
SERTAD1 Sensitizes Breast Cancer Cells to Doxorubicin and Promotes Lysosomal Protein Biosynthesis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials & Methods
2.1. Cell Culture and Treatment Preparation
2.2. Cell Viability Assays
2.3. Western Blot Analysis
2.4. Immunoflorescene (IF) Staining for Cells
2.5. Mitotracker Staining
2.6. Lysotracker Staining
2.7. Acridine Orange (AO) Staining of Lysosomes
2.8. Real-Time Polymerase Chain Reaction (RT-PCR)
2.9. Breast Cancer Gene-Expression Miner v4.5 (bc-GenExMiner v4.5)
2.10. Mice Genotyping and Isolation of Mouse Embryo Fibroblasts (MEFs)
2.11. Xenograft Mice Establishment
2.12. Survival Rate Study
2.13. IF Staining for Tumors
2.14. Statistical Analysis
3. Results
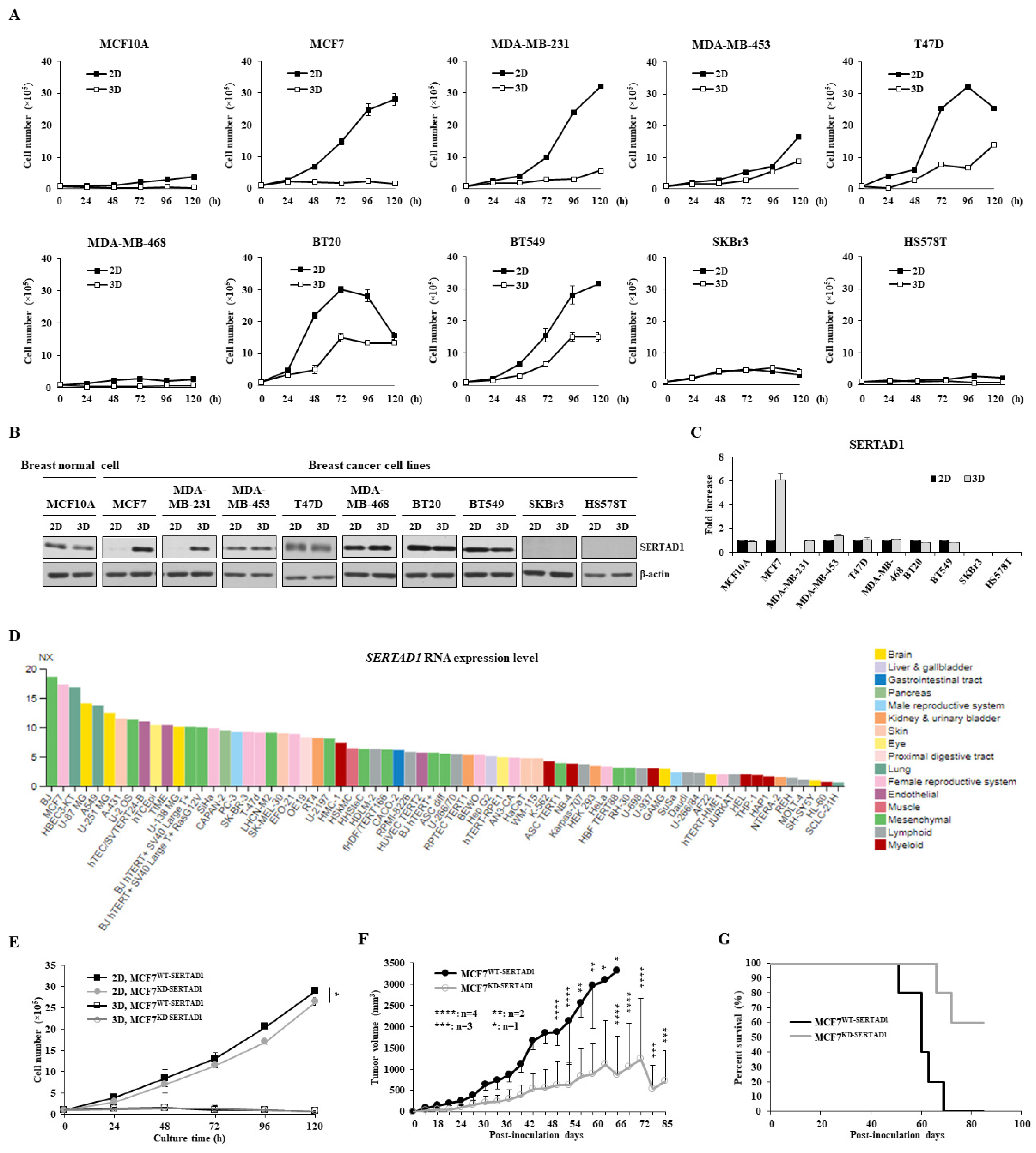
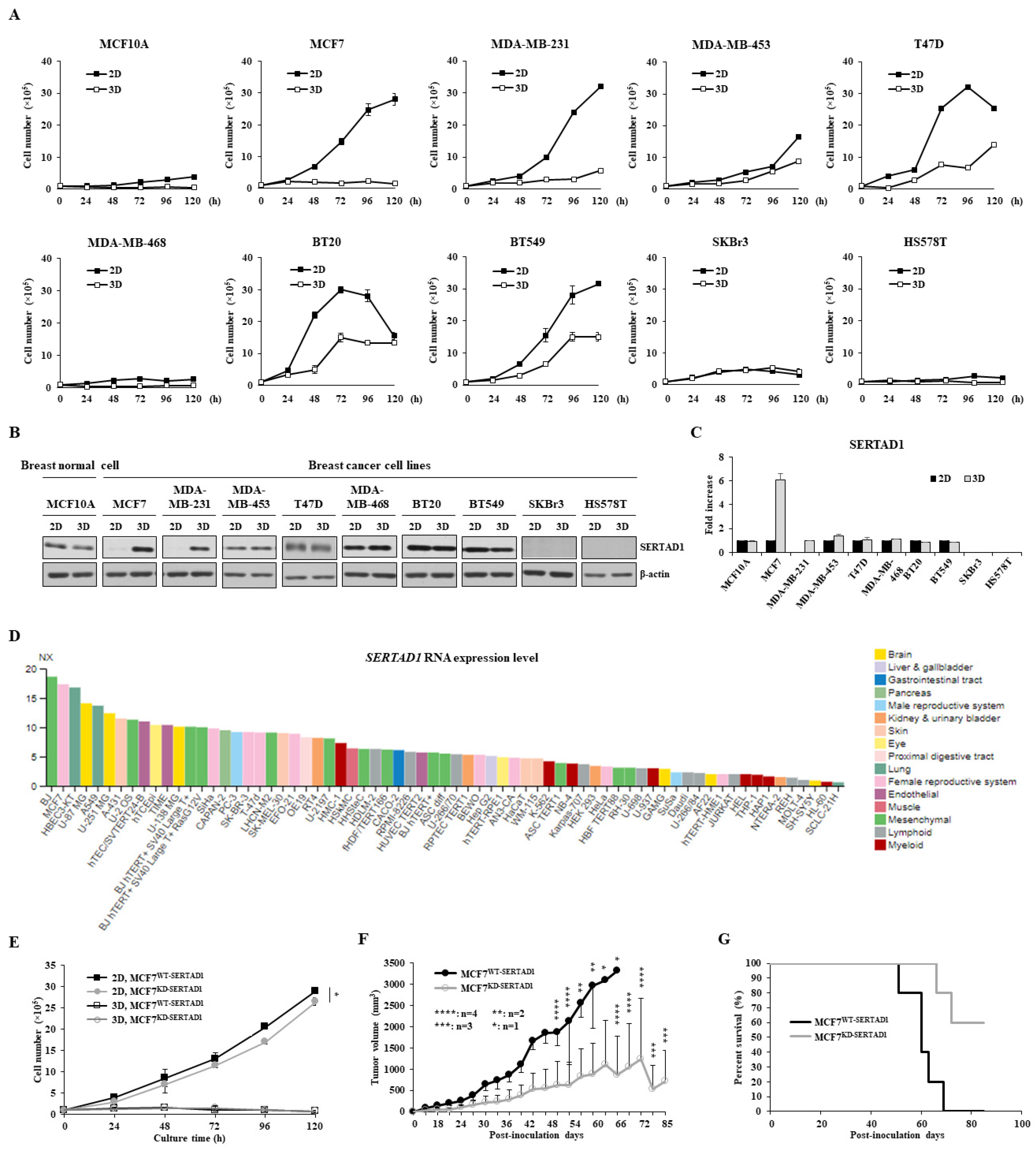
3.1. SERTAD1 Suppresses Anoikis Resistance in Breast Cancer Cells
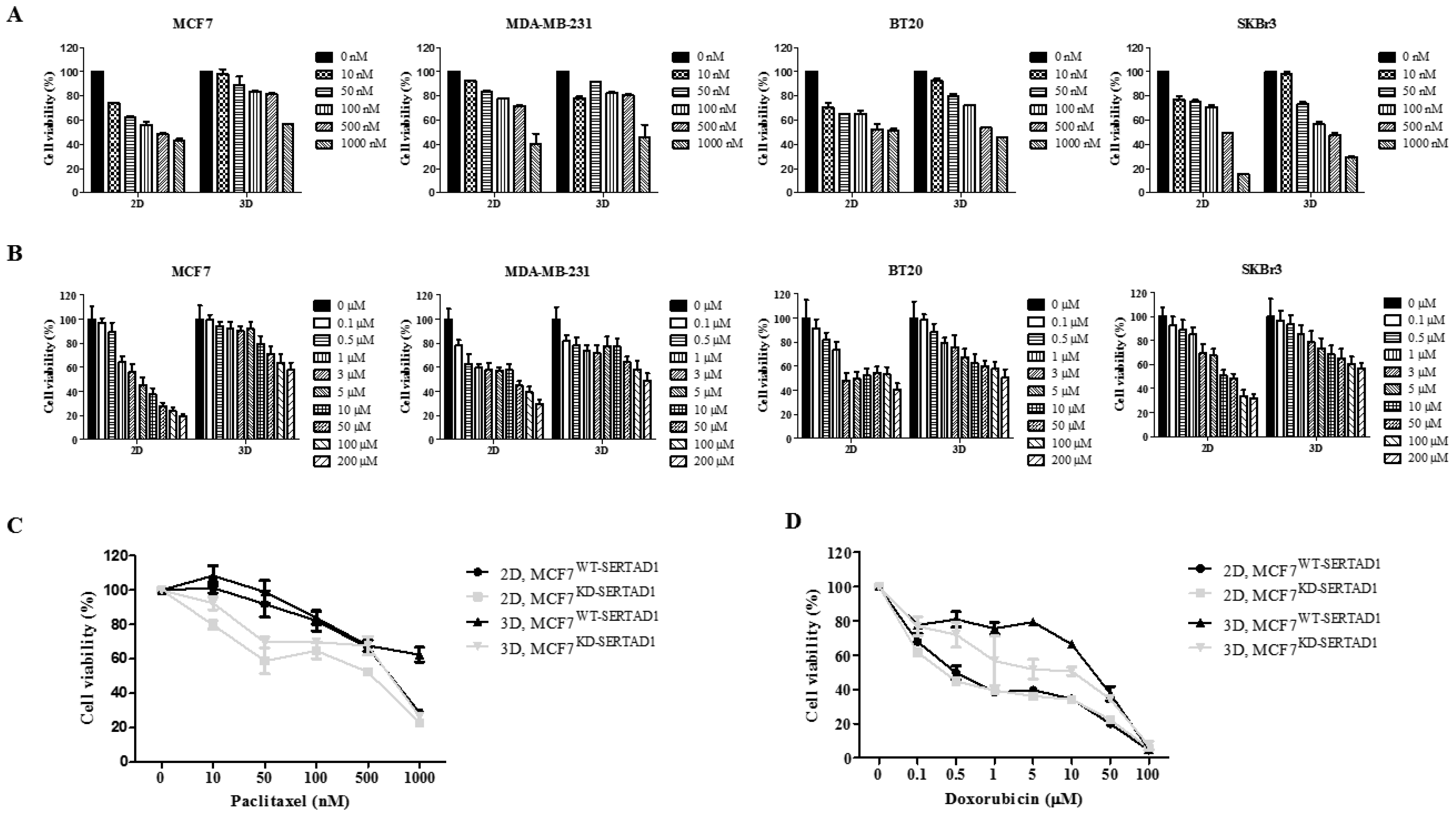
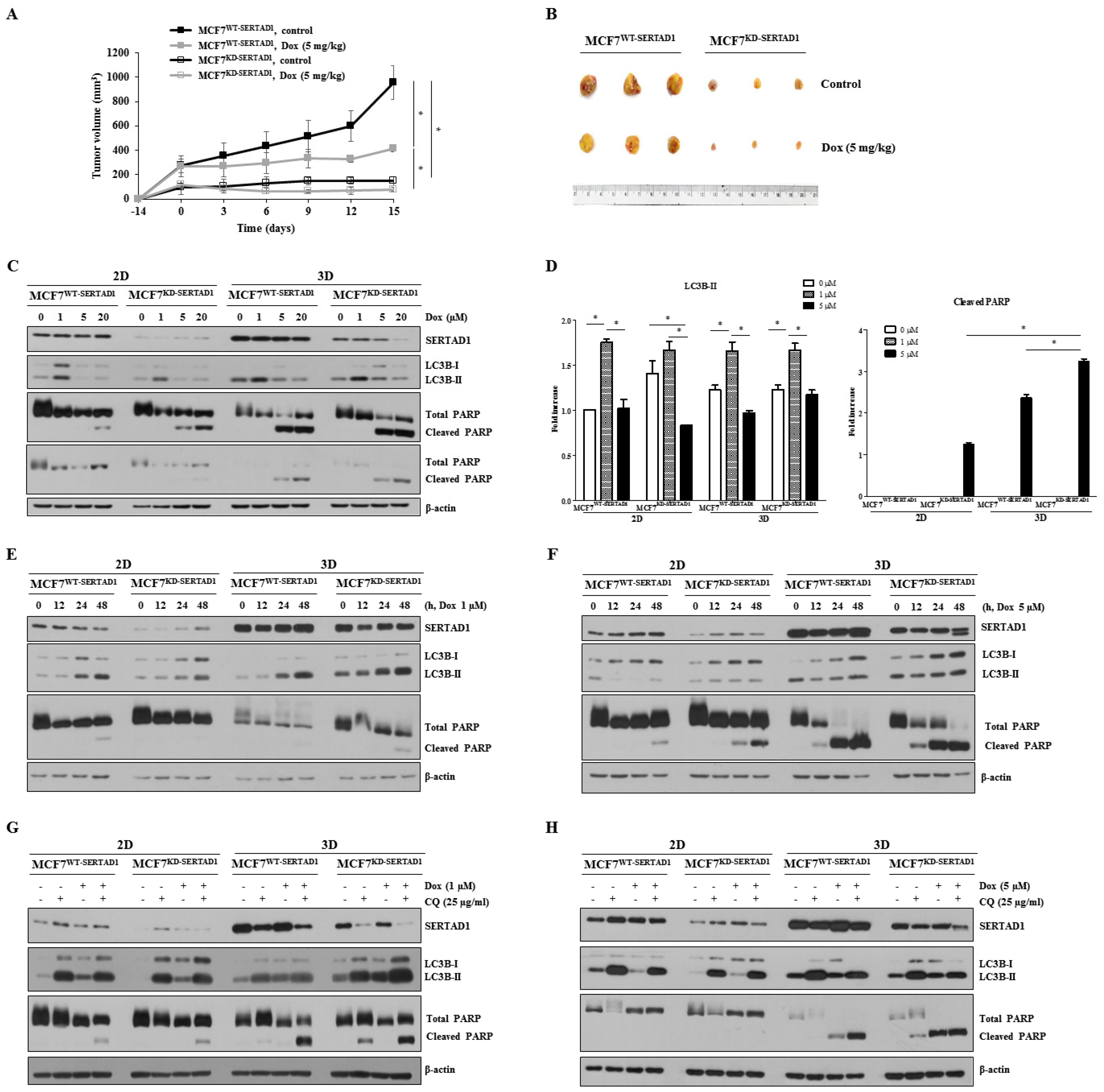
3.2. SERTAD1 Suppression Promotes Sensitivity of Cancer Cells to Anti-Cancer Drugs
