
In Vivo Evaluation of Fibroblast Growth Factor Receptor Inhibition in Mouse Xenograft Models of Gastrointestinal Stromal Tumor

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. GIST Xenografts
2.2. Drugs and Reagents
2.3. Experimental Design
2.4. Histological and Biochemical Assessments
2.5. Statistics
3. Results
3.1. Tumor Volume Assessment
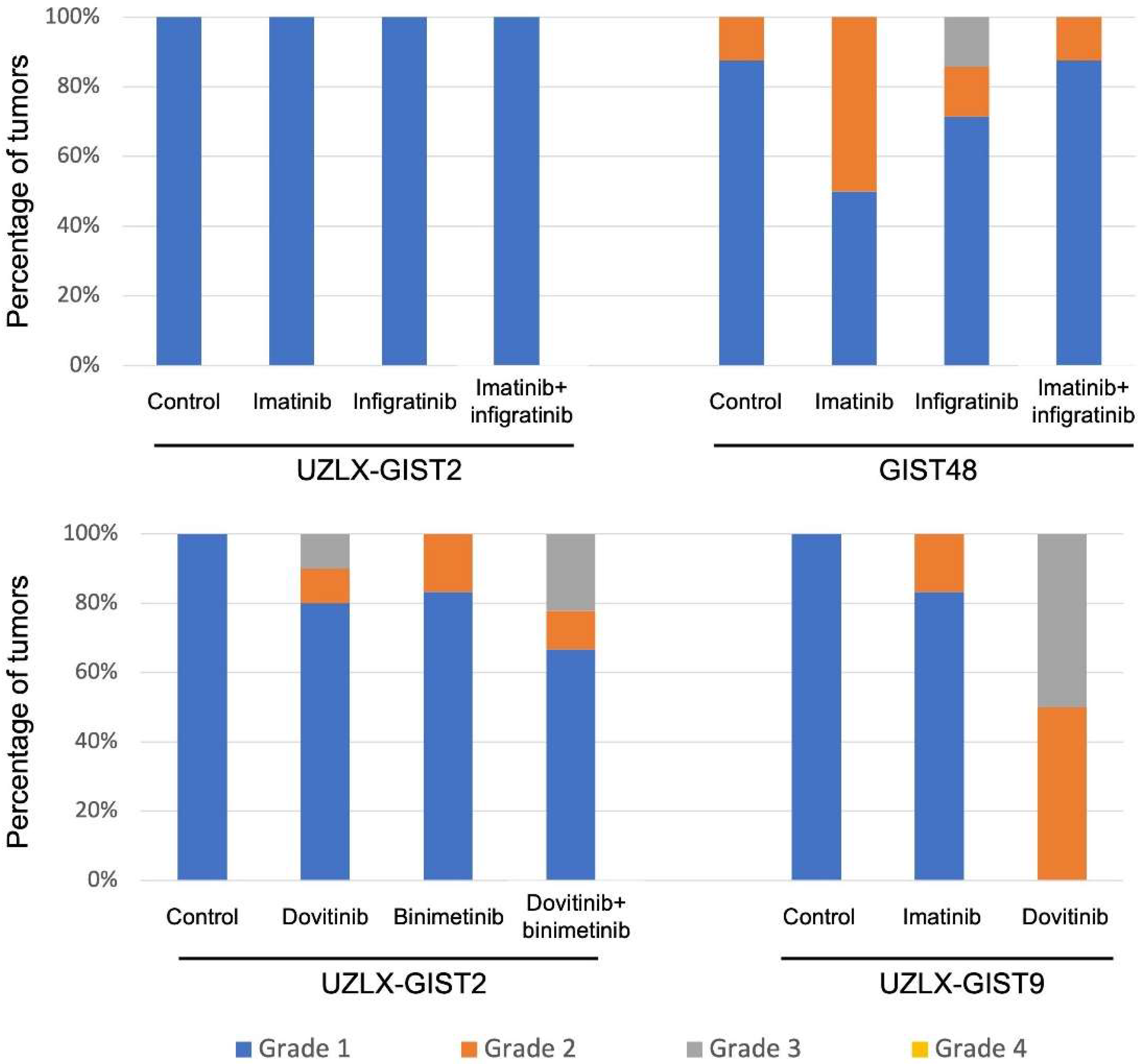
3.2. Histopathological Assessment
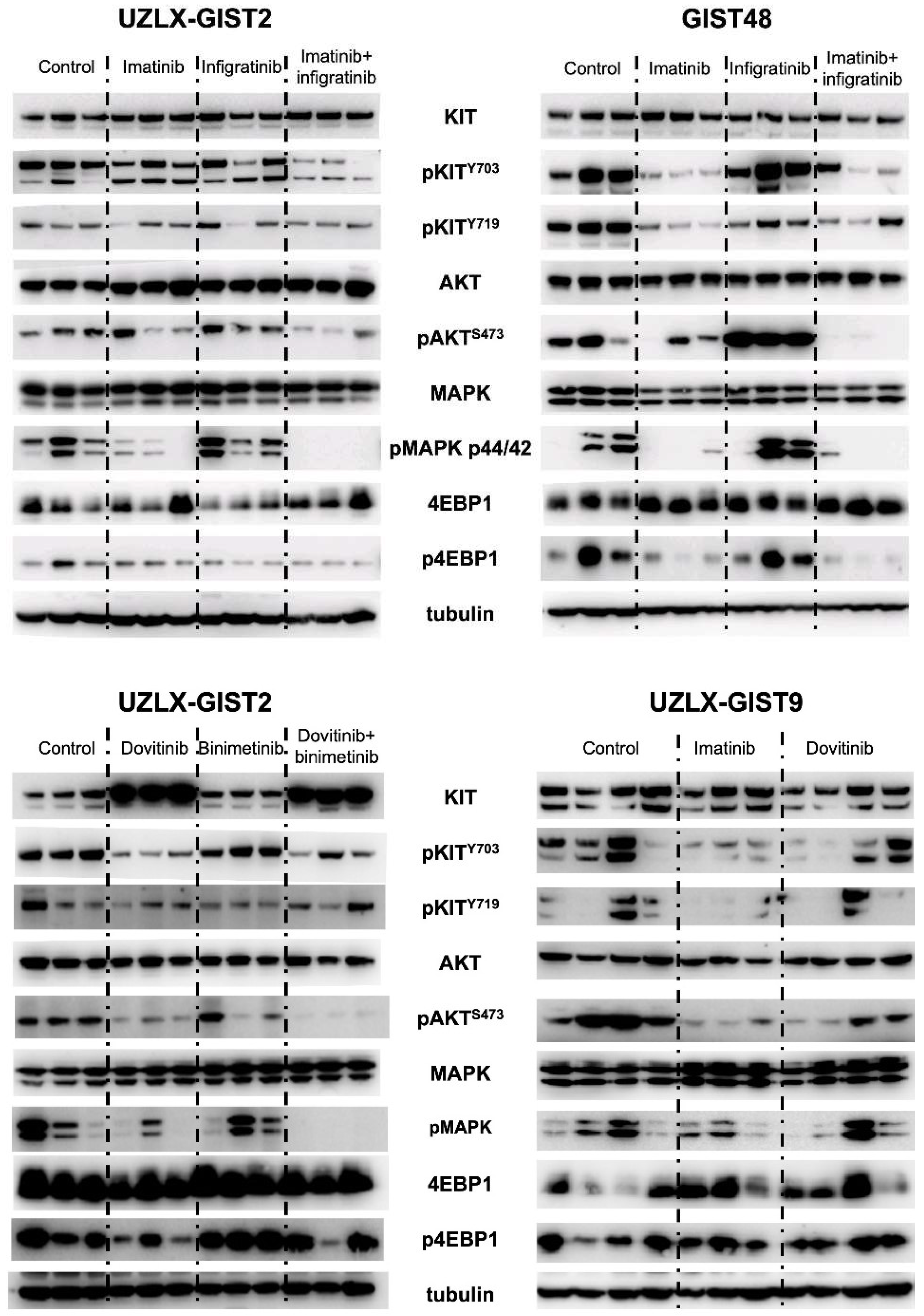
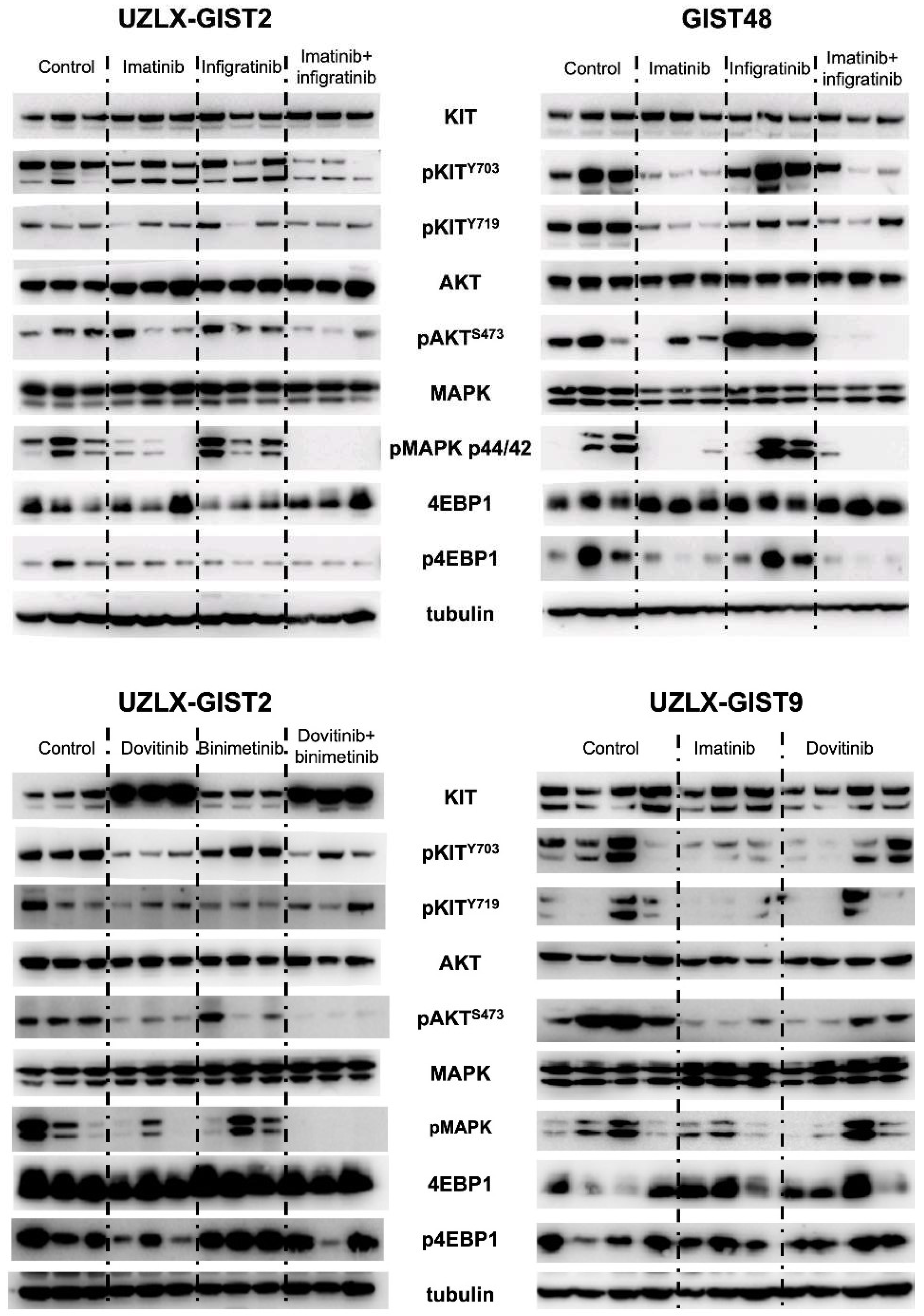
3.3. RTK Signaling Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- von Mehren, M.; Joensuu, H. Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2018, 36, 136–143. [Google Scholar] [CrossRef] [PubMed]
- de Pinieux, G.; Karanian, M.; Le Loarer, F.; Le Guellec, S.; Chabaud, S.; Terrier, P.; Bouvier, C.; Batistella, M.; Neuville, A.; Robin, Y.-M.; et al. Nationwide incidence of sarcomas and connective tissue tumors of intermediate malignancy over four years using an expert pathology review network. PLoS ONE 2021, 16, e0246958. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Besmer, P.; Guo, T.; Arkun, K.; Hom, G.; Koryotowski, B.; Leversha, M.A.; Jeffrey, P.D.; DeSantis, D.; Singer, S.; et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin. Cancer Res. 2005, 11, 4182–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; Von Mehren, M.; Fletcher, C.D.; Sandau, K.; et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef] [PubMed]
- Liegl, B.; Kepten, I.; Le, C.; Zhu, M.; Demetri, G.D.; Heinrich, M.; Fletcher, C.D.M.; Corless, C.L.; Fletcher, J.A. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J. Pathol. 2008, 216, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef] [Green Version]
