, S.; Wainwright, M.S. Depression following traumatic brain injury in mice is associated with down-regulation of hippocampal astrocyte glutamate transporters by thrombin. J. Cereb. Blood Flow. Metab. 2019, 39, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Ben Shimon, M.; Shavit-Stein, E.; Altman, K.; Pick, C.G.; Maggio, N. Thrombin as Key Mediator of Seizure Development Following Traumatic Brain Injury. Front. Pharmacol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Yang, S.H.; Liu, R. Four Decades of Ischemic Penumbra and Its Implication for Ischemic Stroke. Transl. Stroke Res. 2021, 12, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Wilhelmsson, U.; Tatlisumak, T.; Pekna, M. Astrocyte activation and reactive gliosis-A new target in stroke? Neurosci. Lett. 2019, 689, 45–55. [Google Scholar] [CrossRef]
- Aswendt, M.; Wilhelmsson, U.; Wieters, F.; Stokowska, A.; Schmitt, F.J.; Pallast, N.; de Pablo, Y.; Mohammed, L.; Hoehn, M.; Pekna, M.; et al. Reactive astrocytes prevent maladaptive plasticity after ischemic stroke. Prog. NeuroBiol. 2022, 209, 102199. [Google Scholar] [CrossRef]
- Williamson, M.R.; Fuertes, C.J.A.; Dunn, A.K.; Drew, M.R.; Jones, T.A. Reactive astrocytes facilitate vascular repair and remodeling after stroke. Cell Rep. 2021, 35, 109048. [Google Scholar] [CrossRef]
- Ito, U.; Hakamata, Y.; Kawakami, E.; Oyanagi, K. Temporary [corrected] cerebral ischemia results in swollen astrocytic end-feet that compress microvessels and lead to delayed [corrected] focal cortical infarction. J. Cereb. Blood Flow. Metab. 2011, 31, 328–338. [Google Scholar] [CrossRef]
- Begum, G.; Song, S.; Wang, S.; Zhao, H.; Bhuiyan, M.I.H.; Li, E.; Nepomuceno, R.; Ye, Q.; Sun, M.; Calderon, M.J.; et al. Selective knockout of astrocytic Na(+) /H(+) exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia 2018, 66, 126–144. [Google Scholar] [CrossRef]
- Dobolyi, A.; Vincze, C.; Pal, G.; Lovas, G. The neuroprotective functions of transforming growth factor beta proteins. Int. J. Mol. Sci. 2012, 13, 8219–8258. [Google Scholar] [CrossRef]
- Zhu, H.; Gui, Q.; Hui, X.; Wang, X.; Jiang, J.; Ding, L.; Sun, X.; Wang, Y.; Chen, H. TGF-beta1/Smad3 Signaling Pathway Suppresses Cell Apoptosis in Cerebral Ischemic Stroke Rats. Med. Sci. Monit. 2017, 23, 366–376. [Google Scholar] [CrossRef]
- Sugimoto, K.; Nishioka, R.; Ikeda, A.; Mise, A.; Takahashi, H.; Yano, H.; Kumon, Y.; Ohnishi, T.; Tanaka, J. Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-beta1. Glia 2014, 62, 185–198. [Google Scholar] [CrossRef]
- Liu, F.F.; Liu, C.Y.; Li, X.P.; Zheng, S.Z.; Li, Q.Q.; Liu, Q.; Song, L. Neuroprotective effects of SMADs in a rat model of cerebral ischemia/reperfusion. Neural. Regen. Res. 2015, 10, 438–444. [Google Scholar] [CrossRef]
- Doyle, K.P.; Cekanaviciute, E.; Mamer, L.E.; Buckwalter, M.S. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J. Neuroinflamm. 2010, 7, 62. [Google Scholar] [CrossRef]
- Knuckey, N.W.; Finch, P.; Palm, D.E.; Primiano, M.J.; Johanson, C.E.; Flanders, K.C.; Thompson, N.L. Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat hippocampus following transient forebrain ischemia. Brain Res. Mol. Brain Res. 1996, 40, 1–14. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Fathali, N.; Doyle, K.P.; Williams, A.M.; Han, J.; Buckwalter, M.S. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia 2014, 62, 1227–1240. [Google Scholar] [CrossRef]
- Li, D.; Lang, W.; Zhou, C.; Wu, C.; Zhang, F.; Liu, Q.; Yang, S.; Hao, J. Upregulation of Microglial ZEB1 Ameliorates Brain Damage after Acute Ischemic Stroke. Cell Rep. 2018, 22, 3574–3586. [Google Scholar] [CrossRef]
- Salas, I.H.; Burgado, J.; Allen, N.J. Glia: Victims or villains of the aging brain? NeuroBiol. Dis. 2020, 143, 105008. [Google Scholar] [CrossRef]
- Palmer, A.L.; Ousman, S.S. Astrocytes and Aging. Front. Aging Neurosci. 2018, 10, 337. [Google Scholar] [CrossRef]
- Verkerke, M.; Hol, E.M.; Middeldorp, J. Physiological and Pathological Ageing of Astrocytes in the Human Brain. Neurochem. Res. 2021, 46, 2662–2675. [Google Scholar] [CrossRef]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ma, N.; Yu, B.; Zhang, W.; Wan, J. Transcriptomic profiling of microglia and astrocytes throughout aging. J. Neuroinflammation 2020, 17, 97. [Google Scholar] [CrossRef] [PubMed]
- Wruck, W.; Adjaye, J. Meta-analysis of human prefrontal cortex reveals activation of GFAP and decline of synaptic transmission in the aging brain. Acta Neuropathol. Commun. 2020, 8, 26. [Google Scholar] [CrossRef]
- Soreq, L.; Consortium, U.K.B.E.; North American Brain Expression, C.; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; et al. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Augusto-Oliveira, M.; Pivoriunas, A.; Popov, A.; Brazhe, A.; Semyanov, A. Astroglial asthenia and loss of function, rather than reactivity, contribute to the ageing of the brain. Pflug. Arch. 2021, 473, 753–774. [Google Scholar] [CrossRef]
- Werry, E.L.; Enjeti, S.; Halliday, G.M.; Sachdev, P.S.; Double, K.L. Effect of age on proliferation-regulating factors in human adult neurogenic regions. J. Neurochem. 2010, 115, 956–964. [Google Scholar] [CrossRef]
- Tichauer, J.E.; Flores, B.; Soler, B.; Eugenin-von Bernhardi, L.; Ramirez, G.; von Bernhardi, R. Age-dependent changes on TGFbeta1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 2014, 37, 187–196. [Google Scholar] [CrossRef]
- Ueberham, U.; Lange, P.; Ueberham, E.; Bruckner, M.K.; Hartlage-Rubsamen, M.; Pannicke, T.; Rohn, S.; Cross, M.; Arendt, T. Smad2 isoforms are differentially expressed during mouse brain development and aging. Int. J. Dev. Neurosci. 2009, 27, 501–510. [Google Scholar] [CrossRef]
