
A Review of the Recent Development in the Synthesis and Biological Evaluations of Pyrazole Derivatives



Abstract
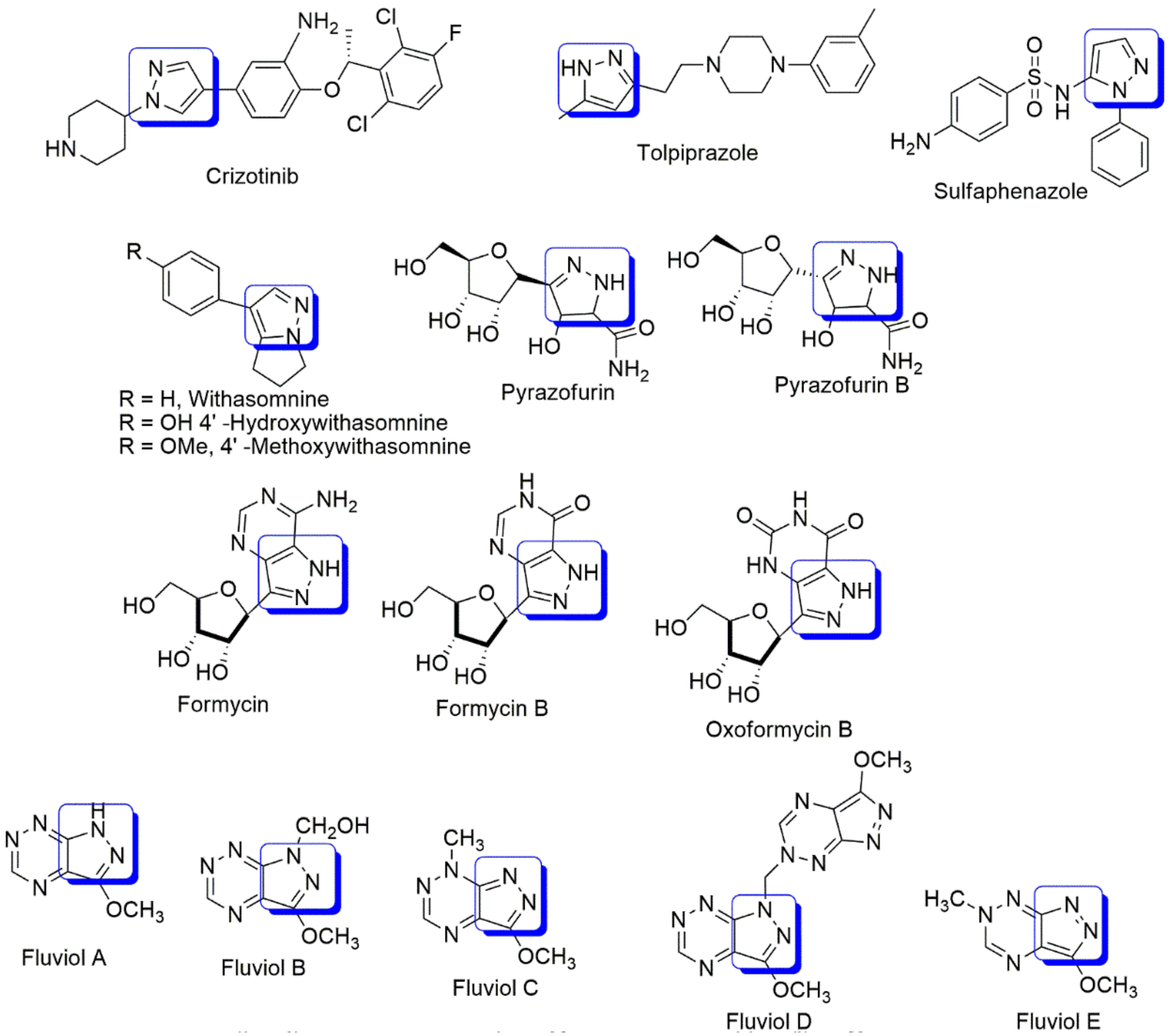
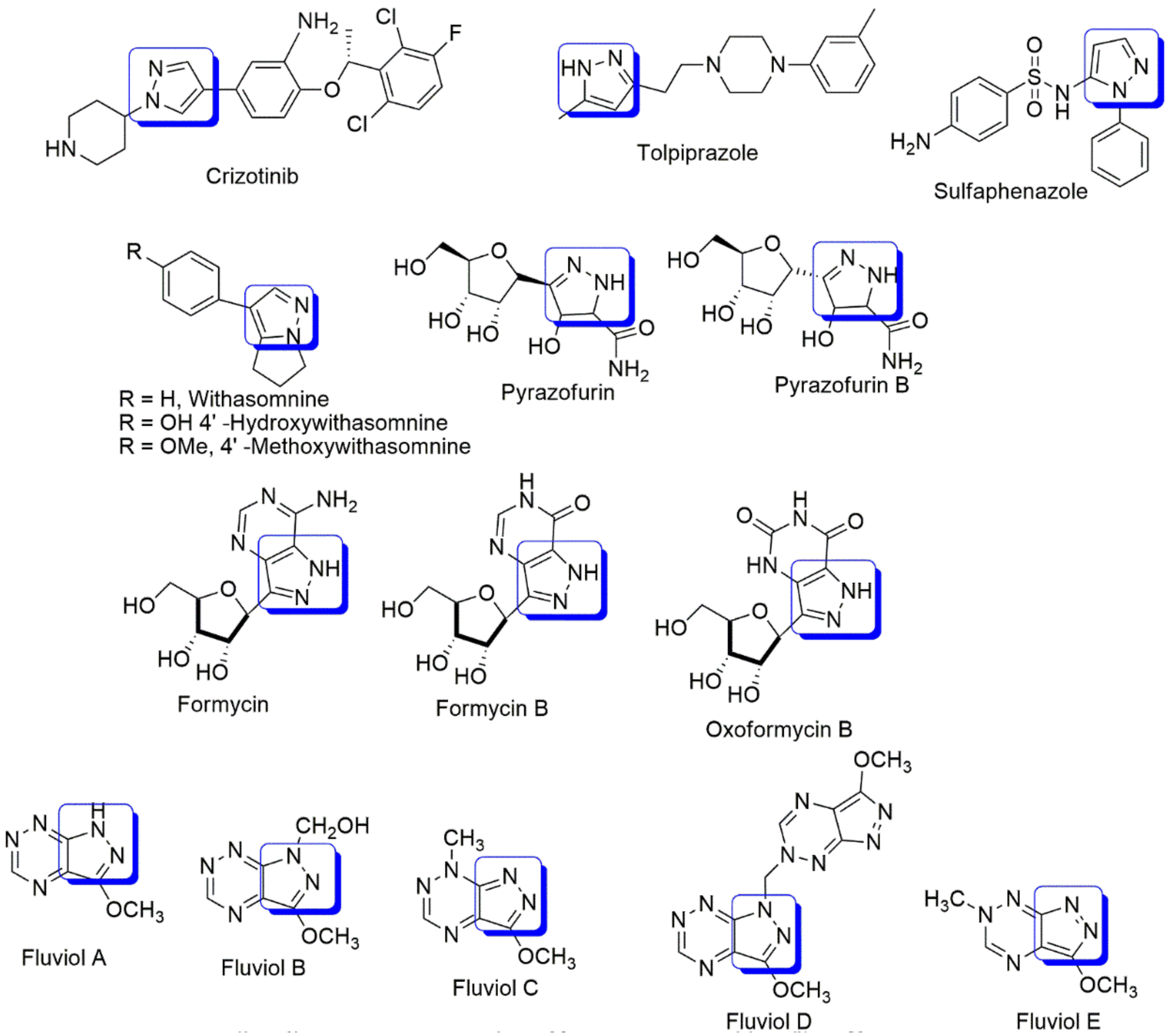
:1. Introduction
2. Synthesis of Pyrazole Derivatives
2.1. Condensation of Hydrazine’s or Similar Nuclei with Carbonyl Functional Group Compounds
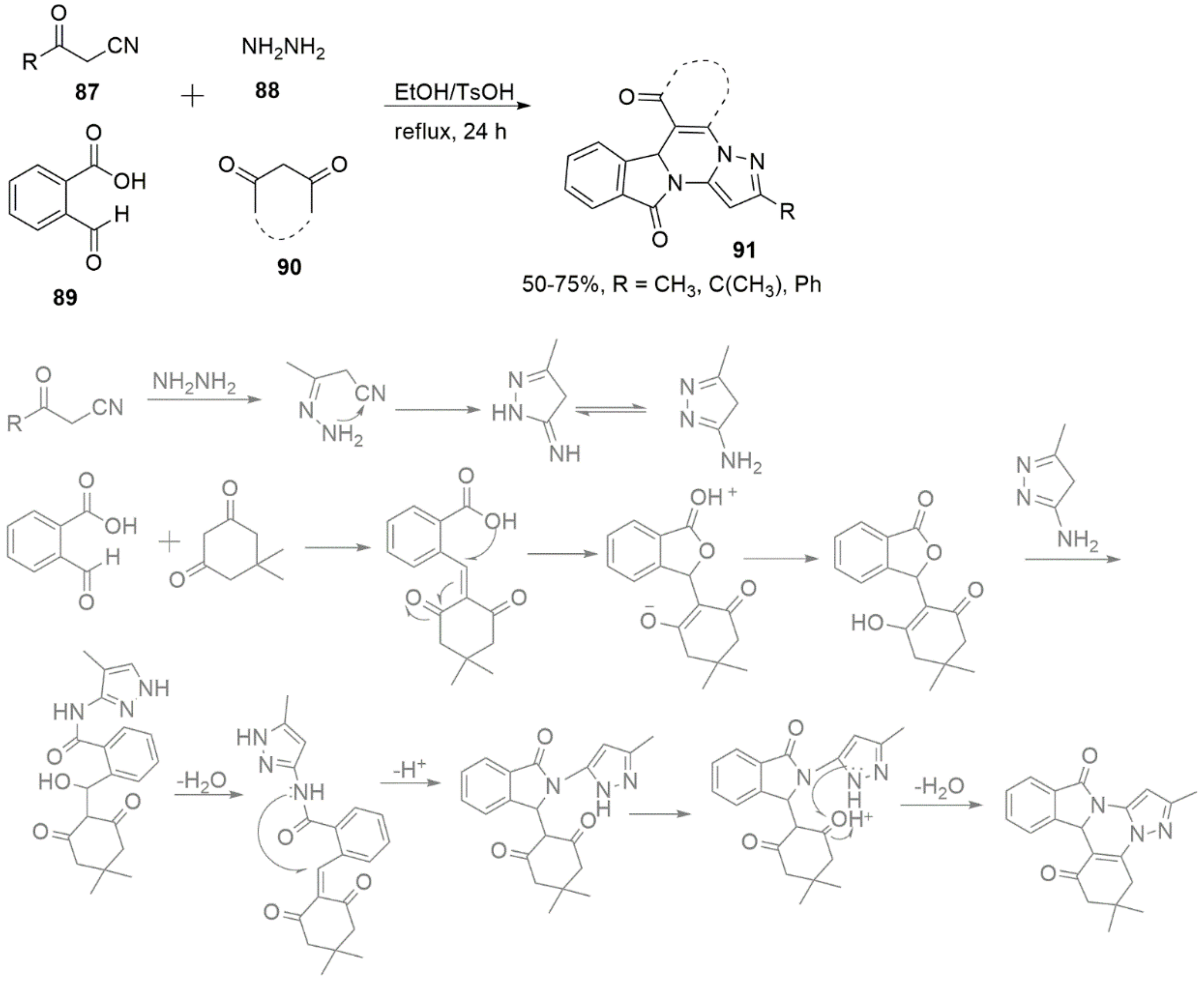
2.1.1. Pyrazoles from Vinyl Ketones
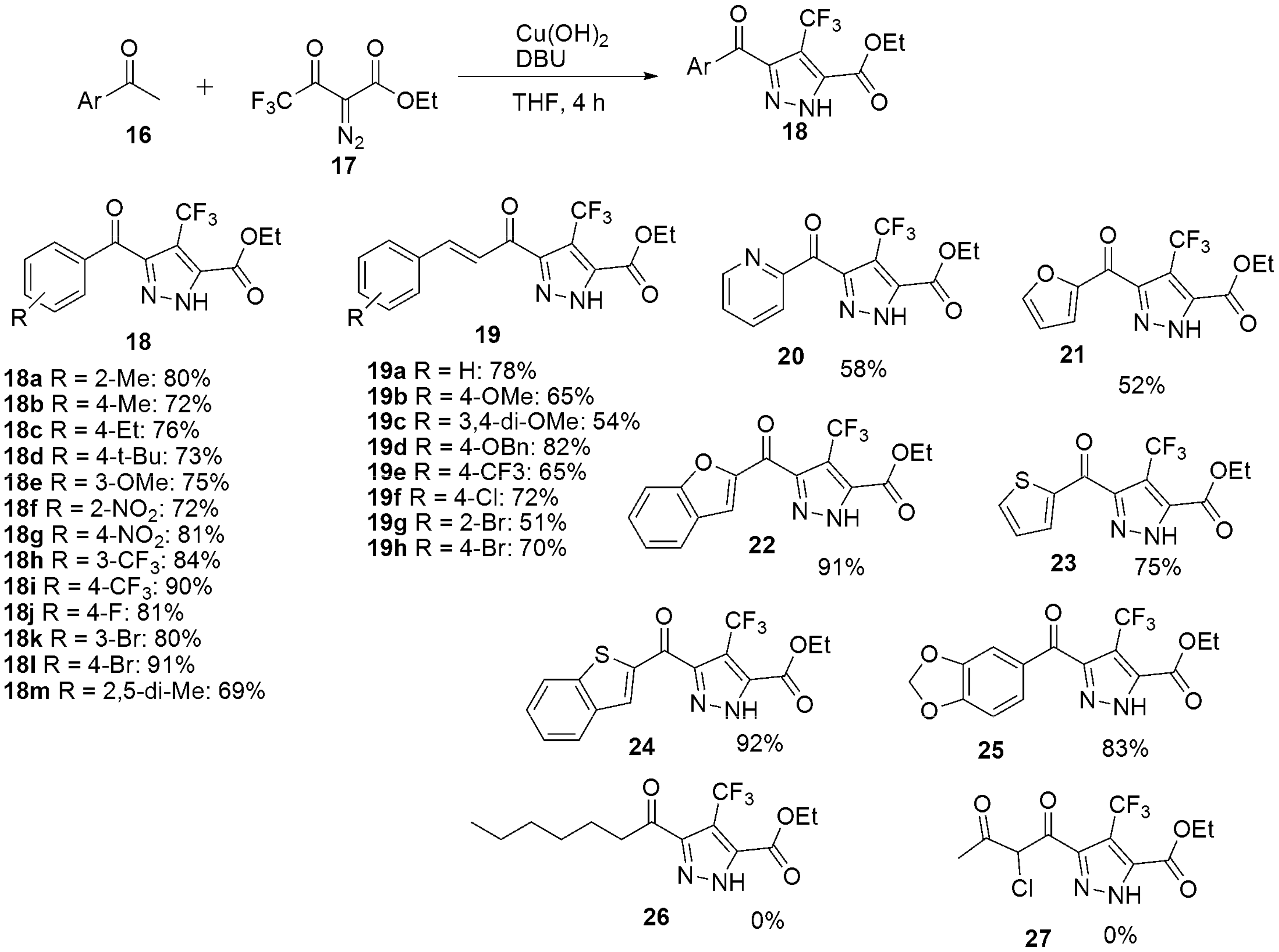
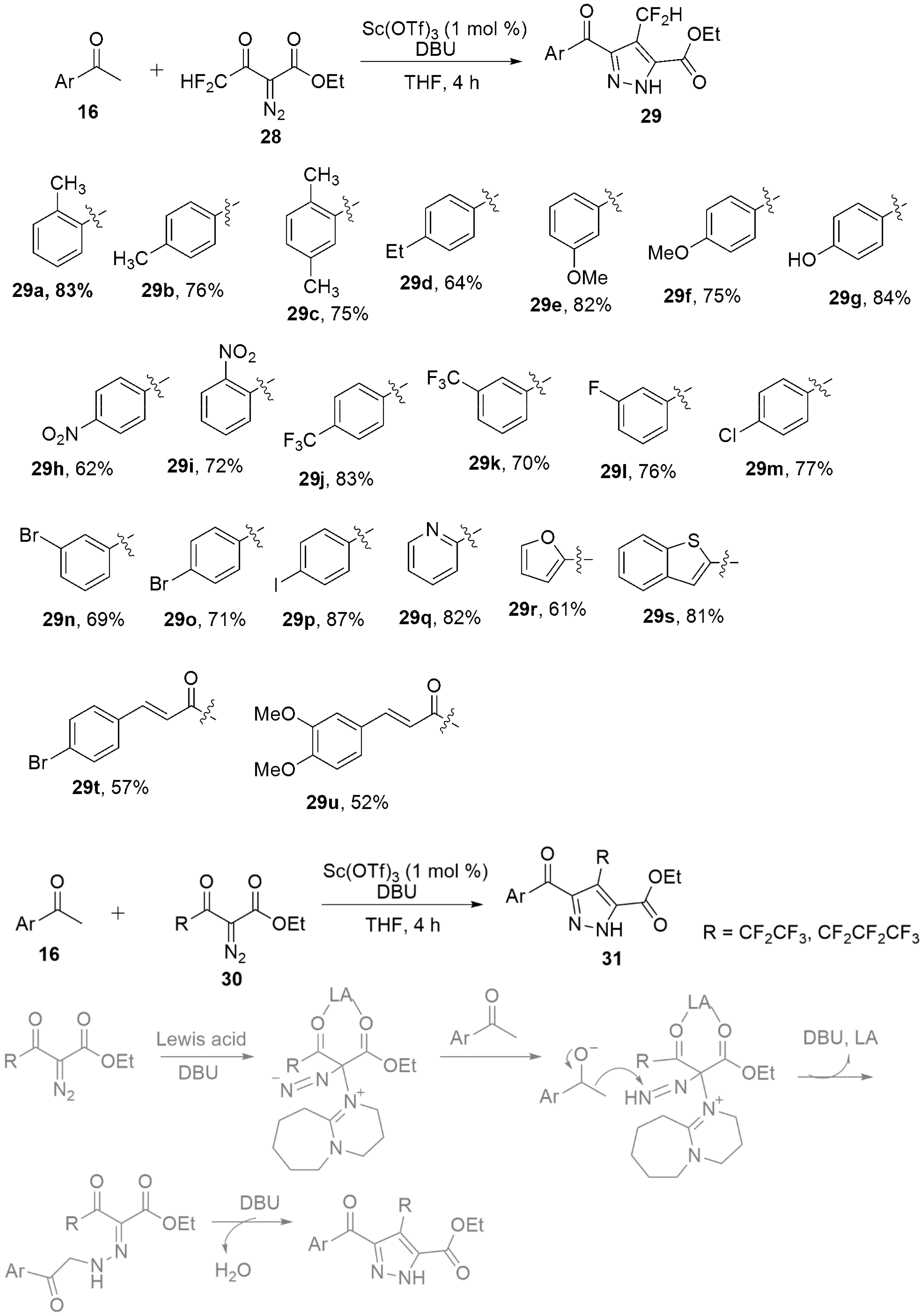
2.1.2. Pyrazoles from 1,3-Diketones
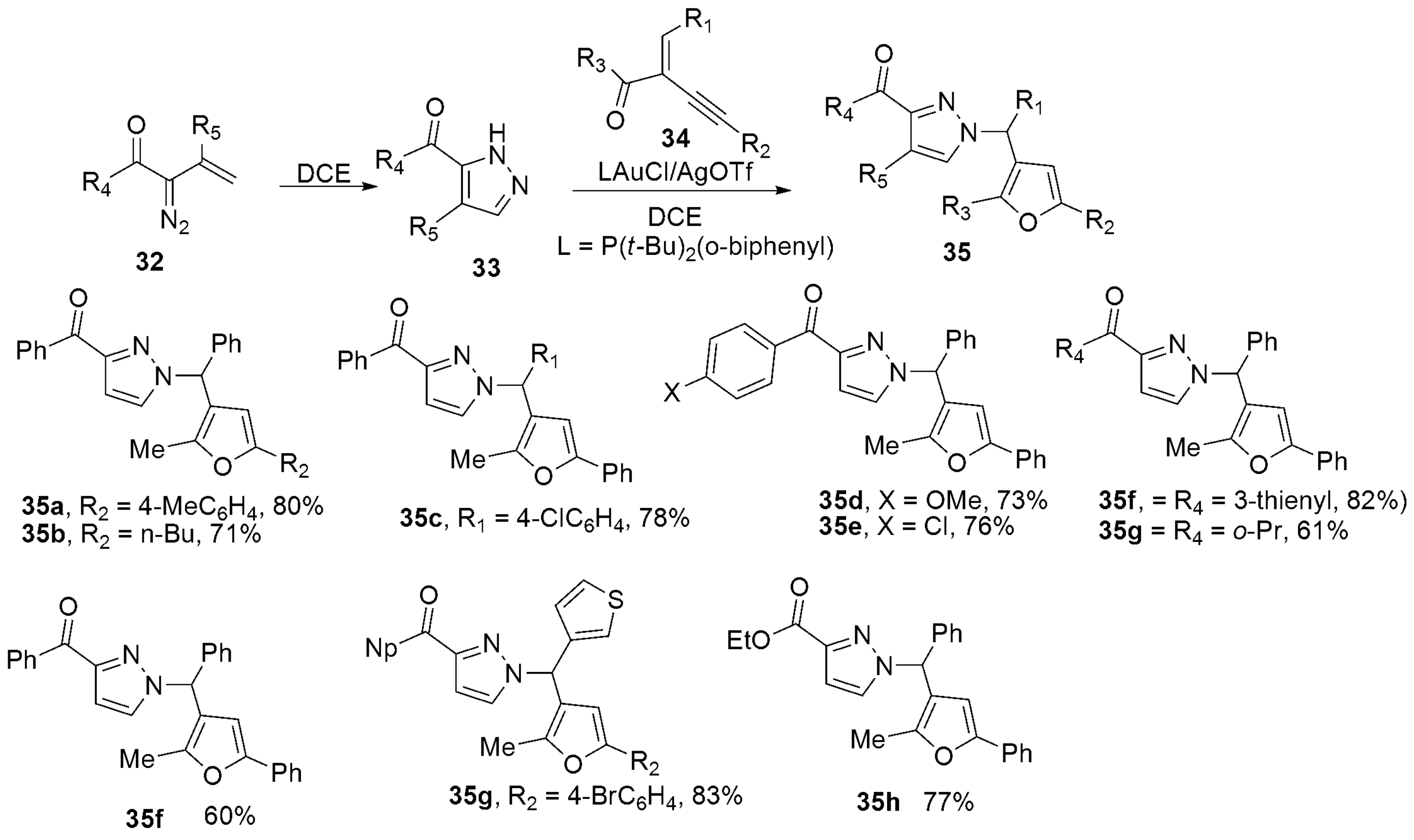
2.1.3. Pyrazoles from Acetylenic Ketones
2.2. Dipolar Cycloadditions
2.2.1. Pyrazoles from Diazoester
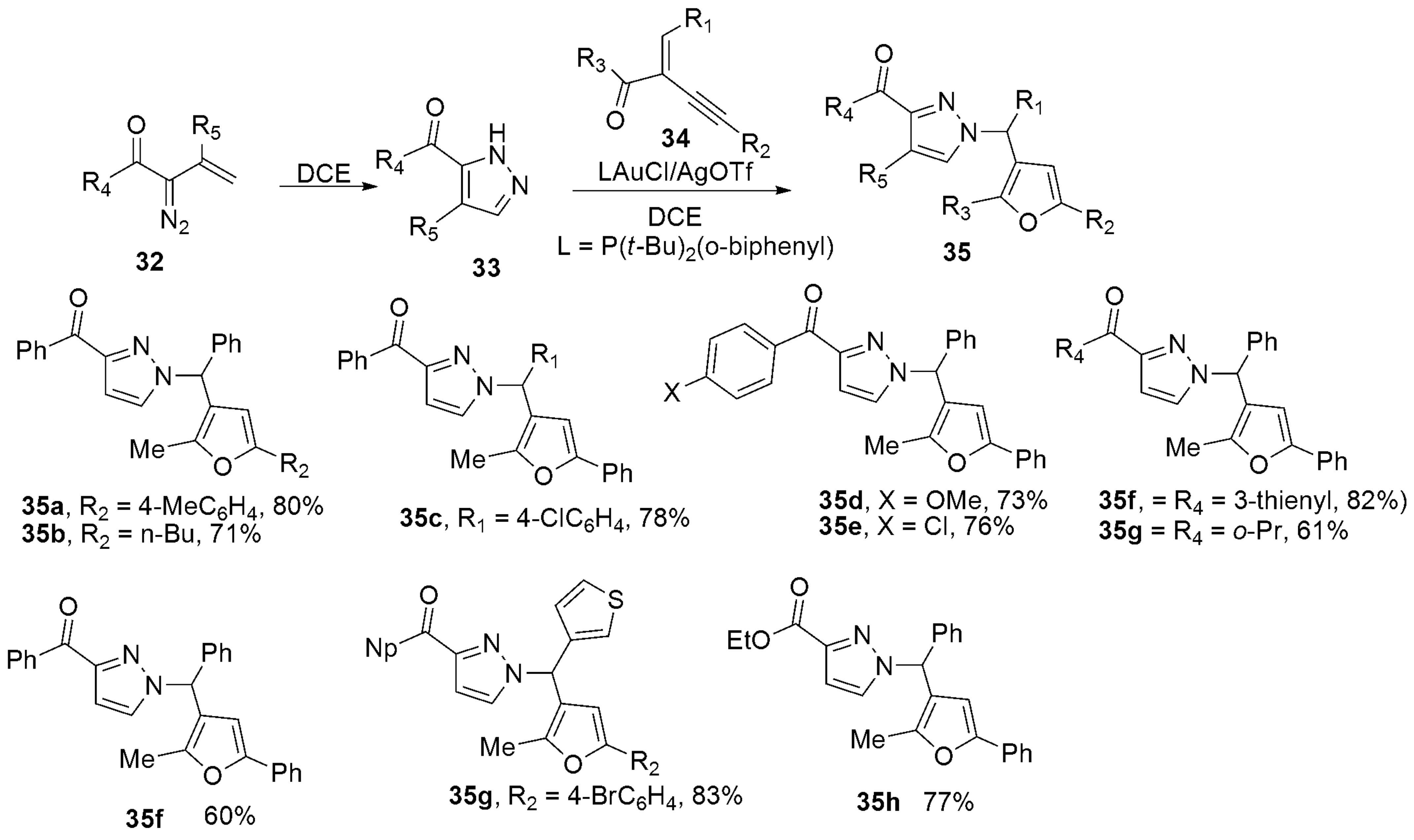
2.2.2. Pyrazoles from Vinyldiazo Ketones
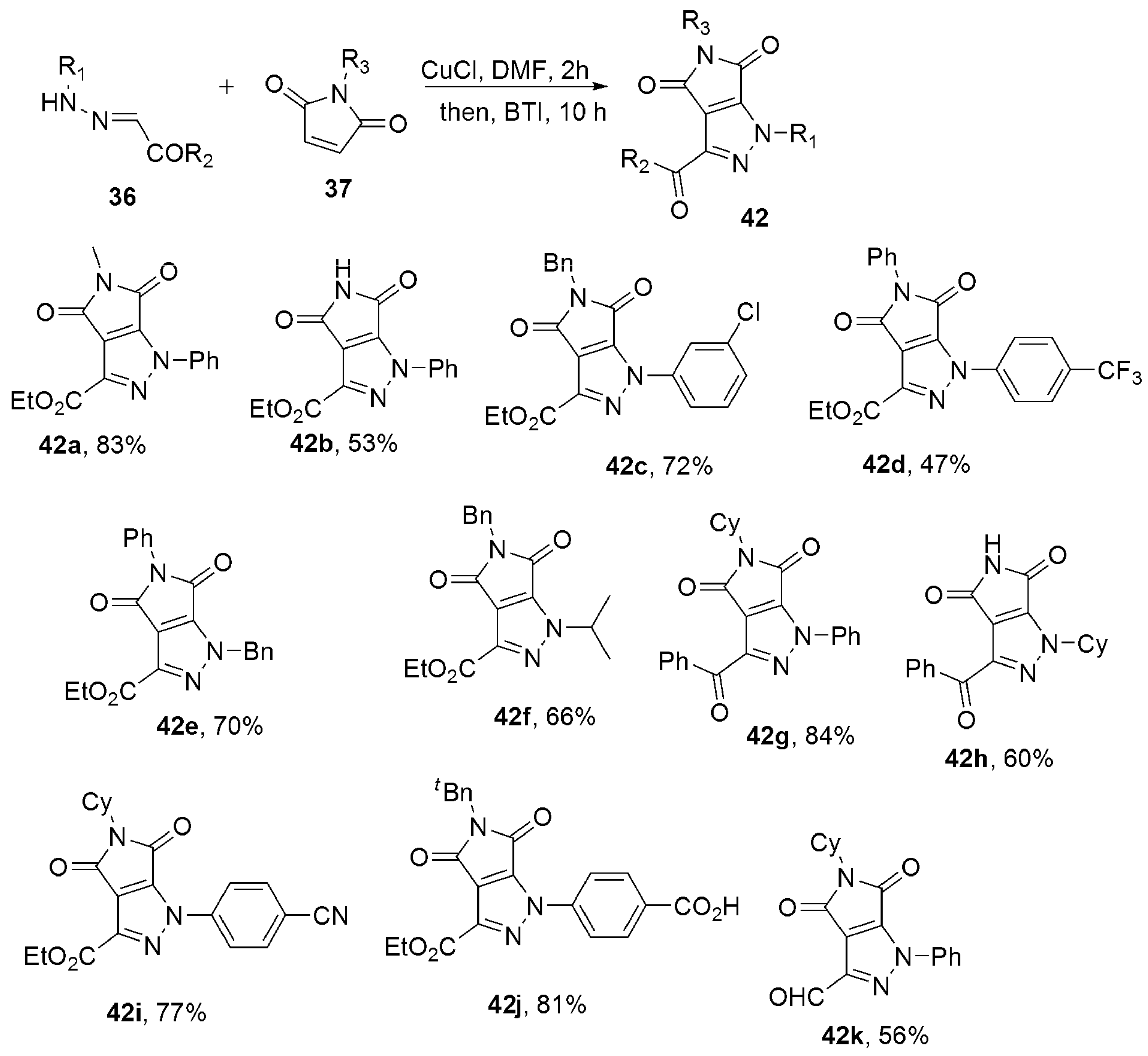
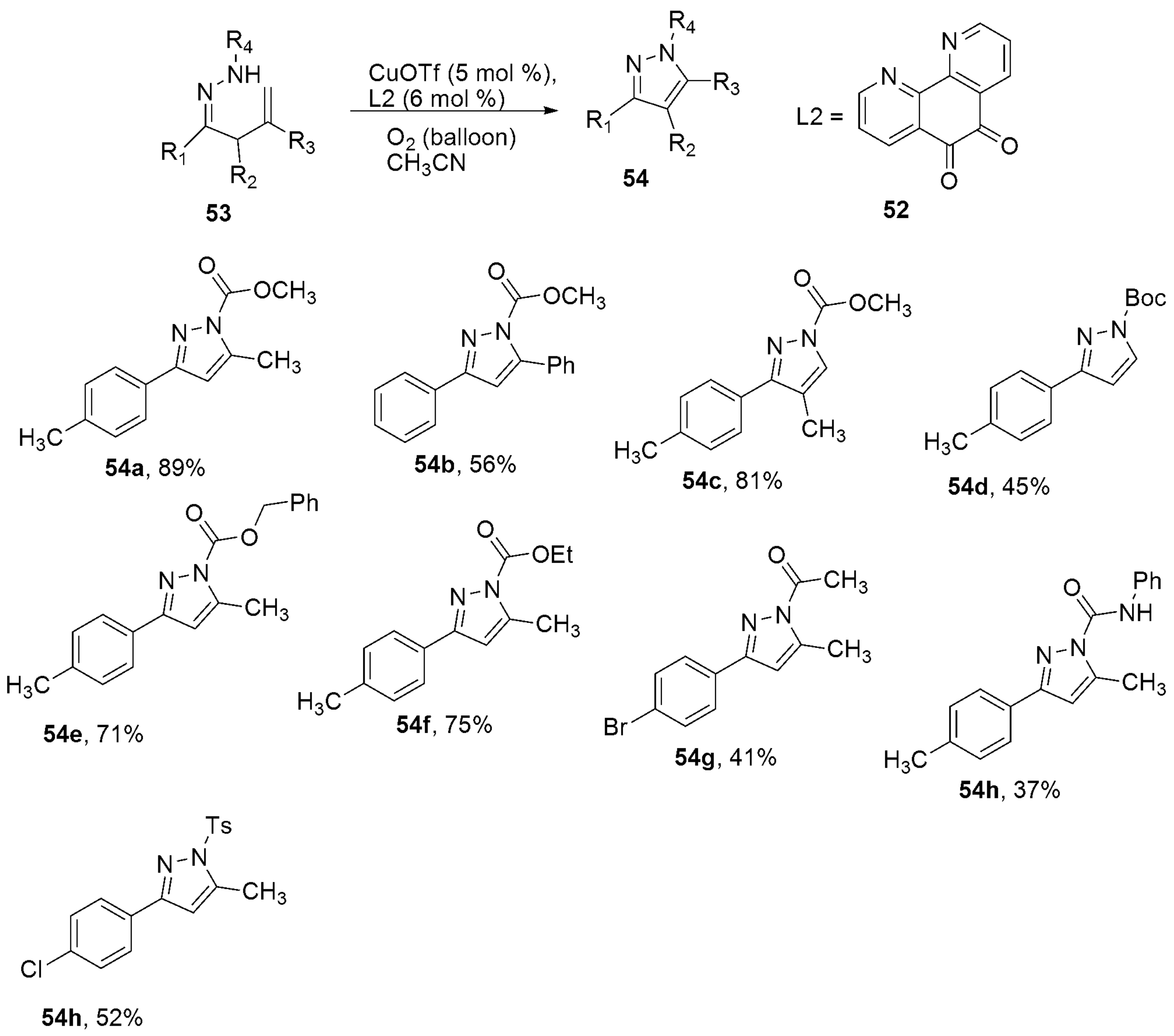
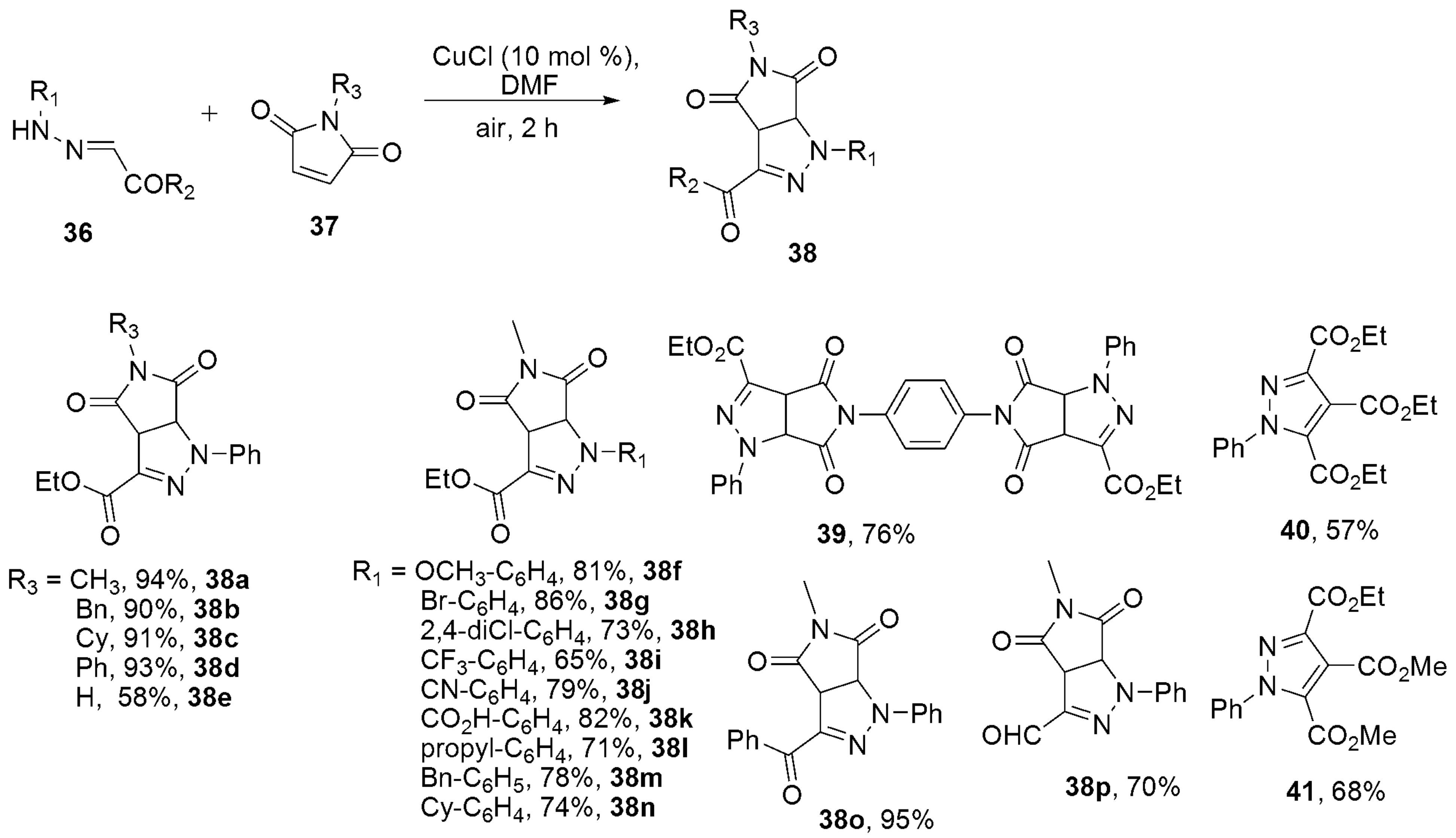
2.2.3. Pyrazoles from Hydrazones
2.2.4. Pyrazoles from Diazo Intermediates and Alkynes
2.2.5. Pyrazoles from Vinyl Sulfone
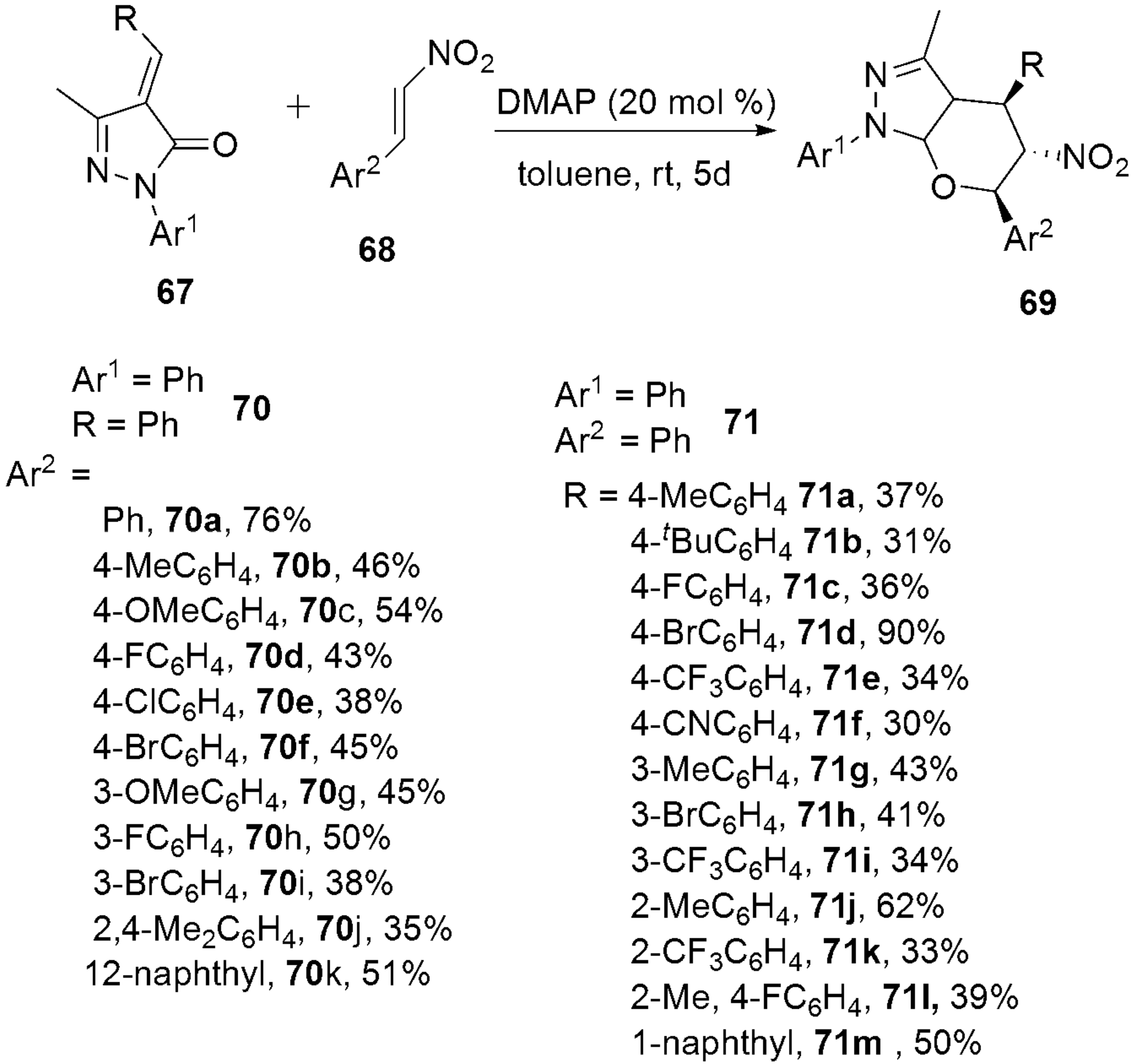
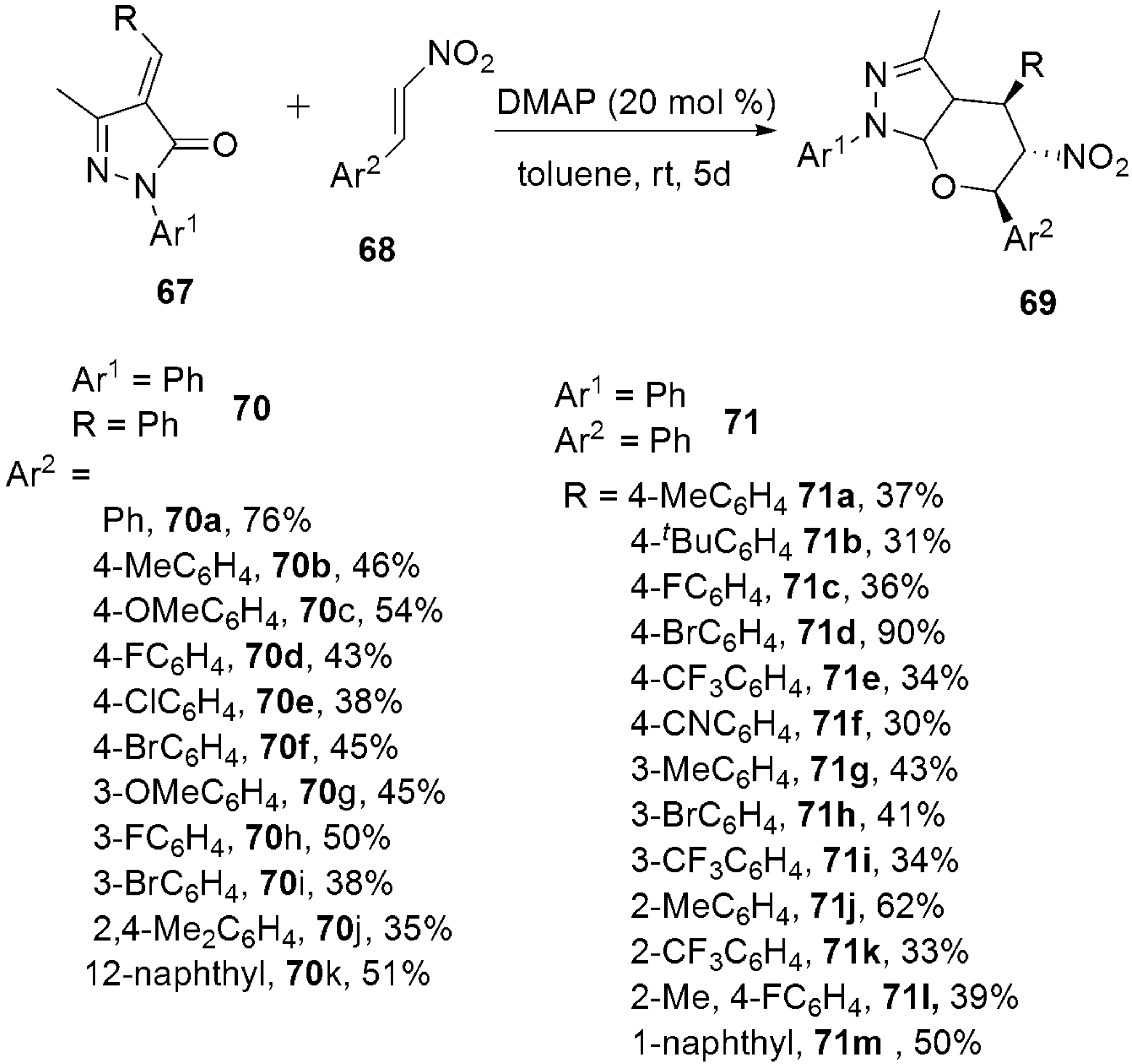
2.2.6. Pyrazoles from Nitro-Olefins
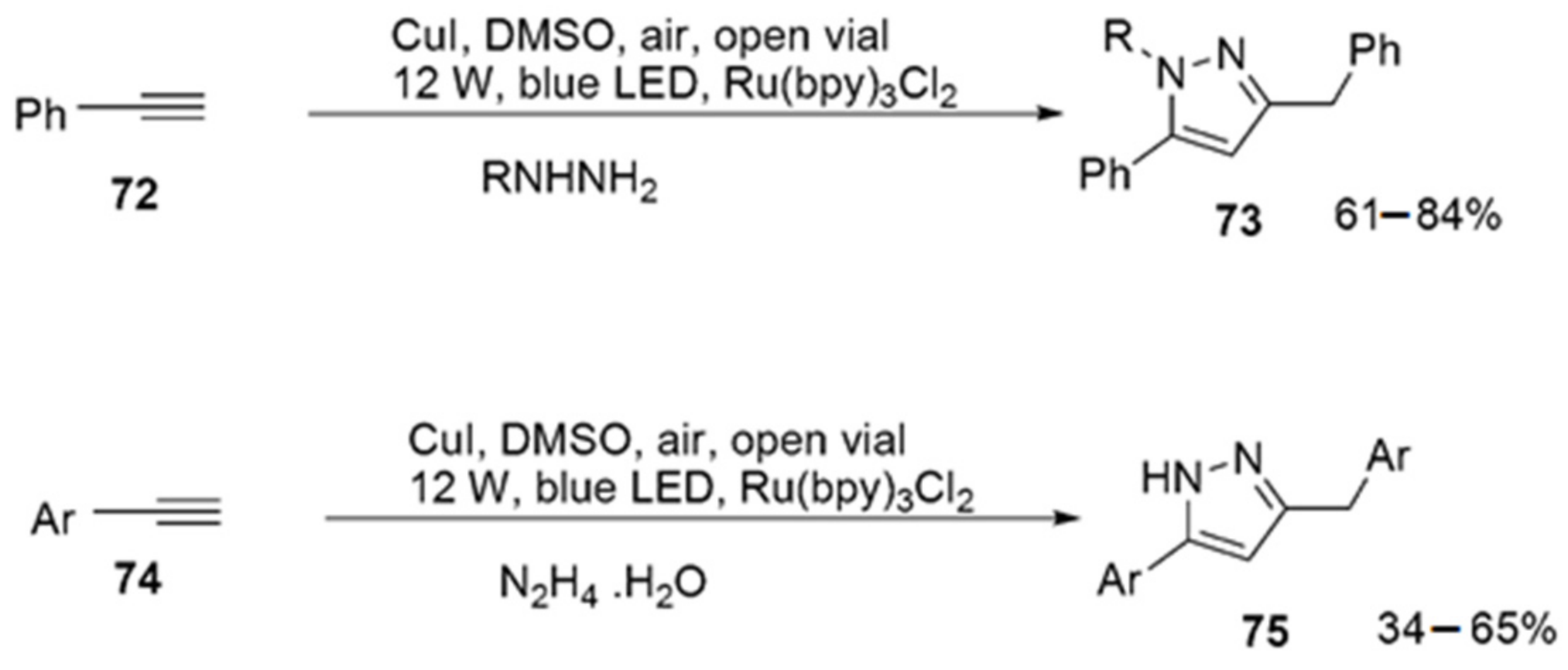
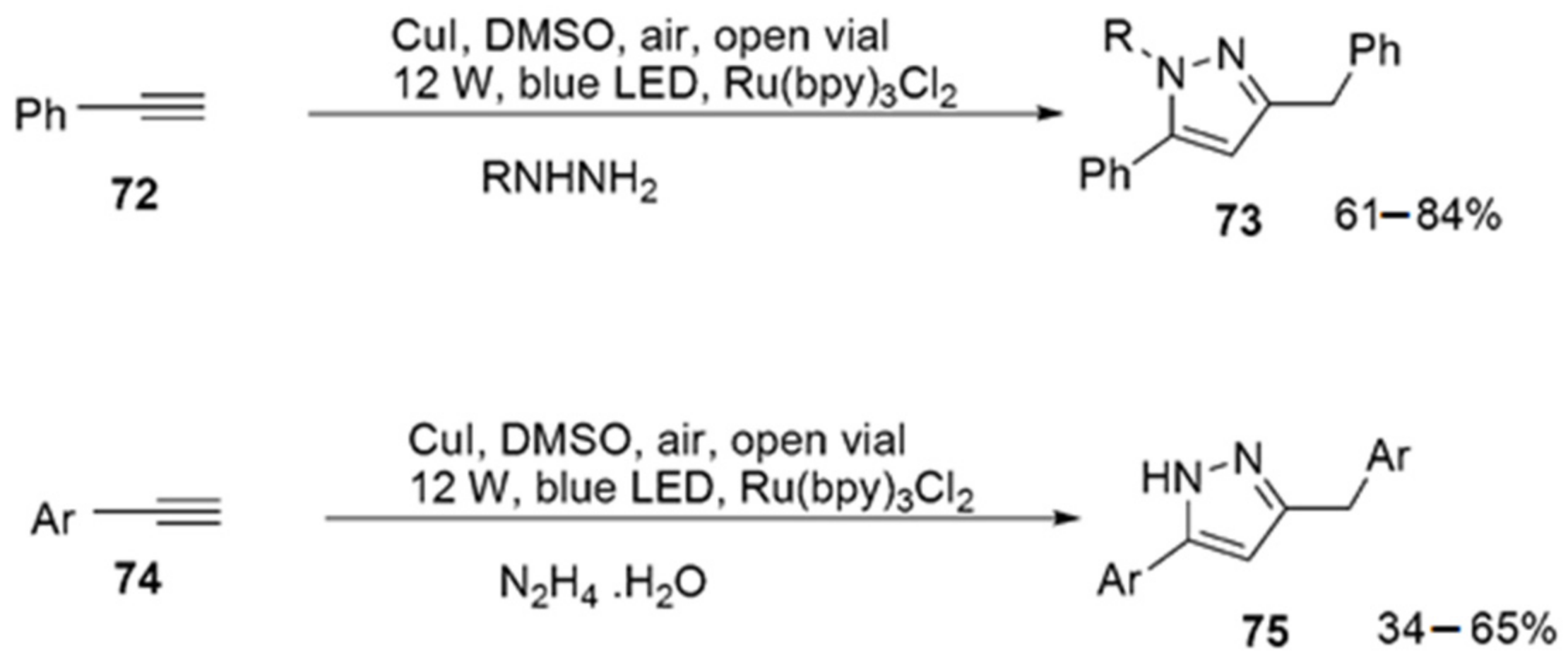
2.2.7. Pyrazoles from Alkynes
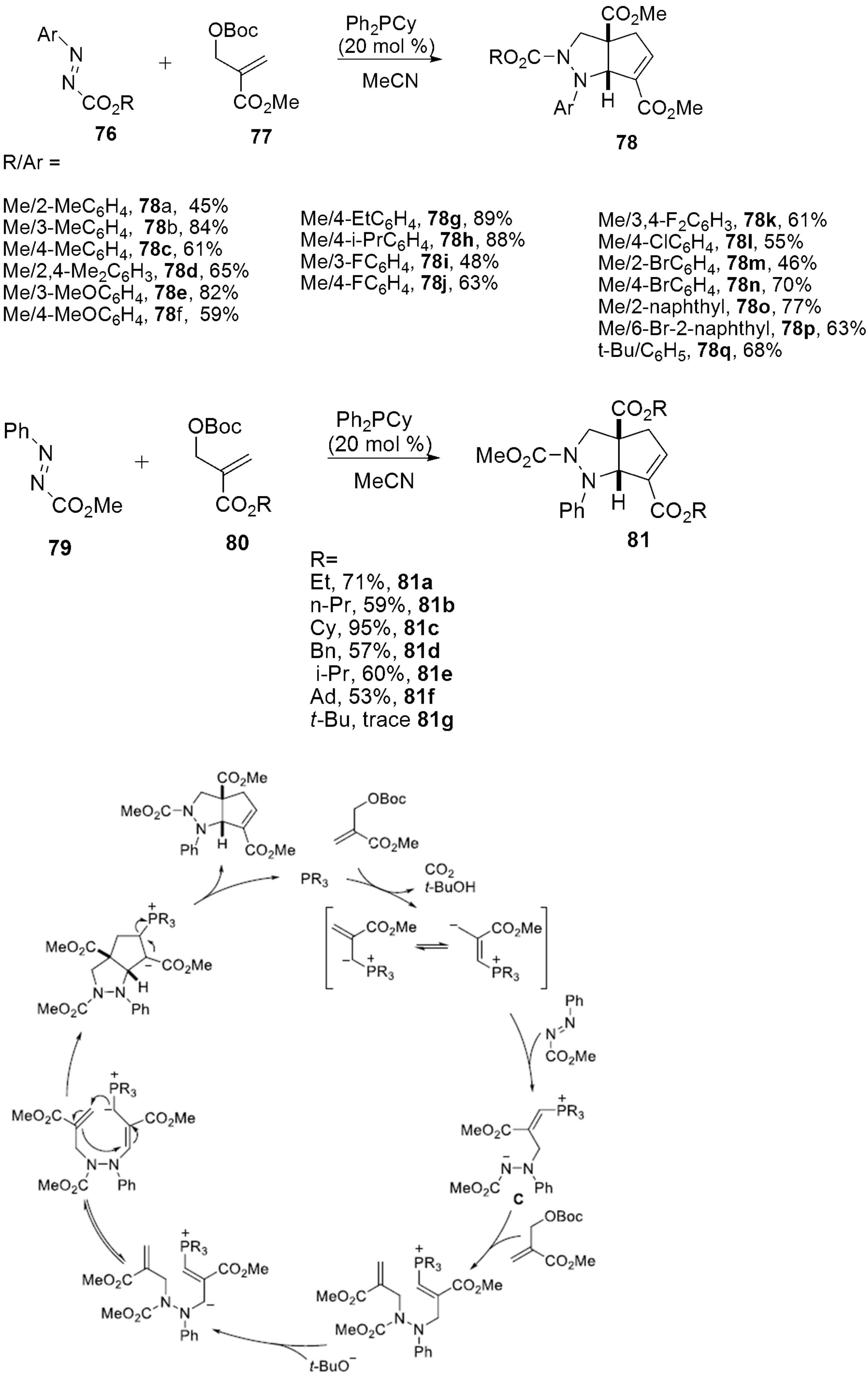
2.2.8. Pyrazoles from Morita−Baylis−Hillman (MBH) Carbonates
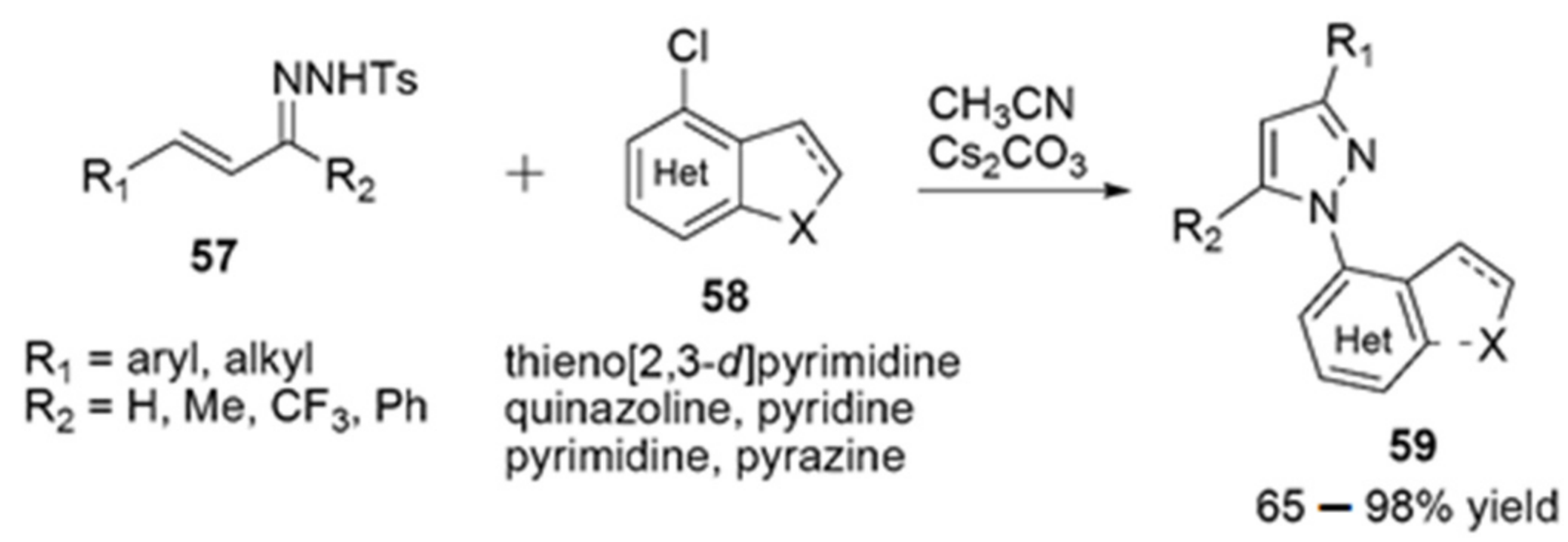
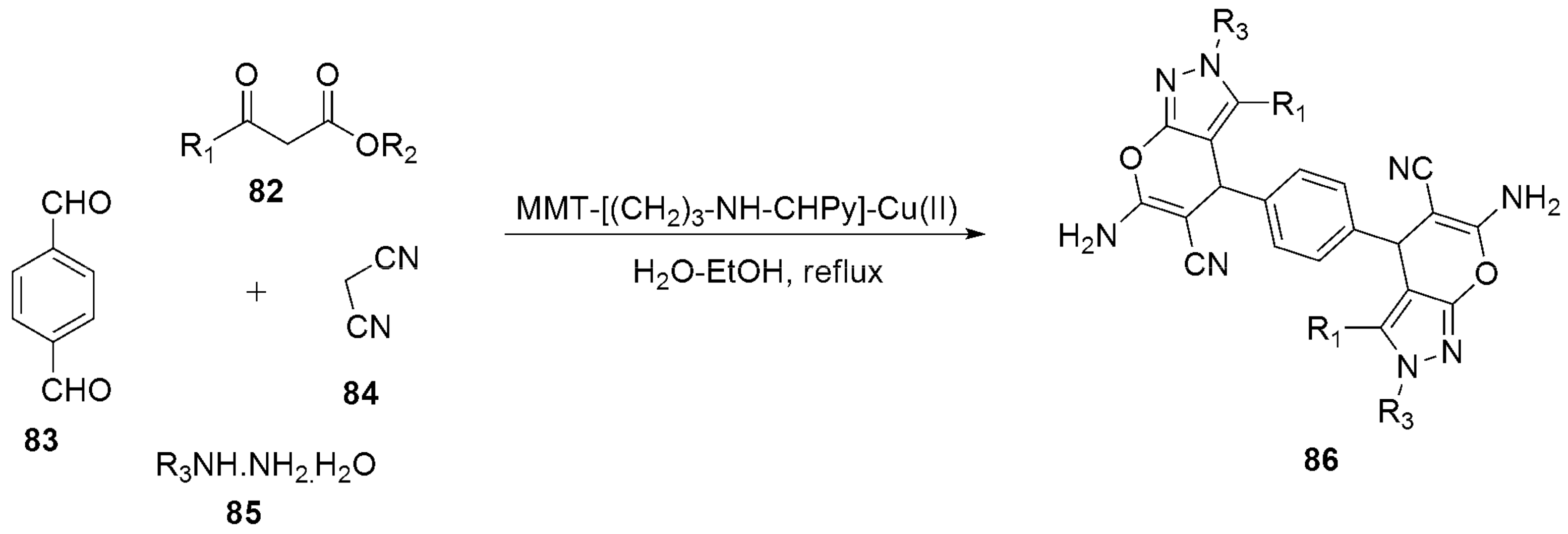
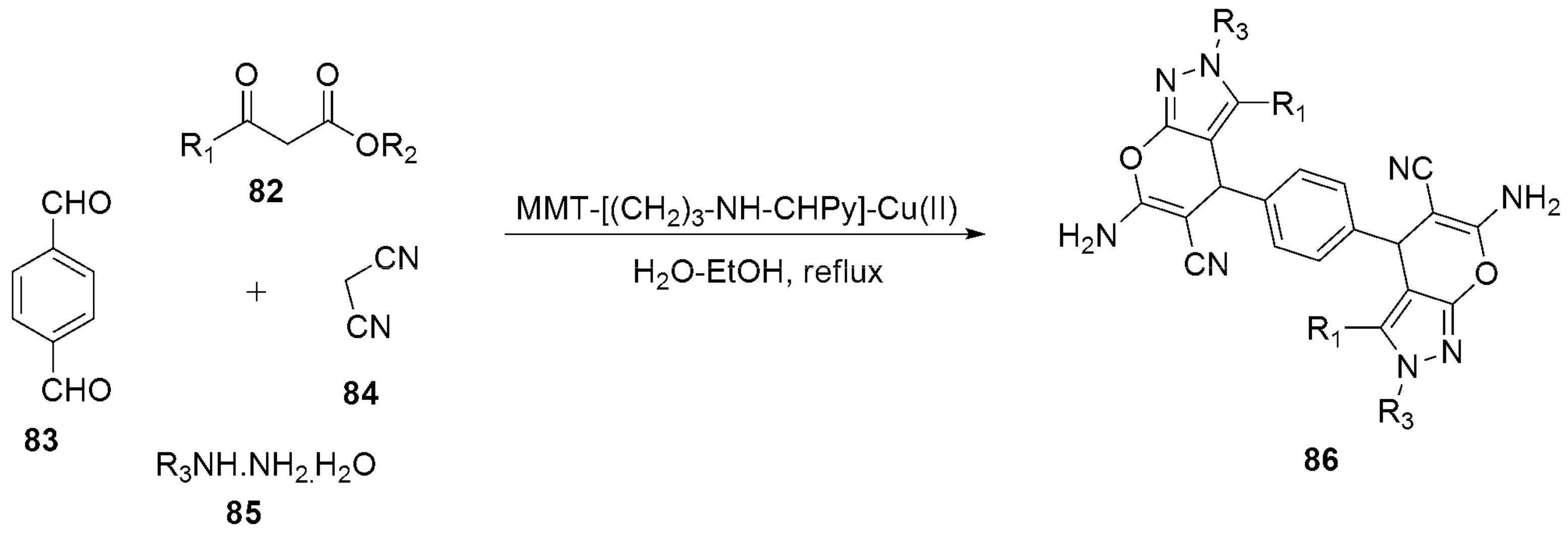
Multicomponent Strategies
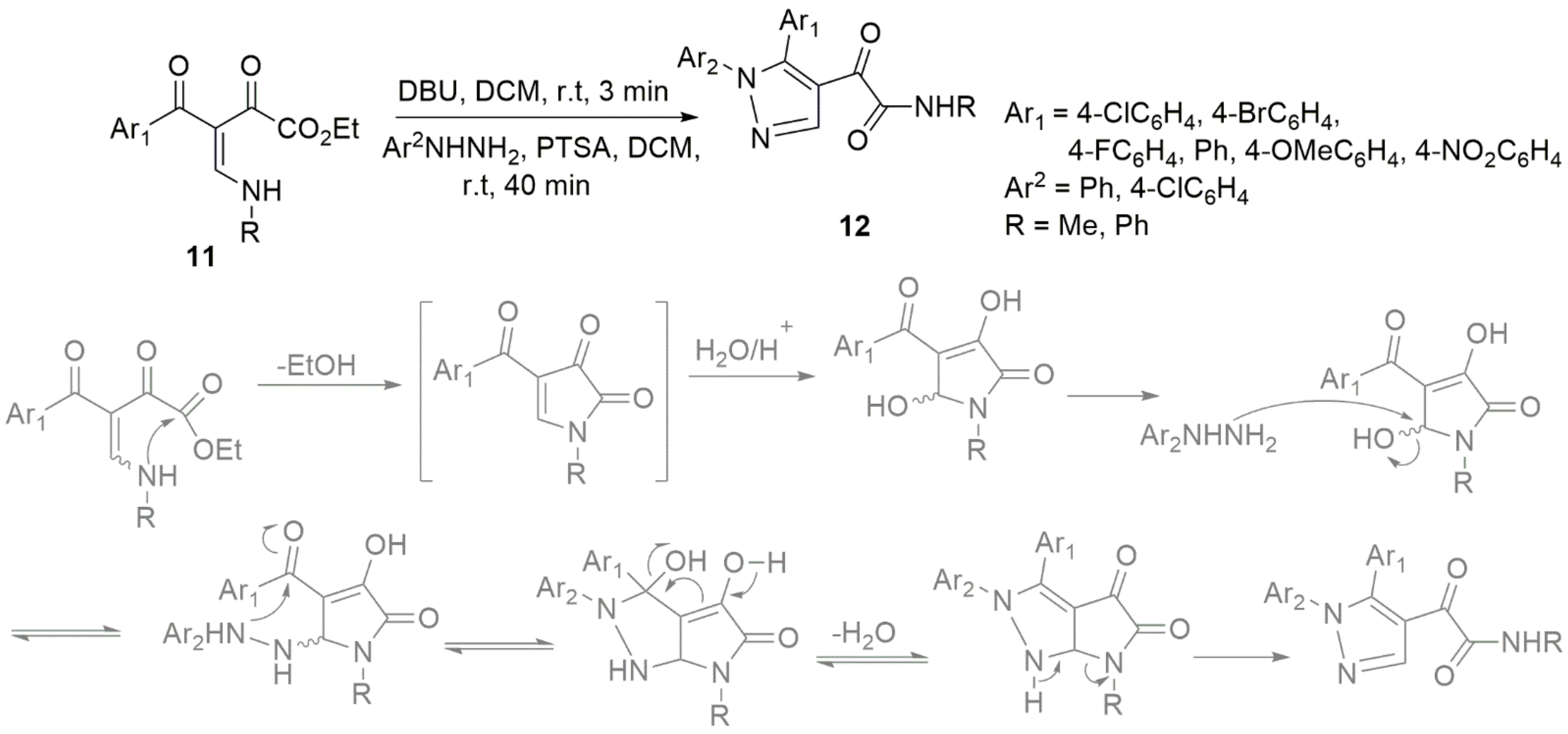
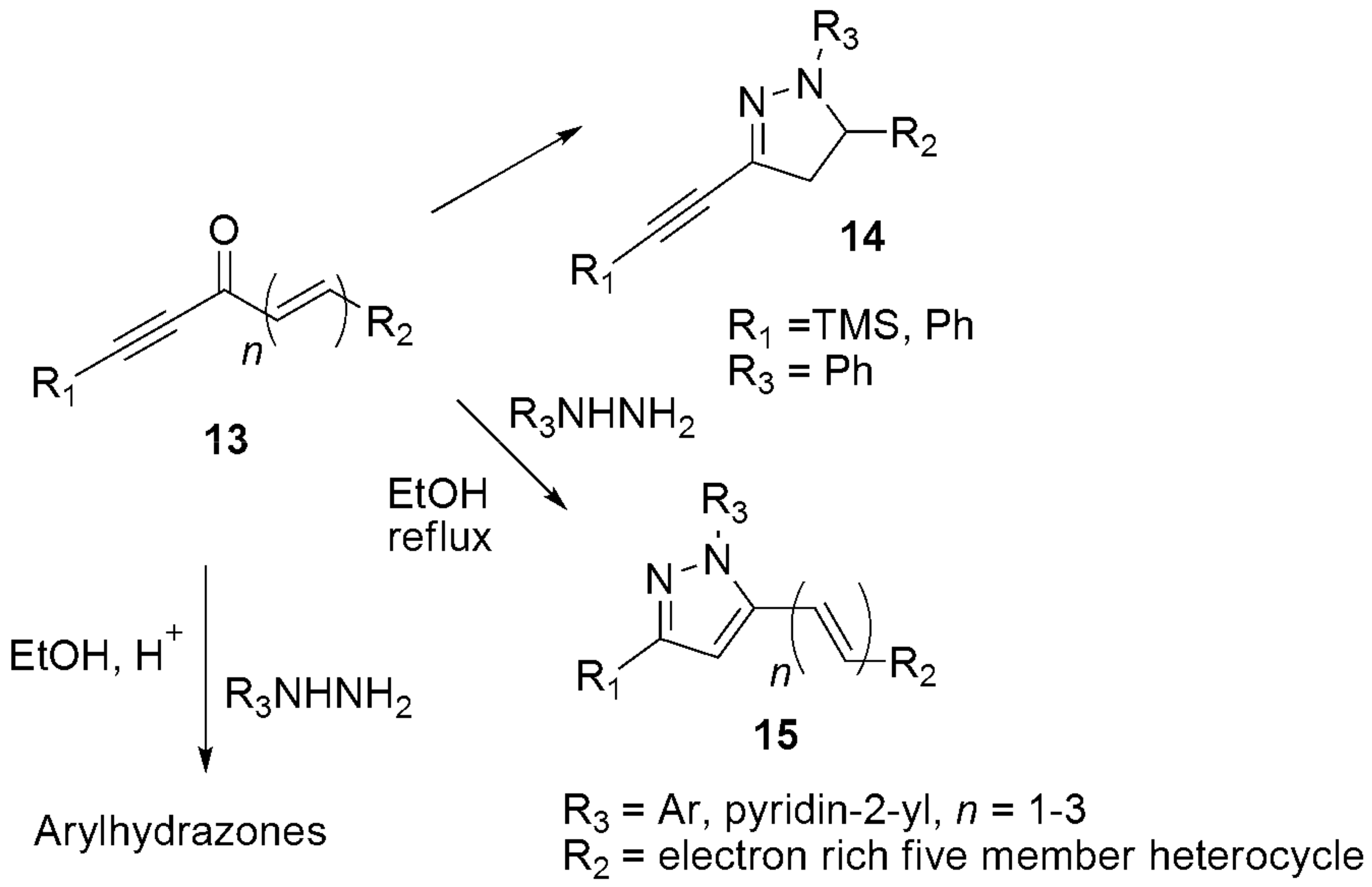
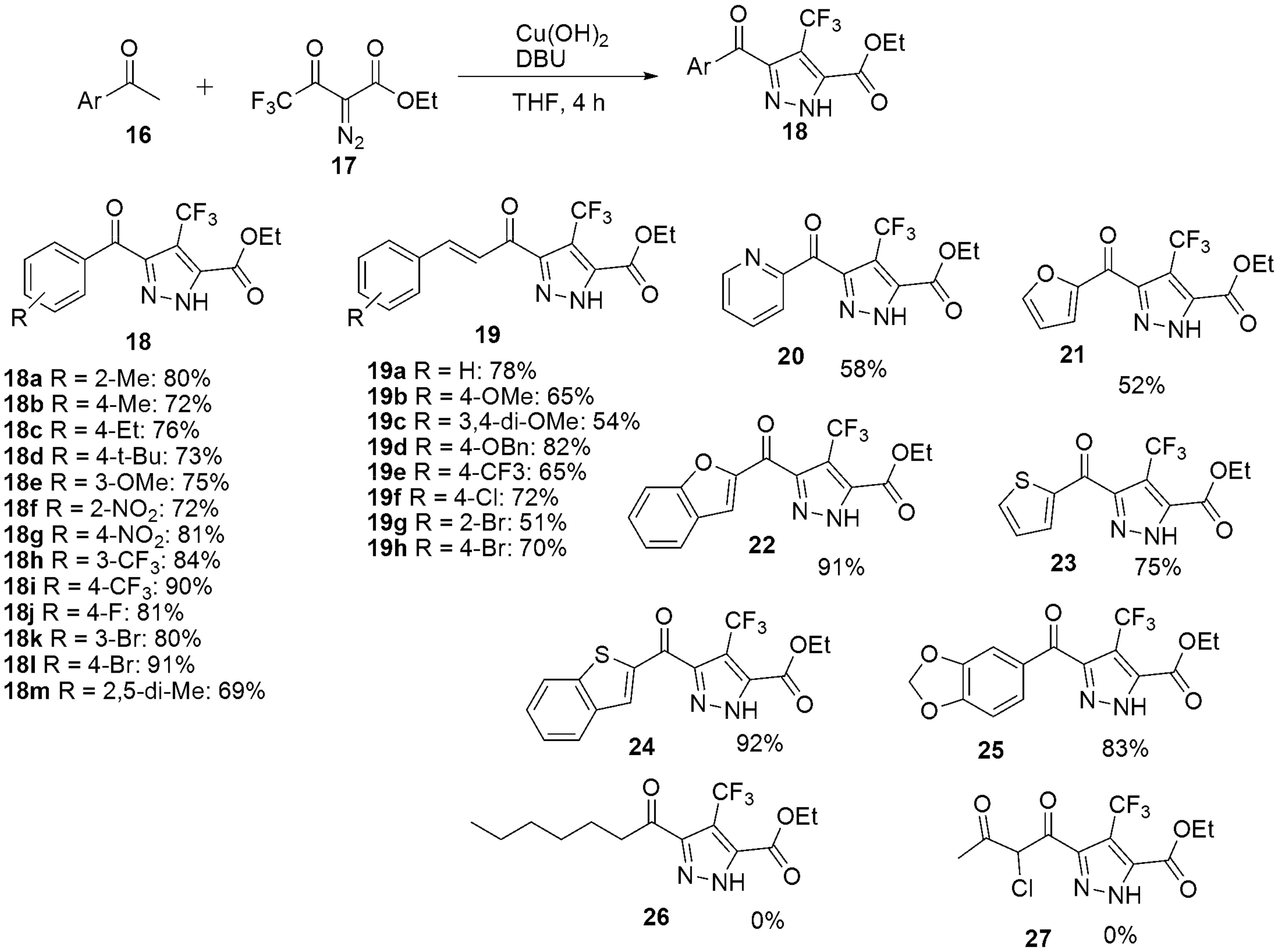
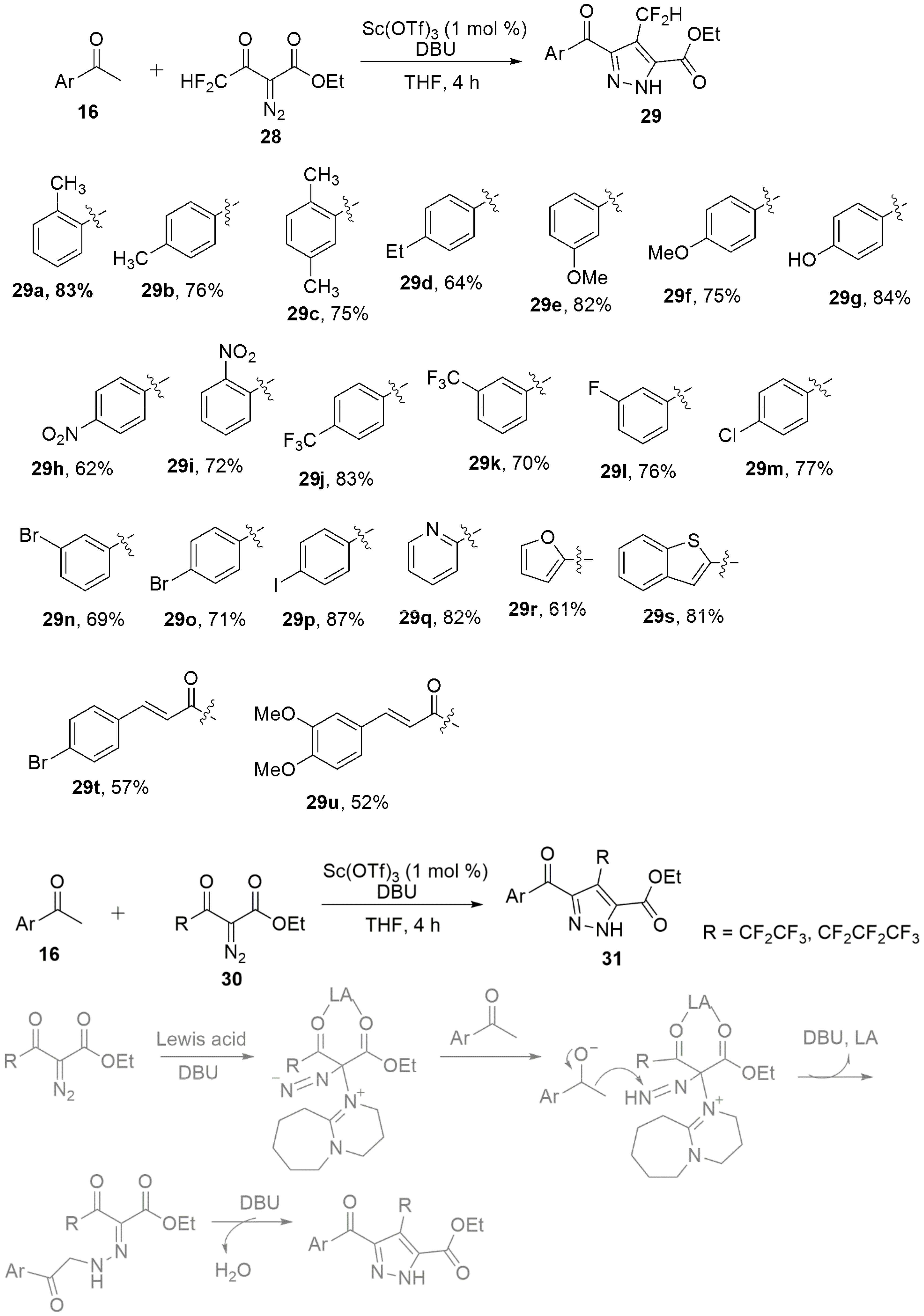
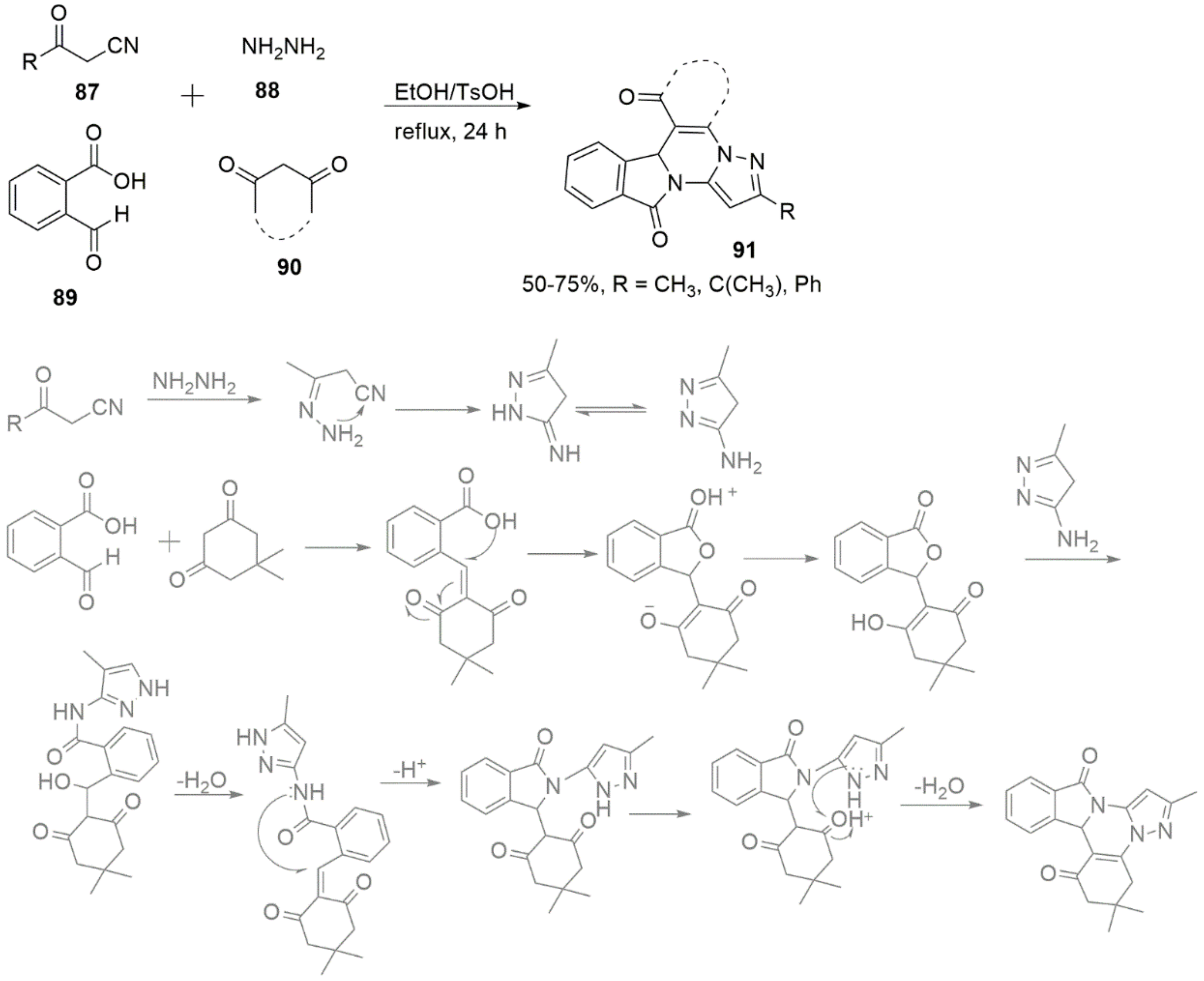
3. Miscellaneous
4. Biological Activity of Pyrazole Derivatives
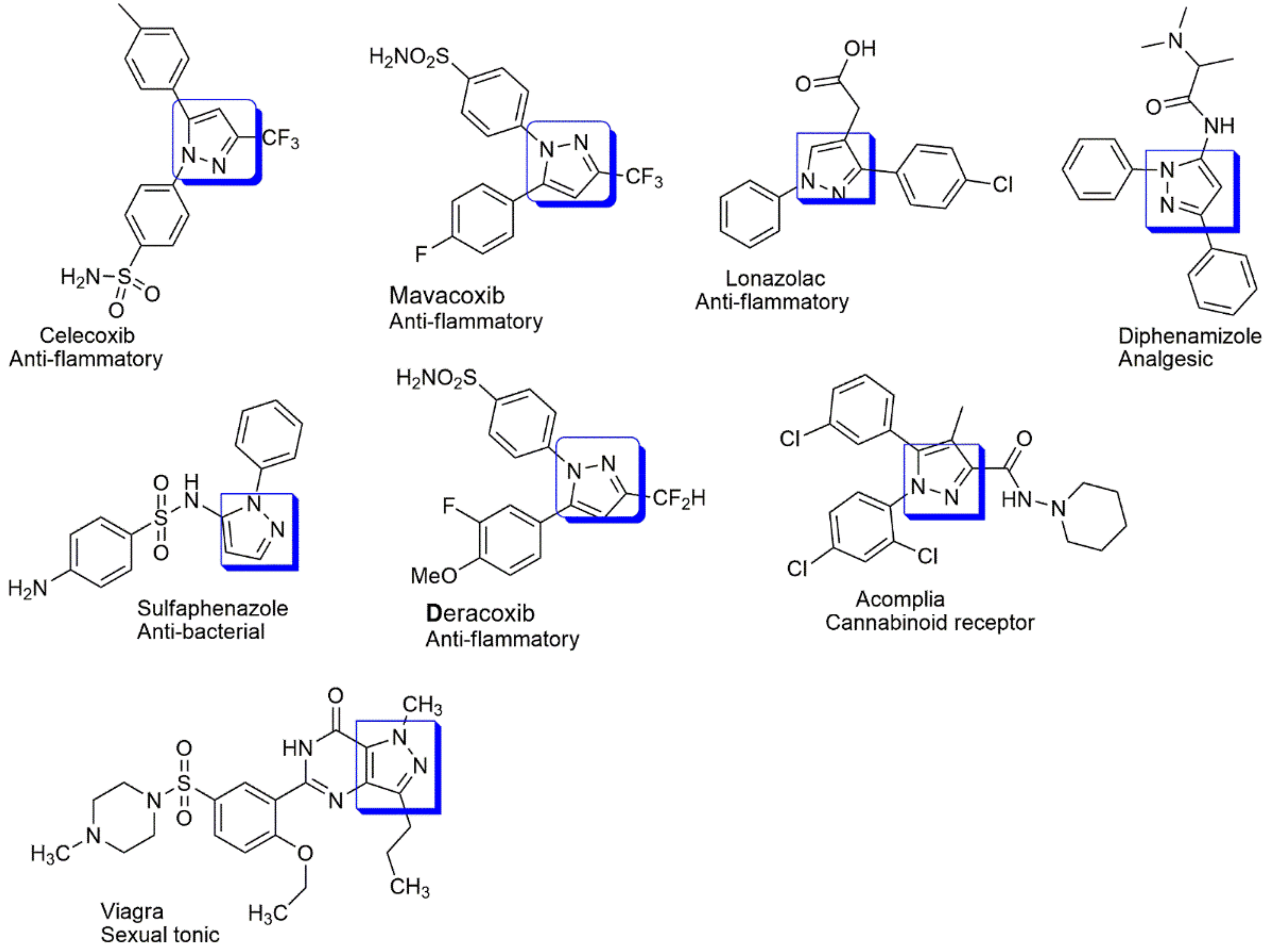
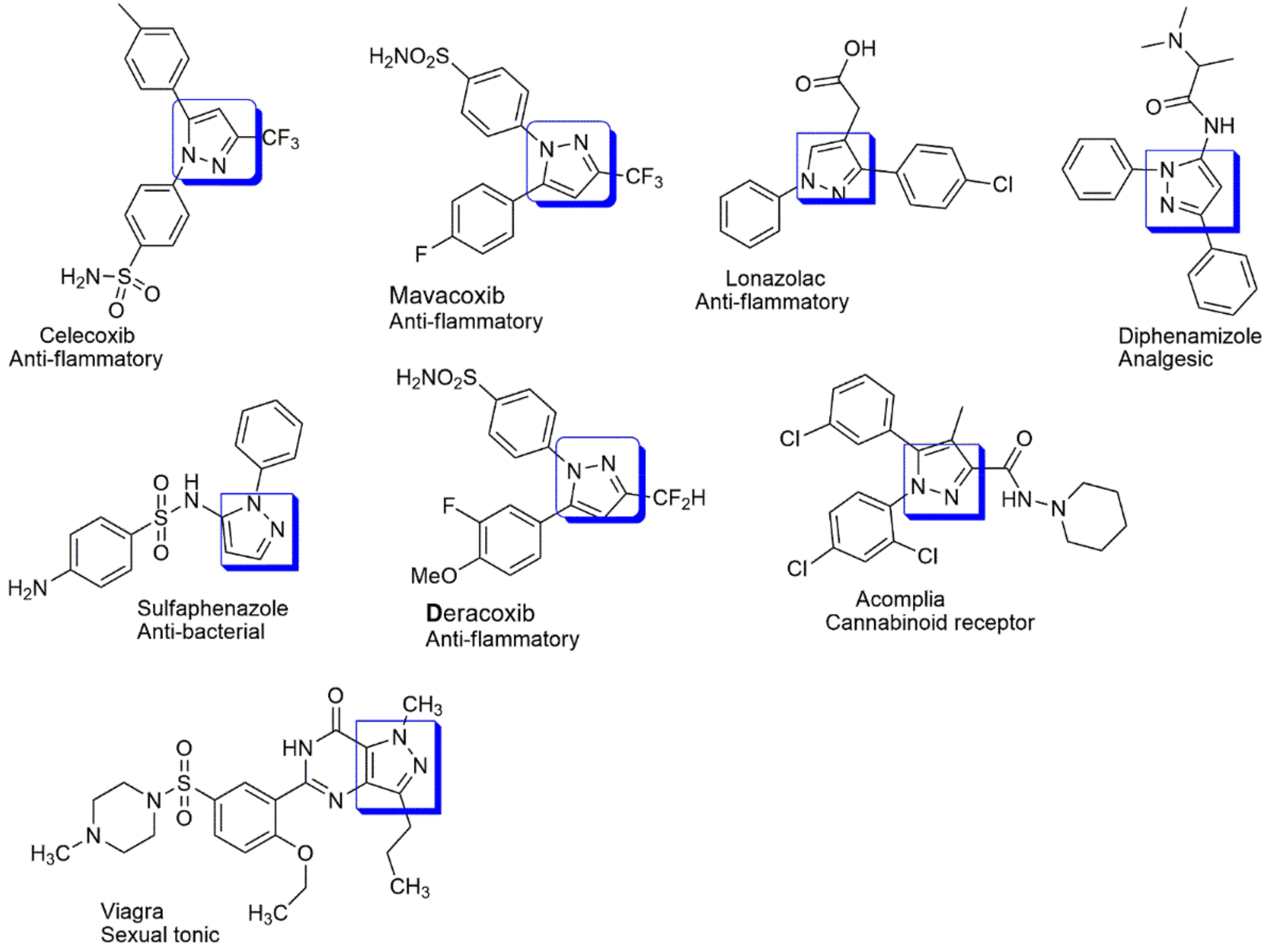
4.1. Anti-Inflammatory
4.2. Anticancer
4.3. Antibacterial
4.4. Antifungal
4.5. Antidiabetics
4.6. Antileishmanial
4.7. Antimalarial
4.8. Antioxidant
4.9. Antituberculosis
4.10. Agrochemical
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ansari, A.; Ali, A.; Asif, M. Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Alam, R.; Wahi, D.; Singh, R.; Sinha, D.; Tandon, V.; Grover, A.; Rahisuddin. Design, synthesis, cytotoxicity, HuTopoIIα inhibitory activity and molecular docking studies of pyrazole derivatives as potential anticancer agents. Bioorg. Chem. 2016, 69, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Gregory, W.A.; Brittelli, D.R.; Wang, C.; Wuonola, M.A.; McRipley, R.J.; Eustice, D.C.; Eberly, V.S.; Slee, A.M.; Forbes, M.; Bartholomew, P. Antibacterials. Synthesis and structure-activity studies of 3-aryl-2-oxooxazolidines. 1. The B group. J. Med. Chem. 1989, 32, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.B.; Sodero, A.C.; Cardoso, M.F.; Lima, E.S.; Kaiser, C.R.; Silva, F.P., Jr.; Ferreira, V.F. Synthesis, biological activity, and molecular modeling studies of 1 h-1, 2, 3-triazole derivatives of carbohydrates as α-glucosidases inhibitors. J. Med. Chem. 2010, 53, 2364–2375. [Google Scholar] [CrossRef] [PubMed]
- Ramtohul, Y.K.; Black, C.; Chan, C.-C.; Crane, S.; Guay, J.; Guiral, S.; Huang, Z.; Oballa, R.; Xu, L.-J.; Zhang, L. SAR and optimization of thiazole analogs as potent stearoyl-CoA desaturase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1593–1597. [Google Scholar] [CrossRef] [PubMed]
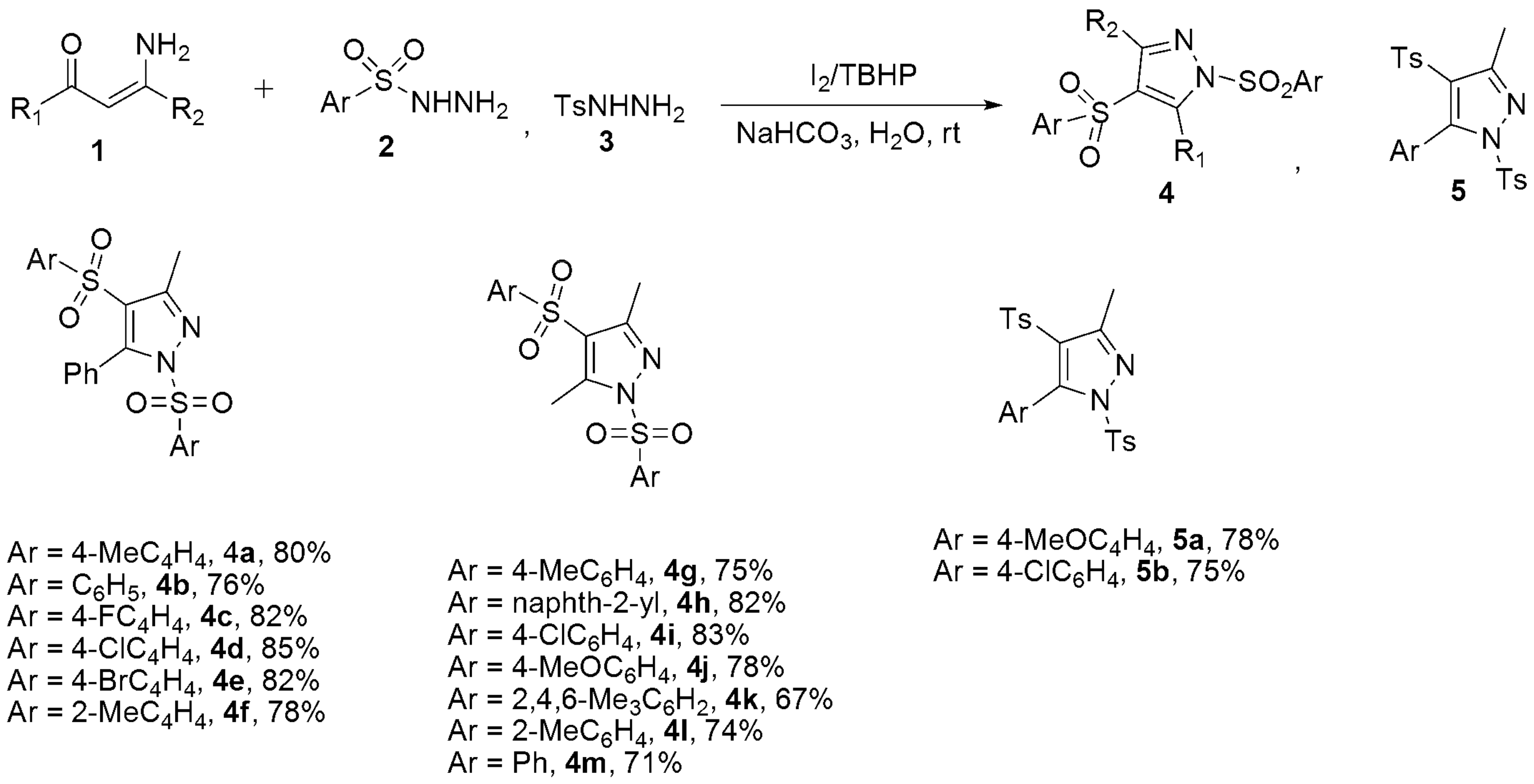
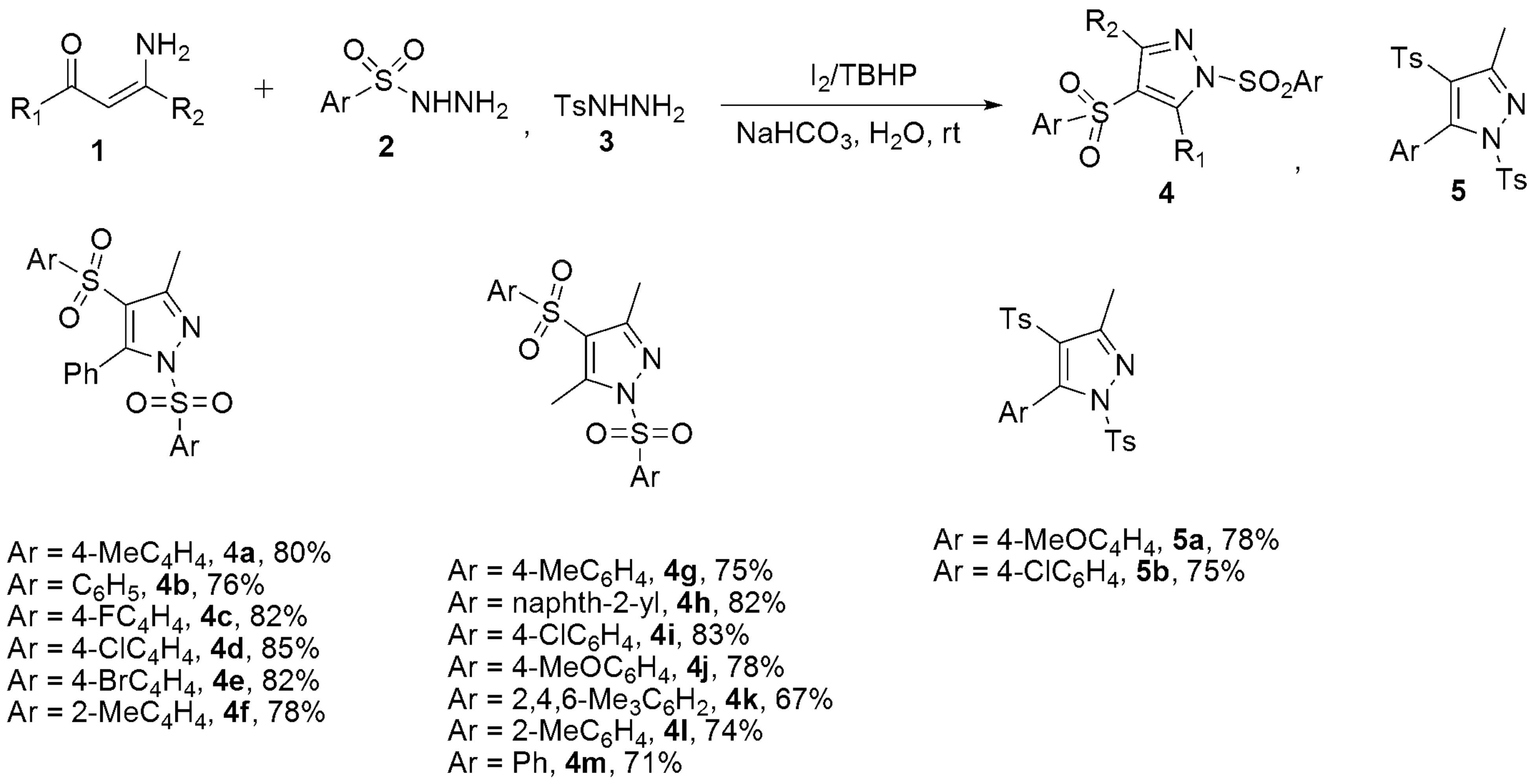
- Guo, Y.H.; Wang, G.D.; Wei, L.; Wan, J.P. Domino C-H Sulfonylation and Pyrazole Annulation for Fully Substituted Pyrazole Synthesis in Water Using Hydrophilic Enaminones. J. Org. Chem. 2019, 84, 2984–2990. [Google Scholar] [CrossRef] [PubMed]
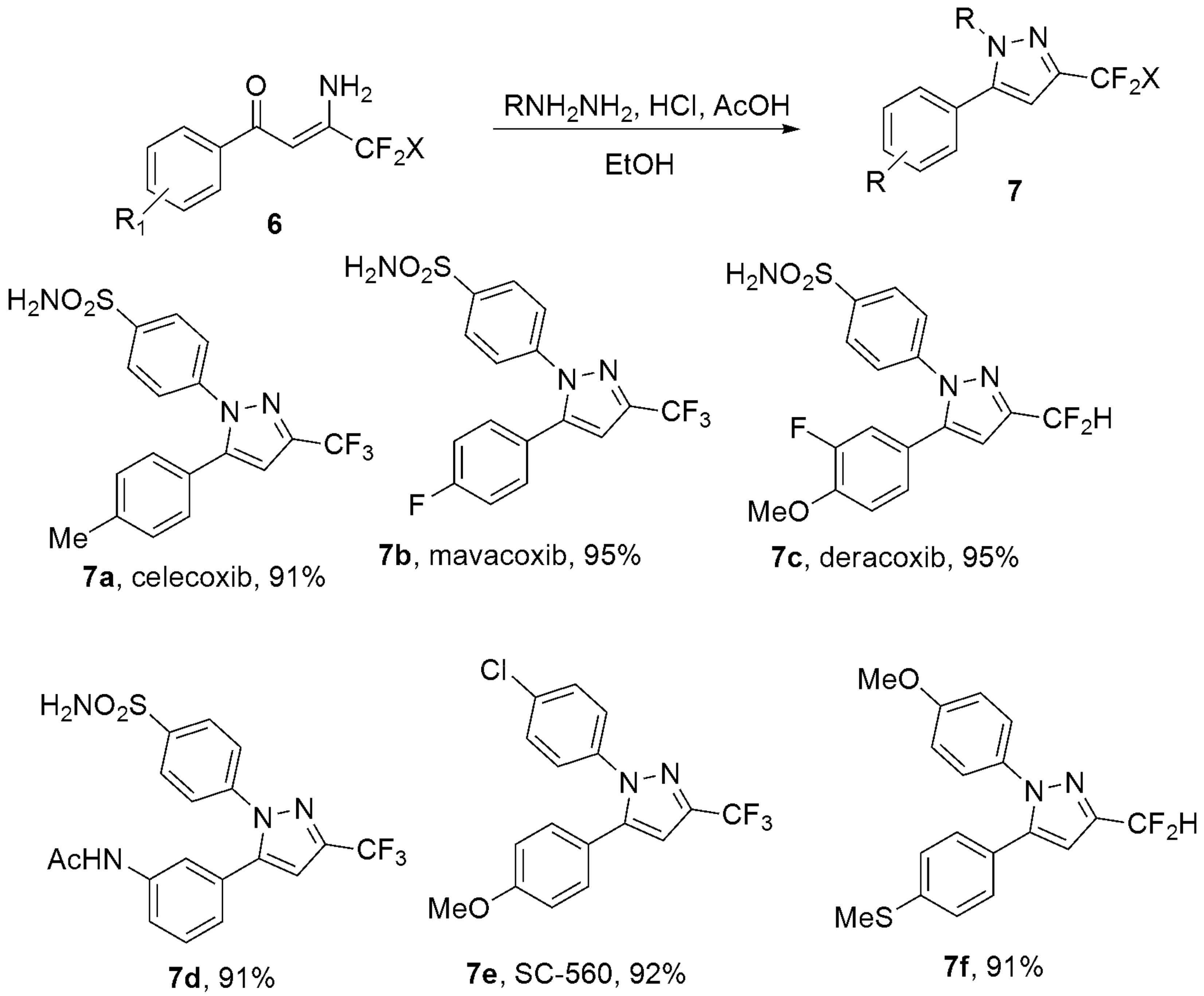
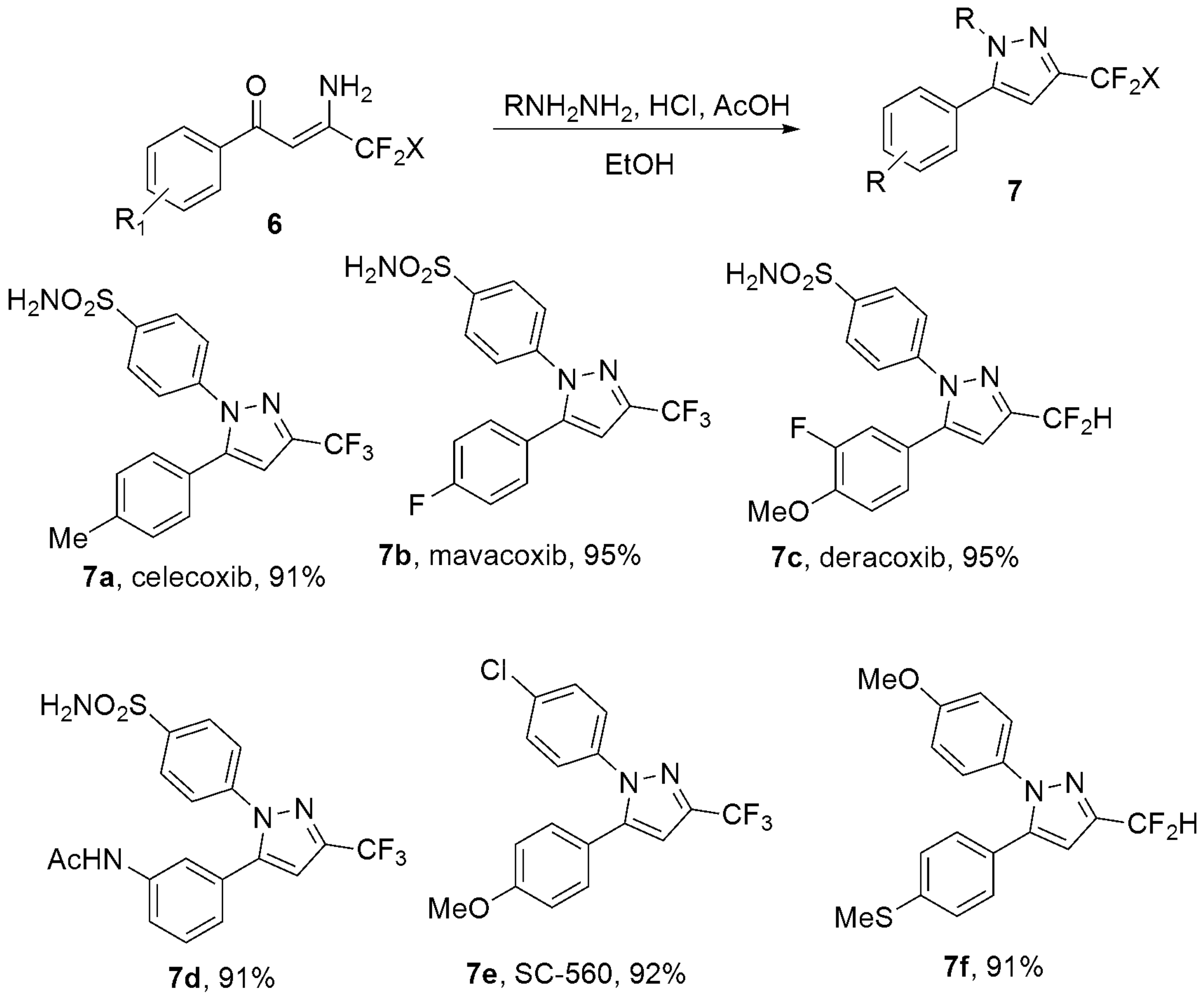
- Wan, C.; Pang, J.Y.; Jiang, W.; Zhang, X.W.; Hu, X.G. Copper-Catalyzed Reductive Ring-Cleavage of Isoxazoles: Synthesis of Fluoroalkylated Enaminones and Application for the Preparation of Celecoxib, Deracoxib, and Mavacoxib. J. Org. Chem. 2021, 86, 4557–4566. [Google Scholar] [CrossRef]
- Zhu, C.; Zeng, H.; Liu, C.; Cai, Y.; Fang, X.; Jiang, H. Regioselective Synthesis of 3-Trifluoromethylpyrazole by Coupling of Aldehydes, Sulfonyl Hydrazides, and 2-Bromo-3,3,3-trifluoropropene. Org. Lett. 2020, 22, 809–813. [Google Scholar] [CrossRef]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef]
- Muzalevskiy, V.M.; Rulev, A.Y.; Romanov, A.R.; Kondrashov, E.V.; Ushakov, I.A.; Chertkov, V.A.; Nenajdenko, V.G. Selective, Metal-Free Approach to 3- or 5-CF(3)-Pyrazoles: Solvent Switchable Reaction of CF(3)-Ynones with Hydrazines. J. Org. Chem. 2017, 82, 7200–7214. [Google Scholar] [CrossRef]
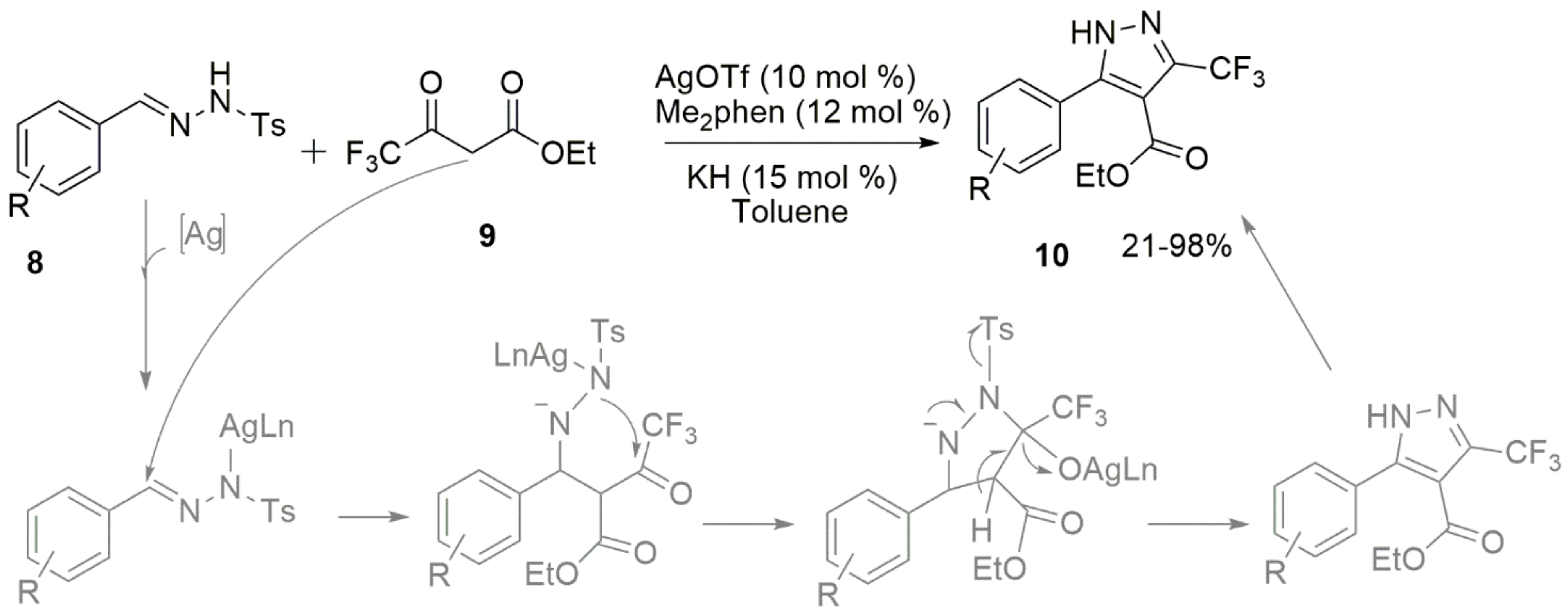
- Xu, Y.H.; Chen, Q.L.; Tian, Y.S.; Wu, W.; You, Y.; Weng, Z.Q. Silver-catalyzed synthesis of 5-aryl-3-trifluoromethyl pyrazoles. Tetrahedron Lett. 2020, 61, 151455. [Google Scholar] [CrossRef]
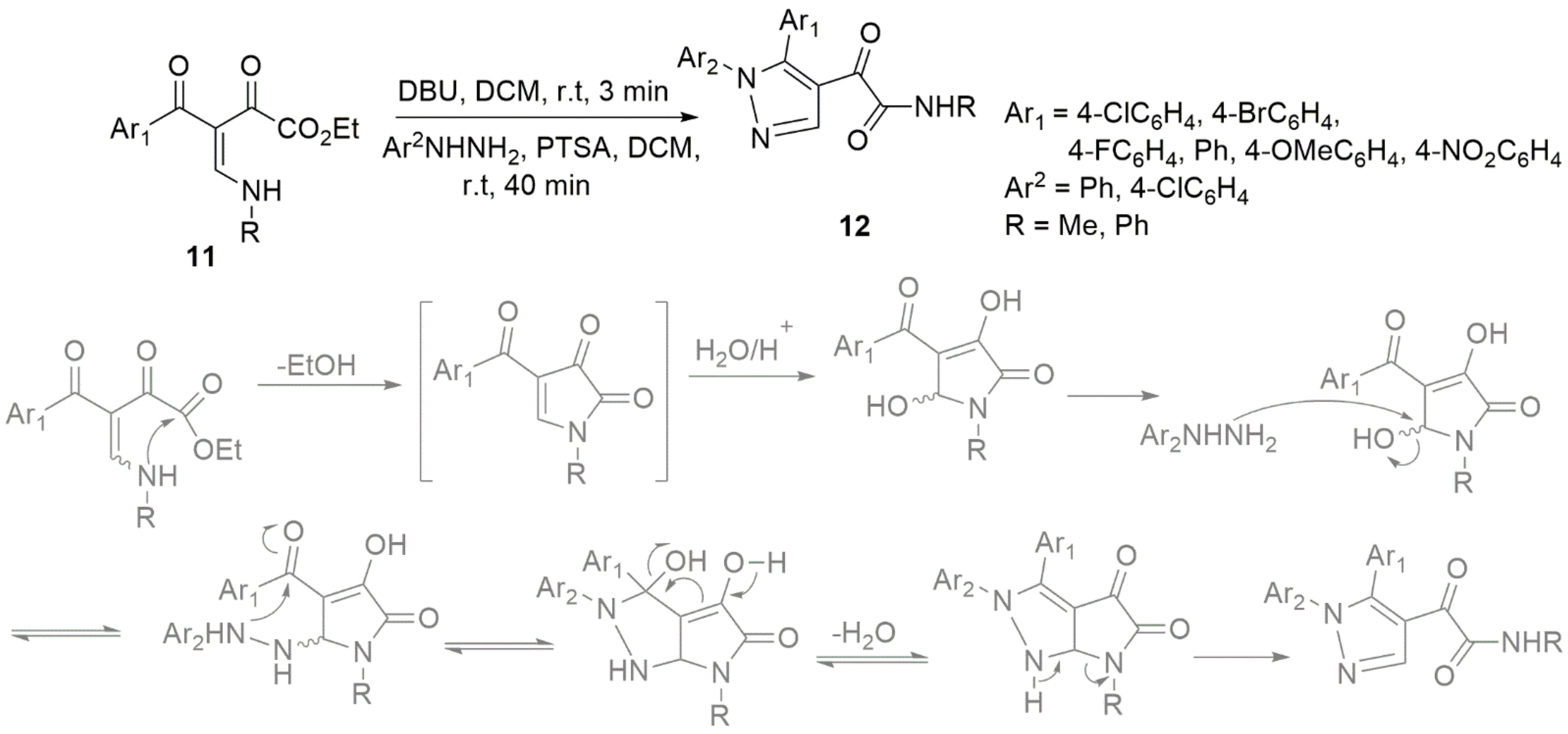
- Poletto, J.; Ribeiro, G.M.; Da Silva, M.J.V.; Jacomini, A.P.; Basso, E.A.; Back, D.F.; Moura, S.; Rosa, F.A. One-Pot Highly Regioselective Synthesis of alpha-Ketoamide N-Arylpyrazoles from Secondary beta-Enamino Diketones. Org. Lett. 2019, 21, 6325–6328. [Google Scholar] [CrossRef] [PubMed]
- Golovanov, A.A.; Odin, I.S.; Gusev, D.M.; Vologzhanina, A.V.; Sosnin, I.M.; Grabovskiy, S.A. Reactivity of Cross-Conjugated Enynones in Cyclocondensations with Hydrazines: Synthesis of Pyrazoles and Pyrazolines. J. Org. Chem. 2021, 86, 7229–7241. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Yin, H.; Lin, L.; Wen, S.; Xie, L.; Huang, Y.; Weng, Z. Collaborative Activation of Trifluoroacetyl Diazoester by a Lewis Acid and Base for the Synthesis of Polysubstituted 4-Trifluoromethylpyrazoles. J. Org. Chem. 2020, 85, 8714–8722. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Wen, S.L.; Cui, Y.B.; Lin, L.; Zhang, H.B.; Fang, Z.; You, Y.; Weng, Z.Q. A method for synthesis of polysubstituted 4-difluoromethyl and perfluoroalkyl pyrazoles. Tetrahedron 2021, 85, 132062. [Google Scholar] [CrossRef]
- Kardile, R.D.; Liu, R.S. Gold(I)-Catalyzed Reactions between 2-(1-Alkynyl)-2-alken-1-ones and Vinyldiazo Ketones for Divergent Synthesis of Nonsymmetric Heteroaryl-Substituted Triarylmethanes: N- versus C-Attack Paths. Org. Lett. 2020, 22, 8229–8233. [Google Scholar] [CrossRef]
- Zhu, J.N.; Wang, W.K.; Jin, Z.H.; Wang, Q.K.; Zhao, S.Y. Pyrrolo 3,4-c pyrazole Synthesis via Copper(I) Chloride-Catalyzed Oxidative Coupling of Hydrazones to Maleimides. Org. Lett. 2019, 21, 5046–5050. [Google Scholar] [CrossRef]