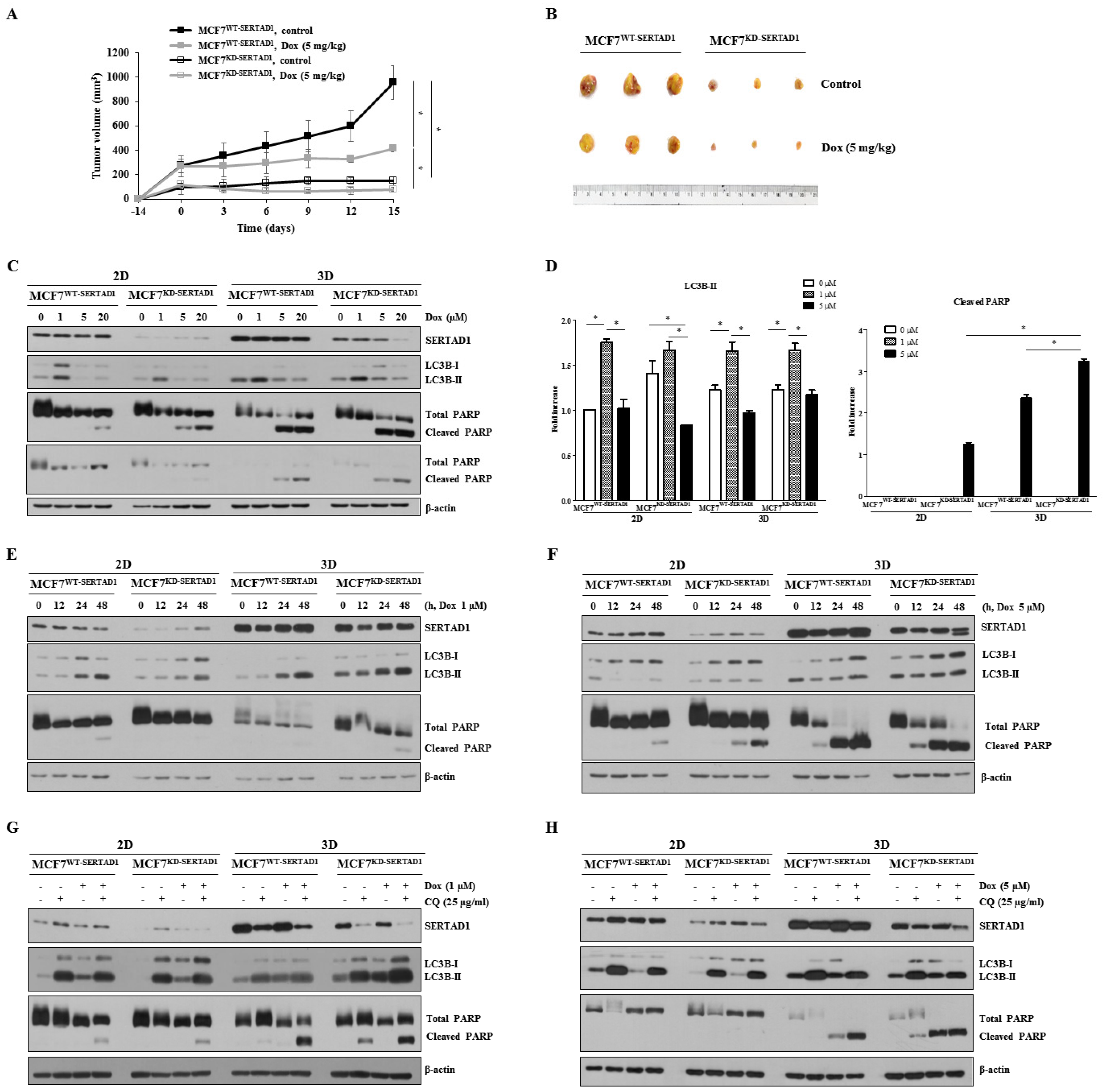
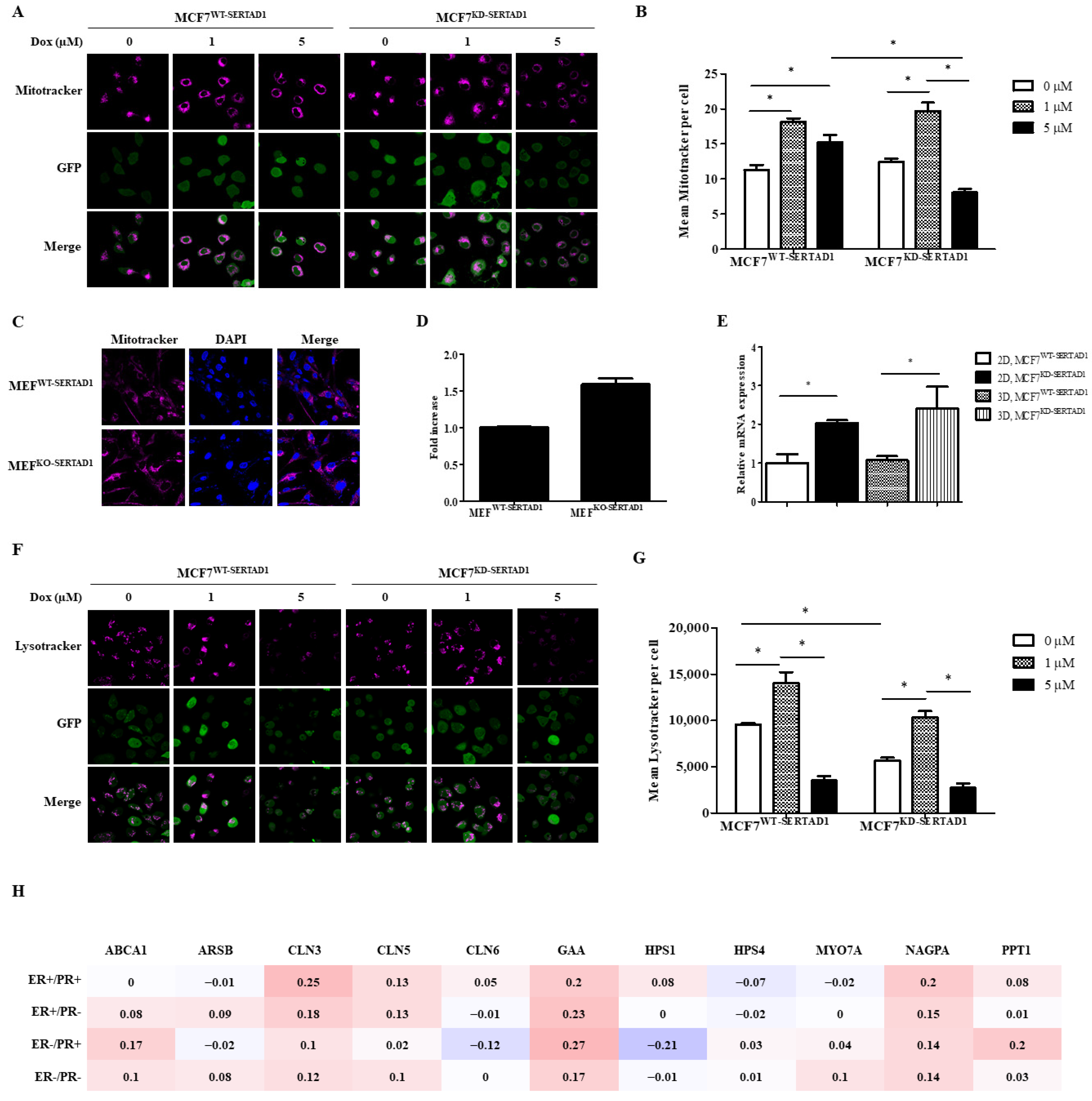
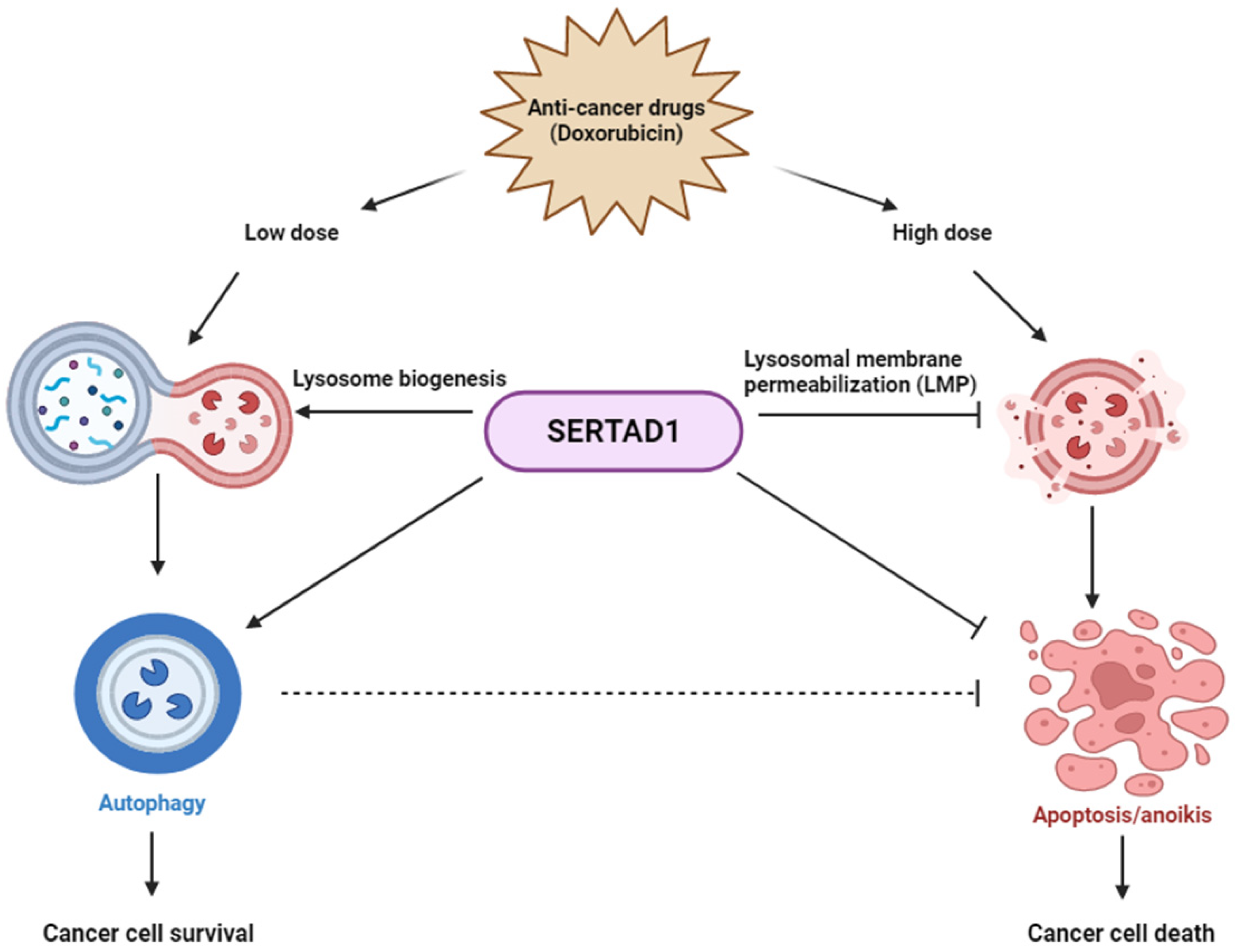
3.3. SERTAD1 Inhibits Dox-Induced Apoptosis/Anoikis by Regulating Mitochondrial and Lysosomal Activity
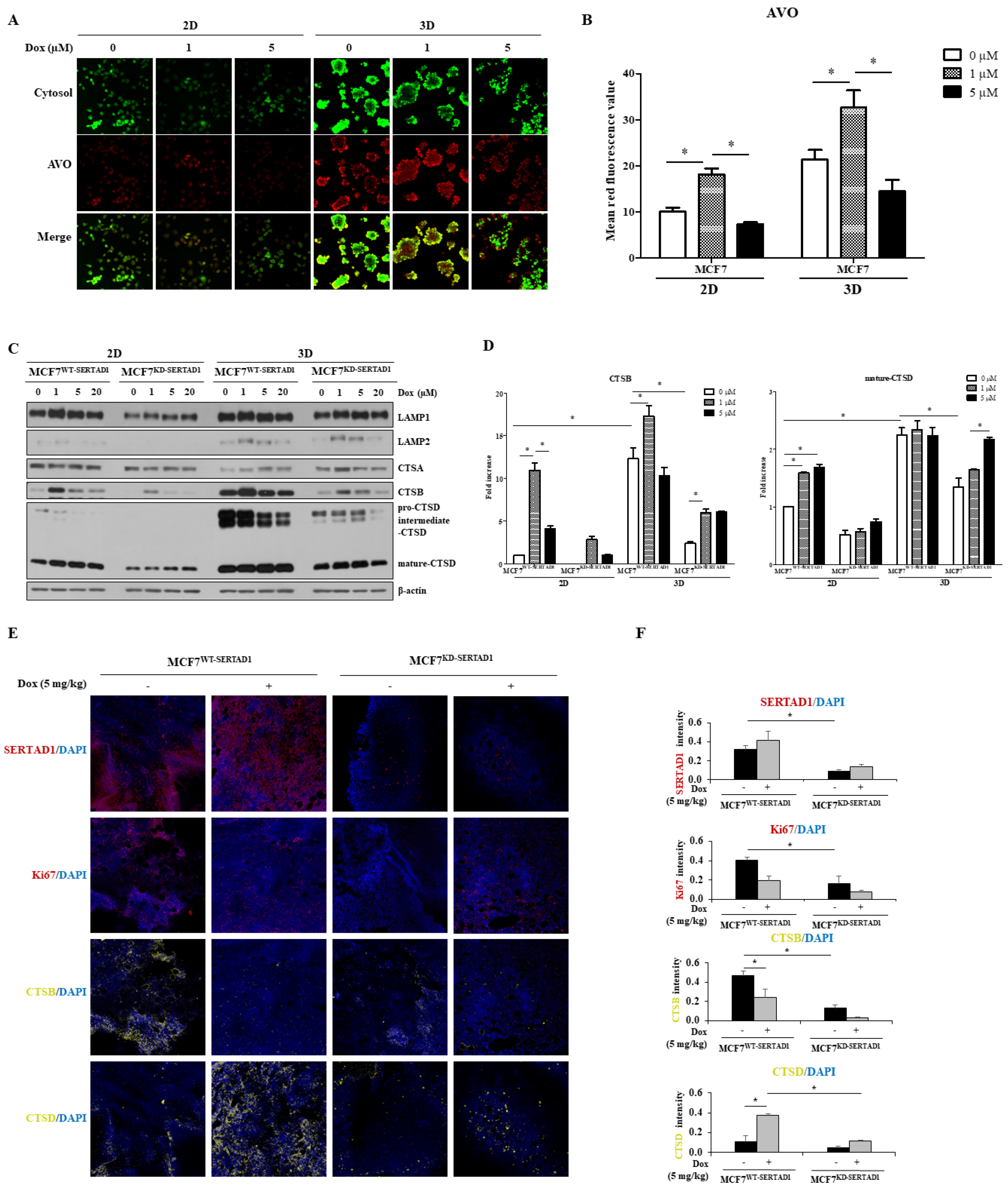
3.4. Dox-Triggered Cell Deaths Are LMP-Dependent and Ablation of SERTAD1 Reduces Protein Expression of Lysosomal Hydrolases
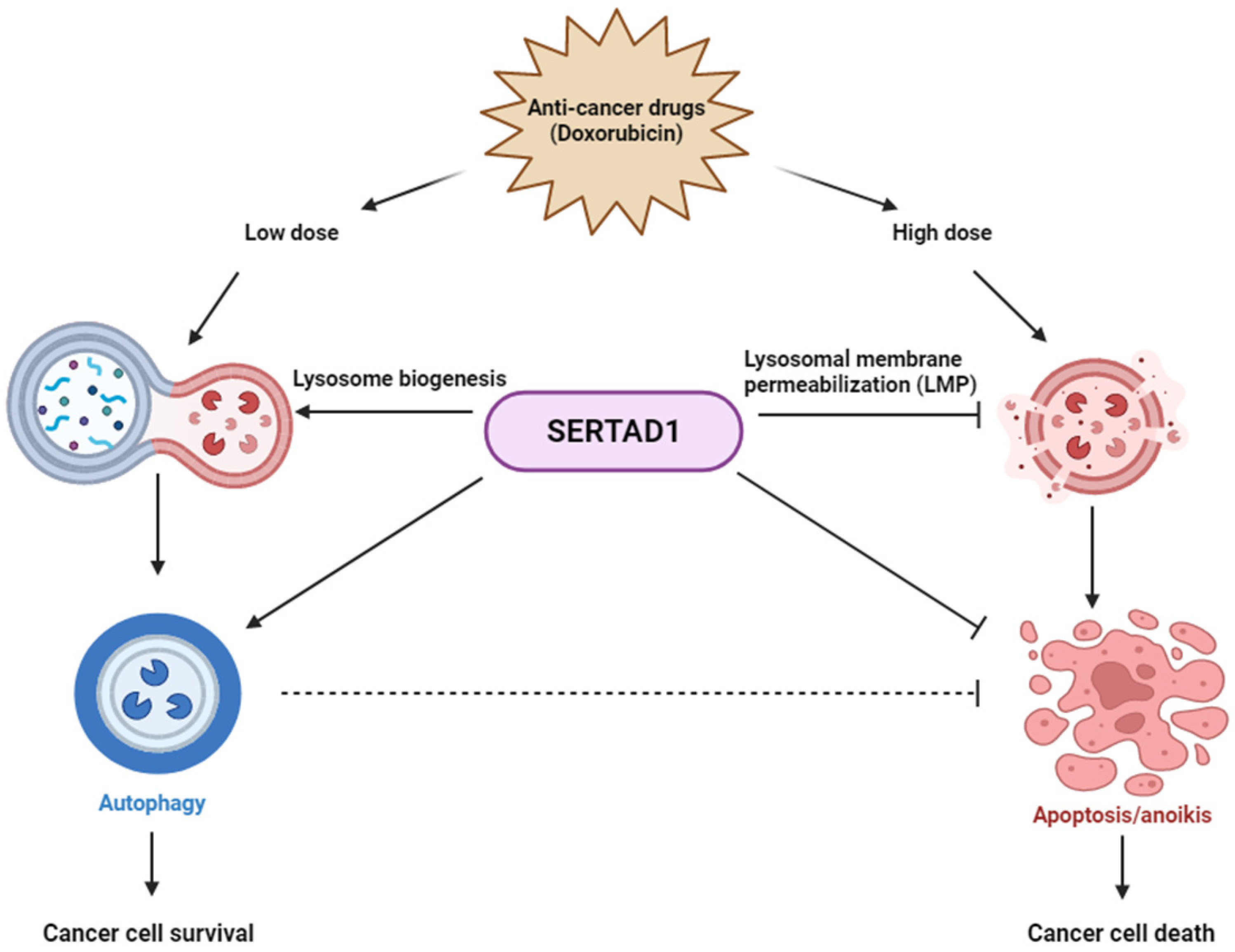
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Fitzwalter, B.E.; Thorburn, A. Recent insights into cell death and autophagy. FEBS J. 2015, 282, 4279–4288. [Google Scholar] [CrossRef] [Green Version]
- Brech, A.; Ahlquist, T.; Lothe, R.A.; Stenmark, H. Autophagy in tumour suppression and promotion. Mol. Oncol. 2009, 3, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Kavčič, N.; Pegan, K.; Turk, B. Lysosomes in programmed cell death pathways: From initiators to amplifiers. Biol. Chem. 2017, 398, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Gómez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Sandag, Z.; Jung, S.; Quynh, N.T.N.; Myagmarjav, D.; Anh, N.H.; Le, D.T.; Lee, B.S.; Mongre, R.K.; Jo, T.; Lee, M. Inhibitory Role of TRIP-Br1/XIAP in Necroptosis under Nutrient/Serum Starvation. Mol. Cells 2020, 43, 236–250. [Google Scholar]
- Jung, S.; Li, C.; Duan, J.; Lee, S.; Kim, K.; Park, Y.; Yang, Y.; Kim, K.I.; Lim, J.S.; Cheon, C.I.; et al. TRIP-Br1 oncoprotein inhibits autophagy, apoptosis, and necroptosis under nutrient/serum-deprived condition. Oncotarget 2015, 6, 29060–29075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.W.; Kim, C.J.; Park, W.S.; Shin, J.S.; Lee, S.D.; Ko, S.G.; Jung, S.I.; Park, I.C.; An, S.K.; Lee, W.K.; et al. p34SEI-1 inhibits apoptosis through the stabilization of the X-linked inhibitor of apoptosis protein: p34SEI-1 as a novel target for anti-breast cancer strategies. Cancer Res. 2009, 69, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.; Li, C.; Jeong, D.; Lee, S.; Ohk, J.; Park, M.; Han, S.; Duan, J.; Kim, C.; Yang, Y.; et al. Oncogenic function of p34SEI-1 via NEDD4-1-mediated PTEN ubiquitination/degradation and activation of the PI3K/AKT pathway. Int. J. Oncol. 