- von Bernhardi, R.; Cornejo, F.; Parada, G.E.; Eugenin, J. Role of TGFbeta signaling in the pathogenesis of Alzheimer’s disease. Front. Cell Neurosci. 2015, 9, 426. [Google Scholar] [CrossRef]
- Babcock, K.R.; Page, J.S.; Fallon, J.R.; Webb, A.E. Adult Hippocampal Neurogenesis in Aging and Alzheimer’s Disease. Stem Cell Rep. 2021, 16, 681–693. [Google Scholar] [CrossRef]
- Wu, X.; Shen, Q.; Zhang, Z.; Zhang, D.; Gu, Y.; Xing, D. Photoactivation of TGFbeta/SMAD signaling pathway ameliorates adult hippocampal neurogenesis in Alzheimer’s disease model. Stem. Cell Res. Ther. 2021, 12, 345. [Google Scholar] [CrossRef]
- Hinks, G.L.; Franklin, R.J. Delayed changes in growth factor gene expression during slow remyelination in the CNS of aged rats. Mol. Cell Neurosci. 2000, 16, 542–556. [Google Scholar] [CrossRef]
- Matias, I.; Diniz, L.P.; Damico, I.V.; Araujo, A.P.B.; Neves, L.D.S.; Vargas, G.; Leite, R.E.P.; Suemoto, C.K.; Nitrini, R.; Jacob-Filho, W.; et al. Loss of lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell 2022, 21, e13521. [Google Scholar] [CrossRef]
- Bellaver, B.; Souza, D.G.; Souza, D.O.; Quincozes-Santos, A. Hippocampal Astrocyte Cultures from Adult and Aged Rats Reproduce Changes in Glial Functionality Observed in the Aging Brain. Mol. NeuroBiol. 2017, 54, 2969–2985. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Goodall, E.F.; Wang, C.; Simpson, J.E.; Baker, D.J.; Drew, D.R.; Heath, P.R.; Saffrey, M.J.; Romero, I.A.; Wharton, S.B. Age-associated changes in the blood-brain barrier: Comparative studies in human and mouse. Neuropathol. Appl. NeuroBiol. 2018, 44, 328–340. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A.M. Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells 2021, 10, 540. [Google Scholar] [CrossRef]
- Sadick, J.S.; Liddelow, S.A. Don’t forget astrocytes when targeting Alzheimer’s disease. Br. J. Pharmacol. 2019, 176, 3585–3598. [Google Scholar] [CrossRef]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S.; et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2(-) production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Begard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Ceyzeriat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef]
- Sekar, S.; McDonald, J.; Cuyugan, L.; Aldrich, J.; Kurdoglu, A.; Adkins, J.; Serrano, G.; Beach, T.G.; Craig, D.W.; Valla, J.; et al. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. NeuroBiol. Aging 2015, 36, 583–591. [Google Scholar] [CrossRef]
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. NeuroBiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Lalakova, J.; Kuhnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e919. [Google Scholar] [CrossRef]
- Lau, S.F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Rodriguez, R.D.; Suemoto, C.K.; et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Viejo, L.; Noori, A.; Merrill, E.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Systematic review of human post-mortem immunohistochemical studies and bioinformatics analyses unveil the complexity of astrocyte reaction in Alzheimer’s disease. Neuropathol. Appl. NeuroBiol. 2022, 48, e12753. [Google Scholar] [CrossRef]
- Luedecking, E.K.; DeKosky, S.T.; Mehdi, H.; Ganguli, M.; Kamboh, M.I. Analysis of genetic polymorphisms in the transforming growth factor-beta1 gene and the risk of Alzheimer’s disease. Hum. Genet. 2000, 106, 565–569. [Google Scholar] [CrossRef]
- van der Wal, E.A.; Gomez-Pinilla, F.; Cotman, C.W. Transforming growth factor-beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport 1993, 4, 69–72. [Google Scholar] [CrossRef]
- Tesseur, I.; Zou, K.; Esposito, L.; Bard, F.; Berber, E.; Can, J.V.; Lin, A.H.; Crews, L.; Tremblay, P.; Mathews, P.; et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J. Clin. Investig. 2006, 116, 3060–3069. [Google Scholar] [CrossRef]
- Ueberham, U.; Hilbrich, I.; Ueberham, E.; Rohn, S.; Glockner, P.; Dietrich, K.; Bruckner, M.K.; Arendt, T. Transcriptional control of cell cycle-dependent kinase 4 by Smad proteins--implications for Alzheimer’s disease. NeuroBiol. Aging 2012, 33, 2827–2840. [Google Scholar] [CrossRef]
- Chalmers, K.A.; Love, S. Neurofibrillary tangles may interfere with Smad 2/3 signaling in neurons. J. Neuropathol. Exp. Neurol. 2007, 66, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Ueda, M.; Zhu, X.; Perry, G.; Smith, M.A. Ectopic expression of phospho-Smad2 in Alzheimer’s disease: Uncoupling of the transforming growth factor-beta pathway? J. Neurosci. Res. 2006, 84, 1856–1861. [Google Scholar] [CrossRef] [PubMed]
- Ueberham, U.; Ueberham, E.; Gruschka, H.; Arendt, T. Altered subcellular location of phosphorylated Smads in Alzheimer’s disease. Eur. J. Neurosci. 2006, 24, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Fessel, J. Ineffective levels of transforming growth factors and their receptor account for old age being a risk factor for Alzheimer’s disease. Alzheimer’s Dement. 2019, 5, 899–905. [Google Scholar] [CrossRef]
- Zheng, J.Y.; Sun, J.; Ji, C.M.; Shen, L.; Chen, Z.J.; Xie, P.; Sun, Y.Z.; Yu, R.T. Selective deletion of apolipoprotein E in astrocytes ameliorates the spatial learning and memory deficits in Alzheimer’s disease (APP/PS1) mice by inhibiting TGF-beta/Smad2/STAT3 signaling. NeuroBiol. Aging 2017, 54, 112–132. [Google Scholar] [CrossRef]
- Blesa, J.; Foffani, G.; Dehay, B.; Bezard, E.; Obeso, J.A. Motor and non-motor circuit disturbances in early Parkinson disease: Which happens first? Nat. Rev. Neurosci. 2022, 23, 115–128. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Li, B.; Zhao, G.; Li, K.; Wang, Z.; Fang, Z.; Wang, X.; Luo, T.; Zhang, Y.; Wang, Y.; Chen, Q.; et al. Characterizing the Expression Patterns of Parkinson’s Disease Associated Genes. Front. Neurosci. 2021, 15, 629156. [Google Scholar] [CrossRef]