- Zhu, J.N.; Wang, W.K.; Zheng, J.; Lin, H.P.; Deng, Y.X.; Zhao, S.Y. Iodine-Catalyzed Regioselective Oxidative Cyclization of Aldehyde Hydrazones with Electron-Deficient Olefins for the Synthesis of Mefenpyr-Diethyl. J. Org. Chem. 2019, 84, 11032–11041. [Google Scholar] [CrossRef]
- Fan, Z.W.; Feng, J.H.; Hou, Y.C.; Rao, M.; Cheng, J.J. Copper-Catalyzed Aerobic Cyclization of ss,gamma-Unsaturated Hydrazones with Concomitant C=C Bond Cleavage. Org. Lett. 2020, 22, 7981–7985. [Google Scholar] [CrossRef]
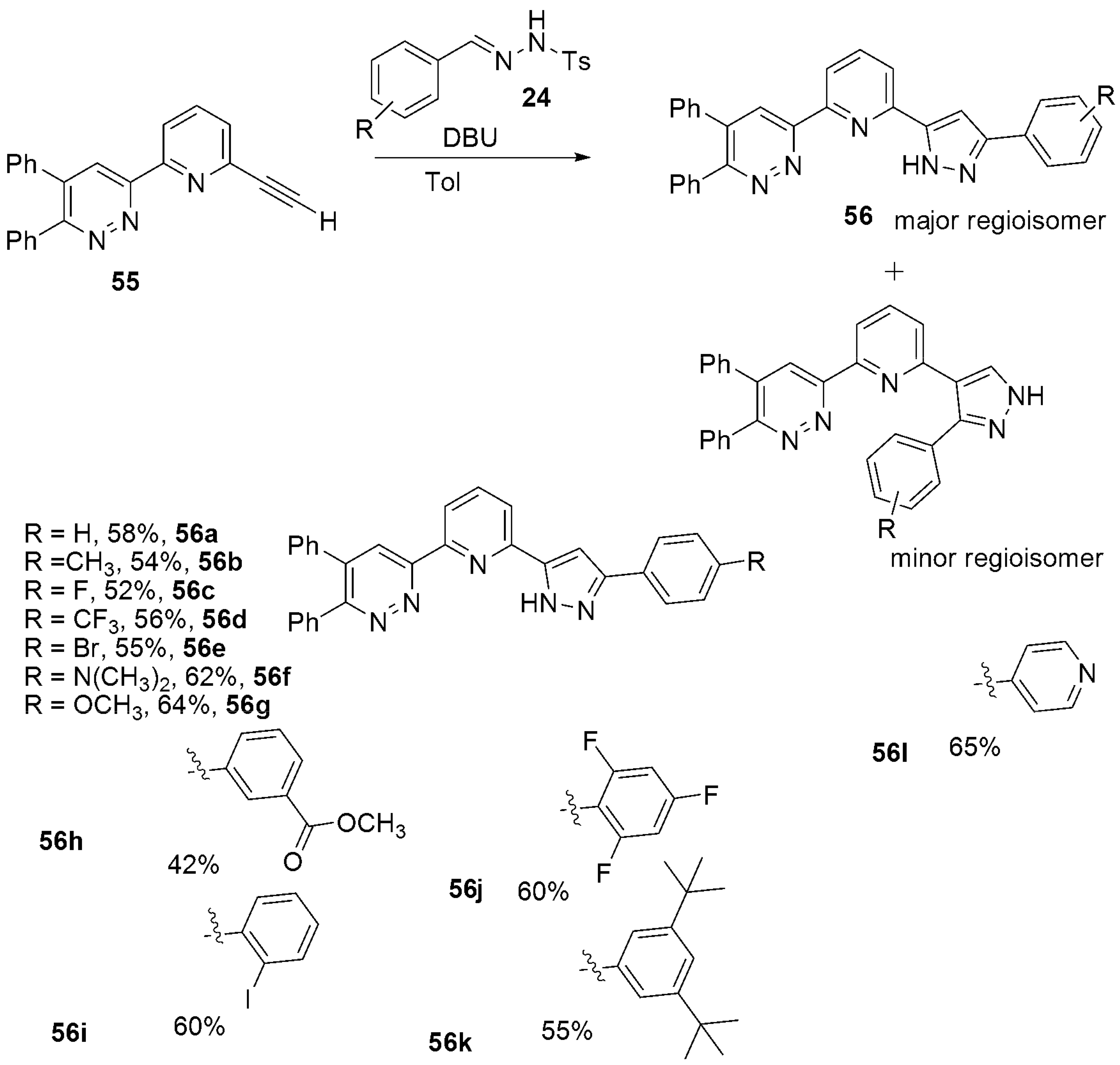
- Veerakanellore, G.B.; Smith, C.M.; Vasiliu, M.; Oliver, A.G.; Dixon, D.A.; Carrick, J.D. Synthesis of 1H-Pyrazol-5-yl-pyridin-2-yl-1,2,4 triazinyl Soft-Lewis Basic Complexants via Metal and Oxidant Free 3+2 Dipolar Cycloaddition of Terminal Ethynyl Pyridines with Tosylhydrazides. J. Org. Chem. 2019, 84, 14558–14570. [Google Scholar] [CrossRef]
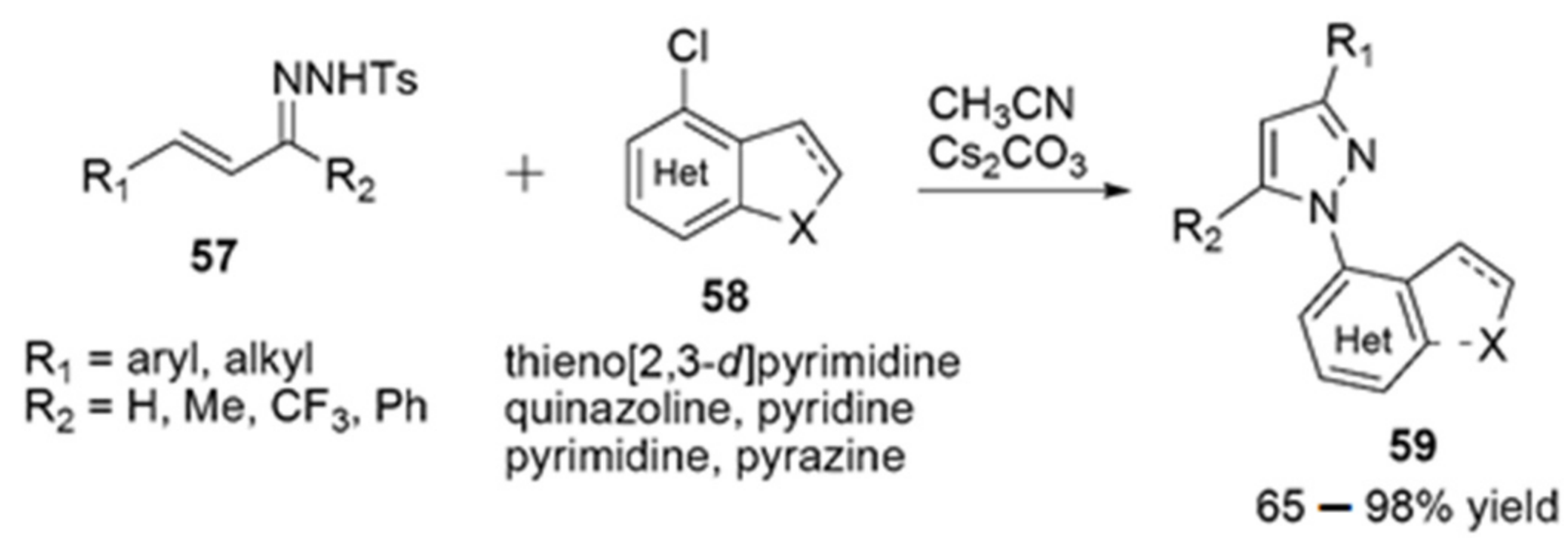
- Zheng, P.F.; Zeng, R.; Jiang, K.; Li, H.W.; Ye, Y.; Mu, C.; Shuai, L.; Quyang, Q.; Chen, Y.C. (3+1) Annulation/Rearrangement Cascade of C,N-Cyclic Azomethine Imines and 3-Chlorooxindoles: Construction of Hexahydroindeno 2,1-c pyrazole Spirooxindole Frameworks. Org. Lett. 2019, 21, 10052–10056. [Google Scholar] [CrossRef] [PubMed]
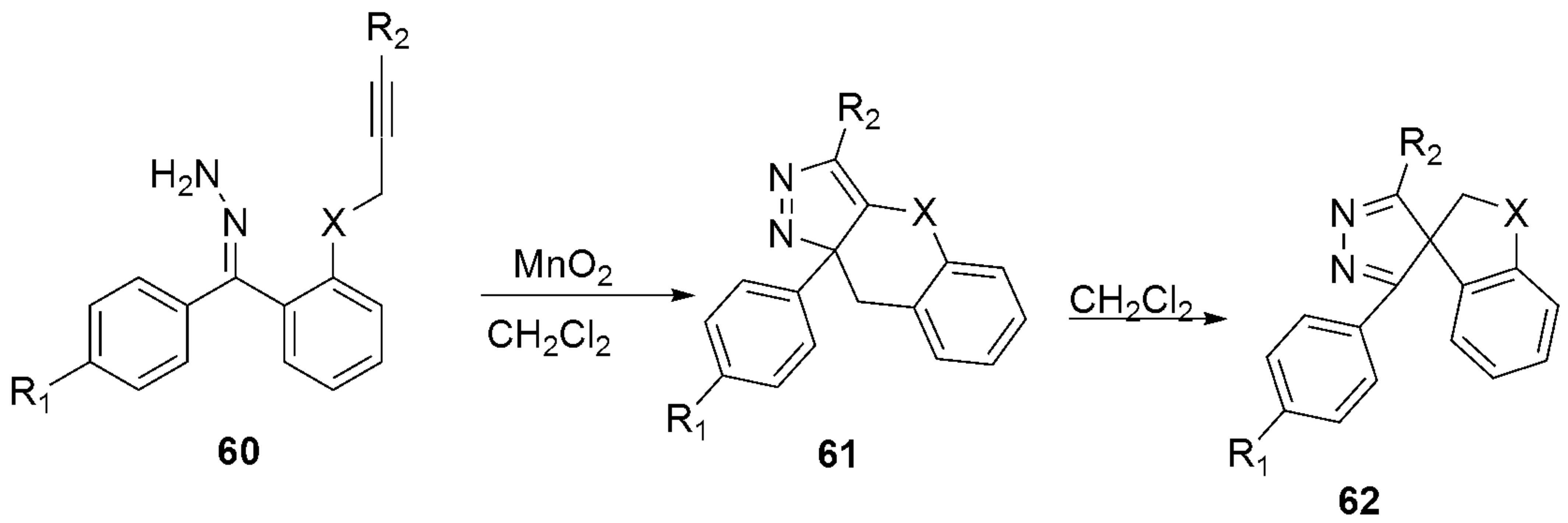
- Dimirjian, C.A.; Reis, M.C.; Balmond, E.I.; Turman, N.C.; Rodriguez, E.P.; Di Maso, M.J.; Fettinger, J.C.; Tantillo, D.J.; Shaw, J.T. Synthesis of Spirobicyclic Pyrazoles by Intramolecular Dipolar Cycloadditions/1s, 5s Sigmatropic Rearrangements. Org. Lett. 2019, 21, 7209–7212. [Google Scholar] [CrossRef] [PubMed]
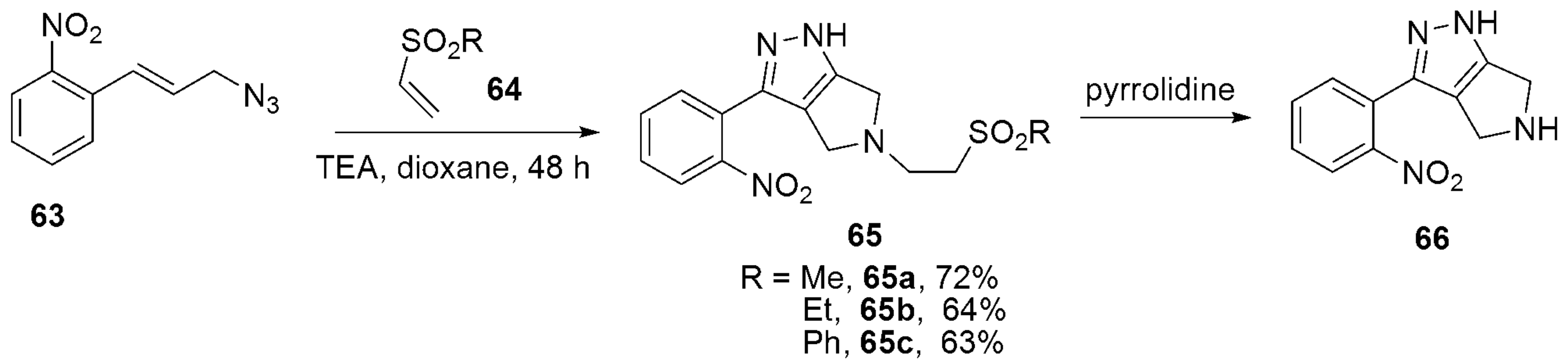
- Carlson, A.S.; Petre, A.M.; Topczewski, J.J. A cascade reaction of cinnamyl azides with vinyl sulfones directly generates dihydro-pyrrolo-pyrazole heterocycles. Tetrahedron Lett. 2021, 67, 152860. [Google Scholar] [CrossRef] [PubMed]
- Bania, N.; Mondal, B.; Ghosh, S.; Pan, S.C. DMAP Catalyzed Domino Rauhut-Currier Cyclization Reaction between Alkylidene Pyrazolones and Nitro-olefins: Access to Tetra hydropyrano 2,3-c pyrazoles. J. Org. Chem. 2021, 86, 4304–4312. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.G.; Zhang, T.; Gong, X.C.; Zhang, M.; Zhu, C.Y. Visible-light promoted one-pot synthesis of pyrazoles from alkynes and hydrazines. Tetrahedron Lett. 2019, 60, 171–174. [Google Scholar] [CrossRef]
- Li, H.X.; Shi, W.Y.; Wang, C.; Liu, H.; Wang, W.; Wu, Y.J.; Guo, H.C. Phosphine-Catalyzed Cascade Annulation of MBH Carbonates and Diazenes: Synthesis of Hexahydrocyclopenta c pyrazole Derivatives. Org. Lett. 2021, 23, 5571–5575. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Sadeghi, M.; Safari, J. Copper(II) Anchored on Amine-Functionalized MMT: A Highly Efficient Catalytic System for the One-Pot Synthesis of Bispyrano 2,3-c pyrazole Derivatives. J. Chem. 2021, 2021, 1784142. [Google Scholar] [CrossRef]
- Alizadeh-Kouzehrash, M.; Rahmati, A. Synthesis of a structure containing three N-fused heterocycles with very high bond-forming through a one-pot reaction. Tetrahedron 2020, 76, 130923. [Google Scholar] [CrossRef]
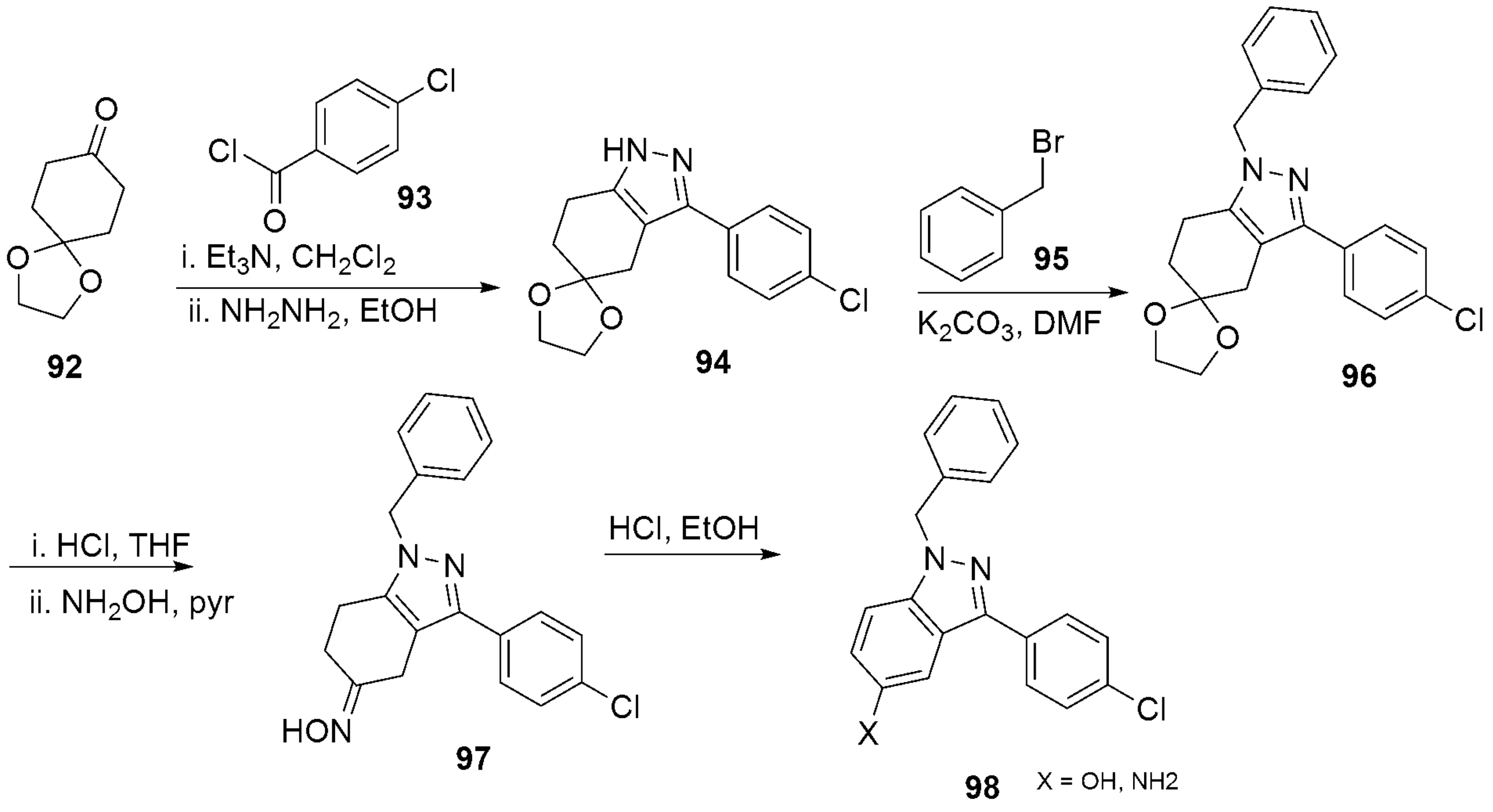
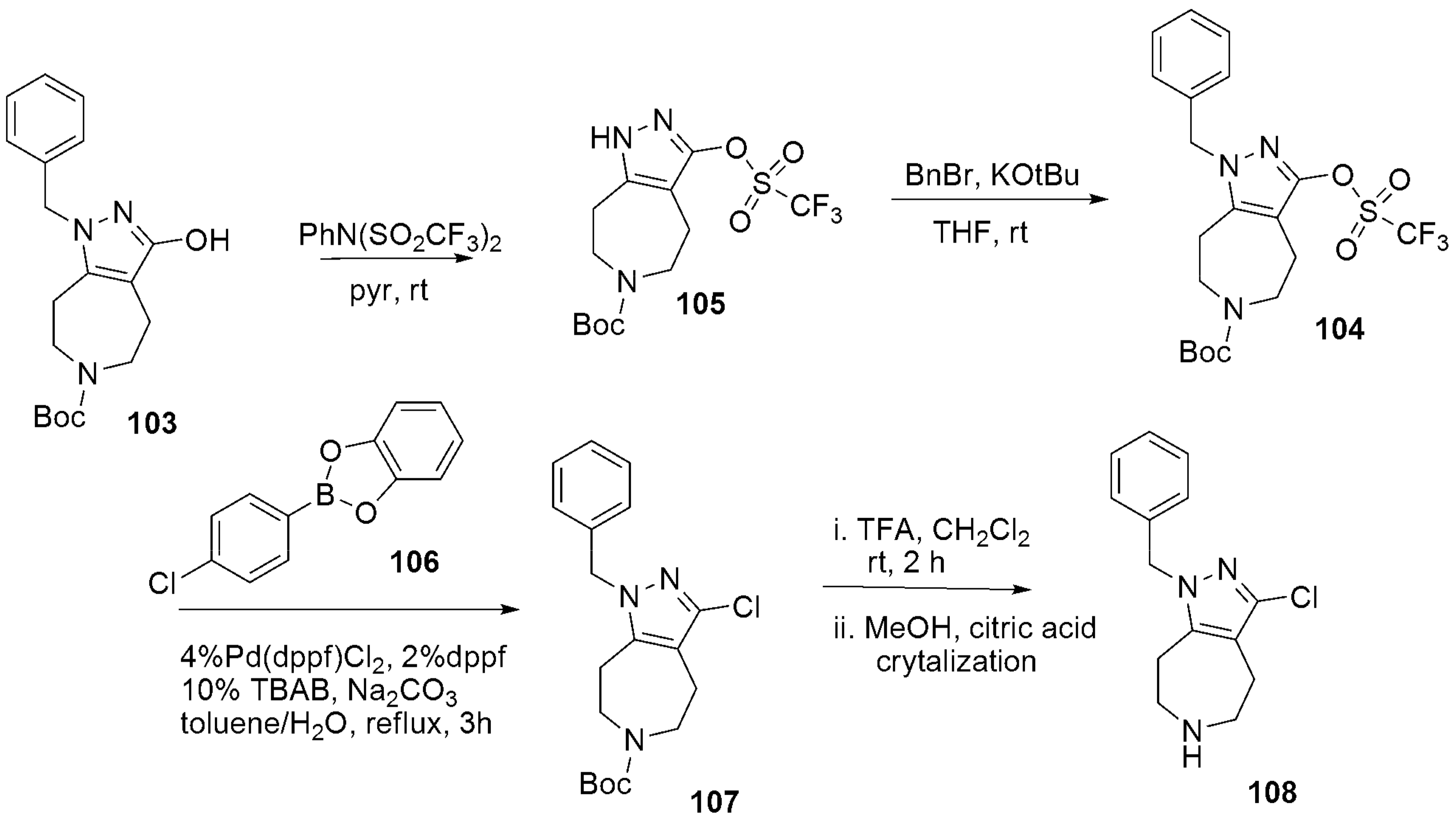
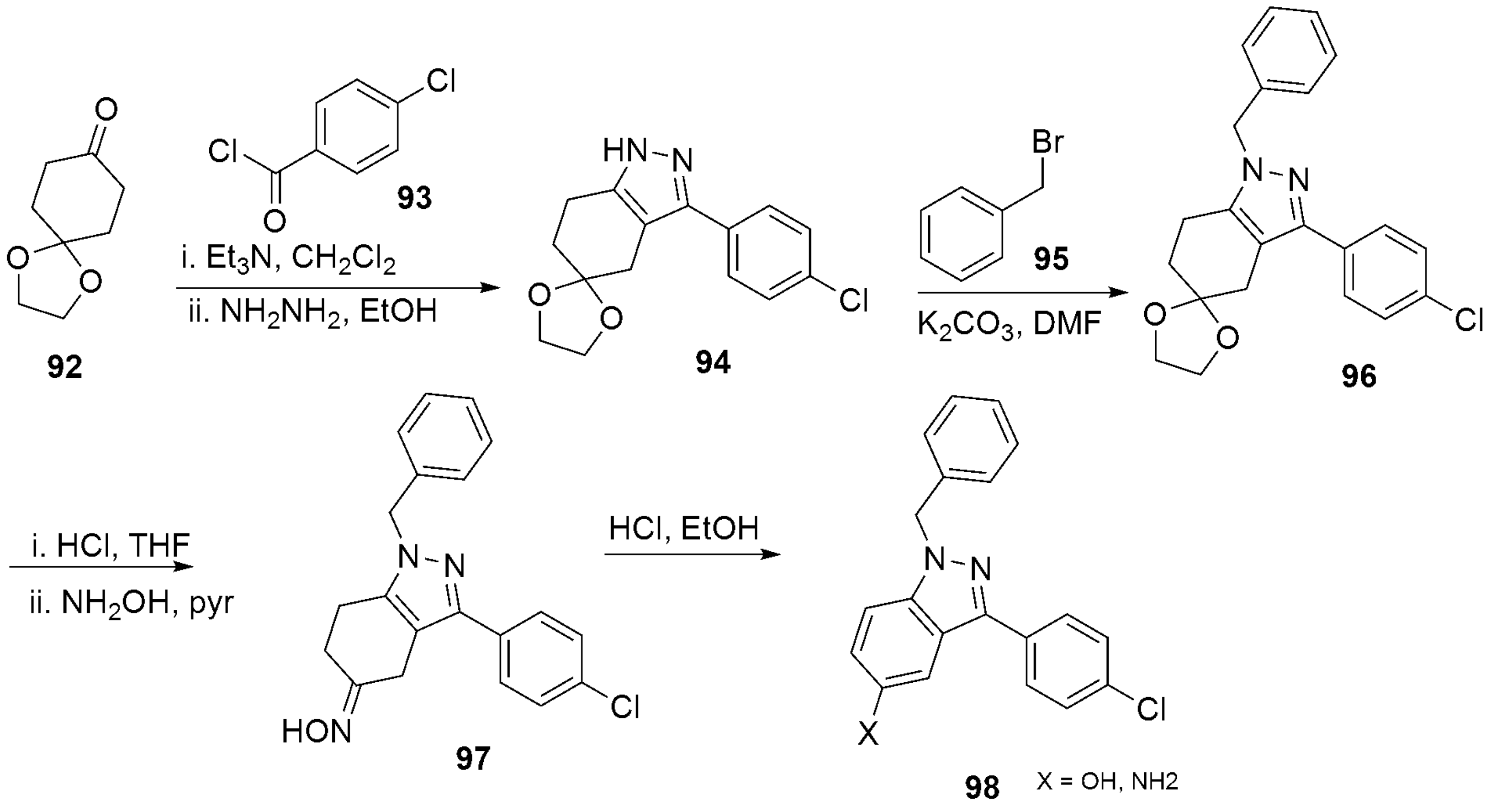
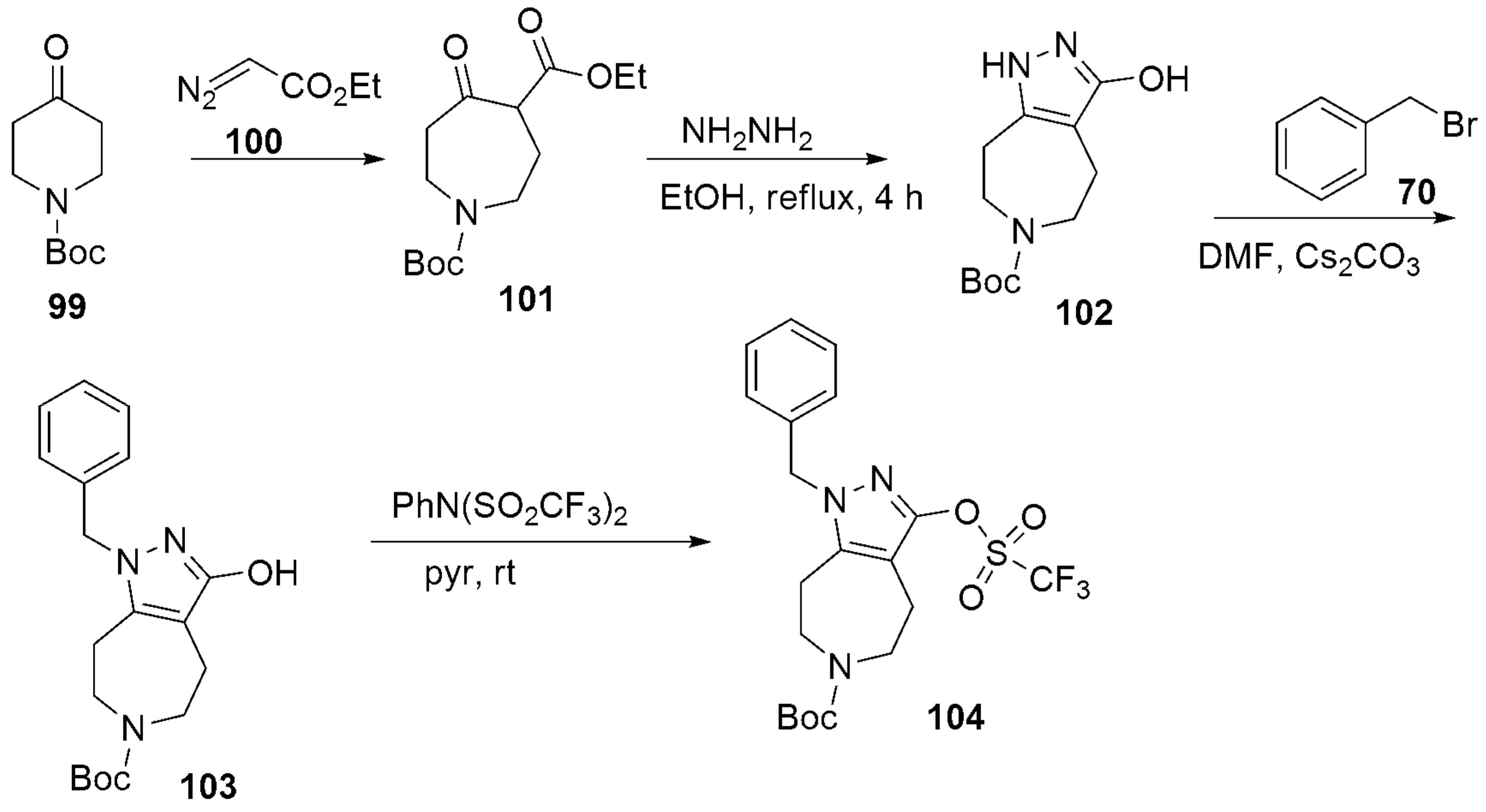
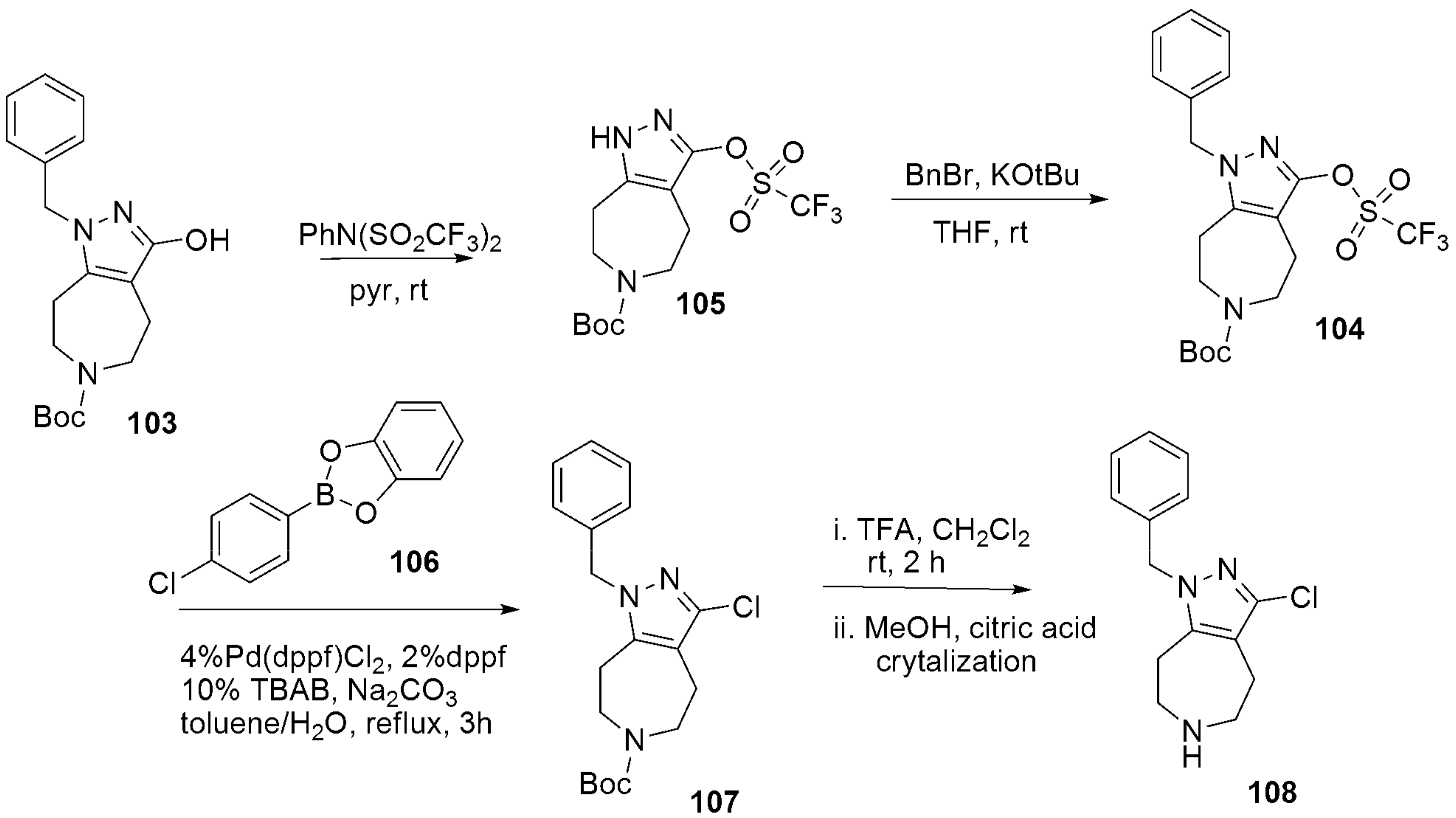
- Dvorak, C.A.; Liang, J.; Mani, N.S.; Carruthers, N.I. Regioselective assembly of fused pyrazole-azepine heterocycles: Synthesis of the 5-HT7 antagonist 1-benzy1-3-(4-chlorophenyl)-1,4,5,6,7,8-hexahydropyrazolo 3,4-d azepine. Tetrahedron Lett. 2021, 67, 152843. [Google Scholar] [CrossRef]
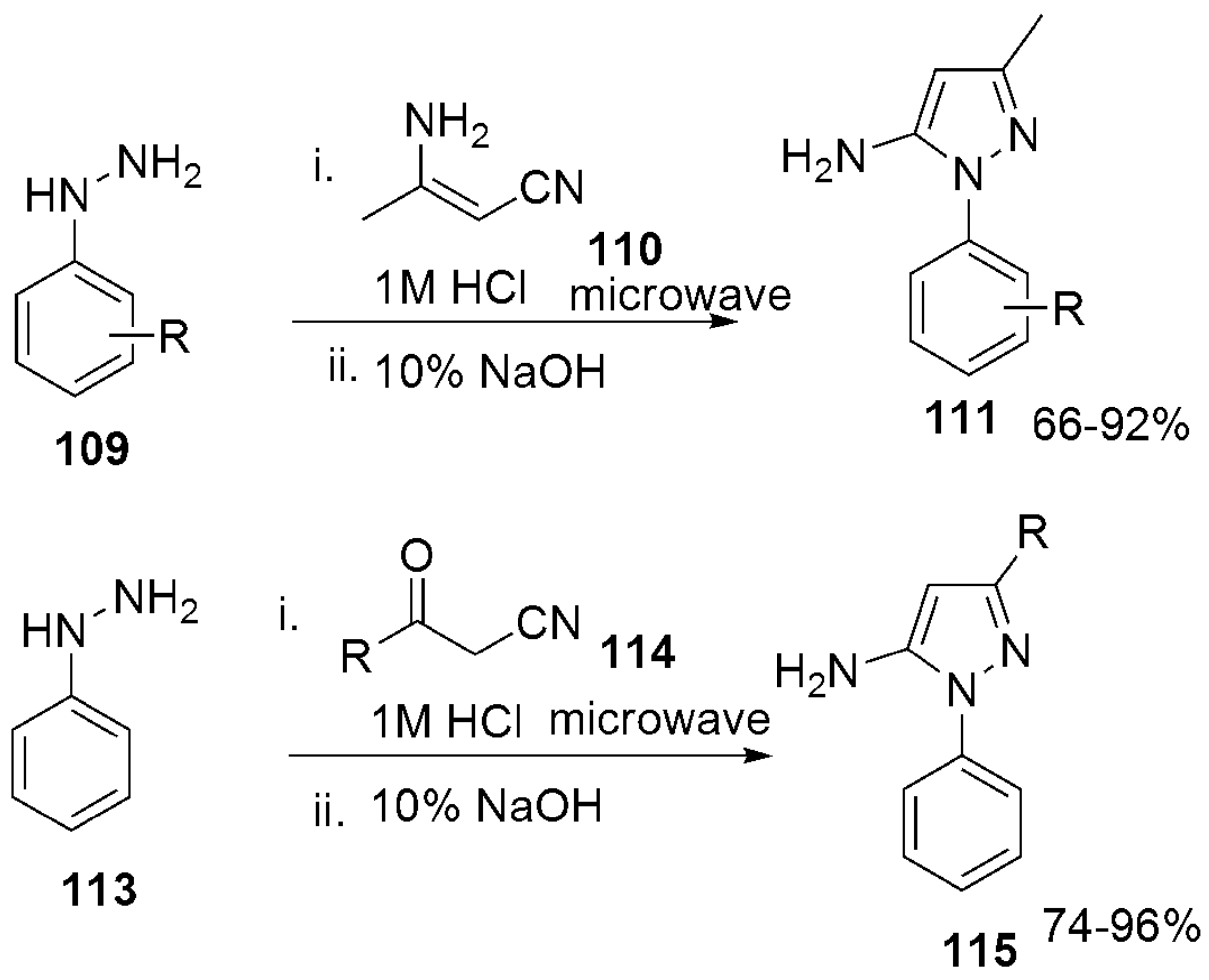
- Everson, N.; Yniguez, K.; Loop, L.; Lazaro, H.; Belanger, B.; Koch, G.; Bach, J.; Manjunath, A.; Schioldager, R.; Law, J.; et al. Microwave synthesis of 1-aryl-1H-pyrazole-5-amines. Tetrahedron Lett. 2019, 60, 72–74. [Google Scholar] [CrossRef]
- Bhale, P.S.; Bandgar, B.P.; Dongare, S.B.; Shringare, S.N.; Sirsat, D.M.; Chavan, H.V. Ketene dithioacetal mediated synthesis of 1,3,4,5-tetrasubstituted pyrazole derivatives and their biological evaluation. Phosphorus Sulfur Relat. Elem. 2019, 194, 843–849. [Google Scholar] [CrossRef]
- Abd El-Karim, S.S.; Mohamed, H.S.; Abdelhameed, M.F.; Amr, A.E.; Almehizia, A.A.; Nossier, E.S. Design, synthesis and molecular docking of new pyrazole-thiazolidinones as potent anti-inflammatory and analgesic agents with TNF-alpha inhibitory activity. Bioorg. Chem. 2021, 111, 104827. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, W.; Marella, A.; Alam, M.M.; Khan, M.F.; Akhtar, M.; Anwer, T.; Khan, F.; Naematullah, M.; Azam, F.; Rizvi, M.A.; et al. Design and synthesis of pyrazole-pyrazoline hybrids as cancer-associated selective COX-2 inhibitors. Archiv. Pharm. 2021, 354, 2000116. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.A.; Abdelall, E.K.A.; Elshemy, H.A.H.; El-Nahass, E.; Abdel-Fattah, M.M.; Abdelgawad, Y.Y.M. New indomethacin analogs as selective COX-2 inhibitors: Synthesis, COX-1/2 inhibitory activity, anti-inflammatory, ulcerogenicity, histopathological, and docking studies. Arch. Pharm. 2021, 354, 2000328. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.B.; Chen, L.Z.; Wang, B.S.; Huang, X.; Jiao, M.M.; Liu, M.M.; Tang, W.J.; Liu, X.H. Novel Pyrazolo 4,3-d pyrimidine as Potent and Orally Active Inducible Nitric Oxide Synthase (iNOS) Dimerization Inhibitor with Efficacy in Rheumatoid Arthritis Mouse Model. J. Med. Chem. 2019, 62, 4013–4031. [Google Scholar] [CrossRef]
- Sivaramakarthikeyan, R.; Shunmugam, I.; Vadivel, S.; Lim, W.M.; Mai, C.W.; Ramalingan, C. Molecular Hybrids Integrated with Benzimidazole and Pyrazole Structural Motifs: Design, Synthesis, Biological Evaluation, and Molecular Docking Studies. ACS Omega 2020, 5, 10089–10098. [Google Scholar] [CrossRef]
- Nayak, S.G.; Poojary, B.; Kamat, V. Novel pyrazole-clubbed thiophene derivatives via Gewald synthesis as antibacterial and anti-inflammatory agents. Arch. Pharm. 2020, 353, 2000103. [Google Scholar] [CrossRef]
- Dimmito, M.P.; Stefanucci, A.; Pieretti, S.; Minosi, P.; Dvoracsko, S.; Tomboly, C.; Zengin, G.; Mollica, A. Discovery of Orexant and Anorexant Agents with Indazole Scaffold Endowed with Peripheral Antiedema Activity. Biomolecules 2019, 9, 492. [Google Scholar] [CrossRef] [Green Version]
- Harras, M.F.; Sabour, R.; Alkamali, O.M. Discovery of new non-acidic lonazolac analogues with COX-2 selectivity as potent anti-inflammatory agents. MedChemComm 2019, 10, 1775–1788. [Google Scholar] [CrossRef]