2013, 43, 1587–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jézéquel, P.; Frénel, J.S.; Campion, L.; Guérin-Charbonnel, C.; Gouraud, W.; Ricolleau, G.; Campone, M. bc-GenExMiner 3.0: New mining module computes breast cancer gene expression correlation analyses. Database J. Biol. Databases Curation 2013, 2013, bas060. [Google Scholar]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Pantoja, C.; Gonzalez-Rodriguez, A.; Martin, N.; Flores, J.M.; Valverde, A.M.; Hara, E.; Serrano, M. Normal proliferation and tumorigenesis but impaired pancreatic function in mice lacking the cell cycle regulator sei1. PLoS ONE 2010, 5, e8744. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D'Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Konorev, E.A.; Kotamraju, S.; Joseph, J.; Kalivendi, S.; Kalyanaraman, B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. intermediacy of H(2)O(2)- and p53-dependent pathways. J. Biol. Chem. 2004, 279, 25535–25543. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Fröhlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhola, P.D.; Letai, A. Mitochondria—Judges and Executioners of Cell Death Sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Mrschtik, M.; Ryan, K.M. Lysosomal proteins in cell death and autophagy. FEBS J. 2015, 282, 1858–1870. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Leist, M.; Gores, G.J. Lysosomes in cell death. Oncogene 2004, 23, 2881–2890. [Google Scholar] [CrossRef] [Green Version]
- Di, Y.Q.; Han, X.L.; Kang, X.L.; Wang, D.; Chen, C.H.; Wang, J.X.; Zhao, X.F. Autophagy triggers CTSD (cathepsin D) maturation and localization inside cells to promote apoptosis. Autophagy 2021, 17, 1170–1192. [Google Scholar] [CrossRef]
- Goldie, J.H.; Coldman, A.J. A mathematic model for relating the drug sensitivity of tumors to their spontaneous mutation rate. Cancer Treat. Rep. 1979, 63, 1727–1733. [Google Scholar]