- Reynolds, R.H.; Botia, J.; Nalls, M.A.; International Parkinson’s Disease Genomics, C.; System Genomics of Parkinson’s, D.; Hardy, J.; Gagliano Taliun, S.A.; Ryten, M. Moving beyond neurons: The role of cell type-specific gene regulation in Parkinson’s disease heritability. NPJ Parkinson’s Dis. 2019, 5, 6. [Google Scholar] [CrossRef]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 2021, 145, 964–978. [Google Scholar] [CrossRef]
- Zhong, J.; Tang, G.; Zhu, J.; Wu, W.; Li, G.; Lin, X.; Liang, L.; Chai, C.; Zeng, Y.; Wang, F.; et al. Single-cell brain atlas of Parkinson’s disease mouse model. J. Genet Genom. 2021, 48, 277–288. [Google Scholar] [CrossRef]
- García-Domínguez, I.; Veselá, K.; García-Revilla, J.; Carrillo-Jiménez, A.; Roca-Ceballos, M.A.; Santiago, M.; de Pablos, R.M.; Venero, J.L. Peripheral Inflammation Enhances Microglia Response and Nigral Dopaminergic Cell Death in an in vivo MPTP Model of Parkinson’s Disease. Front. Cell. Neurosci. 2018, 12, 398. [Google Scholar] [CrossRef]
- Luna-Herrera, C.; Martinez-Davila, I.A.; Soto-Rojas, L.O.; Flores-Martinez, Y.M.; Fernandez-Parrilla, M.A.; Ayala-Davila, J.; Leon-Chavez, B.A.; Soto-Rodriguez, G.; Blanco-Alvarez, V.M.; Lopez-Salas, F.E.; et al. Intranigral Administration of beta-Sitosterol-beta-D-Glucoside Elicits Neurotoxic A1 Astrocyte Reactivity and Chronic Neuroinflammation in the Rat Substantia Nigra. J. Immunol. Res. 2020, 2020, 5907591. [Google Scholar] [CrossRef]
- Song, N.; Zhu, H.; Xu, R.; Liu, J.; Fang, Y.; Zhang, J.; Ding, J.; Hu, G.; Lu, M. Induced Expression of kir6.2 in A1 Astrocytes Propagates Inflammatory Neurodegeneration via Drp1-dependent Mitochondrial Fission. Front. Pharmacol. 2021, 11, 2463. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef]
- Olsen, A.L.; Feany, M.B. Parkinson’s disease risk genes act in glia to control neuronal alpha-synuclein toxicity. NeuroBiol. Dis. 2021, 159, 105482. [Google Scholar] [CrossRef]
- Munoz, M.D.; de la Fuente, N.; Sanchez-Capelo, A. TGF-beta/Smad3 Signalling Modulates GABA Neurotransmission: Implications in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 590. [Google Scholar] [CrossRef]
- Roussa, E.; von Bohlen und Halbach, O.; Krieglstein, K. TGF-beta in dopamine neuron development, maintenance and neuroprotection. Adv. Exp. Med. Biol. 2009, 651, 81–90. [Google Scholar] [PubMed]
- Poulsen, K.T.; Armanini, M.P.; Klein, R.D.; Hynes, M.A.; Phillips, H.S.; Rosenthal, A. TGF beta 2 and TGF beta 3 are potent survival factors for midbrain dopaminergic neurons. Neuron 1994, 13, 1245–1252. [Google Scholar] [CrossRef]
- Goris, A.; Williams-Gray, C.H.; Foltynie, T.; Brown, J.; Maranian, M.; Walton, A.; Compston, D.A.; Barker, R.A.; Sawcer, S.J. Investigation of TGFB2 as a candidate gene in multiple sclerosis and Parkinson’s disease. J. Neurol. 2007, 254, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Zhao, H.; Frugier, T.; Meguro, R.; Grattan, D.R.; Koishi, K.; McLennan, I.S. Transforming growth factor beta2 haploinsufficient mice develop age-related nigrostriatal dopamine deficits. NeuroBiol. Dis. 2006, 21, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, L.; Xu, Y.; Tang, X.; Wang, X. Substantia nigra Smad3 signaling deficiency: Relevance to aging and Parkinson’s disease and roles of microglia, proinflammatory factors, and MAPK. J. Neuroinflamm. 2020, 17, 342. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Gonzalez, S.; Giraldez-Perez, R.M.; Cuartero, M.I.; Casarejos, M.J.; Mena, M.A.; Wang, X.F.; Sanchez-Capelo, A. Dopamine and alpha-synuclein dysfunction in Smad3 null mice. Mol. Neurodegener. 2011, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Rosenblad, C.; Bjorklund, A. Glial cell line-derived neurotrophic factor but not transforming growth factor beta 3 prevents delayed degeneration of nigral dopaminergic neurons following striatal 6-hydroxydopamine lesion. Proc. Natl. Acad. Sci. USA 1995, 92, 8935–8939. [Google Scholar] [CrossRef]
- Sanchez-Capelo, A.; Colin, P.; Guibert, B.; Biguet, N.F.; Mallet, J. Transforming growth factor beta1 overexpression in the nigrostriatal system increases the dopaminergic deficit of MPTP mice. Mol. Cell Neurosci. 2003, 23, 614–625. [Google Scholar] [CrossRef]
- Tesseur, I.; Nguyen, A.; Chang, B.; Li, L.; Woodling, N.S.; Wyss-Coray, T.; Luo, J. Deficiency in Neuronal TGF-beta Signaling Leads to Nigrostriatal Degeneration and Activation of TGF-beta Signaling Protects against MPTP Neurotoxicity in Mice. J. Neurosci. 2017, 37, 4584–4592. [Google Scholar] [CrossRef]
- Luo, S.X.; Timbang, L.; Kim, J.I.; Shang, Y.; Sandoval, K.; Tang, A.A.; Whistler, J.L.; Ding, J.B.; Huang, E.J. TGF-beta Signaling in Dopaminergic Neurons Regulates Dendritic Growth, Excitatory-Inhibitory Synaptic Balance, and Reversal Learning. Cell Rep. 2016, 17, 3233–3245. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Wessely, F.; Connor-Robson, N.; Rinaldi, F.; Vowles, J.; Browne, C.; Evetts, S.G.; Hu, M.T.; Cowley, S.A.; Webber, C.; et al. RNA sequencing reveals MMP2 and TGFB1 downregulation in LRRK2 G2019S Parkinson’s iPSC-derived astrocytes. NeuroBiol. Dis. 2019, 129, 56–66. [Google Scholar] [CrossRef]
- Diniz, L.P.; Matias, I.; Araujo, A.P.B.; Garcia, M.N.; Barros-Aragao, F.G.Q.; Alves-Leon, S.V.; de Souza, J.M.; Foguel, D.; Figueiredo, C.P.; Braga, C.; et al. alpha-synuclein oligomers enhance astrocyte-induced synapse formation through TGF-beta1 signaling in a Parkinson’s disease model. J. Neurochem. 2019, 150, 138–157. [Google Scholar] [CrossRef]
- Diniz, L.P.; Araujo, A.P.B.; Matias, I.; Garcia, M.N.; Barros-Aragao, F.G.Q.; de Melo Reis, R.A.; Foguel, D.; Braga, C.; Figueiredo, C.P.; Romao, L.; et al. Astrocyte glutamate transporters are increased in an early sporadic model of synucleinopathy. Neurochem. Int. 2020, 138, 104758. [Google Scholar] [CrossRef]