- Sivaramakarthikeyan, R.; Iniyaval, S.; Lim, W.M.; Hii, L.W.; Mai, C.W.; Ramalingan, C. Pyrazolylphenanthroimidazole heterocycles: Synthesis, biological and molecular docking studies. New J. Chem. 2020, 44, 19612–19622. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; El-Saadi, M.T.; Elzayat, S.G.; Amin, N.H. New substituted pyrazole derivatives targeting COXs as potential safe anti-inflammatory agents. Future Med. Chem. 2019, 11, 1871–1887. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.A.; Fadaly, W.A.A.; Kamel, G.M.; Elshaier, Y.; El-Magd, M.A. Design, synthesis, modeling studies and biological evaluation of thiazolidine derivatives containing pyrazole core as potential anti-diabetic PPAR-gamma agonists and anti-inflammatory COX-2 selective inhibitors. Bioorg. Chem. 2019, 82, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Abdelall, E.K.A.; Lamie, P.F.; Ahmed, A.K.M.; El-Nahass, E.S. COX-1/COX-2 inhibition assays and histopathological study of the new designed anti-inflammatory agent with a pyrazolopyrimidine core. Bioorg. Chem. 2019, 86, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Thangarasu, P.; Manikandan, A.; Thamaraiselvi, S. Discovery, synthesis and molecular corroborations of medicinally important novel pyrazoles; drug efficacy determinations through in silico, in vitro and cytotoxicity validations. Bioorg. Chem. 2019, 86, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Murahari, M.; Mahajan, V.; Neeladri, S.; Kumar, M.S.; Mayur, Y.C. Ligand based design and synthesis of pyrazole based derivatives as selective COX-2 inhibitors. Bioorg. Chem. 2019, 86, 583–597. [Google Scholar] [CrossRef]
- El-Shoukrofy, M.S.; Abd El Razik, H.A.; AboulWafa, O.M.; Bayad, A.E.; El-Ashmawy, I.M. Pyrazoles containing thiophene, thienopyrimidine and thienotriazolopyrimidine as COX-2 selective inhibitors: Design, synthesis, in vivo anti-inflammatory activity, docking and in silico chemo-informatic studies. Bioorg. Chem. 2019, 85, 541–557. [Google Scholar] [CrossRef]
- Khan, M.F.; Anwer, T.; Bakht, A.; Verma, G.; Akhtar, W.; Alam, M.M.; Rizvi, M.A.; Akhter, M.; Shaquiquzzaman, M. Unveiling novel diphenyl-1H-pyrazole based acrylates tethered to 1,2,3-triazole as promising apoptosis inducing cytotoxic and anti-inflammatory agents. Bioorg. Chem. 2019, 87, 667–678. [Google Scholar] [CrossRef]
- Taher, A.T.; Sarg, M.T.M.; Ali, N.R.E.; Elnagdi, N.H. Design, synthesis, modeling studies and biological screening of novel pyrazole derivatives as potential analgesic and anti-inflammatory agents. Bioorg. Chem. 2019, 89, 103023. [Google Scholar] [CrossRef]
- Mustafa, G.; Angeli, A.; Zia-ur-Rehman, M.; Akbar, N.; Ishtiaq, S.; Supuran, C.T. An efficient method for the synthesis of novel derivatives 4-{5- 4-(4-amino-5-mercapto-4H- 1,2,4 triazol-3-yl)-phenyl -3-trifluorom ethyl-pyrazol-1-yl}-benzenesulfonamide and their anti-inflammatory potential. Bioorg. Chem. 2019, 91, 103110. [Google Scholar] [CrossRef]
- Zabiulla; Gulnaz, A.R.; Mohammed, Y.H.E.; Khanum, S.A. Design, synthesis and molecular docking of benzophenone conjugated with oxadiazole sulphur bridge pyrazole pharmacophores as anti inflammatory and analgesic agents. Bioorg. Chem. 2019, 92, 103220. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; Fadaly, W.A.A.; Mostafa, Y.A.; Zaher, D.M.; Omar, H.A. Thiohydantoin derivatives incorporating a pyrazole core: Design, synthesis and biological evaluation as dual inhibitors of topoisomerase-I and cycloxygenase-2 with anti-cancer and anti-inflammatory activities. Bioorg. Chem. 2019, 91, 103132. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.A.; Abdelall, E.K.A.; Lamie, P.F.; Labib, M.B.; El-Nahaas, E.; Abdelhakeem, M.M. New pyrazole derivatives possessing amino/methanesulphonyl pharmacophore with good gastric safety profile: Design, synthesis, cyclooxygenase inhibition, anti-inflammatory activity and histopathological studies. Bioorg. Chem. 2020, 95, 103540. [Google Scholar] [CrossRef] [PubMed]
- Fadaly, W.A.A.; Elshaier, Y.; Hassanein, E.H.M.; Abdellatif, K.R.A. New 1,2,4-triazole/pyrazole hybrids linked to oxime moiety as nitric oxide donor celecoxib analogs: Synthesis, cyclooxygenase inhibition anti-inflammatory, ulcerogenicity, anti-proliferative activities, apoptosis, molecular modeling and nitric oxide release studies. Bioorg. Chem. 2020, 98, 103752. [Google Scholar] [CrossRef] [PubMed]
- Gedawy, E.M.; Kassab, A.E.; El Kerdawy, A.M. Design, synthesis and biological evaluation of novel pyrazole sulfonamide derivatives as dual COX-2/5-LOX inhibitors. Eur. J. Med. Chem. 2020, 189, 112066. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.Y.; Guo, Q.P.; Wang, M.R.; Wang, R.; Xu, Z.Q. Discovery of pyrazole N-aryl sulfonate: A novel and highly potent cyclooxygenase-2 (COX-2) selective inhibitors. Bioorg. Med. Chem. 2021, 46, 116344. [Google Scholar] [CrossRef]
- Hendawy, O.M.; Gomaa, H.A.M.; Alzarea, S.I.; Alshammari, M.S.; Mohamed, F.A.M.; Mostafa, Y.A.; Abdelazeem, A.H.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G.M. Novel 1,5-diaryl pyrazole-3-carboxamides as selective COX-2/sEH inhibitors with analgesic, anti-inflammatory, and lower cardiotoxicity effects. Bioorg. Chem. 2021, 116, 105302. [Google Scholar] [CrossRef]
- Amer, M.M.K.; Abdellattif, M.H.; Mouneir, S.M.; Zordok, W.A.; Shehab, W.S. Synthesis, DFT calculation, pharmacological evaluation, and catalytic application in the synthesis of diverse pyrano 2,3-c pyrazole derivatives. Bioorg. Chem. 2021, 114, 105136. [Google Scholar] [CrossRef]
- Dawood, D.H.; Nossier, E.S.; Ali, M.M.; Mahmoud, A.E. Synthesis and molecular docking study of new pyrazole derivatives as potent anti-breast cancer agents targeting VEGFR-2 kinase. Bioorg. Chem. 2020, 101, 103916. [Google Scholar] [CrossRef]
- Philoppes, J.N.; Khedr, M.A.; Hassan, M.H.A.; Kamel, G.; Lamie, P.F. New pyrazolopyrimidine derivatives with anticancer activity: Design, synthesis, PIM-1 inhibition, molecular docking study and molecular dynamics. Bioorg. Chem. 2020, 100, 103944. [Google Scholar] [CrossRef]
- Song, Y.L.; Feng, S.R.; Feng, J.J.; Dong, J.J.; Yang, K.; Liu, Z.M.; Qiao, X.Q. Synthesis and biological evaluation of novel pyrazoline derivatives containing indole skeleton as anti-cancer agents targeting topoisomerase II. Eur. J. Med. Chem. 2020, 200, 112459. [Google Scholar] [CrossRef]
- Sener, N.; Ozkinali, S.; Altunoglu, Y.C.; Yerlikaya, S.; Gokce, H.; Zurnaci, M.; Gur, M.; Baloglu, M.C.; Sener, I. Antiproliferative properties and structural analysis of newly synthesized Schiff bases bearing pyrazole derivatives and molecular docking studies. J. Mol. Struct. 2021, 1241, 130520. [Google Scholar] [CrossRef]