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 Promotes Gastrointestinal Stromal Tumor Cell Growth and Drug Resistance. Cancer Res. 2015, 75, 880–891. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Shi, E.; Chmielecki, J.; Tang, C.-M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 399. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; De Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Boichuk, S.; Galembikova, A.; Mikheeva, E.; Bikinieva, F.; Aukhadieva, A.; Dunaev, P.; Khalikov, D.; Petrov, S.; Kurtasanov, R.; Valeeva, E.; et al. Inhibition of FGF2-Mediated Signaling in GIST—Promising Approach for Overcoming Resistance to Imatinib. Cancers 2020, 12, 1674. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huynh, H.T.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. FGFR-Mediated Reactivation of MAPK Signaling Attenuates Antitumor Effects of Imatinib in Gastrointestinal Stromal Tumors. Cancer Discov. 2015, 5, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchinetti, F.; Hollebecque, A.; Bahleda, R.; Loriot, Y.; Olaussen, K.A.; Massard, C.; Friboulet, L. Facts and New Hopes on Selective FGFR Inhibitors in Solid Tumors. Clin. Cancer Res. 2020, 26, 764–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornillie, J.; Wozniak, A.; Li, H.; Wang, Y.; Boeckx, B.; Gebreyohannes, Y.K.; Wellens, J.; Vanleeuw, U.; Hompes, D.; Stas, M.; et al. Establishment and Characterization of Histologically and Molecularly Stable Soft-tissue Sarcoma Xenograft Models for Biological Studies and Preclinical Drug Testing. Mol. Cancer Ther. 2019, 18, 1168–1178. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wozniak, A.; Wellens, J.; Gebreyohannes, Y.K.; Guillén, M.J.; Avilés, P.M.; Debiec-Rychter, M.; Sciot, R.; Schöffski, P. Plocabulin, a novel tubulin inhibitor, has potent antitumor activity in patient-derived xenograft models of gastrointestinal stromal tumors. Transl. Oncol. 2020, 13, 100832. [Google Scholar] [CrossRef]
- Van Looy, T.; Wozniak, A.; Floris, G.; Sciot, R.; Li, H.; Wellens, J.; Vanleeuw, U.; Fletcher, J.A.; Manley, P.W.; Debiec-Rychter, M.; et al. Phosphoinositide 3-kinase inhibitors combined with imatinib in patient-derived xenograft models of gastrointestinal stromal tumors: Rationale and efficacy. Clin. Cancer Res. 2014, 20, 6071–6082. [Google Scholar] [CrossRef] [Green Version]
- Gebreyohannes, Y.K.; Schöffski, P.; Van Looy, T.; Wellens, J.; Vreys, L.; Cornillie, J.; Vanleeuw, U.; Aftab, D.T.; Debiec-Rychter, M.; Sciot, R.; et al. Cabozantinib Is Active against Human Gastrointestinal Stromal Tumor Xenografts Carrying Different KIT Mutations. Mol. Cancer Ther 2016, 15, 2845–2852. [Google Scholar] [CrossRef] [Green Version]
- Van Looy, T.; Gebreyohannes, Y.K.; Wozniak, A.; Cornillie, J.; Wellens, J.; Li, H.; Vanleeuw, U.; Floris, G.; Debiec-Rychter, M.; Sciot, R.; et al. Characterization and assessment of the sensitivity and resistance of a newly established human gastrointestinal stromal tumour xenograft model to treatment with tyrosine kinase inhibitors. Clin. Sarcoma Res. 2014, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Wardelmann, E.; Merkelbach-Bruse, S.; Pauls, K.; Thomas, N.; Schildhaus, H.