- Armstrong, M.J.; McFarland, N. Recognizing and treating atypical Parkinson disorders. Handb. Clin. Neurol. 2019, 167, 301–320. [Google Scholar] [CrossRef]
- Togo, T.; Dickson, D.W. Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol. 2002, 104, 398–402. [Google Scholar] [CrossRef]
- Svenningsson, P. Corticobasal degeneration: Advances in clinicopathology and biomarkers. Curr. Opin. Neurol. 2019, 32, 597–603. [Google Scholar] [CrossRef]
- Ling, H.; Macerollo, A. Is it Useful to Classify PSP and CBD as Different Disorders? Yes. Mov. Disord. Clin. Pract. 2018, 5, 145–148. [Google Scholar] [CrossRef]
- Rini, J.; Asken, B.; Geier, E.; Rankin, K.; Kramer, J.; Boxer, A.; Miller, B.; Yokoyama, J.; Spina, S. Genetic pleiotropy and the shared pathological features of corticobasal degeneration and progressive supranuclear palsy: A case report and a review of the literature. Neurocase 2021, 27, 120–128. [Google Scholar] [CrossRef]
- Krzosek, P.; Madetko, N.; Migda, A.; Migda, B.; Jaguś, D.; Alster, P. Differential Diagnosis of Rare Subtypes of Progressive Supranuclear Palsy and PSP-Like Syndromes—Infrequent Manifestations of the Most Common Form of Atypical Parkinsonism. Front. Aging Neurosci. 2022, 14, 804385. [Google Scholar] [CrossRef]
- Alster, P.; Madetko, N.; Friedman, A. Neutrophil-to-lymphocyte ratio (NLR) at boundaries of Progressive Supranuclear Palsy Syndrome (PSPS) and Corticobasal Syndrome (CBS). Neurol. Neurochir. Polska 2021, 55, 97–101. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Giasson, B.I.; Chakrabarty, P. alpha-Synuclein and astrocytes: Tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol. 2019, 138, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Valdinocci, D.; Radford, R.A.W.; Goulding, M.; Hayashi, J.; Chung, R.S.; Pountney, D.L. Extracellular Interactions of Alpha-Synuclein in Multiple System Atrophy. Int. J. Mol. Sci. 2018, 19, 4129. [Google Scholar] [CrossRef] [PubMed]
- Alster, P.; Madetko, N.; Koziorowski, D.; Friedman, A. Microglial Activation and Inflammation as a Factor in the Pathogenesis of Progressive Supranuclear Palsy (PSP). Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Lippa, C.F.; Smith, T.W.; Flanders, K.C. Transforming growth factor-beta: Neuronal and glial expression in CNS degenerative diseases. Neurodegeneration 1995, 4, 425–432. [Google Scholar] [CrossRef]
- Lippa, C.F.; Flanders, K.C.; Kim, E.S.; Croul, S. TGF-beta receptors-I and -II immunoexpression in Alzheimer’s disease: A comparison with aging and progressive supranuclear palsy. NeuroBiol. Aging 1998, 19, 527–533. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Wang, L.; Gutmann, D.H.; Roos, R.P. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 2011, 20, 286–293. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef]
- Papadeas, S.T.; Kraig, S.E.; O’Banion, C.; Lepore, A.C.; Maragakis, N.J. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 17803–17808. [Google Scholar] [CrossRef]
- Lepore, A.C.; Rauck, B.; Dejea, C.; Pardo, A.C.; Rao, M.S.; Rothstein, J.D.; Maragakis, N.J. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci. 2008, 11, 1294–1301. [Google Scholar] [CrossRef]
- Taha, D.M.; Clarke, B.E.; Hall, C.E.; Tyzack, G.E.; Ziff, O.J.; Greensmith, L.; Kalmar, B.; Ahmed, M.; Alam, A.; Thelin, E.P.; et al. Astrocytes display cell autonomous and diverse early reactive states in familial amyotrophic lateral sclerosis. Brain 2022, 145, 481–489. [Google Scholar] [CrossRef]
- Meyer, K.; Ferraiuolo, L.; Miranda, C.J.; Likhite, S.; McElroy, S.; Renusch, S.; Ditsworth, D.; Lagier-Tourenne, C.; Smith, R.A.; Ravits, J.; et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 829–832. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef]
- Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Messi, E.; Piccolella, M.; Tedesco, B.; Ferrari, V.; Casarotto, E.; Chierichetti, M.; et al. Multiple Roles of Transforming Growth Factor Beta in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 4291. [Google Scholar] [CrossRef]
- Ziff, O.J.; Clarke, B.E.; Taha, D.M.; Crerar, H.; Luscombe, N.M.; Patani, R. Meta-analysis of human and mouse ALS astrocytes reveals multi-omic signatures of inflammatory reactive states. Genome Res. 2022, 32, 71–84. [Google Scholar] [CrossRef]
- Tripathi, P.; Rodriguez-Muela, N.; Klim, J.R.; de Boer, A.S.; Agrawal, S.; Sandoe, J.; Lopes, C.S.; Ogliari, K.S.; Williams, L.A.; Shear, M.; et al. Reactive Astrocytes Promote ALS-like Degeneration and Intracellular Protein Aggregation in Human Motor Neurons by Disrupting Autophagy through TGF-beta1. Stem Cell Rep. 2017, 9, 667–680. [Google Scholar] [CrossRef]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-derived TGF-beta1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 2015, 11, 592–604. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef]
- Bar-Or, A.; Li, R. Cellular immunology of relapsing multiple sclerosis: Interactions, checks, and balances. Lancet Neurol. 2021, 20, 470–483. [Google Scholar] [CrossRef]
- Yi, W.; Schluter, D.; Wang, X. Astrocytes in multiple sclerosis and experimental autoimmune encephalomyelitis: Star-shaped cells illuminating the darkness of CNS autoimmunity. Brain Behav. Immun. 2019, 80, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Eilam, R.; Arnon, R. Astrocytes in Multiple Sclerosis-Essential Constituents with Diverse Multifaceted Functions. Int. J. Mol. Sci. 2021, 22, 5904. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Hwang, S.I.; Roy, D.B.; Lundgren, D.H.; Price, J.V.; Ousman, S.S.; Fernald, G.H.; Gerlitz, B.; Robinson, W.H.; Baranzini, S.E.; et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 2008, 451, 1076–1081. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Quintana, F.J. Regulation of Astrocyte Functions in Multiple Sclerosis. Cold Spring Harb Perspect. Med. 2019, 9. [Google Scholar] [CrossRef]