- Cai, W.X.; Wu, J.W.; Sun, Y.Z.; Liu, A.L.; Wang, R.L.; Ma, Y.; Wang, S.Q.; Dong, W.L. Synthesis, evaluation, molecular dynamics simulation and targets identification of novel pyrazole-containing imide derivatives. J. Biomol. Struct. Dyn. 2021, 39, 2176–2188. [Google Scholar] [CrossRef]
- Zimnitskiy, N.S.; Barkov, A.Y.; Ulitko, M.V.; Kutyashev, I.B.; Korotaev, V.Y.; Sosnovskikh, V.Y. An expedient synthesis of novel spiro indenoquinoxaline-pyrrolizidine-pyrazole conjugates with anticancer activity from 1,5-diarylpent-4-ene-1,3-diones through the 1,3-dipolar cycloaddition/cyclocondensation sequence. New J. Chem. 2020, 44, 16185–16199. [Google Scholar] [CrossRef]
- El Azab, I.H.; El-Sheshtawy, H.S.; Bakr, R.B.; Elkanzi, N.A.A. New 1,2,3-Triazole-Containing Hybrids as Antitumor Candidates: Design, Click Reaction Synthesis, DFT Calculations, and Molecular Docking Study. Molecules 2021, 26, 708. [Google Scholar] [CrossRef] [PubMed]
- Ragab, F.A.; Nissan, Y.M.; Seif, E.M.; Maher, A.; Arafa, R.K. Synthesis and in vitro investigation of novel cytotoxic pyrimidine and pyrazolopyrimidne derivatives showing apoptotic effect. Bioorg. Chem. 2020, 96, 103621. [Google Scholar] [CrossRef]
- Mohamady, S.; Ismail, M.I.; Mogheith, S.M.; Attia, Y.M.; Taylor, S.D. Discovery of 5-aryl-3-thiophen-2-yl-1H-pyrazoles as a new class of Hsp90 inhibitors in hepatocellular carcinoma. Bioorg. Chem. 2020, 94, 103433. [Google Scholar] [CrossRef]
- El Azab, I.H.; Bakr, R.B.; Elkanzi, N.A.A. Facile One-Pot Multicomponent Synthesis of Pyrazolo-Thiazole Substituted Pyridines with Potential Anti-Proliferative Activity: Synthesis, In Vitro and In Silico Studies. Molecules 2021, 26, 3103. [Google Scholar] [CrossRef]
- Wang, G.C.; Liu, W.J.; Peng, Z.Y.; Huang, Y.; Gong, Z.P.; Li, Y.J. Design, synthesis, molecular modeling, and biological evaluation of pyrazole-naphthalene derivatives as potential anticancer agents on MCF-7 breast cancer cells by inhibiting tubulin polymerization. Bioorg. Chem. 2020, 103, 104141. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Saddiq, A.A.; Abdelhamid, I.A. Attacking the mitochondria of colorectal carcinoma by novel 2-cyanoacrylamides linked to ethyl 1,3-diphenylpyrazole-4-carboxylates moiety as a new trend for chemotherapy. Bioorg. Chem. 2020, 103, 104195. [Google Scholar] [CrossRef]
- Burgart, Y.V.; Agafonova, N.A.; Shchegolkov, E.V.; Krasnykh, O.P.; Kushch, S.O.; Evstigneeva, N.P.; Gerasimova, N.A.; Maslova, V.V.; Triandafilova, G.A.; Solodnikov, S.Y.; et al. Multiple biological active 4-aminopyrazoles containing trifluoromethyl and their 4-nitroso-precursors: Synthesis and evaluation. Eur. J. Med. Chem. 2020, 208, 112768. [Google Scholar] [CrossRef]
- Anwer, K.E.; Sayed, G.H. Conventional and microwave reactions of 1,3-diaryl-5,4-enaminonitrile-pyrazole derivative with expected antimicrobial and anticancer activities. J. Heterocycl. Chem. 2020, 57, 2339–2353. [Google Scholar] [CrossRef]
- Hassan, A.Y.; Saleh, N.M.; Kadh, M.S.; Abou-Amra, E.S. New fused pyrazolopyrimidine derivatives; heterocyclic styling, synthesis, molecular docking and anticancer evaluation. J. Heterocycl. Chem. 2020, 57, 2704–2721. [Google Scholar] [CrossRef]
- Kolluri, P.K.; Gurrapu, N.; Subhashini, N.; Putta, S.; Singh, S.S.; Vani, T.; Manga, V. Design, synthesis of novel (Z)-2-(3-(4-((3-benzyl-2, 4-dioxothiazolidin-5-ylidene) methyl)-1-phenyl-1H-pyrazol-3-yl) phenoxy)-N-arylacetamide derivatives: Evaluation of cytotoxic activity and molecular docking studies. J. Mol. Struct. 2020, 1202, 127300. [Google Scholar] [CrossRef]
- Mamidala, S.; Aravilli, R.K.; Ramesh, G.; Khajavali, S.; Chedupaka, R.; Manga, V.; Vedula, R.R. A facile one-pot, three-component synthesis of a new series of thiazolyl pyrazole carbaldehydes: In vitro anticancer evaluation, in silico ADME/T, and molecular docking studies. J. Mol. Struct. 2021, 1236, 130356. [Google Scholar] [CrossRef]
- Raghu, M.S.; Kumar, C.B.P.; Prashanth, M.K.; Kumar, K.Y.; Prathibha, B.S.; Kanthimathi, G.; Alissa, S.A.; Alghulikah, H.A.; Osman, S.M. Novel 1,3,5-triazine-based pyrazole derivatives as potential antitumor agents and EFGR kinase inhibitors: Synthesis, cytotoxicity, DNA binding, molecular docking and DFT studies. New J. Chem. 2021, 45, 13909–13924. [Google Scholar] [CrossRef]
- Mohammed, E.Z.; Mahmoud, W.R.; George, R.F.; Hassan, G.S.; Omar, F.A.; Georgey, H.H. Synthesis, in vitro anticancer activity and in silico studies of certain pyrazole-based derivatives as potential inhibitors of cyclin dependent kinases (CDKs). Bioorg. Chem. 2021, 116, 105347. [Google Scholar] [CrossRef]
- Suryanarayana, K.; Robert, A.R.; Kerru, N.; Pooventhiran, T.; Thomas, R.; Maddila, S.; Jonnalagadda, S.B. Design, synthesis, anticancer activity and molecular docking analysis of novel dinitrophenylpyrazole bearing 1,2,3-triazoles. J. Mol. Struct. 2021, 1243, 130865. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Shi, W.; Wu, C.L.; Wan, L.; Zhao, Y.X.; Zhang, C.L.; Gu, W.; Wang, S.F. Pyrazole ring-containing isolongifolanone derivatives as potential CDK2 inhibitors: Evaluation of anticancer activity and investigation of action mechanism. Biomed. Pharmacother. 2021, 139, 111663. [Google Scholar] [CrossRef]
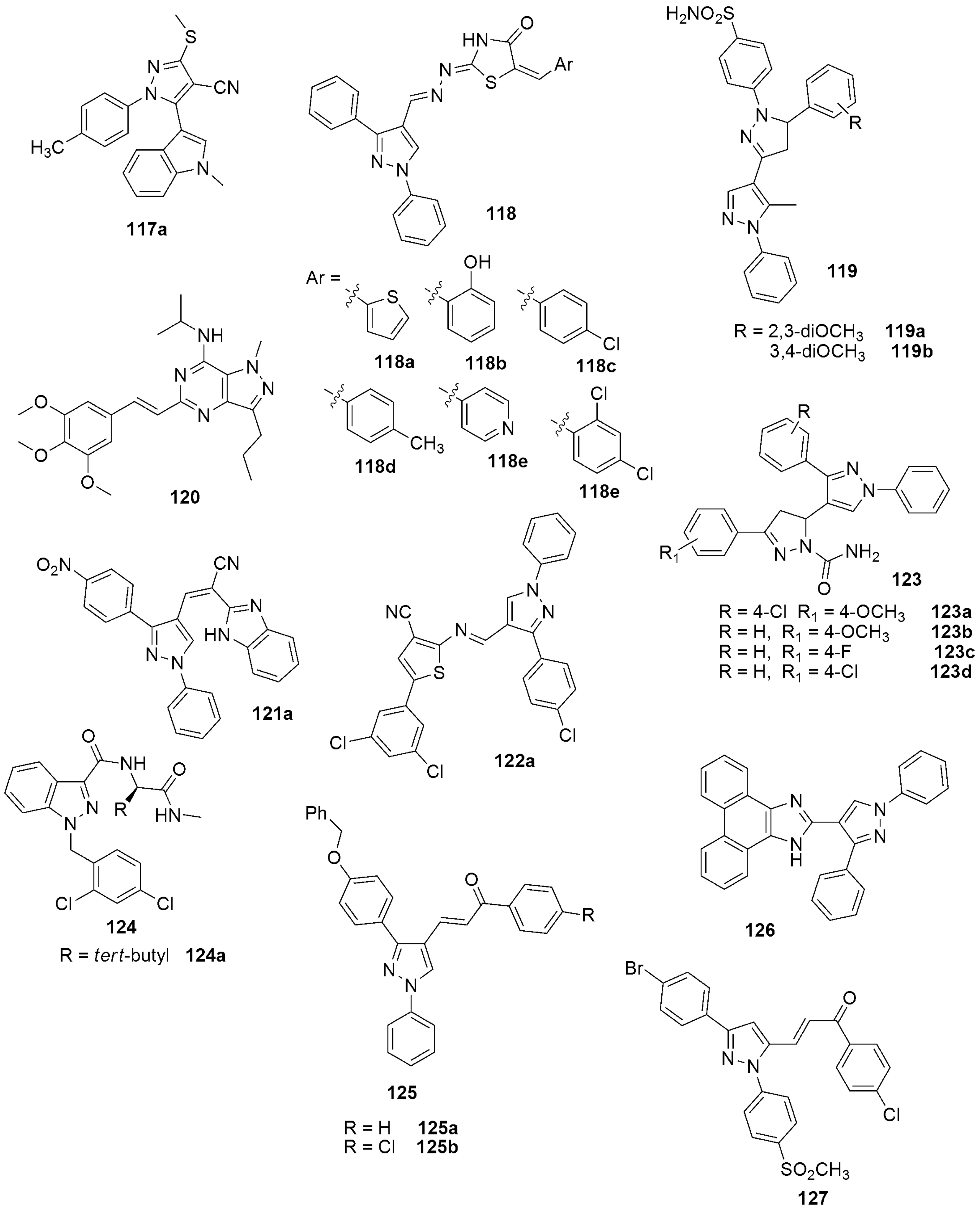
- Signorello, M.G.; Rapetti, F.; Meta, E.; Sidibè, A.; Bruno, O.; Brullo, C. New Series of Pyrazoles and Imidazo-Pyrazoles Targeting Different Cancer and Inflammation Pathways. Molecules 2021, 26, 5735. [Google Scholar] [CrossRef]
- Alfei, S.; Brullo, C.; Caviglia, D.; Piatti, G.; Zorzoli, A.; Marimpietri, D.; Zuccari, G.; Schito, A.M. Pyrazole-Based Water-Soluble Dendrimer Nanoparticles as a Potential New Agent against Staphylococci. Biomedicines 2021, 10, 17. [Google Scholar] [CrossRef]
- Morretta, E.; Sidibè, A.; Spallarossa, A.; Petrella, A.; Meta, E.; Bruno, O.; Monti, M.C.; Brullo, C. Synthesis, functional proteomics and biological evaluation of new 5-pyrazolyl ureas as potential anti-angiogenic compounds. Eur. J. Med. Chem. 2021, 226, 113872. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, O.; Awolade, P.; Koorbanally, N.; Singh, P. New library of pyrazole-imidazo 1,2-alpha pyridine molecular conjugates: Synthesis, antibacterial activity and molecular docking studies. Chem. Biol. Drug Des. 2020, 95, 162–173. [Google Scholar] [CrossRef] [PubMed]
- El-Shershaby, M.H.; El-Gamal, K.M.; Bayoumi, A.H.; El-Adl, K.; Alswah, M.; Ahmed, H.E.A.; Al-Karmalamy, A.A.; Abulkhair, H.S. The antimicrobial potential and pharmacokinetic profiles of novel quinoline-based scaffolds: Synthesis and in silico mechanistic studies as dual DNA gyrase and DHFR inhibitors. New J. Chem. 2021, 45, 13986–14004. [Google Scholar] [CrossRef]