-U.; Heinicke, T.; Speidel, N.; Pietsch, T.; Buettner, R.; Pink, D.; et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin. Cancer Res. 2006, 12, 1743–1749. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; Von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Cohen, N.; Zeng, S.; Seifert, A.M.; Kim, T.; Sorenson, E.C.; Greer, J.B.; Beckman, M.J.; Santamaria-Barria, J.; Crawley, M.H.; Green, B.L.; et al. Pharmacological inhibition of KIT activates MET signaling in gastrointestinal stromal tumors. Cancer Res. 2015, 75, 2061–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; de Menezes, D.L.; Vora, J.; Harris, A.; Ye, H.; Nordahl, L.; Garrett, E.; Samara, E.; Aukerman, S.L.; Gelb, A.B.; et al. In vivo target modulation and biological activity of CHIR-258, a multitargeted growth factor receptor kinase inhibitor, in colon cancer models. Clin. Cancer Res. 2005, 11, 3633–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Growney, J.D.; Li, F.; Qiu, S.; Gorbatcheva, B.; Battalagine, L.; Mestan, J.; Manley, P.; Squires, M.; Cao, A.; Monahan, J.E. Dovitinib has anti-tumor activity in gastrointestinal stromal tumor (GIST) cell lines. Cancer Res. 2013, 73 (Suppl. 8), abstr 1620. [Google Scholar]
- Antonescu, C.R.; Viale, A.; Sarran, L.; Tschernyavsky, S.J.; Gonen, M.; Segal, N.H.; Maki, R.G.; Socci, N.D.; DeMatteo, R.P.; Besmer, P. Gene Expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin. Cancer Res. 2004, 10, 3282–3290. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-K.; Yoo, C.; Ryoo, B.