- Brosnan, C.F.; Raine, C.S. The astrocyte in multiple sclerosis revisited. Glia 2013, 61, 453–465. [Google Scholar] [CrossRef]
- Schirmer, L.; Schafer, D.P.; Bartels, T.; Rowitch, D.H.; Calabresi, P.A. Diversity and Function of Glial Cell Types in Multiple Sclerosis. Trends Immunol. 2021, 42, 228–247. [Google Scholar] [CrossRef]
- Brambilla, R. The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2019, 137, 757–783. [Google Scholar] [CrossRef]
- Tassoni, A.; Farkhondeh, V.; Itoh, Y.; Itoh, N.; Sofroniew, M.V.; Voskuhl, R.R. The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Sci. Rep. 2019, 9, 10010. [Google Scholar] [CrossRef]
- Gharagozloo, M.; Smith, M.D.; Jin, J.; Garton, T.; Taylor, M.; Chao, A.; Meyers, K.; Kornberg, M.D.; Zack, D.J.; Ohayon, J.; et al. Complement component 3 from astrocytes mediates retinal ganglion cell loss during neuroinflammation. Acta Neuropathol. 2021, 142, 899–915. [Google Scholar] [CrossRef]
- Luo, J.; Ho, P.; Steinman, L.; Wyss-Coray, T. Bioluminescence in vivo imaging of autoimmune encephalomyelitis predicts disease. J. Neuroinflamm. 2008, 5, 6. [Google Scholar] [CrossRef]
- Mayo, L.; Trauger, S.A.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisakk, P.; Kallas, K.; et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 2014, 20, 1147–1156. [Google Scholar] [CrossRef]
- Waller, R.; Woodroofe, M.N.; Wharton, S.B.; Ince, P.G.; Francese, S.; Heath, P.R.; Cudzich-Madry, A.; Thomas, R.H.; Rounding, N.; Sharrack, B.; et al. Gene expression profiling of the astrocyte transcriptome in multiple sclerosis normal appearing white matter reveals a neuroprotective role. J. NeuroImmunol. 2016, 299, 139–146. [Google Scholar] [CrossRef]
- Itoh, N.; Itoh, Y.; Tassoni, A.; Ren, E.; Kaito, M.; Ohno, A.; Ao, Y.; Farkhondeh, V.; Johnsonbaugh, H.; Burda, J.; et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, E302–E309. [Google Scholar] [CrossRef]
- Schirmer, L.; Velmeshev, D.; Holmqvist, S.; Kaufmann, M.; Werneburg, S.; Jung, D.; Vistnes, S.; Stockley, J.H.; Young, A.; Steindel, M.; et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019, 573, 75–82. [Google Scholar] [CrossRef]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.P.; et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef]
- Sanmarco, L.M.; Wheeler, M.A.; Gutierrez-Vazquez, C.; Polonio, C.M.; Linnerbauer, M.; Pinho-Ribeiro, F.A.; Li, Z.; Giovannoni, F.; Batterman, K.V.; Scalisi, G.; et al. Gut-licensed IFNgamma(+) NK cells drive LAMP1(+)TRAIL(+) anti-inflammatory astrocytes. Nature 2021, 590, 473–479. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Clark, I.C.; Tjon, E.C.; Li, Z.; Zandee, S.E.J.; Couturier, C.P.; Watson, B.R.; Scalisi, G.; Alkwai, S.; Rothhammer, V.; et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020, 578, 593–599. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Lee, P.W.; Severin, M.E.; Lovett-Racke, A.E. TGF-beta regulation of encephalitogenic and regulatory T cells in multiple sclerosis. Eur. J. Immunol. 2017, 47, 446–453. [Google Scholar] [CrossRef]
- Kuruvilla, A.P.; Shah, R.; Hochwald, G.M.; Liggitt, H.D.; Palladino, M.A.; Thorbecke, G.J. Protective effect of transforming growth factor beta 1 on experimental autoimmune diseases in mice. Proc. Natl. Acad. Sci. USA 1991, 88, 2918–2921. [Google Scholar] [CrossRef] [PubMed]
- De Groot, C.J.; Montagne, L.; Barten, A.D.; Sminia, P.; Van Der Valk, P. Expression of transforming growth factor (TGF)-beta1, -beta2, and -beta3 isoforms and TGF-beta type I and type II receptors in multiple sclerosis lesions and human adult astrocyte cultures. J. Neuropathol. Exp. Neurol. 1999, 58, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Elkjaer, M.L.; Frisch, T.; Reynolds, R.; Kacprowski, T.; Burton, M.; Kruse, T.A.; Thomassen, M.; Baumbach, J.; Illes, Z. Molecular signature of different lesion types in the brain white matter of patients with progressive multiple sclerosis. Acta Neuropathol. Commun. 2019, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic approaches to Huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Wright, G.E.B.; Black, H.F.; Collins, J.A.; Gall-Duncan, T.; Caron, N.S.; Pearson, C.E.; Hayden, M.R. Interrupting sequence variants and age of onset in Huntington’s disease: Clinical implications and emerging therapies. Lancet Neurol. 2020, 19, 930–939. [Google Scholar] [CrossRef]
- Saba, J.; Couselo, F.L.; Bruno, J.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Neuroinflammation in Huntington’s disease: A starring role for astrocyte and microglia. Curr. Neuropharmacol. 2021. [Google Scholar] [CrossRef]
- Khakh, B.S.; Beaumont, V.; Cachope, R.; Munoz-Sanjuan, I.; Goldman, S.A.; Grantyn, R. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci. 2017, 40, 422–437. [Google Scholar] [CrossRef]
- Wilton, D.K.; Stevens, B. The contribution of glial cells to Huntington’s disease pathogenesis. NeuroBiol. Dis. 2020, 143, 104963. [Google Scholar] [CrossRef]
- Onur, T.S.; Laitman, A.; Zhao, H.; Keyho, R.; Kim, H.; Wang, J.; Mair, M.; Wang, H.; Li, L.; Perez, A.; et al. Downregulation of glial genes involved in synaptic function mitigates Huntington’s disease pathogenesis. Elife 2021, 10. [Google Scholar] [CrossRef]
- Birolini, G.; Verlengia, G.; Talpo, F.; Maniezzi, C.; Zentilin, L.; Giacca, M.; Conforti, P.; Cordiglieri, C.; Caccia, C.; Leoni, V.; et al. SREBP2 gene therapy targeting striatal astrocytes ameliorates Huntington’s disease phenotypes. Brain 2021, 144, 3175–3190. [Google Scholar] [CrossRef]
- Polyzos, A.A.; Lee, D.Y.; Datta, R.; Hauser, M.; Budworth, H.; Holt, A.; Mihalik, S.; Goldschmidt, P.; Frankel, K.; Trego, K.; et al. Metabolic Reprogramming in Astrocytes Distinguishes Region-Specific Neuronal Susceptibility in Huntington Mice. Cell Metab. 2019, 29, 1258–1273. [Google Scholar] [CrossRef]