- Berezovskaya, O.; Schimmer, A.D.; Glinskii, A.B.; Pinilla, C.; Hoffman, R.M.; Reed, J.C.; Glinsky, G.V. Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res. 2005, 65, 2378–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Ohk, J.; Jeong, D.; Li, C.; Lee, S.; Duan, J.; Kim, C.; Lim, J.S.; Yang, Y.; Kim, K.I.; et al. Distinct regulatory effect of the p34SEI-1 oncoprotein on cancer metastasis in HER2/neu-positive and -negative cells. Int. J. Oncol. 2014, 45, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Donato, C.; Kunz, L.; Castro-Giner, F.; Paasinen-Sohns, A.; Strittmatter, K.; Szczerba, B.M.; Scherrer, R.; Di Maggio, N.; Heusermann, W.; Biehlmaier, O.; et al. Hypoxia Triggers the Intravasation of Clustered Circulating Tumor Cells. Cell Rep. 2020, 32, 108105. [Google Scholar] [CrossRef]
- Li, C.; Jung, S.; Yang, Y.; Kim, K.-I.; Lim, J.-S.; Cheon, C.-I.; Lee, M.-S. Inhibitory role of TRIP-Br1 oncoprotein in hypoxia-induced apoptosis in breast cancer cell lines. Int. J. Oncol. 2016, 48, 2639–2646. [Google Scholar] [CrossRef]