- Hansa, R.K.C.; Khan, M.M.K.; Frangie, M.M.; Gilmore, D.F.; Shelton, R.S.; Savenka, A.V.; Basnakian, A.G.; Shuttleworth, S.L.; Smeltzer, M.S.; Alam, M.A. 4-4-(Anilinomethyl)-3-4-(trifluoromethyl)phenyl-1H-pyrazol-1-ylbenzoic acid derivatives as potent anti-gram-positive bacterial agents. Eur. J. Med. Chem. 2021, 219, 113402. [Google Scholar] [CrossRef]
- Liu, H.; Chu, Z.W.; Xia, D.G.; Cao, H.Q.; Lv, X.H. Discovery of novel multi-substituted benzo-indole pyrazole schiff base derivatives with antibacterial activity targeting DNA gyrase. Bioorg. Chem. 2020, 99, 103807. [Google Scholar] [CrossRef]
- Patel, B.; Zunk, M.; Grant, G.; Rudrawar, S. Design, synthesis and bioactivity evaluation of novel pyrazole linked phenylthiazole derivatives in context of antibacterial activity. Bioorg. Med. Chem. Lett. 2021, 39, 127853. [Google Scholar] [CrossRef]
- Ebenezer, O.; Singh-Pillay, A.; Koorbanally, N.A.; Singh, P. Antibacterial evaluation and molecular docking studies of pyrazole-thiosemicarbazones and their pyrazole-thiazolidinone conjugates. Mol. Divers. 2021, 25, 191–204. [Google Scholar] [CrossRef]
- Bhirud, J.D.; Patil, R.D.; Narkhede, H.P. Sulfamic acid catalyzed synthesis of new 3,5- (sub)phenyl -1H-pyrazole bearing N-1-isonicotinoyl: And their pharmacological activity evaluation. Bioorg. Med. Chem. Lett. 2020, 30, 127558. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Fayed, E.A.; Rizk, H.F.; Desouky, S.E.; Ragab, A. Hydrazonoyl bromide precursors as DHFR inhibitors for the synthesis of bis-thiazolyl pyrazole derivatives; antimicrobial activities, antibiofilm, and drug combination studies against MRSA. Bioorg. Chem. 2021, 116, 105339. [Google Scholar] [CrossRef]
- Desai, N.C.; Vaja, D.V.; Monapara, J.D.; Manga, V.; Vani, T. Synthesis, biological evaluation, and molecular docking studies of novel pyrazole, pyrazoline-clubbed pyridine as potential antimicrobial agents. J. Heterocycl. Chem. 2021, 58, 737–750. [Google Scholar] [CrossRef]
- Saber, A.F.; Zaki, R.M.; El-Dean, A.M.K.; Radwan, S.M. Synthesis, reactions, and spectral characterization of some new biologically active compounds derived from thieno 2,3-c pyrazole-5-carboxamide. J. Heterocycl. Chem. 2020, 57, 238–247. [Google Scholar] [CrossRef]
- Othman, I.M.M.; Gad-Elkareem, M.A.M.; Amr, A.E.; Al-Omar, M.A.; Nossier, E.S.; Elsayed, E.A. Novel heterocyclic hybrids of pyrazole targeting dihydrofolate reductase: Design, biological evaluation andinsilicostudies. J. Enzyme Inhib. Med. Chem. 2020, 35, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zhou, S.; Cao, L.; Hao, Z.; Yang, D.; Guo, X.; Zhang, N.; Bakulev, V.A.; Fan, Z. Design, Synthesis, and Evaluation of the Antifungal Activity of Novel Pyrazole–Thiazole Carboxamides as Succinate Dehydrogenase Inhibitors. J. Agric. Food Chem. 2020, 68, 7093–7102. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Wang, A.; Qu, L.L.; Chen, M.; Lu, A.M.; Li, G.H.; Yang, C.L.; Xue, W. Expedient Discovery for Novel Antifungal Leads Targeting Succinate Dehydrogenase: Pyrazole-4-formylhydrazide Derivatives Bearing a Diphenyl Ether Fragment. J. Agric. Food Chem. 2020, 68, 14426–14437. [Google Scholar] [CrossRef]
- Zhao, Y.T.; Yang, N.; Deng, Y.M.; Tao, K.; Jin, H.; Hou, T.P. Mechanism of Action of Novel Pyrazole Carboxamide Containing a Diarylamine Scaffold against Rhizoctonia solani. J. Agric. Food Chem. 2020, 68, 11068–11076. [Google Scholar] [CrossRef]
- Tan, F.S.; Su, S.N.; Liu, Z.Z. Green synthesis and antifungal activities of novel 3-(trifluoromethyl)-4,5-dihydro-1H-furo 2,3-c pyrazole derivatives. J. Heterocycl. Chem. 2020, 57, 2904–2910. [Google Scholar] [CrossRef]
- Kaddouri, Y.; Abrigach, F.; Ouahhoud, S.; Benabbes, R.; El Kodadi, M.; Alsalme, A.; Al-Zaqri, N.; Warad, I.; Touzani, R. Synthesis, characterization, reaction mechanism prediction and biological study of mono, bis and tetrakis pyrazole derivatives against Fusarium oxysporum f. sp. Albedinis with conceptual DFT and ligand-protein docking studies. Bioorg. Chem. 2021, 110, 104696. [Google Scholar] [CrossRef]
- Masaret, G.S. A New Approach for the Synthesis and Biological Activities of Novel Thiazolyl-Pyrazole Derivatives. ChemistrySelect 2021, 6, 974–982. [Google Scholar] [CrossRef]
- Phougat, H.; Devi, V.; Rai, S.; Reddy, T.S.; Singh, K. Urea derivatives of piperazine doped with pyrazole-4-carboxylic acids: Synthesis and antimicrobial evaluation. J. Heterocycl. Chem. 2021, 58, 1992–1999. [Google Scholar] [CrossRef]
- Dong, C.T.; Gao, W.; Li, X.T.; Sun, S.S.; Huo, J.Q.; Wang, Y.E.; Ren, D.; Zhang, J.L.; Chen, L. Synthesis of pyrazole-4-carboxamides as potential fungicide candidates. Mol. Divers. 2021, 25, 2379–2388. [Google Scholar] [CrossRef]
- Makhanya, T.R.; Gengan, R.M.; Kasumbwe, K. Synthesis of Fused Indolo-Pyrazoles and Their Antimicrobial and Insecticidal Activities against Anopheles arabiensis Mosquito. ChemistrySelect 2020, 5, 2756–2762. [Google Scholar] [CrossRef]
- Bayazeed, A.; Alshehrei, F.; Muhammad, Z.A.; Alfahmi, G.; El-Metwally, N.; Farghaly, T.A. Synthesis of Coumarin-Analogues: Analytical, Spectral, Conformational, MOE-Docking and Antimicrobial Studies. ChemistrySelect 2020, 5, 3874–3891. [Google Scholar] [CrossRef]
- Wang, X.B.; Wang, M.Q.; Han, L.; Jin, F.; Jiao, J.; Chen, M.; Yang, C.L.; Xue, W. Novel Pyrazole-4-acetohydrazide Derivatives Potentially Targeting Fungal Succinate Dehydrogenase: Design, Synthesis, Three-Dimensional Quantitative Structure-Activity Relationship, and Molecular Docking. J. Agric. Food Chem. 2021, 69, 9557–9570. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.B.; Park, H.Y.; Xie, D.W.; Yang, J.X.; Hou, S.T.; Shahzad, N.; Kim, C.K.; Yang, S. Synthesis, Biological Evaluation, and 3D-QSAR Studies of N-(Substituted pyridine-4-yl)-1-(substituted phenyl)-5-trifluoromethyl-1H-pyrazole-4-carboxamide Derivatives as Potential Succinate Dehydrogenase Inhibitors. J. Agric. Food Chem. 2021, 69, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.G.; Cheng, X.; Liu, X.H.; Zhang, C.Q.; Wang, Y.X.; Liu, Q.Y.; Zeng, Q.; Huang, N.Q.; Cheng, Y.; Lv, X.H. Discovery of Novel Pyrazole Carboxylate Derivatives Containing Thiazole as Potential Fungicides. J. Agric. Food Chem. 2021, 69, 8358–8365. [Google Scholar] [CrossRef] [PubMed]
- Kattimani, P.P.; Somagond, S.M.; Bayannavar, P.K.; Kamble, R.R.; Bijjaragi, S.C.; Hunnur, R.K.; Joshi, S.D. Novel 5-(1-aryl-1H-pyrazol-3-yl)-1H-tetrazoles as glycogen phosphorylase inhibitors: An in vivo antihyperglycemic activity study. Drug Dev. Res. 2020, 81, 70–84. [Google Scholar] [CrossRef]