- Al-Dalahmah, O.; Sosunov, A.A.; Shaik, A.; Ofori, K.; Liu, Y.; Vonsattel, J.P.; Adorjan, I.; Menon, V.; Goldman, J.E. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 2020, 8, 19. [Google Scholar] [CrossRef]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Yu, X.; Nagai, J.; Marti-Solano, M.; Soto, J.S.; Coppola, G.; Babu, M.M.; Khakh, B.S. Context-Specific Striatal Astrocyte Molecular Responses Are Phenotypically Exploitable. Neuron 2020, 108, 1146–1162.e1110. [Google Scholar] [CrossRef]
- Abjean, L.; Ben Haim, L.; Riquelme-Perez, M.; Gipchtein, P.; Derbois, C.; Palomares, M.A.; Petit, F.; Herard, A.S.; Gaillard, M.C.; Guillermier, M.; et al. Reactive astrocytes promote proteostasis in Huntington’s disease through the JAK2-STAT3 pathway. Brain 2022. [Google Scholar] [CrossRef]
- Bowles, K.R.; Jones, L. Kinase signalling in Huntington’s disease. J. Huntington’s Dis. 2014, 3, 89–123. [Google Scholar] [CrossRef]
- Kandasamy, M.; Aigner, L. Reactive Neuroblastosis in Huntington’s Disease: A Putative Therapeutic Target for Striatal Regeneration in the Adult Brain. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Di Pardo, A.; Alberti, S.; Maglione, V.; Amico, E.; Cortes, E.P.; Elifani, F.; Battaglia, G.; Busceti, C.L.; Nicoletti, F.; Vonsattel, J.P.; et al. Changes of peripheral TGF-beta1 depend on monocytes-derived macrophages in Huntington disease. Mol. Brain 2013, 6, 55. [Google Scholar] [CrossRef]
- Chang, K.H.; Wu, Y.R.; Chen, Y.C.; Chen, C.M. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav. Immun. 2015, 44, 121–127. [Google Scholar] [CrossRef]
- Battaglia, G.; Cannella, M.; Riozzi, B.; Orobello, S.; Maat-Schieman, M.L.; Aronica, E.; Busceti, C.L.; Ciarmiello, A.; Alberti, S.; Amico, E.; et al. Early defect of transforming growth factor beta1 formation in Huntington’s disease. J. Cell Mol. Med. 2011, 15, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Plinta, K.; Plewka, A.; Wojcik-Pedziwiatr, M.; Zmarzly, N.; Rudzinski, M.; Rudzinska-Bar, M. Is TGF-beta1 a Biomarker of Huntington’s Disease Progression? J. Clin. Med. 2021, 10, 3001. [Google Scholar] [CrossRef] [PubMed]
- An, M.C.; Zhang, N.; Scott, G.; Montoro, D.; Wittkop, T.; Mooney, S.; Melov, S.; Ellerby, L.M. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 2012, 11, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Bowles, K.R.; Stone, T.; Holmans, P.; Allen, N.D.; Dunnett, S.B.; Jones, L. SMAD transcription factors are altered in cell models of HD and regulate HTT expression. Cell Signal. 2017, 31, 1–14. [Google Scholar] [CrossRef]
- Ring, K.L.; An, M.C.; Zhang, N.; O’Brien, R.N.; Ramos, E.M.; Gao, F.; Atwood, R.; Bailus, B.J.; Melov, S.; Mooney, S.D.; et al. Genomic Analysis Reveals Disruption of Striatal Neuronal Development and Therapeutic Targets in Human Huntington’s Disease Neural Stem Cells. Stem Cell Rep. 2015, 5, 1023–1038. [Google Scholar] [CrossRef]
- Ament, S.A.; Pearl, J.R.; Cantle, J.P.; Bragg, R.M.; Skene, P.J.; Coffey, S.R.; Bergey, D.E.; Wheeler, V.C.; MacDonald, M.E.; Baliga, N.S.; et al. Transcriptional regulatory networks underlying gene expression changes in Huntington’s disease. Mol. Syst. Biol. 2018, 14, e7435. [Google Scholar] [CrossRef]
- Scarpa, J.R.; Jiang, P.; Losic, B.; Readhead, B.; Gao, V.D.; Dudley, J.T.; Vitaterna, M.H.; Turek, F.W.; Kasarskis, A. Systems Genetic Analyses Highlight a TGFbeta-FOXO3 Dependent Striatal Astrocyte Network Conserved across Species and Associated with Stress, Sleep, and Huntington’s Disease. PLoS Genet. 2016, 12, e1006137. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Binder, D.K.; Steinhauser, C. Astrocytes and Epilepsy. Neurochem. Res. 2021, 46, 2687–2695. [Google Scholar] [CrossRef]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef]
- Rosch, R.E.; Samarut, E. Epileptic Seizures: Glia-Neuron Interactions For Better or For Worse. Curr. Biol. 2019, 29, R1248–R1251. [Google Scholar] [CrossRef]
- Coulter, D.A.; Steinhauser, C. Role of astrocytes in epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022434. [Google Scholar] [CrossRef]
- Chen, M.; Edwards, S.R.; Reutens, D.C. Complement in the Development of Post-Traumatic Epilepsy: Prospects for Drug Repurposing. J. Neurotrauma 2020, 37, 692–705. [Google Scholar] [CrossRef]
- Schartz, N.D.; Wyatt-Johnson, S.K.; Price, L.R.; Colin, S.A.; Brewster, A.L. Status epilepticus triggers long-lasting activation of complement C1q-C3 signaling in the hippocampus that correlates with seizure frequency in experimental epilepsy. NeuroBiol. Dis. 2018, 109, 163–173. [Google Scholar] [CrossRef]
- Gage, M.; Gard, M.; Thippeswamy, T. Characterization of Cortical Glial Scars in the Diisopropylfluorophosphate (DFP) Rat Model of Epilepsy. Front. Cell Dev. Biol. 2022, 10, 867949. [Google Scholar] [CrossRef]
- Jiang, G.T.; Shao, L.; Kong, S.; Zeng, M.L.; Cheng, J.J.; Chen, T.X.; Han, S.; Yin, J.; Liu, W.H.; He, X.H.; et al. Complement C3 Aggravates Post-epileptic Neuronal Injury Via Activation of TRPV1. Neurosci. Bull. 2021, 37, 1427–1440. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, T.; Bosco, D.B.; Xie, M.; Zheng, J.; Dheer, A.; Ying, Y.; Wu, Q.; Lennon, V.A.; Wu, L.J. The complement C3-C3aR pathway mediates microglia-astrocyte interaction following status epilepticus. Glia 2021, 69, 1155–1169. [Google Scholar] [CrossRef]