- Bhoopathi, P.; Chetty, C.; Gujrati, M.; Dinh, D.H.; Rao, J.S.; Lakka, S. Cathepsin B facilitates autophagy-mediated apoptosis in SPARC overexpressed primitive neuroectodermal tumor cells. Cell Death Differ. 2010, 17, 1529–1539. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Madden, D.T.; Egger, L.; Bredesen, D.E. A calpain-like protease inhibits autophagic cell death. Autophagy 2007, 3, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Rudzińska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A., Jr. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar] [CrossRef]
- Martínez-Fábregas, J.; Prescott, A.; van Kasteren, S.; Pedrioli, D.L.; McLean, I.; Moles, A.; Reinheckel, T.; Poli, V.; Watts, C. Lysosomal protease deficiency or substrate overload induces an oxidative-stress mediated STAT3-dependent pathway of lysosomal homeostasis. Nat. Commun. 2018, 9, 5343. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.A.; Vu, S.H.; Jung, S.; Lee, B.S.; Nguyen, T.N.Q.; Lee, H.; Lee, H.-g.; Myagmarjav, D.; Jo, T.; Choi, Y.; et al. SERTAD1 Sensitizes Breast Cancer Cells to Doxorubicin and Promotes Lysosomal Protein Biosynthesis. Biomedicines 2022, 10, 1148. https://doi.org/10.3390/biomedicines10051148