- Bansal, G.; Singh, S.; Monga, V.; Thanikachalam, P.V.; Chawla, P. Synthesis and biological evaluation of thiazolidine-2,4-dione-pyrazole conjugates as antidiabetic, anti-inflammatory and antioxidant agents. Bioorg. Chem. 2019, 92, 103271. [Google Scholar] [CrossRef]
- Pogaku, V.; Gangarapu, K.; Basavoju, S.; Tatapudi, K.K.; Katragadda, S.B. Design, synthesis, molecular modelling, ADME prediction and antihyperglycemic evaluation of new pyrazole-triazolopyrimidine hybrids as potent alpha-glucosidase inhibitors. Bioorg. Chem. 2019, 93, 103307. [Google Scholar] [CrossRef]
- Karrouchi, K.; Brandan, S.A.; Sert, Y.; El-marzouqi, H.; Radi, S.; Ferbinteanu, M.; Faouzi, M.E.; Garcia, Y.; Ansar, M. Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N′-(4-(dimethylamino)benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2020, 1219, 128541. [Google Scholar] [CrossRef]
- Singh, P.; Mothilal, S.; Kerru, N.; Singh-Pillay, A.; Gummidi, L.; Erukainure, O.L.; Islam, M.S. Comparative -glucosidase and -amylase inhibition studies of rhodanine-pyrazole conjugates and their simple rhodanine analogues. Med. Chem. Res. 2019, 28, 143–159. [Google Scholar] [CrossRef]
- Jo, J.; Lee, D.; Park, Y.H.; Choi, H.; Han, J.; Park, D.H.; Choi, Y.K.; Kwak, J.; Yang, M.K.; Yoo, J.W.; et al. Discovery and optimization of novel 3-benzyl-N-phenyl-1H-pyrazole-5-carboxamides as bifunctional antidiabetic agents stimulating both insulin secretion and glucose uptake. Eur. J. Med. Chem. 2021, 217, 113325. [Google Scholar] [CrossRef] [PubMed]
- Pogaku, V.; Krishnan, R.; Basavoju, S. Synthesis and biological evaluation of new benzo d 1,2,3 triazol-1-yl-pyrazole-based dihydro- 1,2,4 triazolo 4,3-a pyrimidines as potent antidiabetic, anticancer and antioxidant agents. Res. Chem. Intermed. 2021, 47, 551–571. [Google Scholar] [CrossRef]
- Kaur, R.; Palta, K.; Kumar, M. Hybrids of Isatin-Pyrazole as Potential α-Glucosidase Inhibitors: Synthesis, Biological Evaluations and Molecular Docking Studies. ChemistrySelect 2019, 4, 13219–13227. [Google Scholar] [CrossRef]
- Kausar, N.; Ullah, S.; Khan, M.A.; Zafar, H.; Atia tul, W.; Choudhary, M.I.; Yousuf, S. Celebrex derivatives: Synthesis, α-glucosidase inhibition, crystal structures and molecular docking studies. Bioorg. Chem. 2021, 106, 104499. [Google Scholar] [CrossRef] [PubMed]
- Taj, S.; Ahmad, M.; Alshammari, A.; Alghamdi, A.; Ali Ashfaq, U. Exploring the therapeutic potential of benzothiazine-pyrazole hybrid molecules against alpha-glucosidase: Pharmacological and molecular modelling based approach. Saudi J. Biol. Sci. 2021, 29, 1416–1421. [Google Scholar] [CrossRef]
- Shen, J.; Deng, X.; Sun, R.; Tavallaie, M.S.; Wang, J.; Cai, Q.; Lam, C.; Lei, S.; Fu, L.; Jiang, F. Structural optimization of pyrazolo[1,5-a]pyrimidine derivatives as potent and highly selective DPP-4 inhibitors. Eur. J. Med. Chem. 2020, 208, 112850. [Google Scholar] [CrossRef]
- Karrouchi, K.; Fettach, S.; Anouar, E.H.; Tüzün, B.; Radi, S.; Alharthi, A.I.; Ghabbour, H.A.; Mabkhot, Y.N.; Faouzi, M.E.A.; Ansar, M.; et al. Synthesis, crystal structure, DFT, α-glucosidase and α-amylase inhibition and molecular docking studies of (E)-N′-(4-chlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2021, 1245, 131067. [Google Scholar] [CrossRef]
- Verma, G.; Khan, M.F.; Nainwal, L.M.; Ishaq, M.; Akhter, M.; Bakht, A.; Anwer, T.; Afrin, F.; Islamuddin, M.; Husain, I.; et al. Targeting malaria and leishmaniasis: Synthesis and pharmacological evaluation of novel pyrazole-1,3,4-oxadiazole hybrids. Part II. Bioorg. Chem. 2019, 89, 102986. [Google Scholar] [CrossRef]
- Camargo, J.D.A.; Pianoski, K.E.; Dos Santos, M.G.; Lazarin-Bidoia, D.; Volpato, H.; Moura, S.; Nakamura, C.V.; Rosa, F.A. Antiparasitic Behavior of Trifluoromethylated Pyrazole 2-Amino-1,3,4-thiadiazole Hybrids and Their Analogues: Synthesis and Structure-Activity Relationship. Front. Pharmacol. 2020, 11, 591570. [Google Scholar] [CrossRef]
- Da Silva, M.J.V.; Jacomini, A.P.; Goncalves, D.S.; Pianoski, K.E.; Poletto, J.; Lazarin-Bidoia, D.; Volpato, H.; Nakamura, C.V.; Rosa, F.A. Discovery of 1,3,4,5-tetrasubstituted pyrazoles as anti-trypanosomatid agents: Identification of alterations in flagellar structure of L. amazonensis. Bioorg. Chem. 2021, 114, 105082. [Google Scholar] [CrossRef]
- Akolkar, H.N.; Dengale, S.G.; Deshmukh, K.K.; Karale, B.K.; Darekar, N.R.; Khedkar, V.M.; Shaikh, M.H. Design, Synthesis and Biological Evaluation of Novel Furan & Thiophene Containing Pyrazolyl Pyrazolines as Antimalarial Agents. Polycycl. Aromat. Compd. 2020, 11, 1–3. [Google Scholar] [CrossRef]
- Strasek, N.; Lavrencic, L.; Ostrek, A.; Slapsak, D.; Groselj, U.; Klemencic, M.; Zugelj, H.B.; Wagger, J.; Novinec, M.; Svete, J. Tetrahydro-1H,5H-pyrazolo 1,2-a pyrazole-1-carboxylates as inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. Bioorg. Chem. 2019, 89, 102982. [Google Scholar] [CrossRef] [PubMed]
- Gogoi, P.; Shakya, A.; Ghosh, S.K.; Gogoi, N.; Gahtori, P.; Singh, N.; Bhattacharyya, D.R.; Singh, U.P.; Bhat, H.R. In silico study, synthesis, and evaluation of the antimalarial activity of hybrid dimethoxy pyrazole 1,3,5-triazine derivatives. J. Biochem. Mol. Toxicol. 2021, 35, 22682. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.E.; Bakhotmah, D.A.; Assiri, M.A. Synthesis of some new functionalized pyrano 2,3-c pyrazoles and pyrazolo 4′,3′:5,6 pyrano 2,3-d pyrimidines bearing a chromone ring as antioxidant agents. Synth. Commun. 2020, 50, 3314–3325. [Google Scholar] [CrossRef]
- Ali, S.A.; Awad, S.M.; Said, A.M.; Mahgoub, S.; Taha, H.; Ahmed, N.M. Design, synthesis, molecular modelling and biological evaluation of novel 3-(2-naphthyl)-1-phenyl-1H-pyrazole derivatives as potent antioxidants and 15-Lipoxygenase inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 847–863. [Google Scholar] [CrossRef]
- De Oliveira, D.H.; Sousa, F.S.S.; Birmann, P.T.; Pesarico, A.P.; Alves, D.; Jacob, R.G.; Savegnago, L. Evaluation of antioxidant activity and toxicity of sulfur- or selenium-containing 4-(arylchalcogenyl)-1H-pyrazoles. Can. J. Physiol. Pharmacol. 2020, 98, 441–448. [Google Scholar] [CrossRef]
- El-Badawy, A.A.; Elgubbi, A.S.; El-Helw, E.A.E. Acryloyl isothiocyanate skeleton as a precursor for synthesis of some novel pyrimidine, triazole, triazepine, thiadiazolopyrimidine and acylthiourea derivatives as antioxidant agents. J. Sulphur Chem. 2021, 42, 295–307. [Google Scholar] [CrossRef]