- Yu, W.; Zou, Y.; Du, Y.; Luo, J.; Zhang, M.; Yang, W.; Wang, X.; Lu, Y. Altered cerebrospinal fluid concentrations of TGFbeta1 in patients with drug-resistant epilepsy. Neurochem. Res. 2014, 39, 2211–2217. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, T.; Yuan, J.; Li, Y.; Wu, Y.; Xi, Z.; Xiao, Z.; Chen, Y.; Wang, X. Increased expression of TGFbeta type I receptor in brain tissues of patients with temporal lobe epilepsy. Clin. Sci. 2009, 117, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Yan, Y.; Guo, Q.; Wang, L.; Han, X.; Liu, S. Genetic Interaction of H19 and TGFBR1 Polymorphisms with Risk of Epilepsy in a Chinese Population. Pharmgenomics Pers. Med. 2021, 14, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kotlarz, D.; Marquardt, B.; Baroy, T.; Lee, W.S.; Konnikova, L.; Hollizeck, S.; Magg, T.; Lehle, A.S.; Walz, C.; Borggraefe, I.; et al. Human TGF-beta1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat. Genet. 2018, 50, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Du, Y.; Zou, Y.; Luo, J.; Lu, Y.; Yu, W. Smad Anchor for Receptor Activation and Phospho-Smad3 Were Upregulated in Patients with Temporal Lobe Epilepsy. J. Mol. Neurosci. 2019, 68, 91–98. [Google Scholar] [CrossRef]
- Rusina, E.; Bernard, C.; Williamson, A. The Kainic Acid Models of Temporal Lobe Epilepsy. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Veno, M.T.; Reschke, C.R.; Morris, G.; Connolly, N.M.C.; Su, J.; Yan, Y.; Engel, T.; Jimenez-Mateos, E.M.; Harder, L.M.; Pultz, D.; et al. A systems approach delivers a functional microRNA catalog and expanded targets for seizure suppression in temporal lobe epilepsy. Proc. Natl. Acad. Sci. USA 2020, 117, 15977–15988. [Google Scholar] [CrossRef]
- Fu, Y.; Wu, Z.; Guo, Z.; Chen, L.; Ma, Y.; Wang, Z.; Xiao, W.; Wang, Y. Systems-level analysis identifies key regulators driving epileptogenesis in temporal lobe epilepsy. Genomics 2020, 112, 1768–1780. [Google Scholar] [CrossRef]
- Mukherjee, S.; Arisi, G.M.; Mims, K.; Hollingsworth, G.; O’Neil, K.; Shapiro, L.A. Neuroinflammatory mechanisms of post-traumatic epilepsy. J. Neuroinflamm. 2020, 17, 193. [Google Scholar] [CrossRef]
- Fordington, S.; Manford, M. A review of seizures and epilepsy following traumatic brain injury. J. Neurol. 2020, 267, 3105–3111. [Google Scholar] [CrossRef]
- Golub, V.M.; Reddy, D.S. Post-Traumatic Epilepsy and Comorbidities: Advanced Models, Molecular Mechanisms, Biomarkers, and Novel Therapeutic Interventions. Pharmacol. Rev. 2022, 74, 387–438. [Google Scholar] [CrossRef]
- Wang, F.; Wang, X.; Shapiro, L.A.; Cotrina, M.L.; Liu, W.; Wang, E.W.; Gu, S.; Wang, W.; He, X.; Nedergaard, M.; et al. NKCC1 up-regulation contributes to early post-traumatic seizures and increased post-traumatic seizure susceptibility. Brain Struct. Funct. 2017, 222, 1543–1556. [Google Scholar] [CrossRef]
- Heinemann, U.; Kaufer, D.; Friedman, A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia 2012, 60, 1251–1257. [Google Scholar] [CrossRef]
- Loscher, W.; Friedman, A. Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? Int. J. Mol. Sci. 2020, 21, 591. [Google Scholar] [CrossRef]
- Ivens, S.; Kaufer, D.; Flores, L.P.; Bechmann, I.; Zumsteg, D.; Tomkins, O.; Seiffert, E.; Heinemann, U.; Friedman, A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 2007, 130, 535–547. [Google Scholar] [CrossRef]
- Dey, A.; Kang, X.; Qiu, J.; Du, Y.; Jiang, J. Anti-Inflammatory Small Molecules To Treat Seizures and Epilepsy: From Bench to Bedside. Trends Pharmacol. Sci. 2016, 37, 463–484. [Google Scholar] [CrossRef]
- Weissberg, I.; Wood, L.; Kamintsky, L.; Vazquez, O.; Milikovsky, D.Z.; Alexander, A.; Oppenheim, H.; Ardizzone, C.; Becker, A.; Frigerio, F.; et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-beta/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. NeuroBiol. Dis. 2015, 78, 115–125. [Google Scholar] [CrossRef]
- Vila Verde, D.; de Curtis, M.; Librizzi, L. Seizure-Induced Acute Glial Activation in the in vitro Isolated Guinea Pig Brain. Front. Neurol. 2021, 12. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Cacheaux, L.P.; Kamintsky, L.; Prager, O.; Weissberg, I.; Schoknecht, K.; Cheng, P.; Kim, S.Y.; Wood, L.; Heinemann, U.; et al. Losartan prevents acquired epilepsy via TGF-beta signaling suppression. Ann. Neurol. 2014, 75, 864–875. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Lublinsky, S.; Kamintsky, L.; Noyman, I.; Veksler, R.; Dalipaj, H.; Senatorov, V.V., Jr.; Swissa, E.; Rosenbach, D.; Elazary, N.; et al. Imaging blood-brain barrier dysfunction as a biomarker for epileptogenesis. Brain 2017, 140, 1692–1705. [Google Scholar] [CrossRef]
- Lukawski, K.; Czuczwar, S.J. Emerging therapeutic targets for epilepsy: Preclinical insights. Expert. Opin. Ther. Targets 2022, 26, 193–206. [Google Scholar] [CrossRef]
- Milikovsky, D.Z.; Ofer, J.; Senatorov, V.V., Jr.; Friedman, A.R.; Prager, O.; Sheintuch, L.; Elazari, N.; Veksler, R.; Zelig, D.; Weissberg, I.; et al. Paroxysmal slow cortical activity in Alzheimer’s disease and epilepsy is associated with blood-brain barrier dysfunction. Sci. Transl. Med. 2019, 11, eaaw8954. [Google Scholar] [CrossRef] [PubMed]
- Spradling, K.D.; Lumley, L.A.; Robison, C.L.; Meyerhoff, J.L.; Dillman, J.F., 3rd. Transcriptional analysis of rat piriform cortex following exposure to the organophosphonate anticholinesterase sarin and induction of seizures. J. Neuroinflamm. 2011, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xu, G.; Du, H.; Yi, M.; Li, C. Integrative analysis of gene expression associated with epilepsy in human epilepsy and animal models. Mol. Med. Rep. 2016, 13, 4920–4926. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cukovic, D.; Bagla, S.; Ukasik, D.; Stemmer, P.M.; Jena, B.P.; Naik, A.R.; Sood, S.; Asano, E.; Luat, A.; Chugani, D.C.; et al. Exosomes in Epilepsy of Tuberous Sclerosis Complex: Carriers of Pro-Inflammatory MicroRNAs. Noncoding RNA 2021, 7, 40. [Google Scholar] [CrossRef]