-Y.; Lee, J.J.; Tan, E.; Park, I.; Park, J.H.; Choi, Y.J.; Jo, J.; Ryu, J.-S.; et al. Phase II study of dovitinib in patients with metastatic and/or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib. Br. J. Cancer 2013, 109, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Blay, J.-Y.; Comandone, A.; Martin-Broto, J.; Fumagalli, E.; Grignani, G.; Del Muro, X.G.; Adenis, A.; Valverde, C.; Pousa, A.L.; et al. Dovitinib in patients with gastrointestinal stromal tumour refractory and/or intolerant to imatinib. Br. J. Cancer 2017, 117, 1278–1285. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.; Ryu, M.-H.; Na, Y.S.; Ryoo, B.-Y.; Park, S.R.; Kang, Y.-K. Analysis of serum protein biomarkers, circulating tumor DNA, and dovitinib activity in patients with tyrosine kinase inhibitor-refractory gastrointestinal stromal tumors. Ann. Oncol. 2014, 25, 2272–2277. [Google Scholar] [CrossRef]
- Javle, M.; Roychowdhury, S.; Kelley, R.K.; Sadeghi, S.; Macarulla, T.; Weiss, K.H.; Waldschmidt, D.-T.; Goyal, L.; Borbath, I.; El-Khoueiry, A.; et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: Mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol. Hepatol. 2021, 6, 803–815. [Google Scholar] [CrossRef]
- Bauer, S.; Duensing, A.; Demetri, G.D.; Fletcher, J.A. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene 2007, 26, 7560–7568. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, E.; Kelly, C.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Movva, S.; Condy, M.; Adamson, T.; Mcfadyen, C.R.; et al. A Phase I Study of Binimetinib (MEK162) Combined with Pexidartinib (PLX3397) in Patients with Advanced Gastrointestinal Stromal Tumor. Oncologist 2019, 24, 1309-e983. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Montero, C.M.; Mao, F.J.; Barnard, J.; Parker, Y.; Zamanian-Daryoush, M.; Pink, J.J.; Finke, J.H.; Rini, B.I.; Lindner, D.J. MEK inhibition abrogates sunitinib resistance in a renal cell carcinoma patient-derived xenograft model. Br. J. Cancer 2016, 115, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.-H.; Lee, J.-H.; Chang, Y.-J.; Tsai, H.-H.; Lin, Y.-L.; Lin, A.M.-Y.; Yang, J.C.-H. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol. Oncol. 2013, 7, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Dunaev, P.; Galembikova, A.; Mustafin, I.; Valeeva, E. Inhibition of fibroblast growth factor receptor-signaling sensitizes imatinib-resistant gastrointestinal stromal tumors to low doses of topoisomerase II inhibitors. Anti-Cancer Drugs 2018, 29, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.M.; Shoushtari, A.N.; Qin, L.-X.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Mcfadyen, C.; Sjoberg, A.; Singer, S.; et al. A phase Ib study of BGJ398, a pan-FGFR kinase inhibitor in combination with imatinib in patients with advanced gastrointestinal stromal tumor. Investig. New Drugs 2019, 37, 282–290. [Google Scholar] [CrossRef] [PubMed]
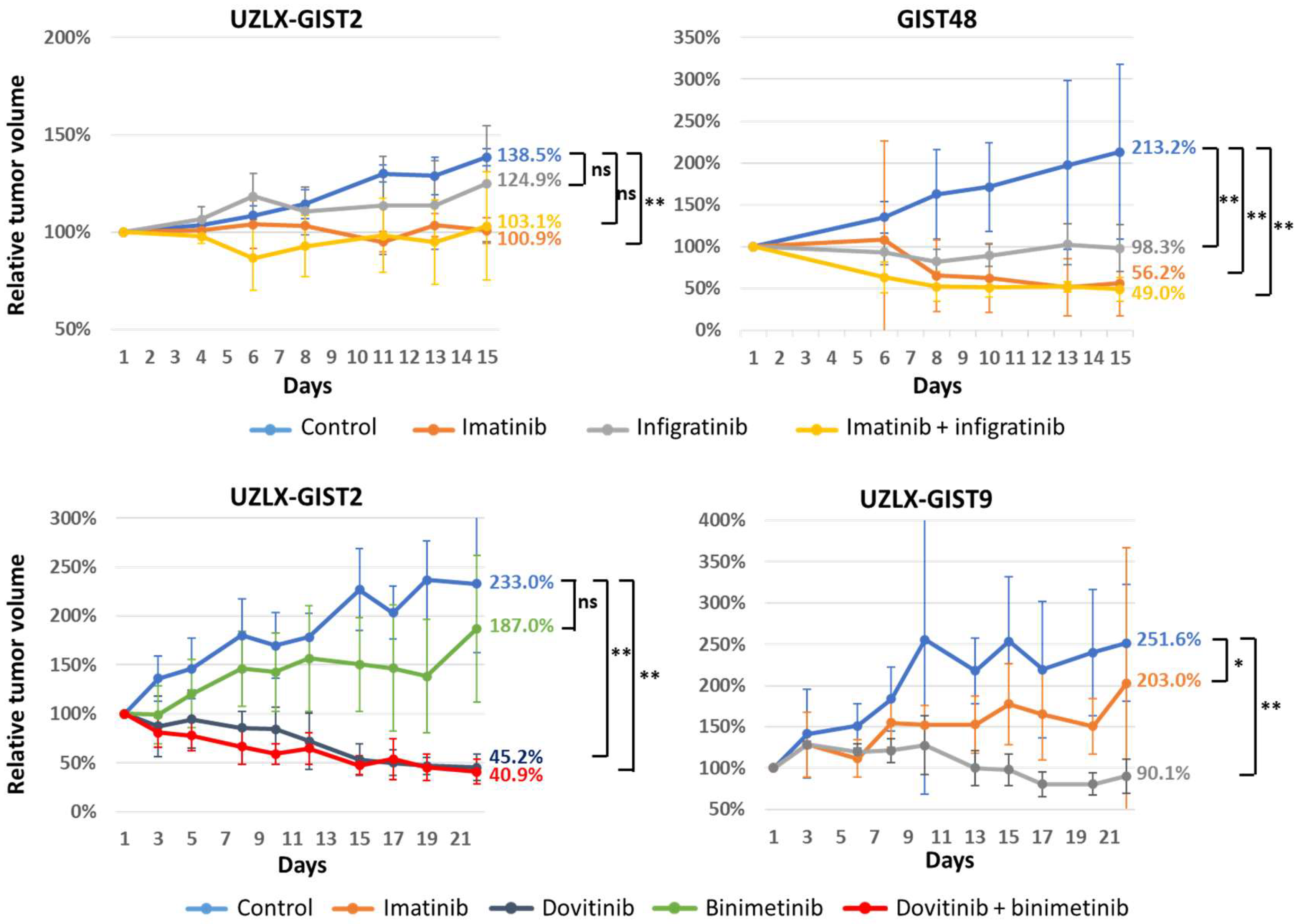


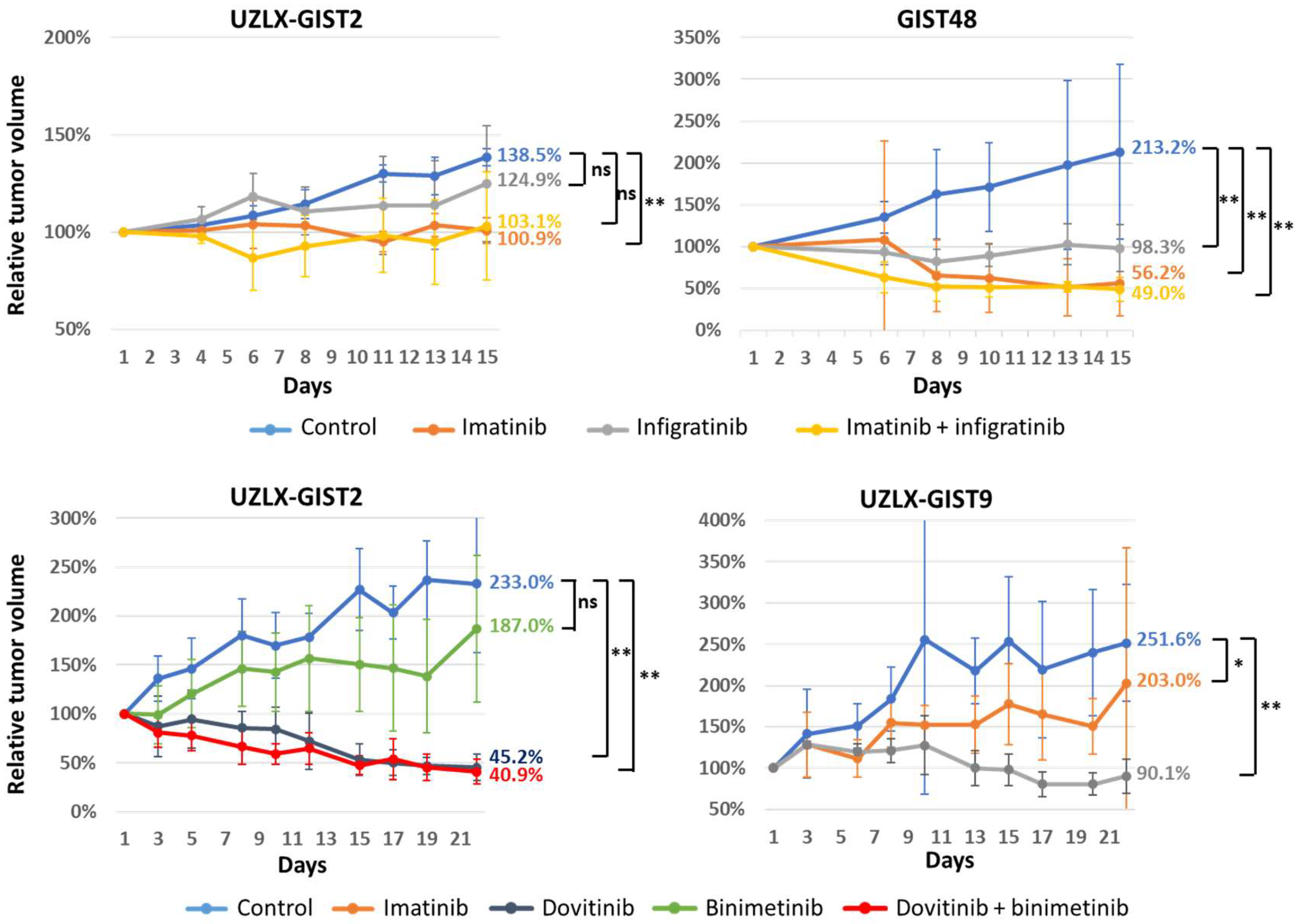{kind=link}
{kind=link}
{kind=link}
| Xenograft Model | Histopathological Characteristics | KIT Mutation | In Vivo Sensitivity to Standard TKI | |
|---|---|---|---|---|
| UZLX-GIST2 | Patient-derived | Spindle cells KIT(+), DOG1(+) | p.A502_Y503dup | Imatinib dose-dependent sensitive Sunitinib sensitive |