- El-Borai, M.A.; Rizk, H.F.; Ibrahim, S.A.; Fares, A.K.; El-Tahawy, M.M.T.; Beltagy, D.M. Assessment of anti-hemolytic, cytotoxicity, antioxidant activities and molecular docking study based on thienopyrazole scaffold as pharmacophore. J. Mol. Struct. 2021, 1240, 130602. [Google Scholar] [CrossRef]
- Elnagdy, H.M.F.; Gogoi, N.G.; Handique, J.G.; Sarma, D. CuO-NPs/TFA: A New Catalytic System to Synthesize a Novel Series of Pyrazole Imines with High Antioxidant Properties. Bionanoscience 2021, 11, 929–938. [Google Scholar] [CrossRef]
- Kaddouri, Y.; Abrigach, F.; Yousfi, E.; El Kodadi, M.; Touzani, R. New thiazole, pyridine and pyrazole derivatives as antioxidant candidates: Synthesis, DFT calculations and molecular docking study. Heliyon 2020, 6, E03185. [Google Scholar] [CrossRef] [Green Version]
- Naveen, S.; Kumara, K.; Kumar, A.D.; Kumar, K.A.; Zarrouk, A.; Warad, I.; Lokanath, N.K. Synthesis, characterization, crystal structure, Hirshfeld surface analysis, antioxidant properties and DFT calculations of a novel pyrazole derivative: Ethyl 1-(2,4-dimethylphenyl)-3-methy1-5-phenyl-1H-pyrazole-4-carboxylate. J. Mol. Struct. 2021, 1226, 129350. [Google Scholar] [CrossRef]
- Tabarsaei, N.; Hamedani, N.F.; Shafiee, S.; Khandan, S.; Hossaini, Z. Catalyst-free green synthesis and study of antioxidant activity of new pyrazole derivatives. J. Heterocycl. Chem. 2020, 57, 2945–2954. [Google Scholar] [CrossRef]
- Kumara, K.; Prabhudeva, M.G.; Vagish, C.B.; Vivek, H.K.; Rai, K.M.L.; Lokanath, N.K.; Kumar, K.A. Design, synthesis, characterization, and antioxidant activity studies of novel thienyl-pyrazoles. Heliyon 2021, 7, E07592. [Google Scholar] [CrossRef] [PubMed]
- Patila, P.; Yadava, A.; Bavkar, L.; Nippu, B.N.; Satyanarayanc, N.D.; Maned, A.; Gurava, A.; Hangirgekara, S.; Sankpala, S. MerDABCO-SO3H Cl catalyzed synthesis, antimicrobial and antioxidant evaluation and molecular docking study of pyrazolopyranopyrimidines. J. Mol. Struct. 2021, 1242, 130672. [Google Scholar] [CrossRef]
- Popova, S.A.; Pavlova, E.V.; Shevchenko, O.G.; Chukicheva, I.Y.; Kutchin, A.V. Isobornylchalcones as Scaffold for the Synthesis of Diarylpyrazolines with Antioxidant Activity. Molecules 2021, 26, 3579. [Google Scholar] [CrossRef]
- Jagadale, S.M.; Abhale, Y.K.; Pawar, H.R.; Shinde, A.; Bobade, V.D.; Chavan, A.P.; Sarkar, D.; Mhaske, P.C. Synthesis of New Thiazole and Pyrazole Clubbed 1,2,3-Triazol Derivatives as Potential Antimycobacterial and Antibacterial Agents. Polycycl. Aromat. Compd. 2020, 28, 1–22. [Google Scholar] [CrossRef]
- Reddy, G.S.; Snehalatha, A.V.; Edwin, R.K.; Hossain, K.A.; Giliyaru, V.B.; Hariharapura, R.C.; Gautham Shenoy, G.; Misra, P.; Pal, M. Synthesis of 3-indolylmethyl substituted (pyrazolo/benzo)triazinone derivatives under Pd/Cu-catalysis: Identification of potent inhibitors of chorismate mutase (CM). Bioorg. Chem. 2019, 91, 103155. [Google Scholar] [CrossRef]
- Ray, P.C.; Huggett, M.; Turner, P.A.; Taylor, M.; Cleghorn, L.A.T.; Early, J.; Kumar, A.; Bonnett, S.A.; Flint, L.; Joerss, D.; et al. Spirocycle MmpL3 Inhibitors with Improved hERG and Cytotoxicity Profiles as Inhibitors of Mycobacterium tuberculosis Growth. ACS Omega 2021, 6, 2284–2311. [Google Scholar] [CrossRef]
- Hu, X.; Wan, B.; Liu, Y.; Shen, J.; Franzblau, S.G.; Zhang, T.; Ding, K.; Lu, X. Identification of Pyrazolo[1,5-a]pyridine-3-carboxamide Diaryl Derivatives as Drug Resistant Antituberculosis Agents. ACS Med. Chem. Lett. 2019, 10, 295–299. [Google Scholar] [CrossRef]
- Parikh, P.H.; Timaniya, J.B.; Patel, M.J.; Patel, K.P. Microwave-assisted synthesis of pyrano 2,3-c-pyrazole derivatives and their anti-microbial, anti-malarial, anti-tubercular, and anti-cancer activities. J. Mol. Struct. 2022, 1249, 131605. [Google Scholar] [CrossRef]
- Desai, N.C.; Bhatt, K.; Monapara, J.; Pandit, U.; Khedkar, V.M. Conventional and Microwave-Assisted Synthesis, Antitubercular Activity, and Molecular Docking Studies of Pyrazole and Oxadiazole Hybrids. ACS Omega 2021, 6, 28270–28284. [Google Scholar] [CrossRef] [PubMed]
- Shaik, A.B.; Bhandar, R.R.; Nissankararao, S.; Edis, Z.; Tangirala, N.R.; Shahanaaz, S.; Rahman, M.M. Design, facile synthesis and characterization of dichloro substituted chalcones and dihydropyrazole derivatives for their antifungal, antitubercular and antiproliferative activities. Molecules 2020, 25, 3188. [Google Scholar] [CrossRef] [PubMed]
- Pola, S.; Banoth, K.K.; Sankaranarayanan, M.; Ummani, R.; Garlapati, A. Design, synthesis, in silico studies, and evaluation of novel chalcones and their pyrazoline derivatives for antibacterial and antitubercular activities. Med. Chem. Res. 2020, 29, 1819–1835. [Google Scholar] [CrossRef]
- Patel, D.M.; Sharma, M.G.; Vala, R.M.; Lagunes, I.; Puerta, A.; Padron, J.M.; Rajani, D.P.; Patel, H.M. Hydroxyl alkyl ammonium ionic liquid assisted green and one-pot regioselective access to functionalized pyrazolodihydropyridine core and their pharmacological evaluation. Bioorg. Chem. 2019, 86, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Modi, P.; Patel, S.; Chhabria, M. Structure-based design, synthesis and biological evaluation of a newer series of pyrazolo[1,5-a]pyrimidine analogues as potential anti-tubercular agents. Bioorg. Chem. 2019, 87, 240–251. [Google Scholar] [CrossRef]
- Pogaku, V.; Krishna, V.S.; Sriram, D.; Rangan, K.; Basavoju, S. Ultrasonication-ionic liquid synergy for the synthesis of new potent anti-tuberculosis 1,2,4-triazol-1-yl-pyrazole based spirooxindolopyrrolizidines. Bioorg. Med. Chem. Lett. 2019, 29, 1682–1687. [Google Scholar] [CrossRef]
- Brahmbhatt, G.C.; Sutariya, T.R.; Atara, H.D.; Parmar, N.J.; Gupta, V.K.; Lagunes, I.; Padrón, J.M.; Murumkar, P.R.; Yadav, M.R. New pyrazolyl-dibenzo[b,e][1,4]diazepinones: Room temperature one-pot synthesis and biological evaluation. Mol. Divers. 2020, 24, 355–377. [Google Scholar] [CrossRef]
- Kishk, S.M.; McLean, K.J.; Sood, S.; Smith, D.; Evans, J.W.D.; Helal, M.A.; Gomaa, M.S.; Salama, I.; Mostafa, S.M.; De Carvalho, L.P.S.; et al. Design and Synthesis of Imidazole and Triazole Pyrazoles as Mycobacterium Tuberculosis CYP121A1 Inhibitors. ChemistryOpen 2019, 8, 995–1011. [Google Scholar] [CrossRef] [Green Version]
- Vavaiya, B.; Patel, S.; Pansuriya, V.; Marvaniya, V.; Patel, P. Synthesis, anti-tubercular evaluation and molecular docking studies of nitrogen-rich piperazine-pyrimidine-pyrazole hybrid motifs. Curr. Chem. Lett. 2022, 11, 95–104. [Google Scholar] [CrossRef]