- Cava, C.; Manna, I.; Gambardella, A.; Bertoli, G.; Castiglioni, I. Potential Role of miRNAs as Theranostic Biomarkers of Epilepsy. Mol. Ther. Nucleic Acids 2018, 13, 275–290. [Google Scholar] [CrossRef]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Bergamo Araujo, A.P.; Melo, H.M.; Seixas da Silva, G.S.; Alves-Leon, S.V.; de Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Abeta Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797–6809. [Google Scholar] [CrossRef]
- Gernert, M.; Feja, M. Bypassing the Blood–Brain Barrier: Direct Intracranial Drug Delivery in Epilepsies. Pharmaceutics 2020, 12, 1134. [Google Scholar] [CrossRef]
- Ozdas, M.S.; Shah, A.S.; Johnson, P.M.; Patel, N.; Marks, M.; Yasar, T.B.; Stalder, U.; Bigler, L.; von der Behrens, W.; Sirsi, S.R.; et al. Non-invasive molecularly-specific millimeter-resolution manipulation of brain circuits by ultrasound-mediated aggregation and uncaging of drug carriers. Nat. Commun. 2020, 11, 4929. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- O’Carroll, S.J.; Cook, W.H.; Young, D. AAV Targeting of Glial Cell Types in the Central and Peripheral Nervous System and Relevance to Human Gene Therapy. Front. Mol. Neurosci. 2021, 13. [Google Scholar] [CrossRef]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef]
- Grasso, M.; Caruso, G.; Godos, J.; Bonaccorso, A.; Carbone, C.; Castellano, S.; Currenti, W.; Grosso, G.; Musumeci, T.; Caraci, F. Improving Cognition with Nutraceuticals Targeting TGF-beta1 Signaling. Antioxidants 2021, 10, 1075. [Google Scholar] [CrossRef]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFbeta: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Budi, E.H.; Schaub, J.R.; Decaris, M.; Turner, S.; Derynck, R. TGF-beta as a driver of fibrosis: Physiological roles and therapeutic opportunities. J. Pathol. 2021, 254, 358–373. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFbeta biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Kim, B.G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel therapies emerging in oncology to target the TGF-beta pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef]
- Ruwanpura, S.M.; Thomas, B.J.; Bardin, P.G. Pirfenidone: Molecular Mechanisms and Potential Clinical Applications in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2020, 62, 413–422. [Google Scholar] [CrossRef]
- Castro-Torres, R.D.; Chaparro-Huerta, V.; Flores-Soto, M.E.; Jave-Suarez, L.; Camins, A.; Armendariz-Borunda, J.; Beas-Zarate, C.; Mena-Munguia, S. Pirfenidone Attenuates Microglial Reactivity and Reduces Inducible Nitric Oxide Synthase mRNA Expression After Kainic Acid-Mediated Excitotoxicity in Pubescent Rat Hippocampus. J. Mol. Neurosci. 2015, 56, 245–254. [Google Scholar] [CrossRef]
- Bozkurt, I.; Ozturk, Y.; Guney, G.; Arslan, B.; Gulbahar, O.; Guvenc, Y.; Senturk, S.; Yaman, M.E. Effects of pirfenidone on experimental head injury in rats. Int. J. Clin. Exp. Pathol. 2022, 15, 20–28. [Google Scholar]
- Nozaki, T.; Ura, H.; Takumi, I.; Kobayashi, S.; Maru, E.; Morita, A. The angiotensin II type I receptor antagonist losartan retards amygdala kindling-induced epileptogenesis. Brain Res. 2018, 1694, 121–128. [Google Scholar] [CrossRef]
- Tchekalarova, J.D.; Ivanova, N.; Atanasova, D.; Pechlivanova, D.M.; Lazarov, N.; Kortenska, L.; Mitreva, R.; Lozanov, V.; Stoynev, A. Long-Term Treatment with Losartan Attenuates Seizure Activity and Neuronal Damage Without Affecting Behavioral Changes in a Model of Co-morbid Hypertension and Epilepsy. Cell. Mol. Neurobiol. 2016, 36, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; JianCheng, H.; JiaWen, W.; ShuQin, Z.; GuiLian, Z.; HaiQin, W.; Ru, Z.; Zhen, G.; HongWei, R. Losartan inhibits development of spontaneous recurrent seizures by preventing astrocyte activation and attenuating blood-brain barrier permeability following pilocarpine-induced status epilepticus. Brain Res. Bull. 2019, 149, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Hanael, E.; Chai, O.; Konstanitin, L.; Gibeon, L.; Rapaport, K.; Ruggeri, M.; Friedman, A.; Shamir, M.H. Telmisartan as an add-on treatment for dogs with refractory idiopathic epilepsy: A nonrandomized, uncontrolled, open-label clinical trial. J. Am. Vet. Med. Assoc. 2022, 36, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Garcia, S.Z.; Scorza, C.A.; Ortiz-Villatoro, N.N.; Cavalheiro, E.A. Losartan fails to suppress epileptiform activity in brain slices from resected tissues of patients with drug resistant epilepsy. J. Neurol. Sci. 2019, 397, 169–171. [Google Scholar] [CrossRef]
- Klein, P.; Friedman, A.; Hameed, M.Q.; Kaminski, R.M.; Bar-Klein, G.; Klitgaard, H.; Koepp, M.; Jozwiak, S.; Prince, D.A.; Rotenberg, A.; et al. Repurposed molecules for antiepileptogenesis: Missing an opportunity to prevent epilepsy? Epilepsia 2020, 61, 359–386. [Google Scholar] [CrossRef]
- Villarreal, A.; Vogel, T. Different Flavors of Astrocytes: Revising the Origins of Astrocyte Diversity and Epigenetic Signatures to Understand Heterogeneity after Injury. Int. J. Mol. Sci. 2021, 22, 6867. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J. TGF-β as a Key Modulator of Astrocyte Reactivity: Disease Relevance and Therapeutic Implications. Biomedicines 2022, 10, 1206. https://doi.org/10.3390/biomedicines10051206
Luo J. TGF-β as a Key Modulator of Astrocyte Reactivity: Disease Relevance and Therapeutic Implications. Biomedicines. 2022; 10(5):1206. https://doi.org/10.3390/biomedicines10051206
Chicago/Turabian StyleLuo, Jian. 2022. "TGF-β as a Key Modulator of Astrocyte Reactivity: Disease Relevance and Therapeutic Implications" Biomedicines 10, no. 5: 1206. https://doi.org/10.3390/biomedicines10051206
APA StyleLuo, J. (2022). TGF-β as a Key Modulator of Astrocyte Reactivity: Disease Relevance and Therapeutic Implications. Biomedicines, 10(5), 1206. https://doi.org/10.3390/biomedicines10051206