Nguyen HA, Vu SH, Jung S, Lee BS, Nguyen TNQ, Lee H, Lee H-g, Myagmarjav D, Jo T, Choi Y, et al. SERTAD1 Sensitizes Breast Cancer Cells to Doxorubicin and Promotes Lysosomal Protein Biosynthesis. Biomedicines. 2022; 10(5):1148. https://doi.org/10.3390/biomedicines10051148
Chicago/Turabian StyleNguyen, Hai Anh, Son Hai Vu, Samil Jung, Beom Suk Lee, Thi Ngoc Quynh Nguyen, Hyojeong Lee, Hye-gyeong Lee, Davaajargal Myagmarjav, Taeyeon Jo, Yeongseon Choi, and et al. 2022. "SERTAD1 Sensitizes Breast Cancer Cells to Doxorubicin and Promotes Lysosomal Protein Biosynthesis" Biomedicines 10, no. 5: 1148. https://doi.org/10.3390/biomedicines10051148
APA StyleNguyen, H. A., Vu, S. H., Jung, S., Lee, B. S., Nguyen, T. N. Q., Lee, H., Lee, H.-g., Myagmarjav, D., Jo, T., Choi, Y., & Lee, M.-S. (2022). SERTAD1 Sensitizes Breast Cancer Cells to Doxorubicin and Promotes Lysosomal Protein Biosynthesis. Biomedicines, 10(5), 1148. https://doi.org/10.3390/biomedicines10051148