| UZLX-GIST9 | Patient-derived | Spindle cells KIT(+), DOG1(+) | p.P577del;W557LfsX5;D820G | Imatinib resistant Sunitinib resistant |
| GIST48 | Cell line-derived | Spindle cells KIT(+), DOG1(+) | p.V560D;D820A | Imatinib sensitive Sunitinib sensitive |
| Model Name | Passage | Number of Mice/Tumors Per Treatment Group | ||||||
|---|---|---|---|---|---|---|---|---|
| Control (Vehicle) | Imatinib (50 mg/kg BID) | Dovitinib (30 mg/kg QD) | Binimetinib (3.5 mg/kg BID) | Dovitinib + Binimetinib * | Infigratinib (30 mg/kg QD) | Imatinib + Infigratinib * | ||
| UZLX-GIST2 | 12 | 2/4 | 2/4 | n/a | n/a | n/a | 3/6 | 4/7 |
| 17 | 6/12 | n/a | 5/10 | 5/6 | 5/9 | n/a | n/a | |
| UZLX-GIST9 | 4 | 7/8 | 6/6 | 7/7 | n/a | n/a | n/a | n/a |
| GIST48 | 10 | 4/8 | 3/6 | n/a | n/a | n/a | 4/7 | 4/8 |
| Mitotic and Proliferative Activity | Apoptotic Activity | Microvessel Density | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Xenograft Model | Treatment Group | H&E | pHH3 | Ki67 | H&E | Cleaved PARP | CD31 | ||||||
| UZLX-GIST2 | |||||||||||||
| Imatinib | = | = | = | ↑ 1.8 | * | = | = | ||||||
| Infigratinib | = | = | = | = | = | = | |||||||
| Imatinib+infigratinib | = | = | ↓ 1.8 | * | = | = | = | ||||||
| Dovitinib | ↓↓↓ | ** | ↓ 44.8 | ** | ↓↓↓ | ** | ↓ 2.4 | ** | ↓ 5.3 | ** | ↓ 2.0 | ** | |
| Binimetinib | = | = | ↓ 1.9 | ** | = | = | ↓ 1.2 | * | |||||
| Dovitinib+binimetinib | ↓↓↓ | ** | ↓↓↓ | ** | ↓↓↓ | ** | ↓ 3.9 | ** | ↓ 5.4 | ** | ↓ 2.4 | ** | |
| GIST48 | |||||||||||||
| Imatinib | ↓ 14.8 | ** | ↓ 7.1 | ** | ↓ 12.1 | ** | = | = | = | ||||
| Infigratinib | = | = | ↓ 1.3 | * | = | ↑ 1.4 | * | = | |||||
| Imatinib+infigratinib | ↓ 23.0 | ** | ↓ 13.5 | ** | ↓↓↓ | ** | = | = | ↓ 1.3 | ** | |||
| UZLX-GIST9 | |||||||||||||
| Imatinib | = | = | = | = | = | = | |||||||
| Dovitinib | ↑ 1.4 | * | ↑ 1.5 | * | ↑ 1.3 | * | = | = | = | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schöffski, P.; Gebreyohannes, Y.; Van Looy, T.; Manley, P.; Growney, J.D.; Squires, M.; Wozniak, A. In Vivo Evaluation of Fibroblast Growth Factor Receptor Inhibition in Mouse Xenograft Models of Gastrointestinal Stromal Tumor. Biomedicines 2022, 10, 1135. https://doi.org/10.3390/biomedicines10051135
Schöffski P, Gebreyohannes Y, Van Looy T, Manley P, Growney JD, Squires M, Wozniak A. In Vivo Evaluation of Fibroblast Growth Factor Receptor Inhibition in Mouse Xenograft Models of Gastrointestinal Stromal Tumor. Biomedicines. 2022; 10(5):1135. https://doi.org/10.3390/biomedicines10051135
Chicago/Turabian StyleSchöffski, Patrick, Yemarshet Gebreyohannes, Thomas Van Looy, Paul Manley, Joseph D. Growney, Matthew Squires, and Agnieszka Wozniak. 2022. "In Vivo Evaluation of Fibroblast Growth Factor Receptor Inhibition in Mouse Xenograft Models of Gastrointestinal Stromal Tumor" Biomedicines 10, no. 5: 1135. https://doi.org/10.3390/biomedicines10051135
APA StyleSchöffski, P., Gebreyohannes, Y., Van Looy, T., Manley, P., Growney, J. D., Squires, M., & Wozniak, A. (2022). In Vivo Evaluation of Fibroblast Growth Factor Receptor Inhibition in Mouse Xenograft Models of Gastrointestinal Stromal Tumor. Biomedicines, 10(5), 1135. https://doi.org/10.3390/biomedicines10051135