- Wong, K.T.; Osman, H.; Parumasivam, T.; Supratman, U.; Che Omar, M.T.; Azmi, M.N. Synthesis, characterization and biological evaluation of new 3,5-disubstituted-pyrazoline derivatives as potential anti-Mycobacterium tuberculosis H37Ra compounds. Molecules 2021, 26, 2081. [Google Scholar] [CrossRef]
- Chen, H.; Wang, B.; Li, P.; Yan, H.; Li, G.; Huang, H.H.; Lu, Y. The optimization and characterization of functionalized sulfonamides derived from sulfaphenazole against Mycobacterium tuberculosis with reduced CYP 2C9 inhibition. Bioorg. Med. Chem. Lett. 2021, 40, 127924. [Google Scholar] [CrossRef] [PubMed]
- Takate, S.J.; Shinde, A.D.; Karale, B.K.; Akolkar, H.; Nawale, L.; Sarkar, D.; Mhaske, P.C. Thiazolyl-pyrazole derivatives as potential antimycobacterial agents. Bioorg. Med. Chem. Lett. 2019, 29, 1199–1202. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Dong, J.; Lin, H.Y.; Wang, M.Y.; Li, X.K.; Zheng, B.F.; Chen, Q.; Hao, G.F.; Yang, W.C.; Yang, G.F. Pyrazole-Isoindoline-1,3-dione Hybrid: A Promising Scaffold for 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors. J. Agric. Food Chem. 2019, 67, 10844–10852. [Google Scholar] [CrossRef]
- Jiang, B.B.; Guo, B.B.; Cui, J.L.; Dong, Y.W.; Cui, L.; Zhang, L.; Yang, Q.; Yang, X.L. New lead discovery of insect growth regulators based on the scaffold hopping strategy. Bioorg. Med. Chem. Lett. 2020, 30, 127500. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Li, H.G.; Sun, P.W.; Gao, L.; Liu, J.B.; Zhou, S.; Xiong, L.X.; Yang, N.; Li, Y.X.; Li, Z.M. Synthesis, biological activities, and SAR studies of novel 1-(2-chloro-4,5-difluorophenyl)-1H-pyyrazole derivatives. Bioorg. Med. Chem. Lett. 2020, 30, 127535. [Google Scholar] [CrossRef] [PubMed]
- Judge, N.R.; Chacktas, G.; Ma, L.; Schink, A.; Buckpesch, R.; Schmutzler, D.; Machettira, A.B.; Dietrich, H.; Asmus, E.; Bierer, D.; et al. Flexible Synthesis and Herbicidal Activity of Fully Substituted 3-Hydroxypyrazoles. Eur. J. Org. Chem. 2021, 2021, 5677–5684. [Google Scholar] [CrossRef]
- Khallaf, A.; Wang, P.; Zhuo, S.P.; Zhu, H.J.; Liu, H. Synthesis, insecticidal activities, and structure-activity relationships of 1,3,4-oxadiazole-ring-containing pyridylpyrazole-4-carboxamides as novel insecticides of the anthranilic diamide family. J. Heterocycl. Chem. 2021, 58, 2189–2202. [Google Scholar] [CrossRef]









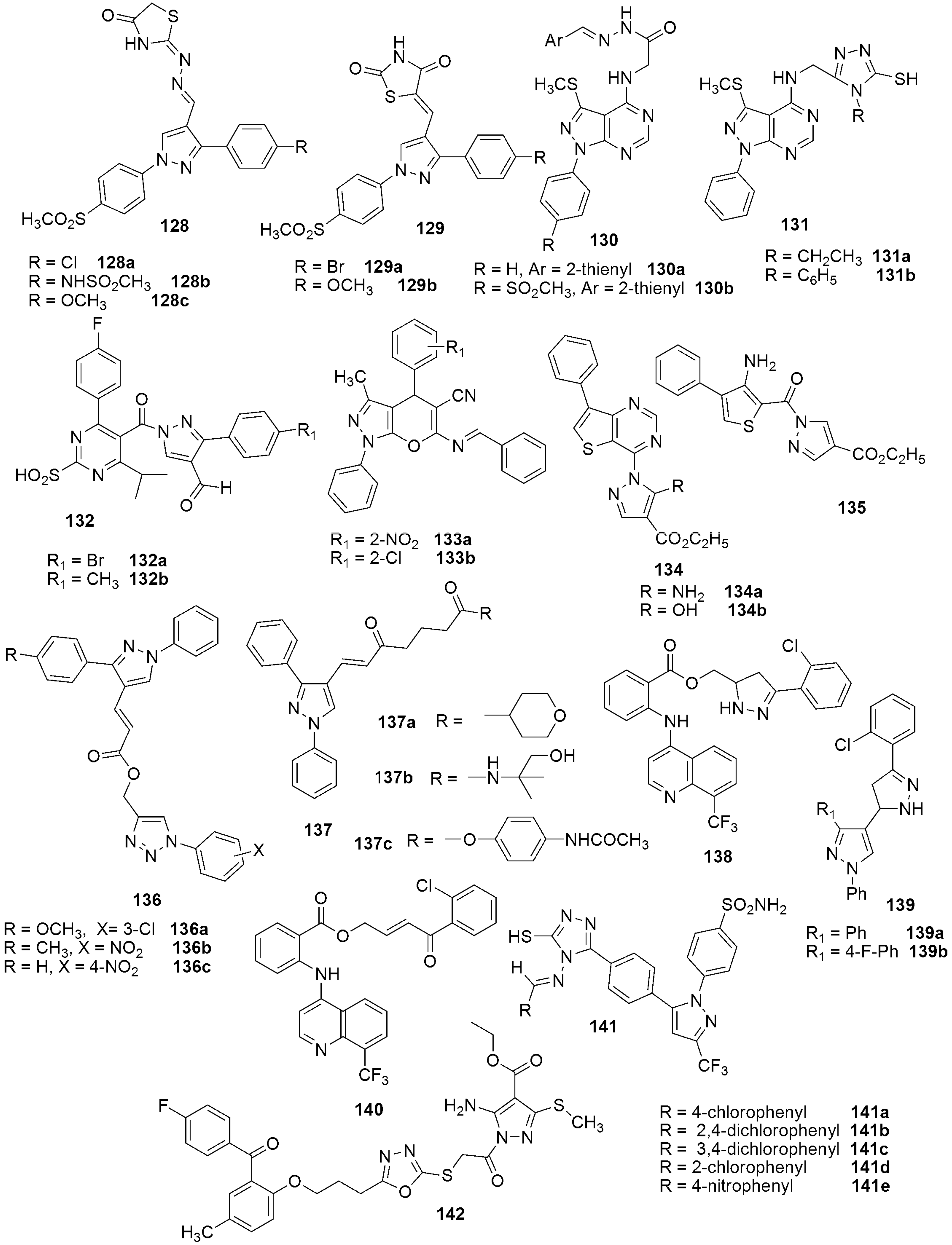

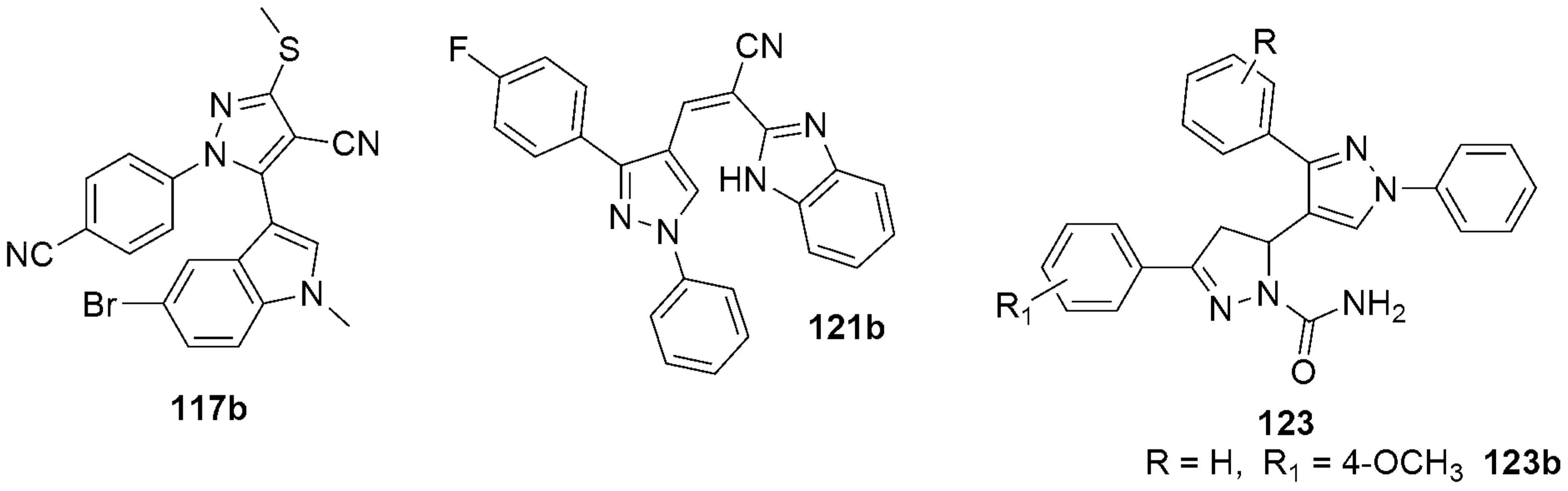


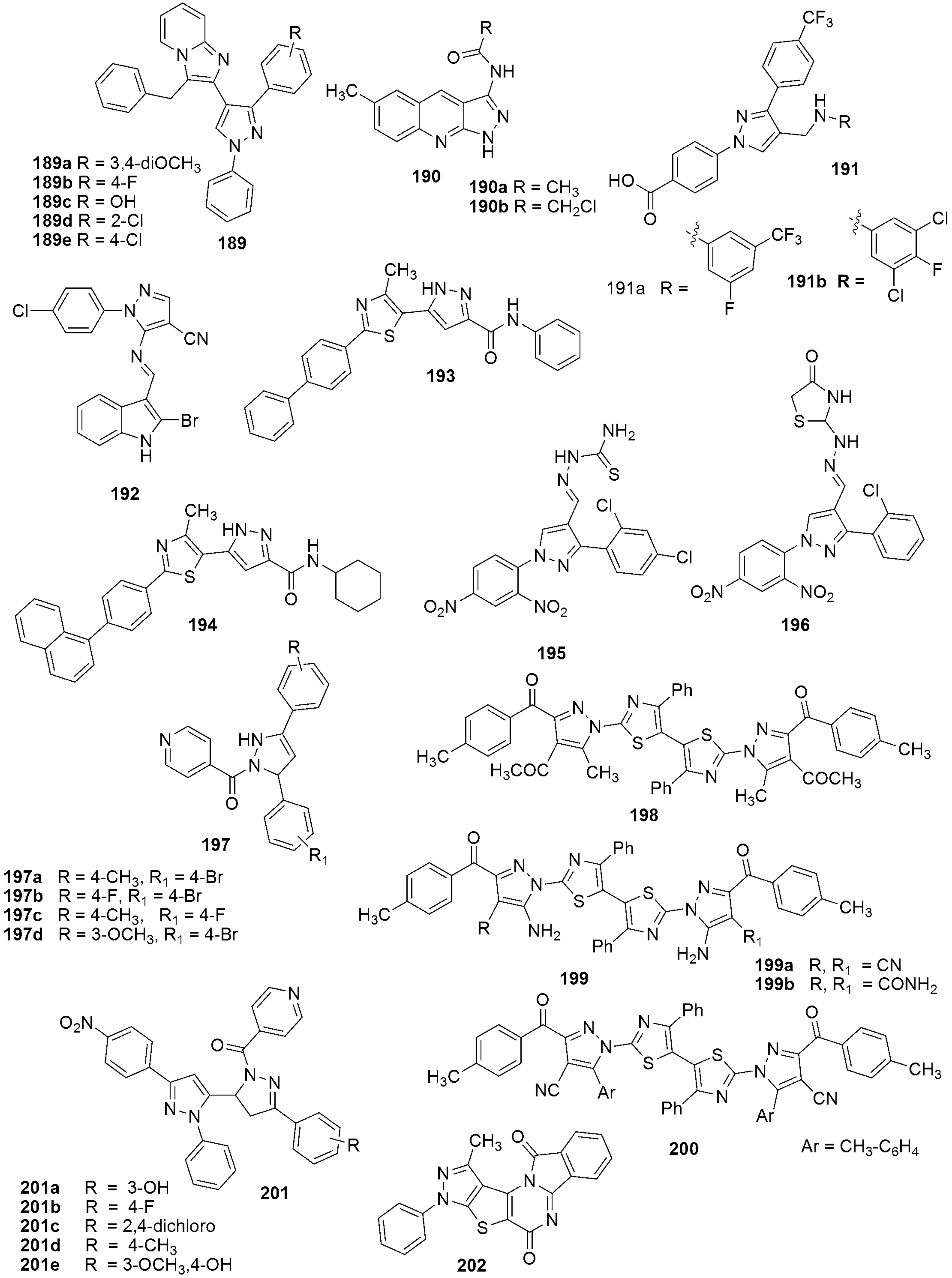
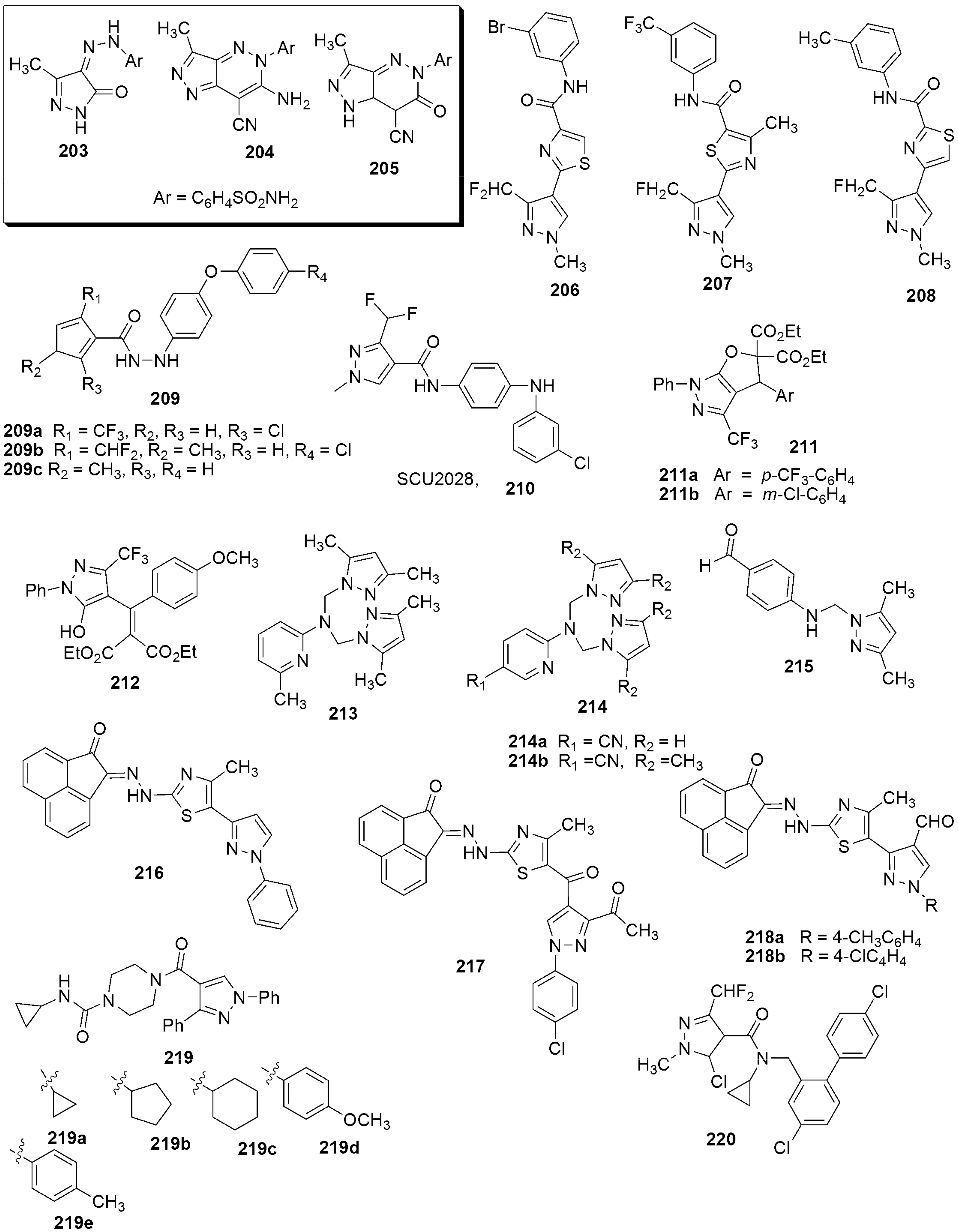
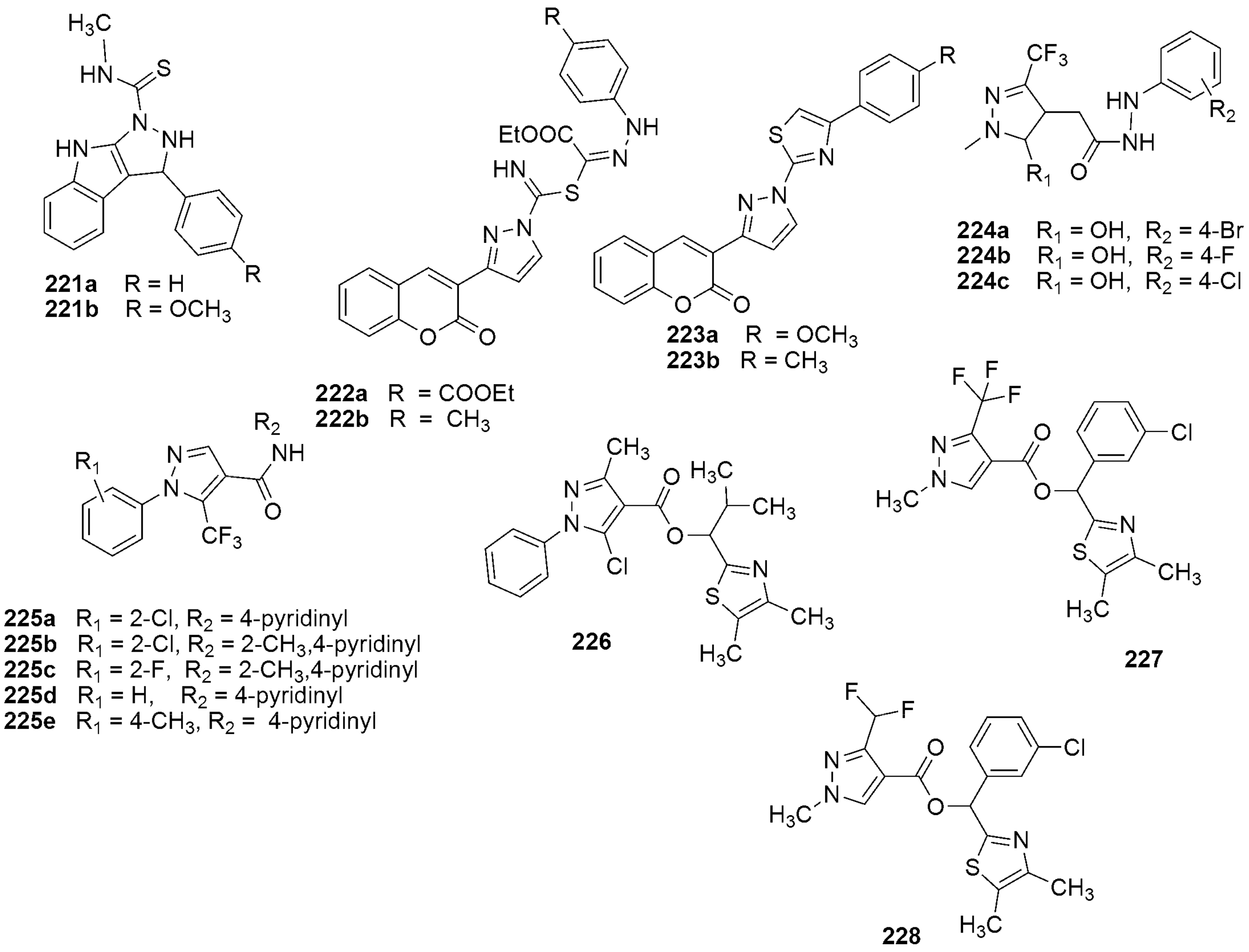
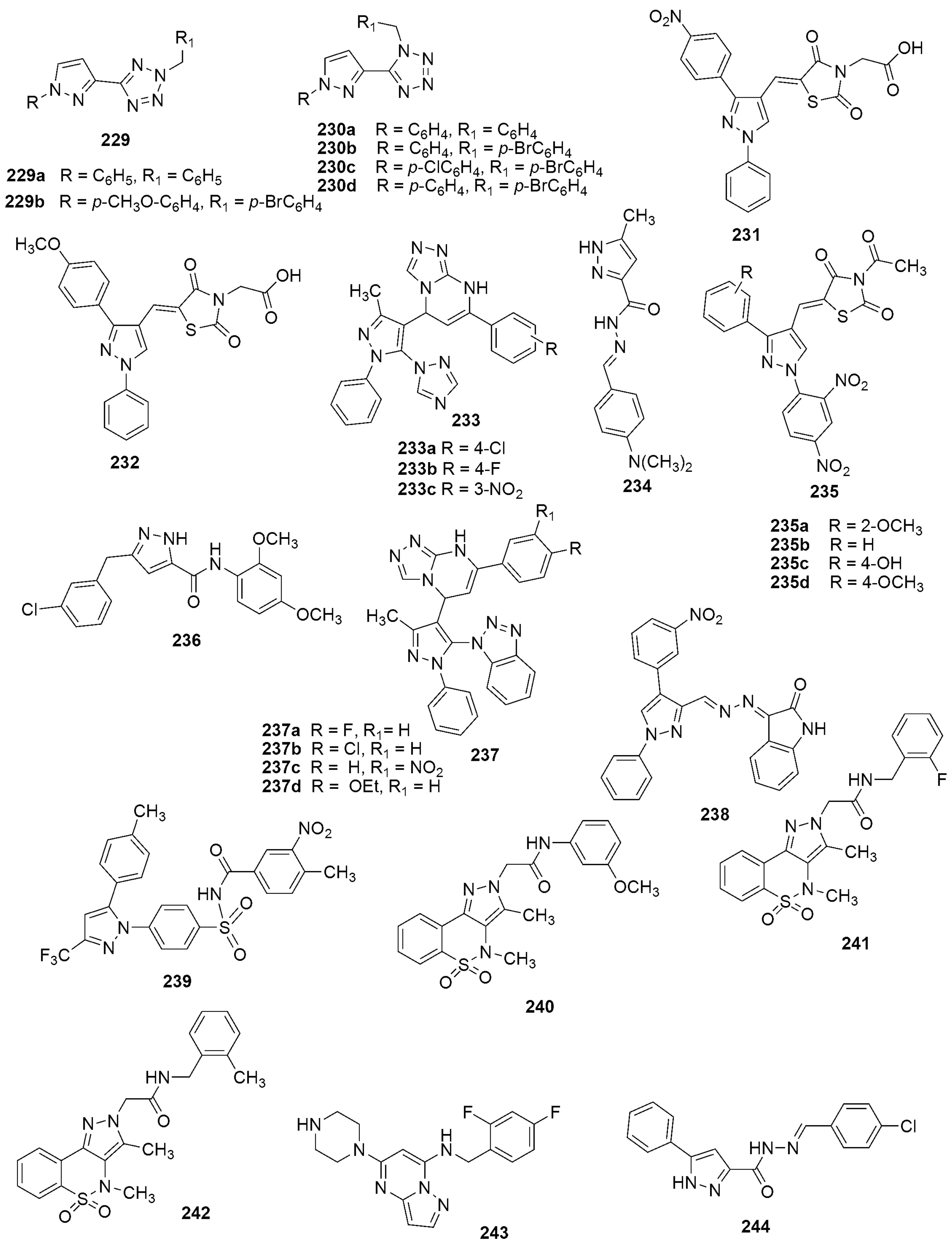

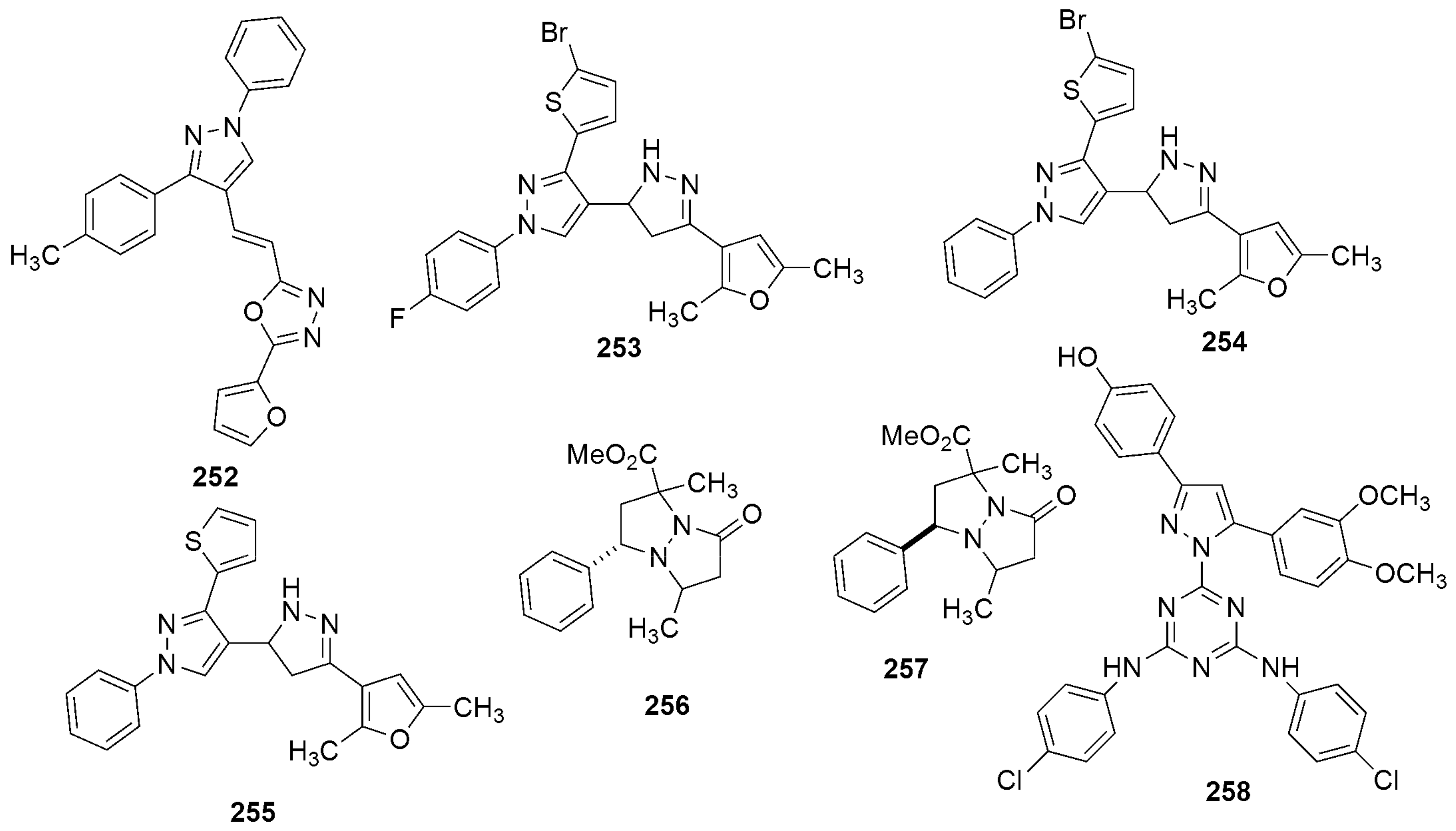

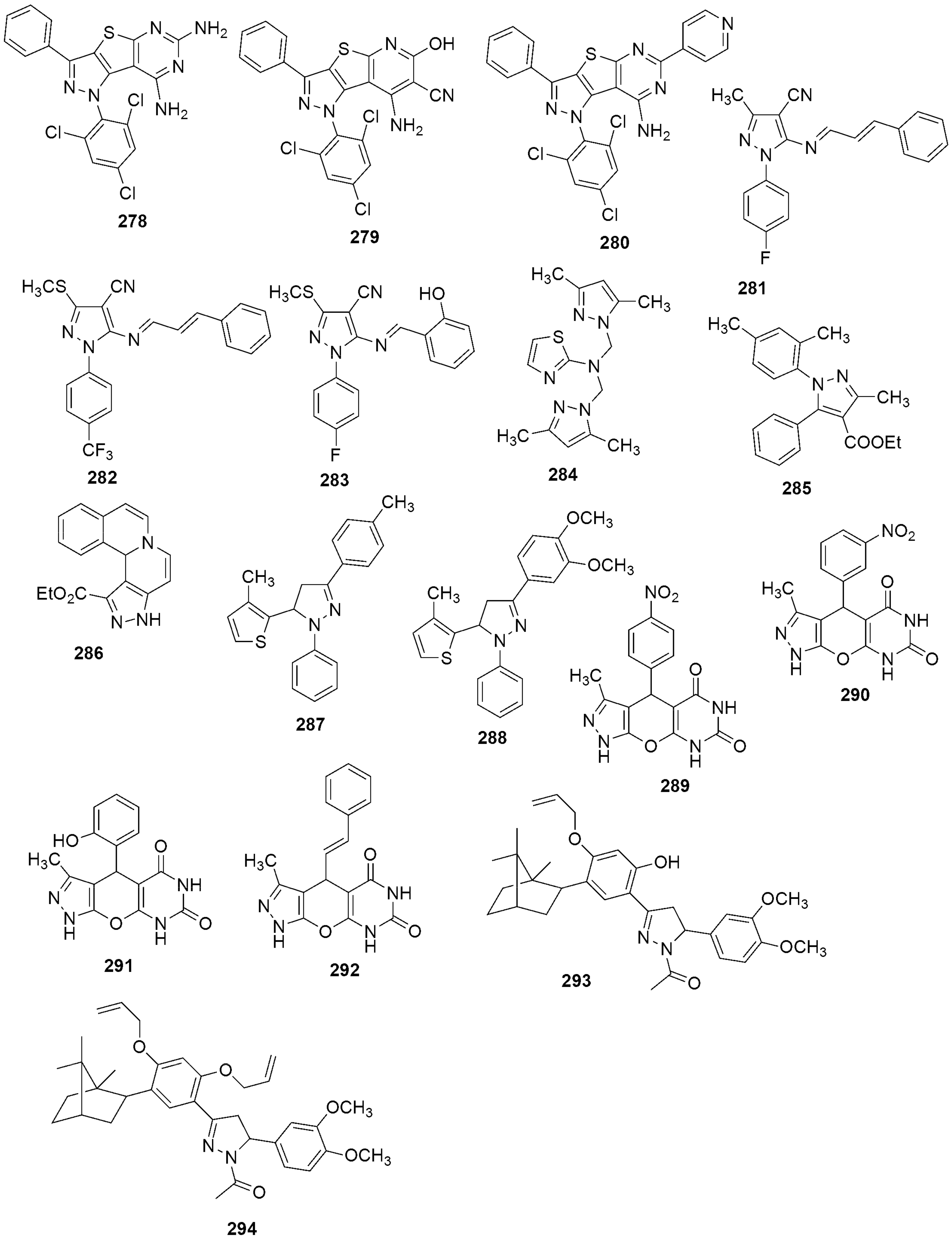

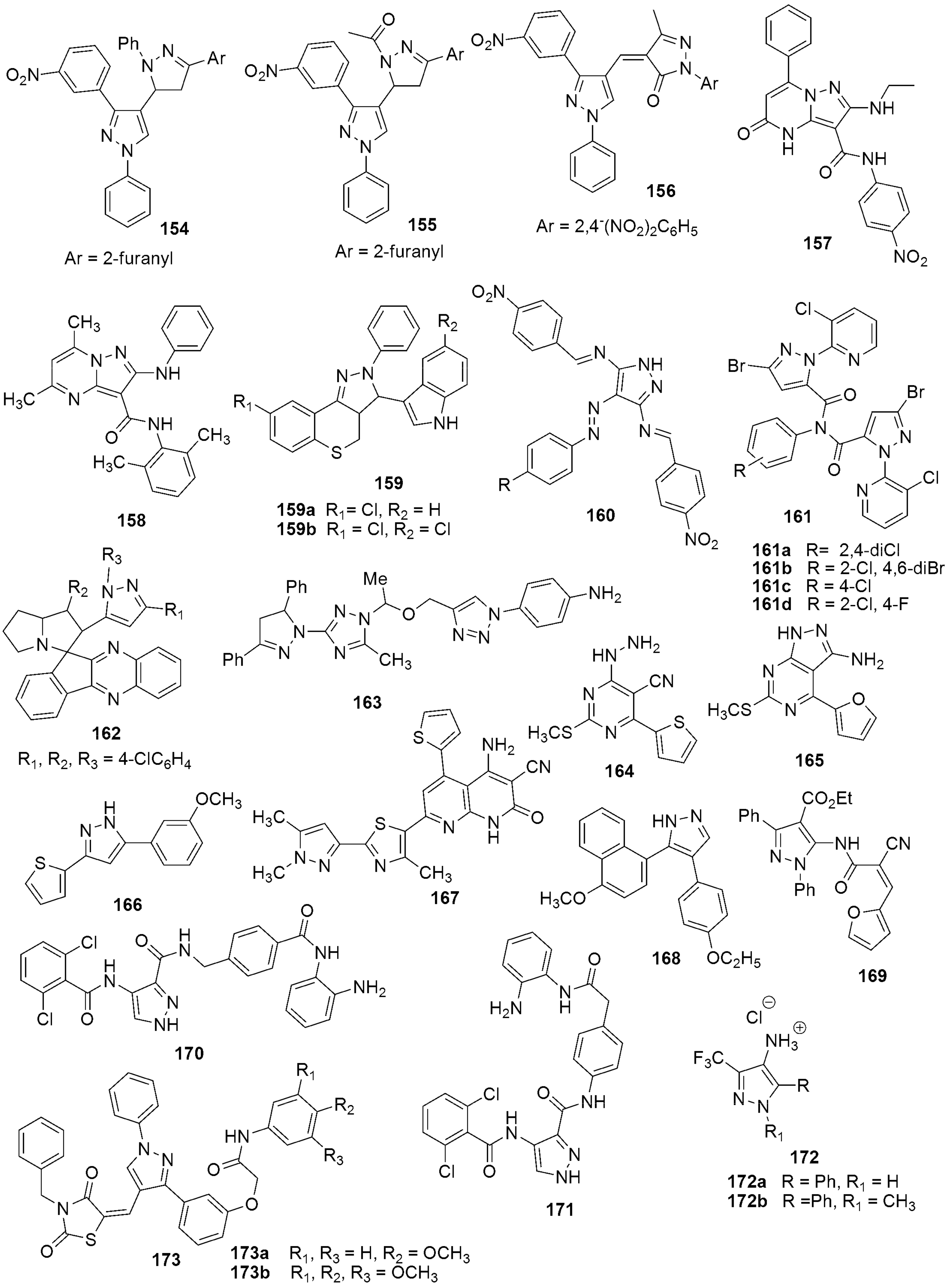
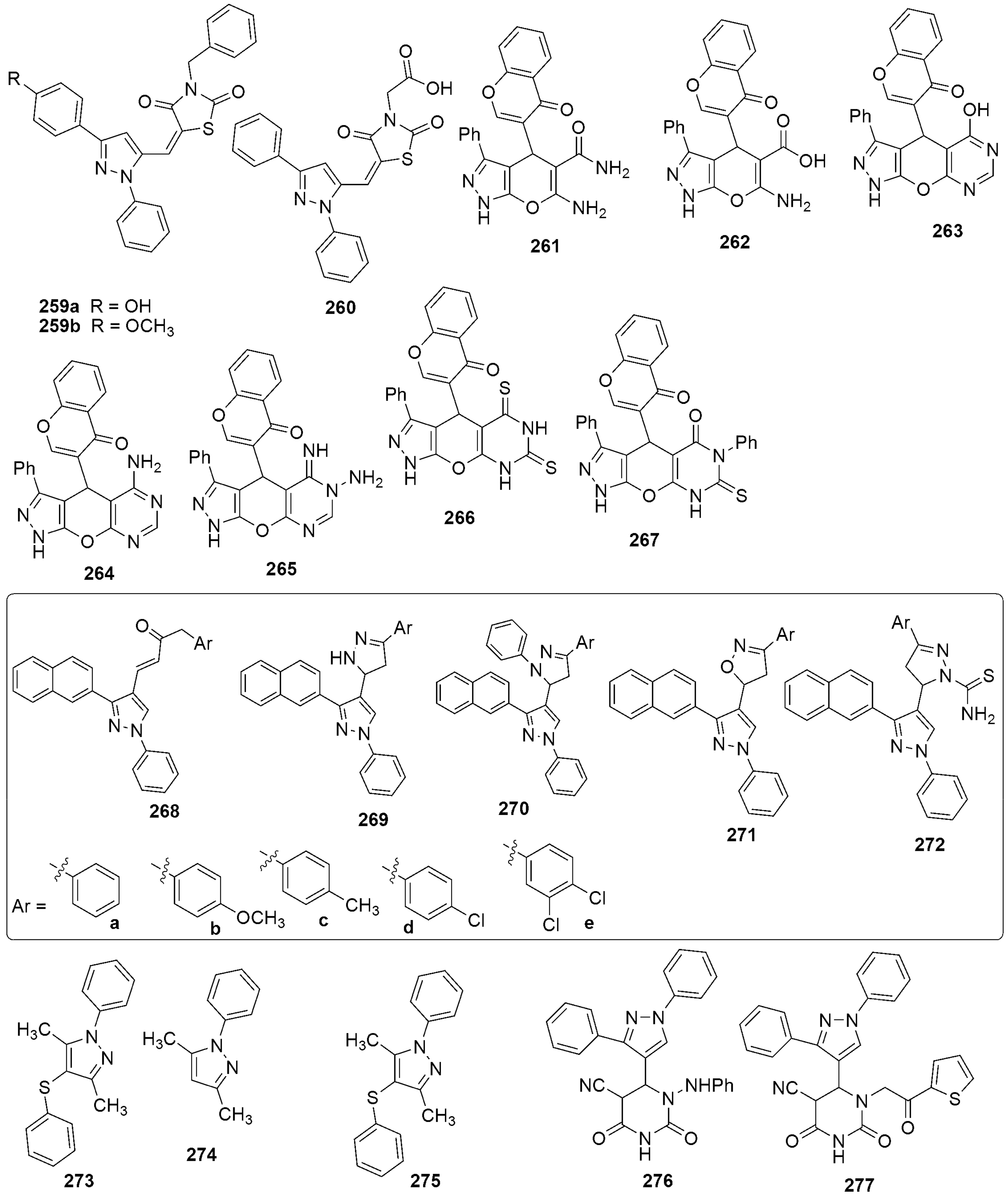
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
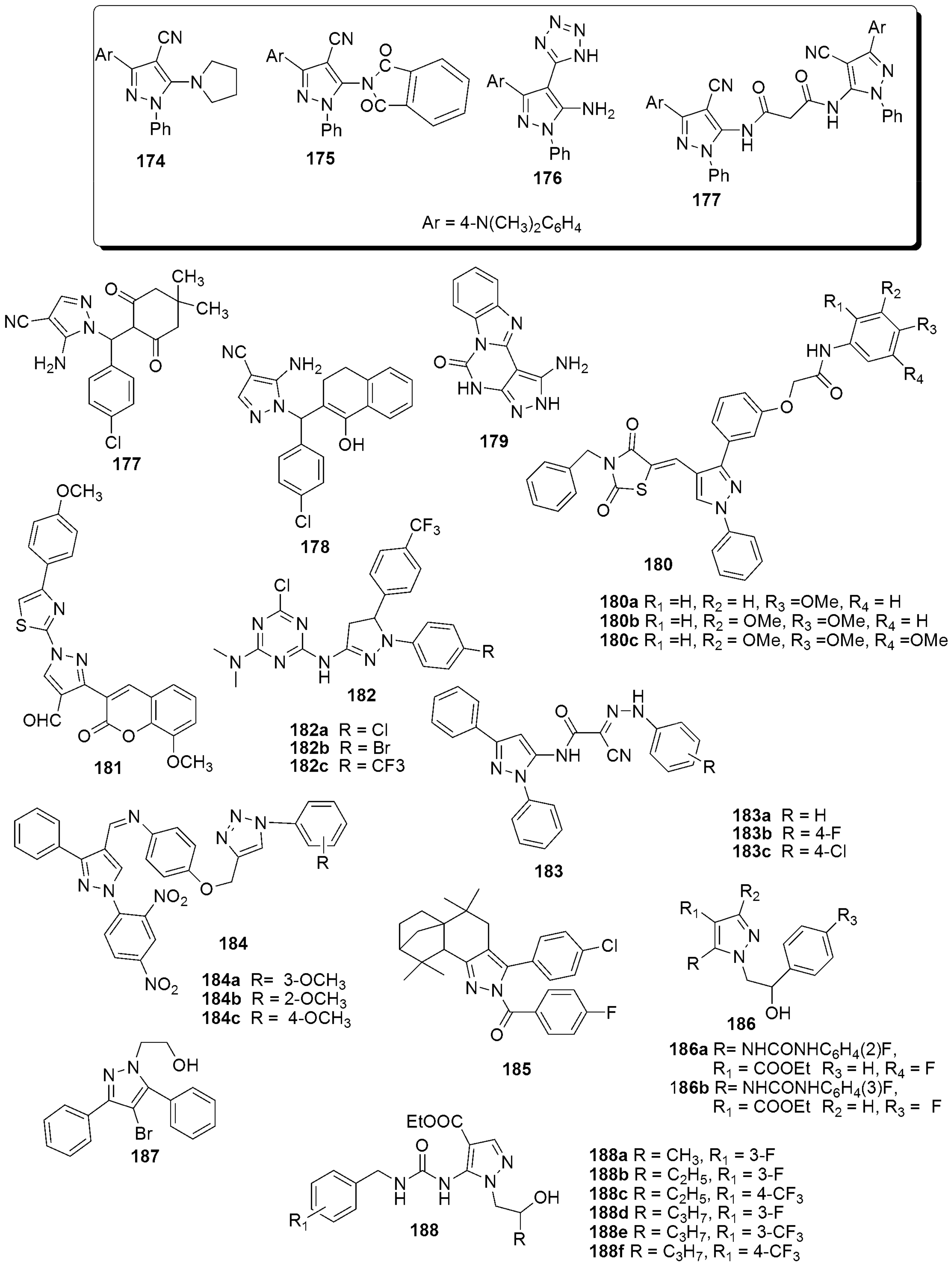
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
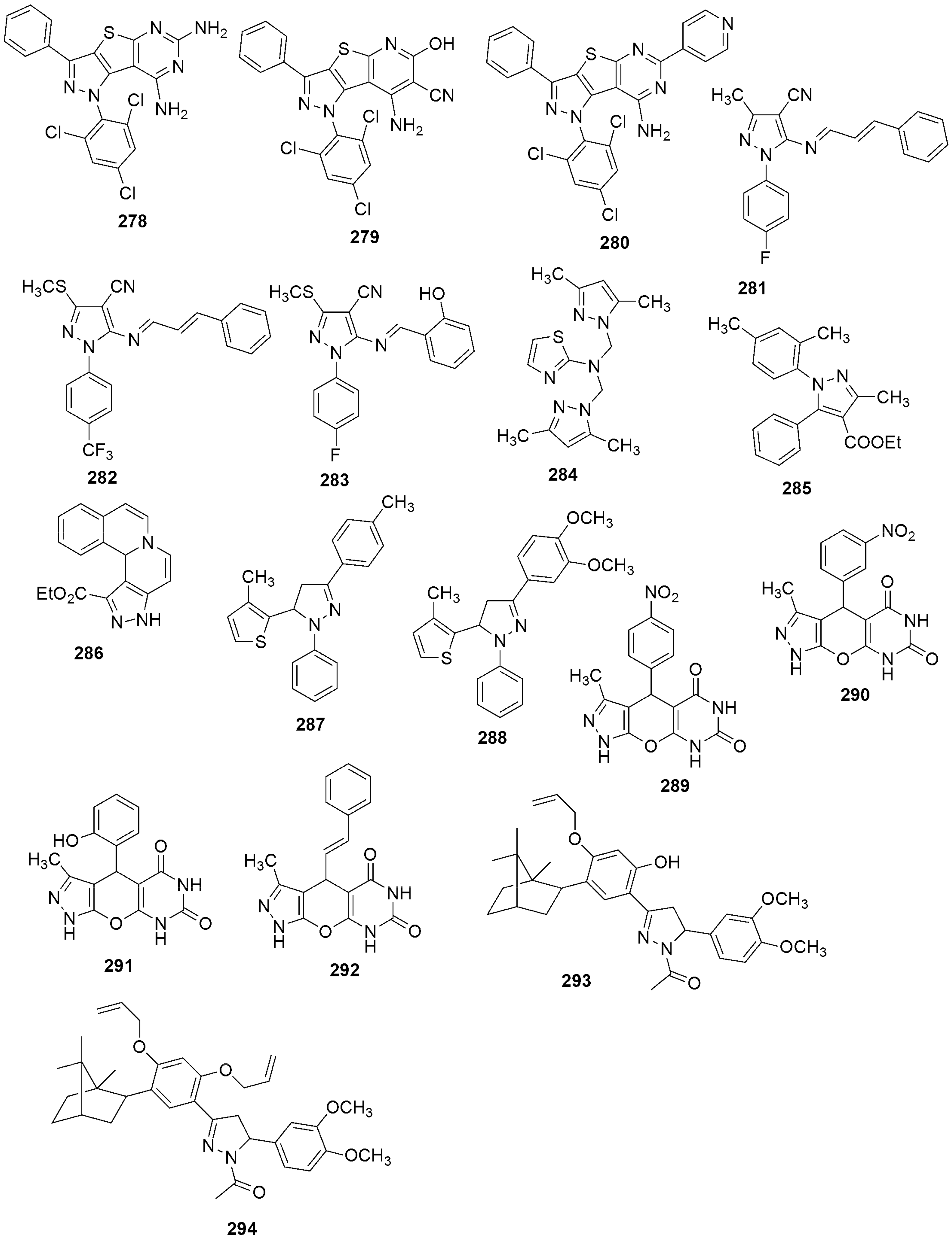{kind=link}
{kind=link}
{kind=link}
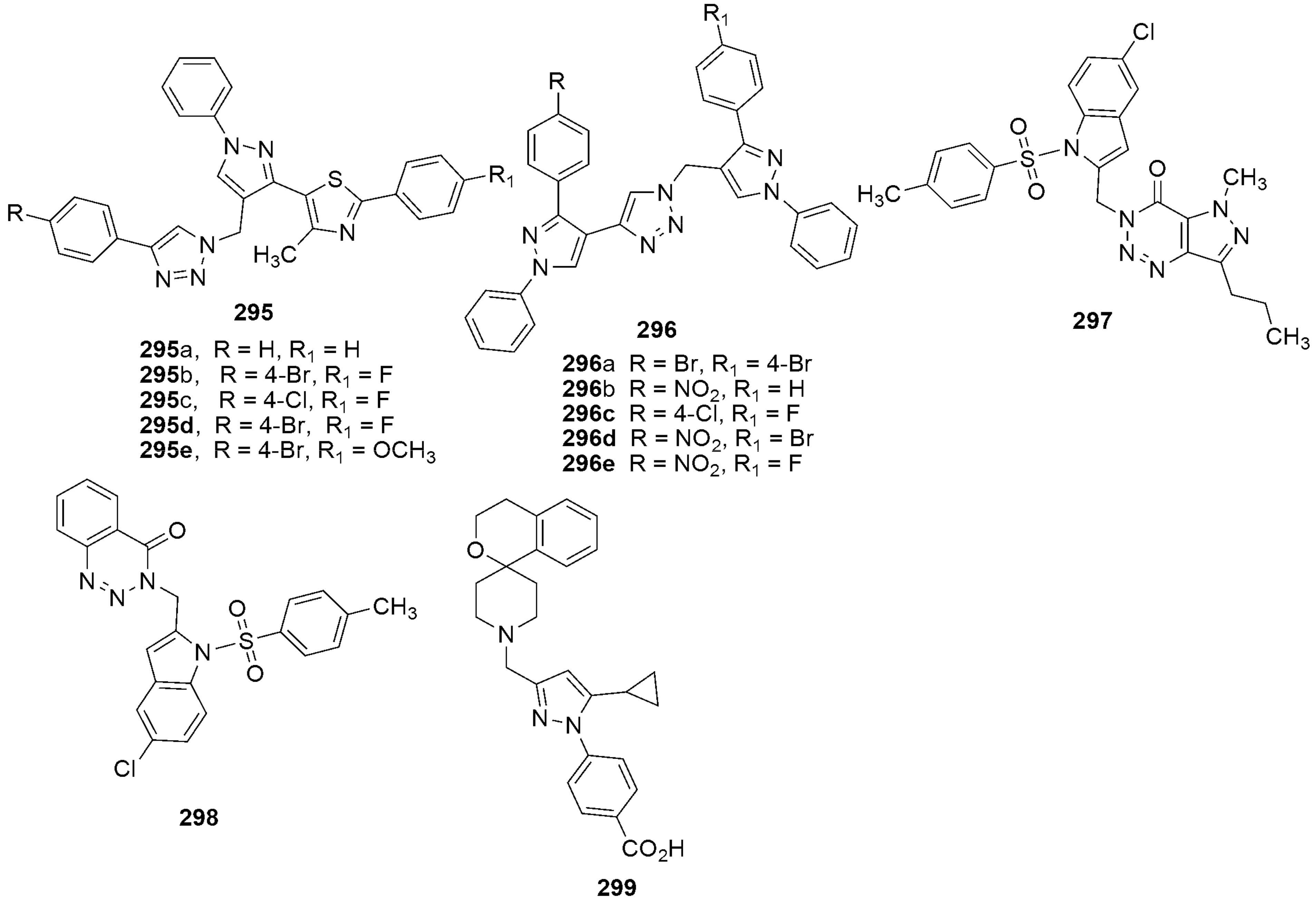
{kind=link}
{kind=link}
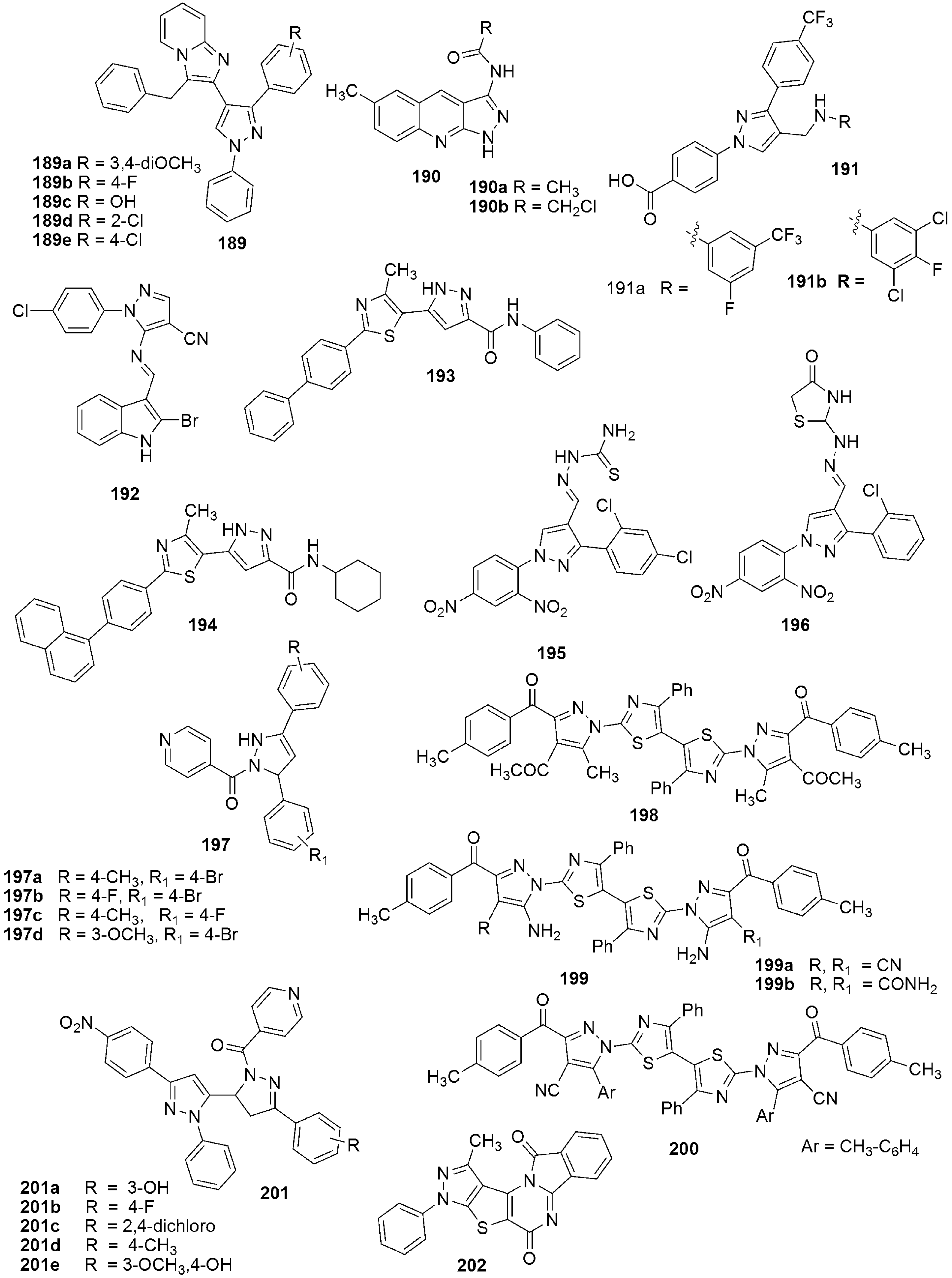
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
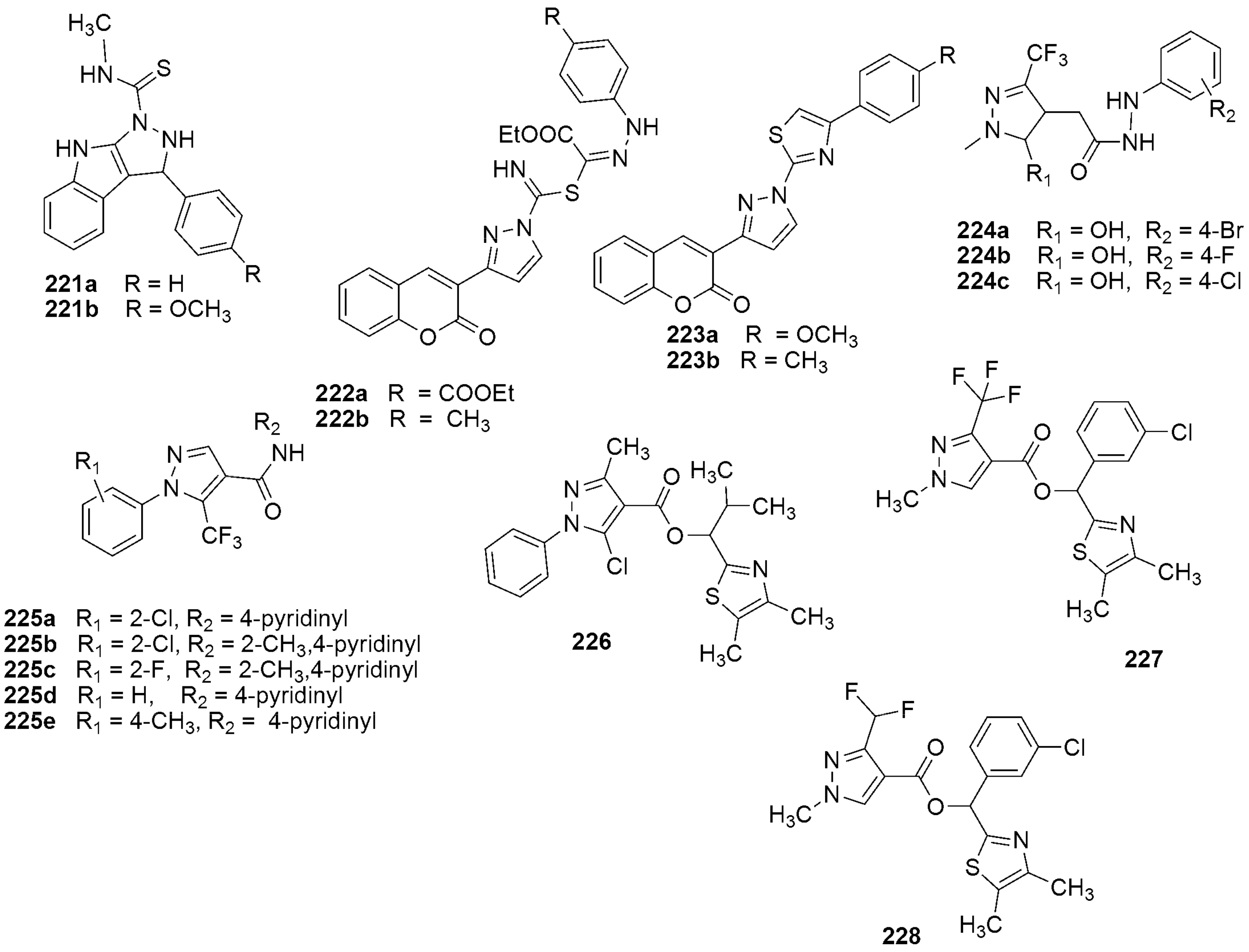
{kind=link}
{kind=link}
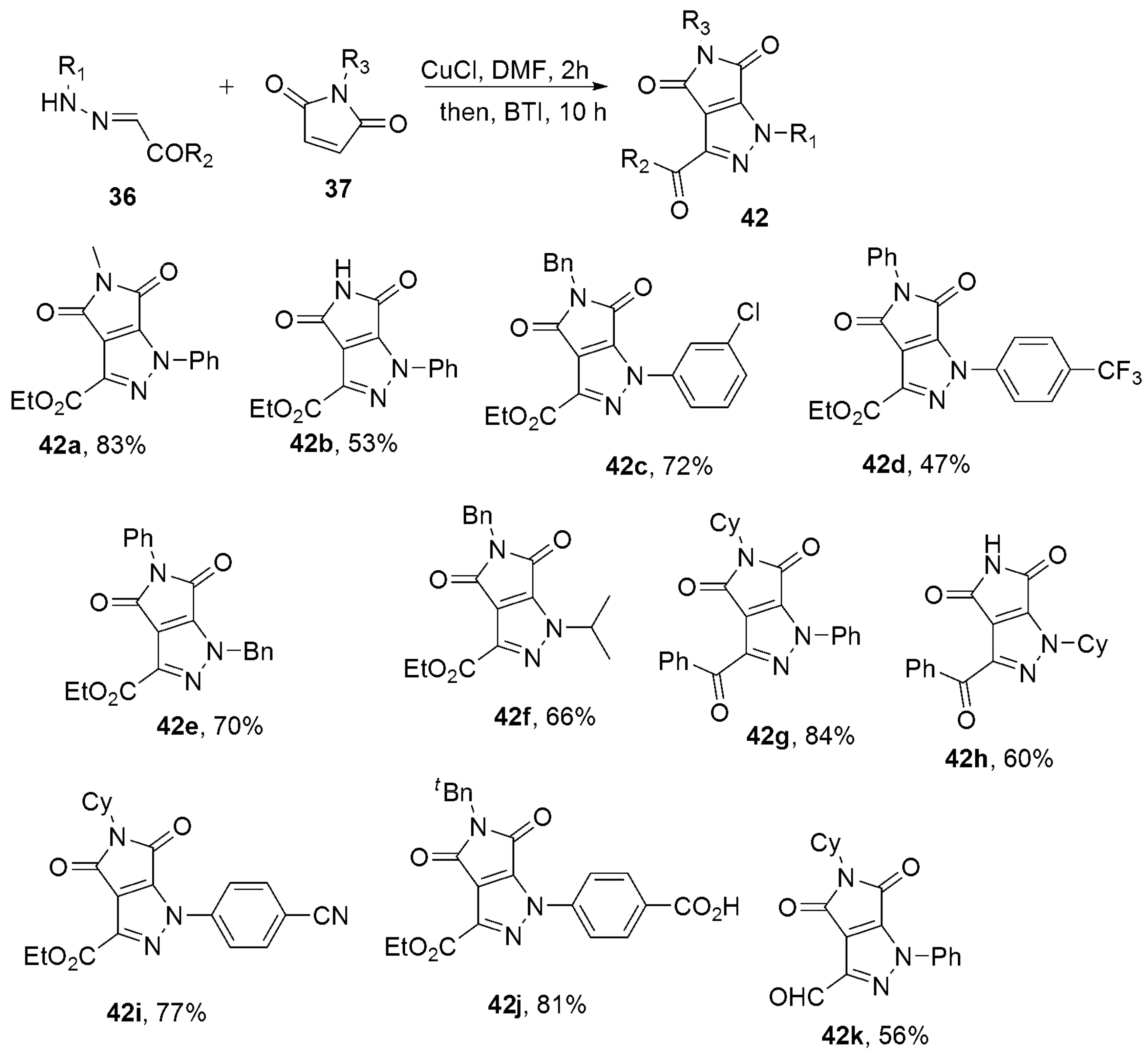{kind=link}
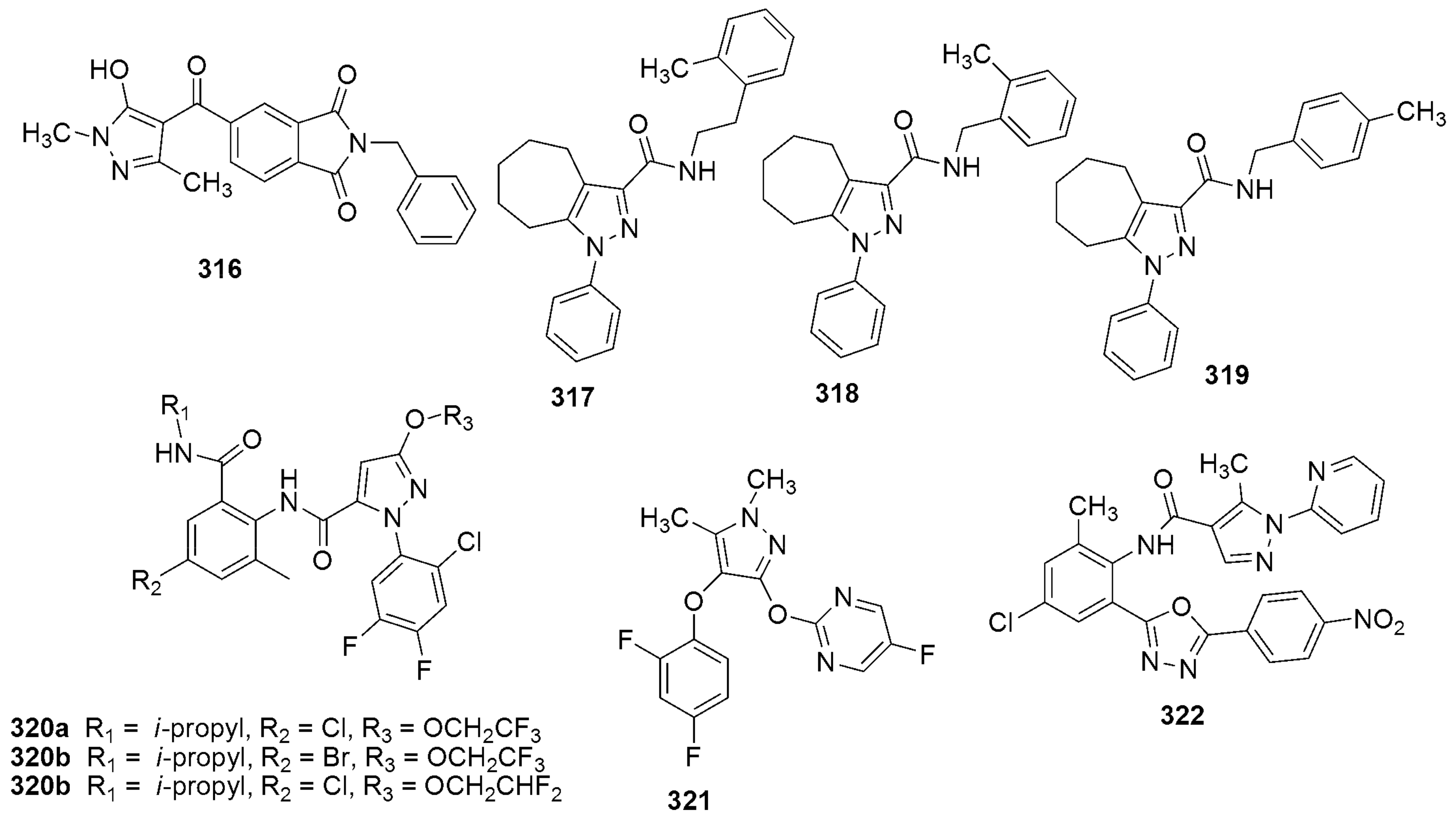
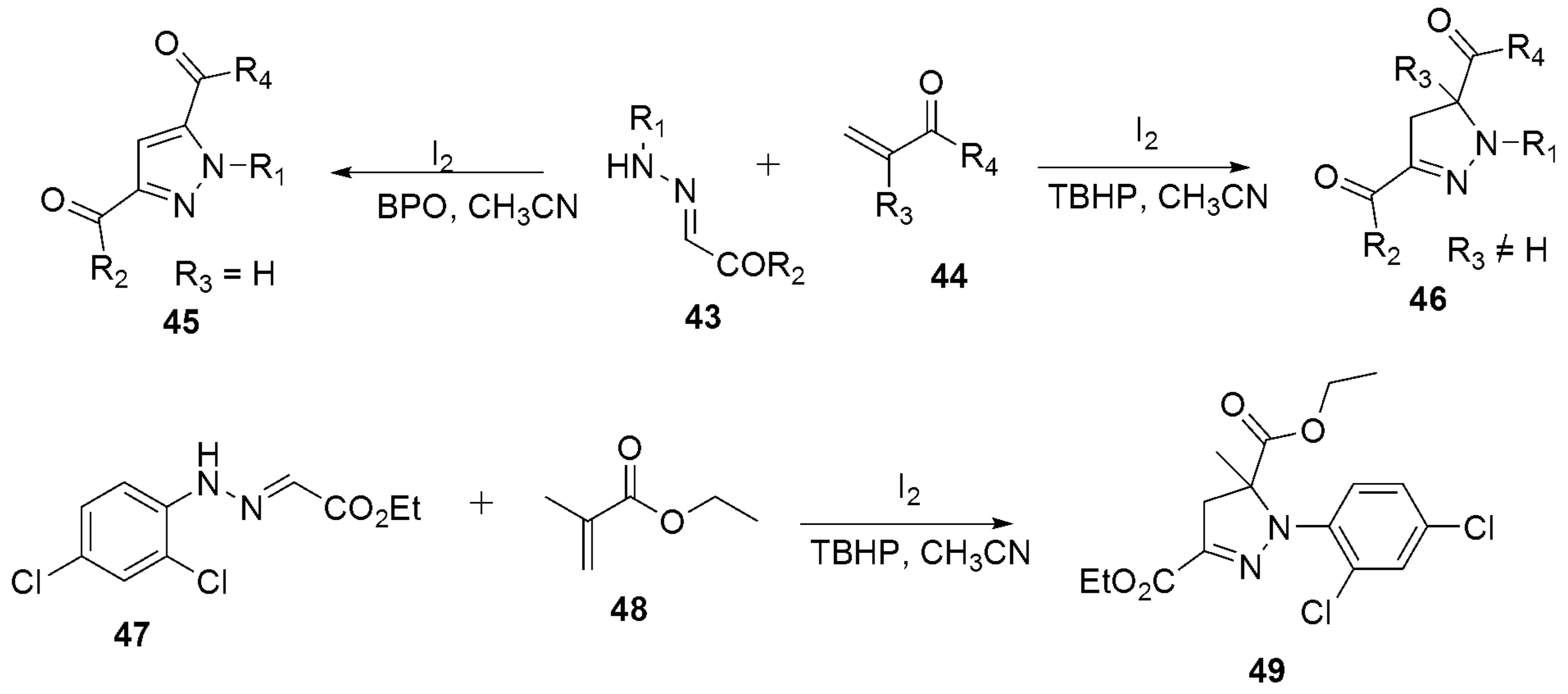{kind=link}
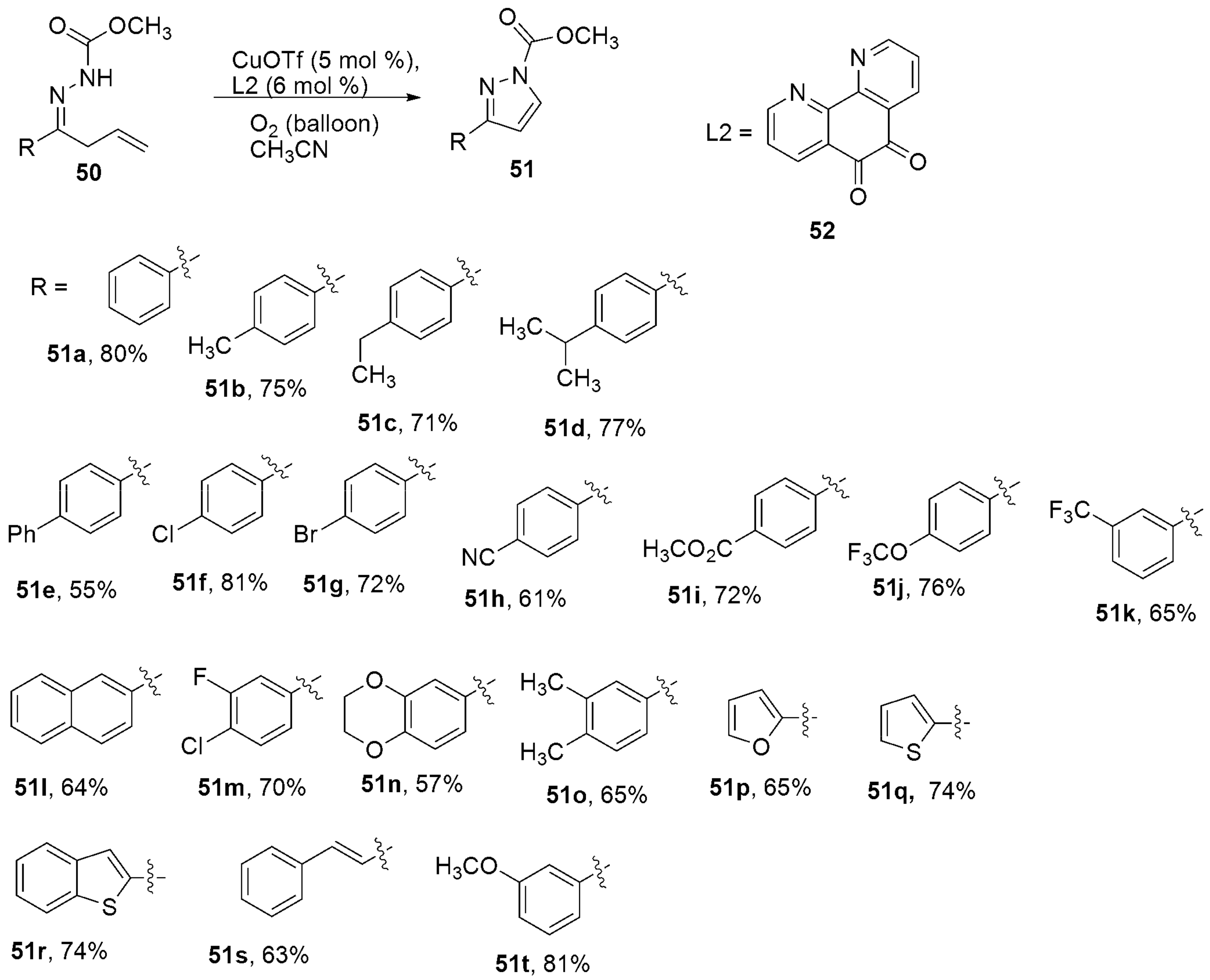{kind=link}
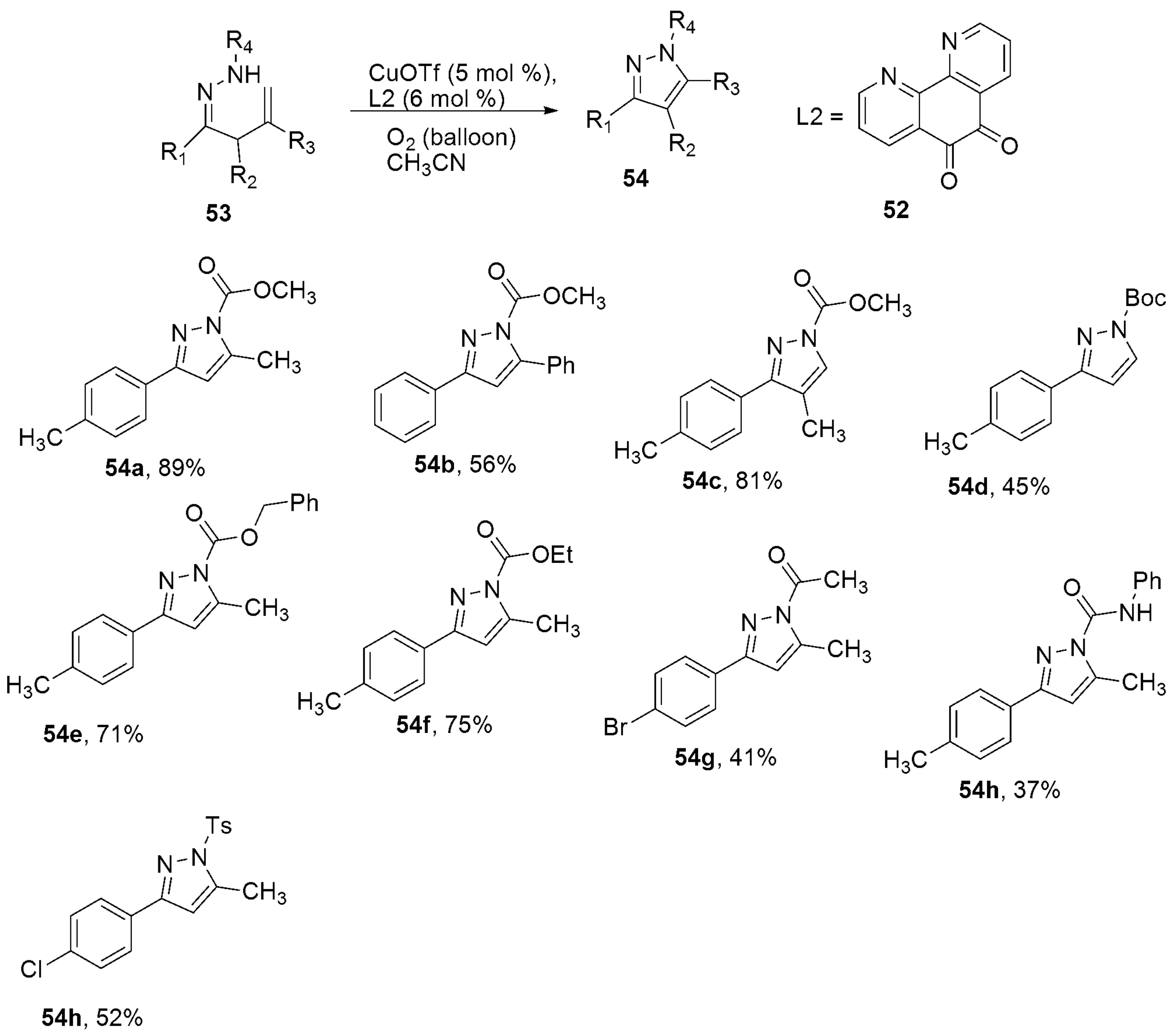{kind=link}
{kind=link}
{kind=link}
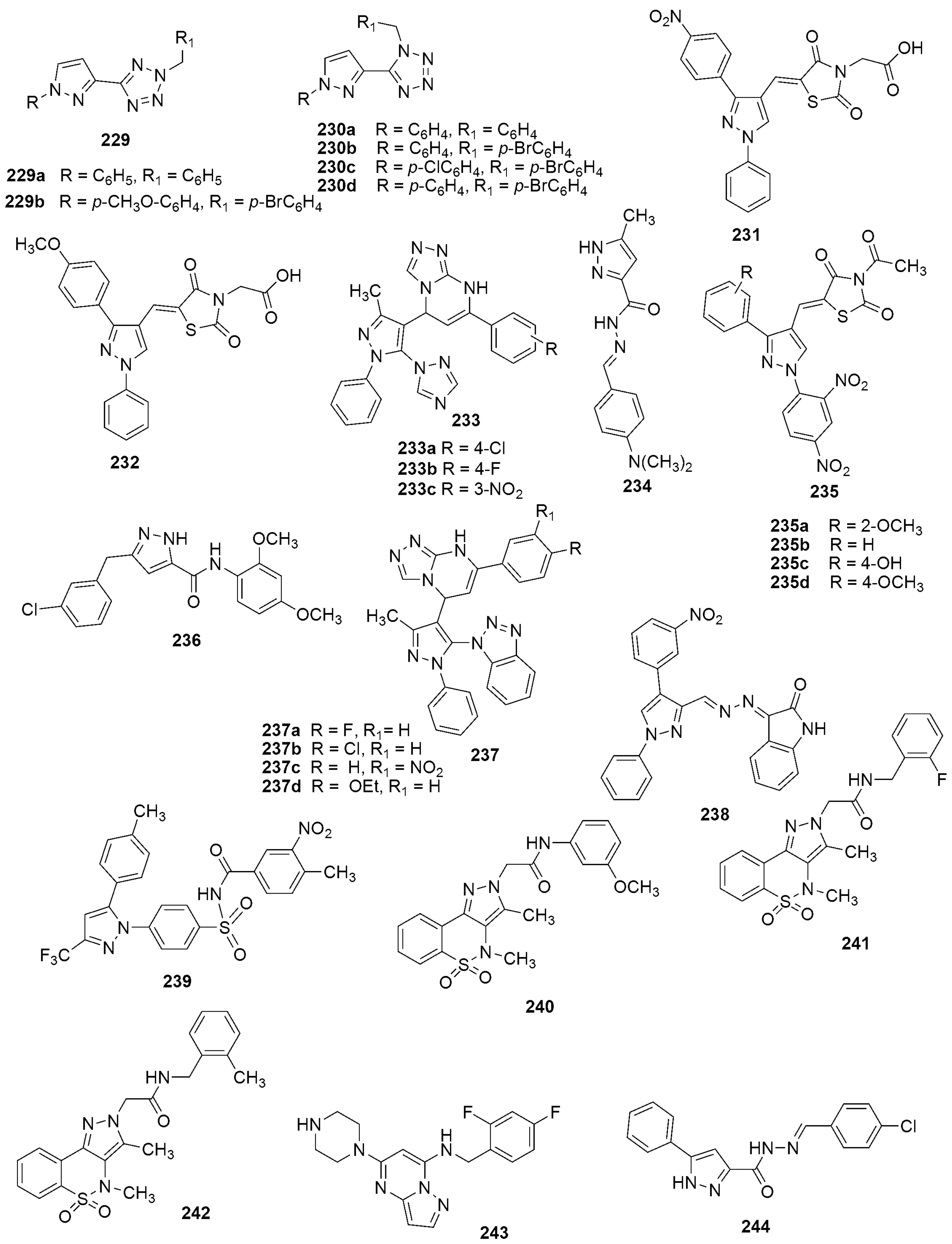
{kind=link}
{kind=link}
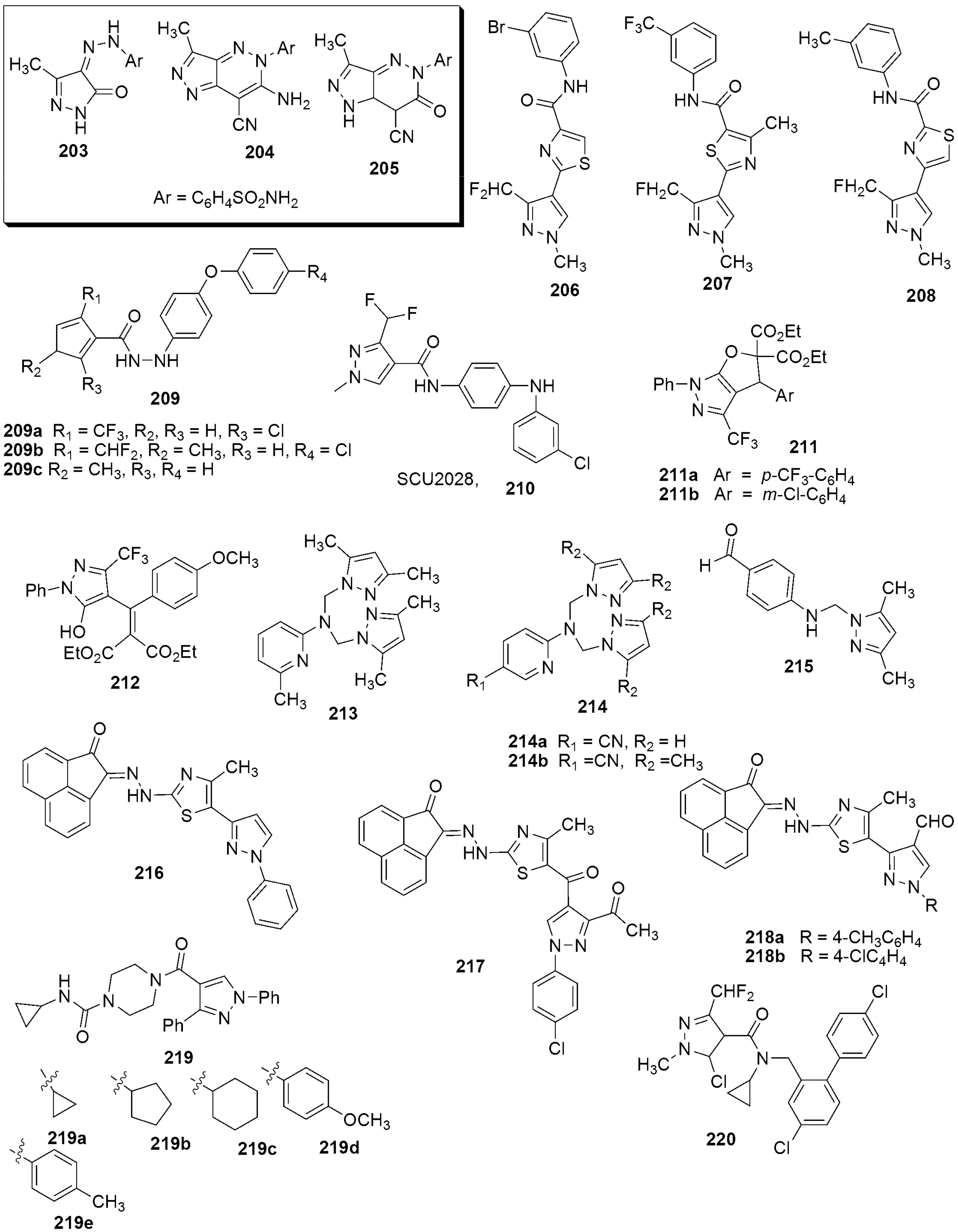
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 301 301MIC (μg/mL) = 25 against H37RV strain | [140] | |||
 302 302 | [141] | |||
| IC50 (μg/mL) | ||||
| H37Ra | M. bovis BCG | |||
| 302a R = 4-NO2-C6H4 | 0.52 | 302f R = 3-CH3-C6H4 | 0.92 | |
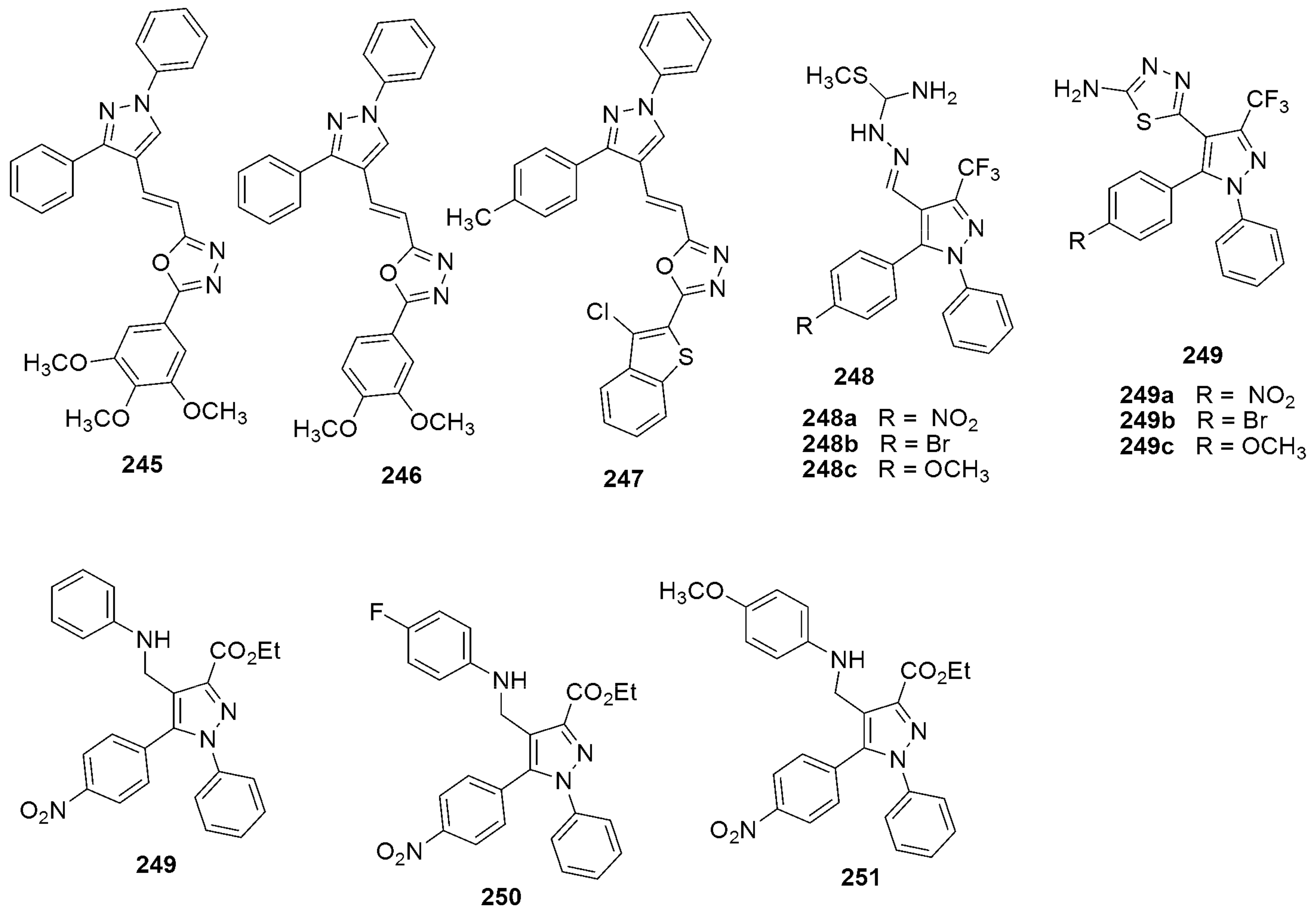
| 302b R = 4-OCH3-C6H4 | 0.50 | 302g R = 4-CH3-C6H4 | 0.97 | |
| 302c R = 4-Cl-C6H4 | 0.79 | 302e R = CH2-O-C6H4-2-Cl | 0.62 | |
| 302d R = CH2-O-C6H4-2-NO2 | 1.48 | |||
| 302e R = CH2-O-C6H4-2-Cl | 0.53 | |||
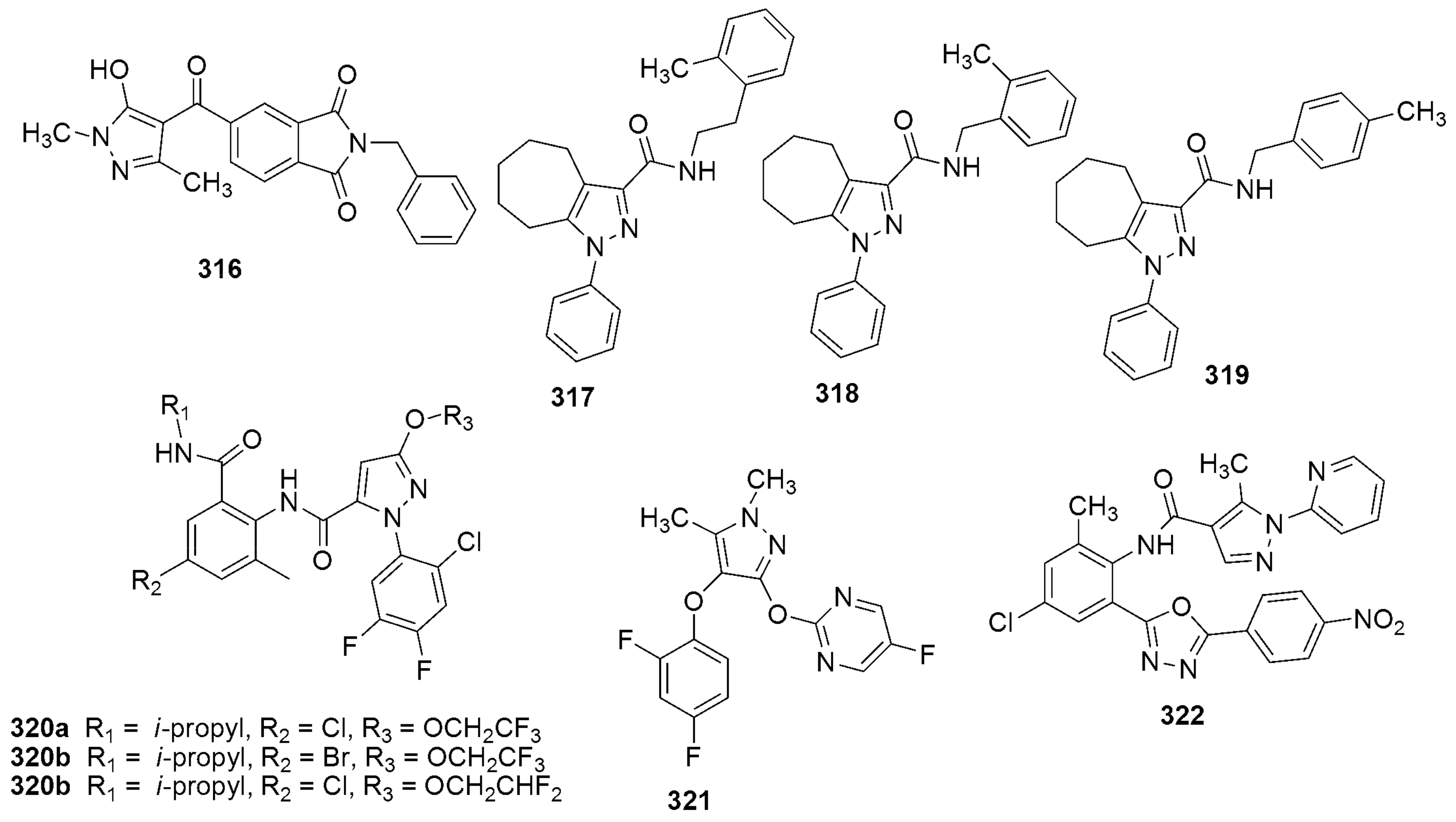
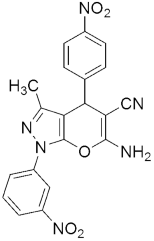
 303a 303b MIC(µM) = 3.96 and 3.67 µM against H37RV | [142] | |||
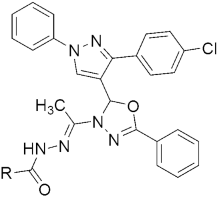
 304 304MIC(µM) = 6.25, against H37RV | [143] | |||
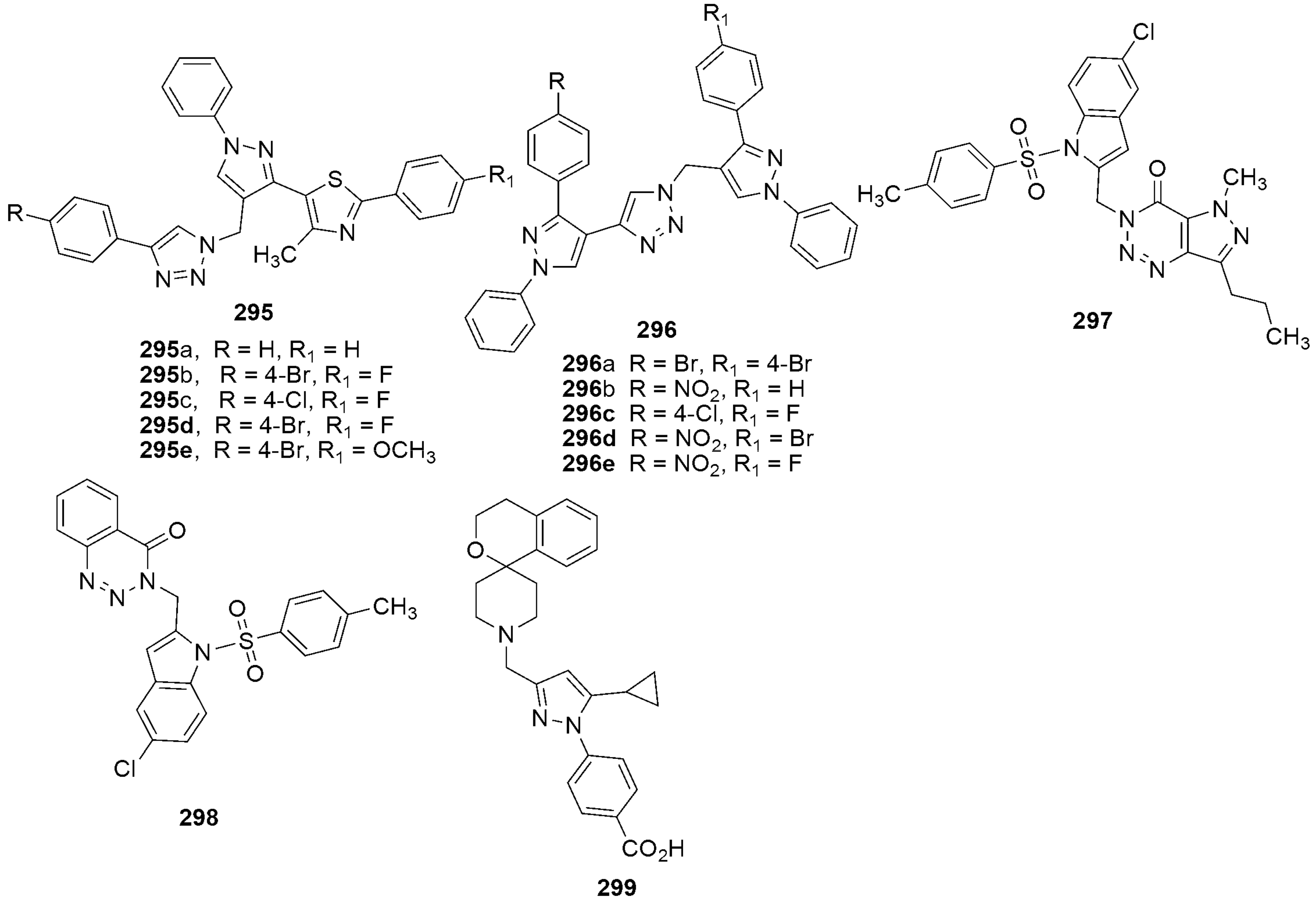
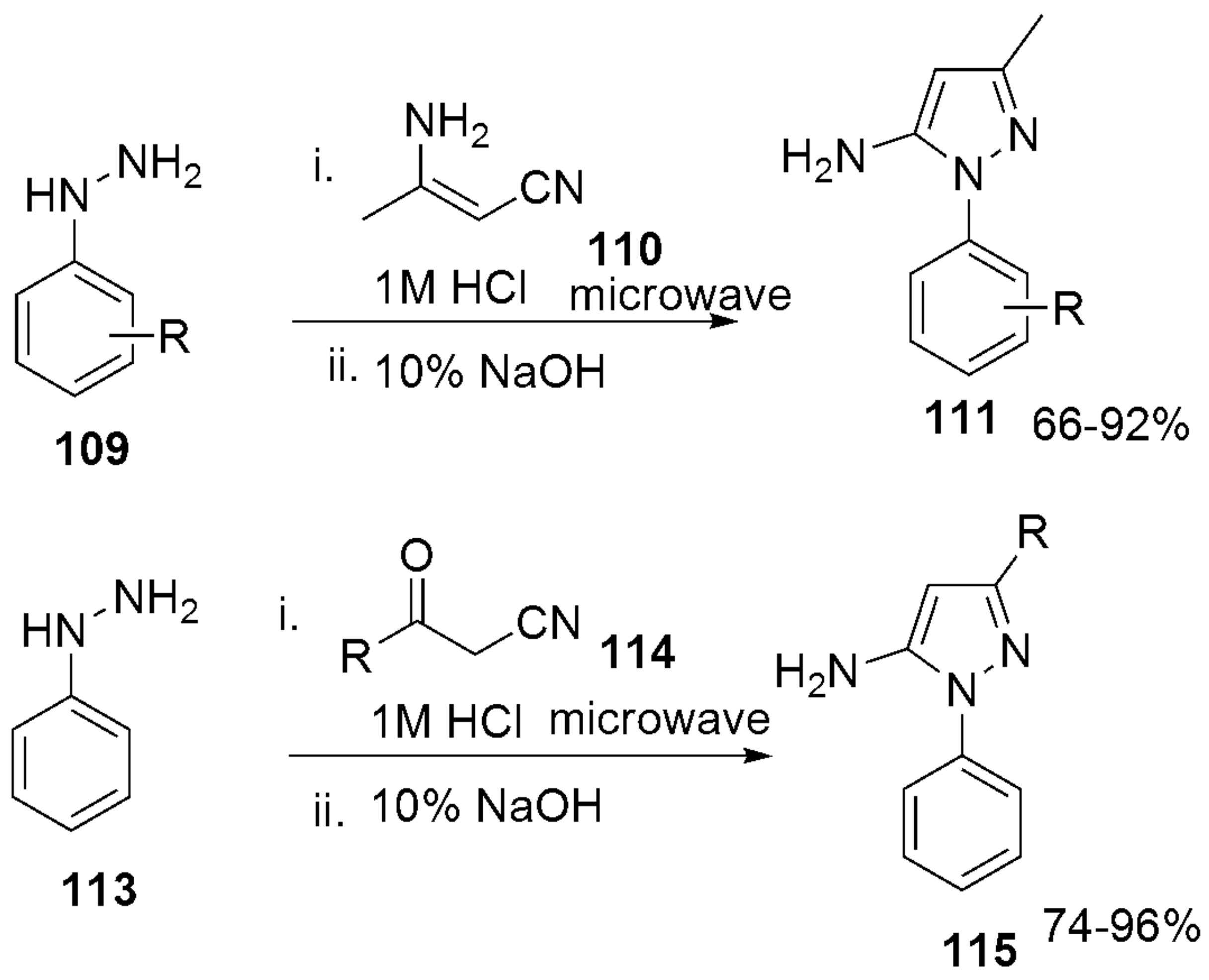
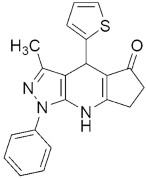 305 305MIC (μg/mL) = 25 against H37RV | [144] | |||
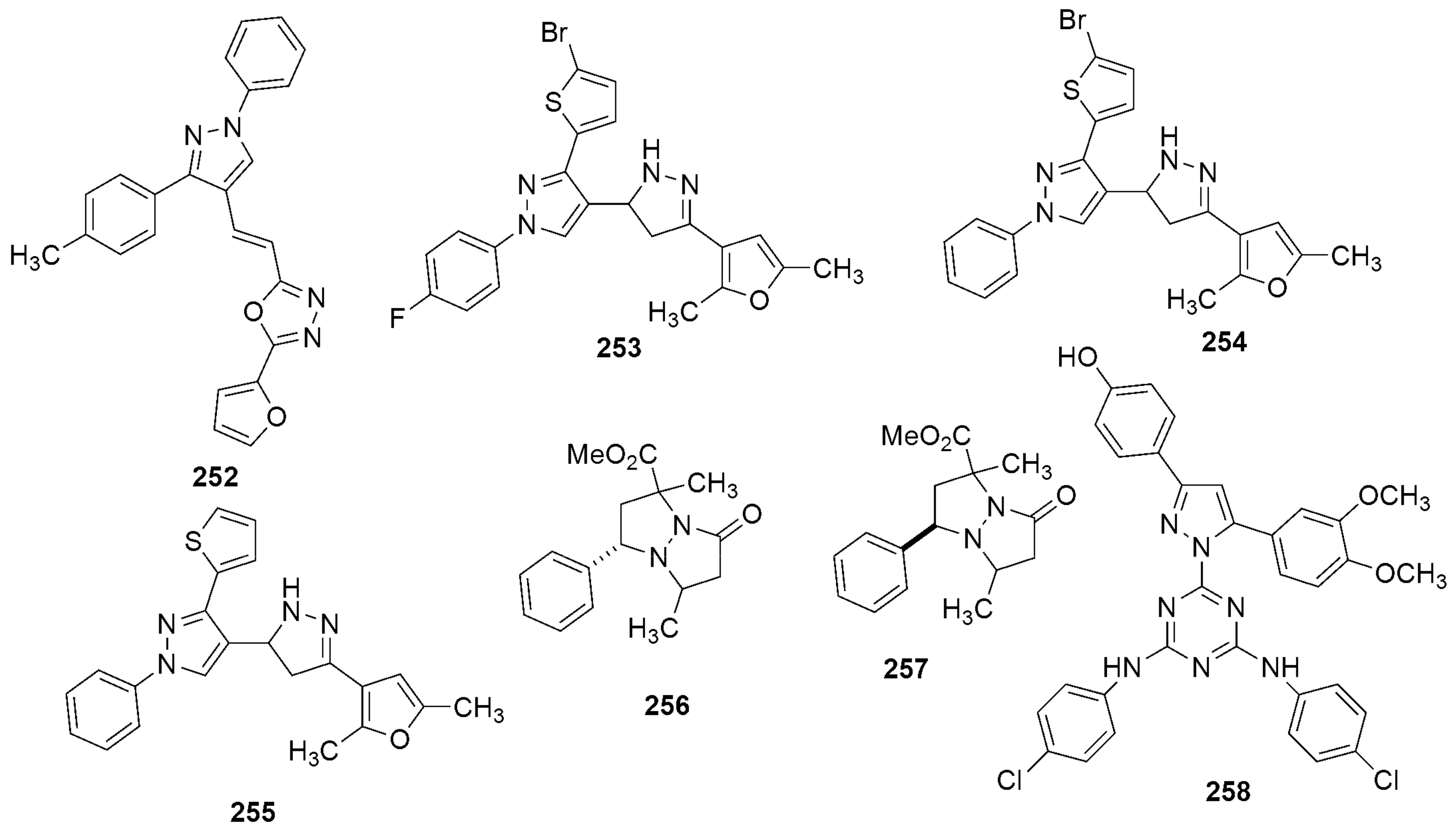
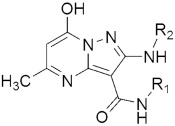 306 306H37RV MIC(μg/mL) | [145] | |||
| 306a R1 = 4-F-C6H5, R2 = 3,4-di-F-C6H4 | 0.8 | |||
| 306b R1 = 4-CH3-C6H5, R2 = 4-Cl-C6H5 | 3.12 | |||
| 306c R1 = 2-OCH3-C6H5, R2 = 3-CH3-C6H5 | 3.12 | |||
| 306d R1 = H, R2 = 3,4-di-Cl-C6H4 | 6.25 | |||
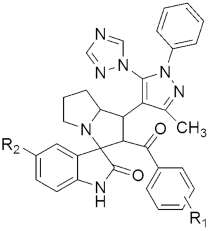 307 307MIC(μg/mL) H37RV | [146] | |||
| 307a R1 = H, R2 = Cl | 0.78 | |||
| 307b R1 = 4-Br, R2 = H | 1.56 | |||
| 307c R1 = 4-Cl, R2 = Cl | 1.56 | |||
| 307d R1 = 4-Br, R2 = Cl | 1.56 | |||
| 307e R1 = 4-Cl, R2 = Cl | 1.56 | |||
| 307f R1 = 4-Cl, R2 = Br | 1.56 | |||
| 307g R1 = Br, R2 = Br | 1.56 | |||
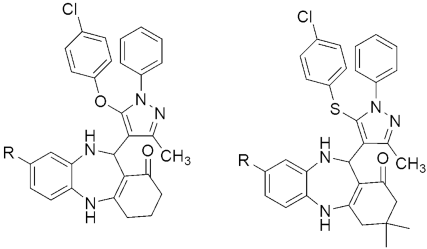 308 309 Growth Inhibition (%GI) H37RV | [147] | |||
| 308a R = H | 86 | |||
| 308b R = COPh | 85 | |||
| 309a R = H | 90 | |||
| 309b R = COPh | 88 | |||
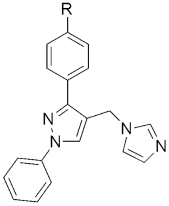 310 310MIC(μg/mL) H37RV | [148] | |||
| 310a R = isobutyl | 1.562 | |||
| 310b R = tert-butyl | 1.562 | |||
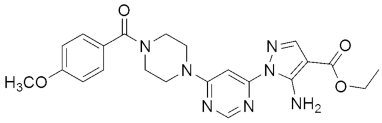 311 311MIC(μg/mL) = 1.6, against H37RV | [149] | |||
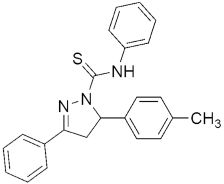 312 312MIC(μM) = 17, and MBC(μM) = 34 against H37Ra | [150] | |||
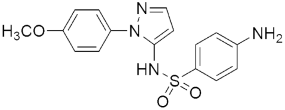 313 313MIC(μg/mL) = 5.96, H37RV MIC(μg/mL) = 10.62, DR-TB | [151] | |||
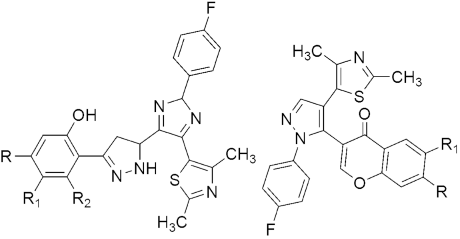 314 315 MIC(μg/mL) | [152] | |||
| H37Ra | M. bovis BCG | |||
| 314a R = H, R1 = Br, R2 = H | 2.96 | 0.20 | ||
| 314b R = H, R1 = Cl, R2 = H | 1.16 | 0.72 | ||
| 314c R = CH3, R1 = H, R2 = H | 4.65 | 2.3 | ||
| 314d R = CH3, R1 = Cl, R2 = CH3 | 2.5 | 2.26 | ||
| 315a R = H, R1 = Cl | 2.54 | 0.51 | ||
| 315b R = CH3, R1 = Cl | 1.72 | 1.33 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebenezer, O.; Shapi, M.; Tuszynski, J.A. A Review of the Recent Development in the Synthesis and Biological Evaluations of Pyrazole Derivatives. Biomedicines 2022, 10, 1124. https://doi.org/10.3390/biomedicines10051124
Ebenezer O, Shapi M, Tuszynski JA. A Review of the Recent Development in the Synthesis and Biological Evaluations of Pyrazole Derivatives. Biomedicines. 2022; 10(5):1124. https://doi.org/10.3390/biomedicines10051124
Chicago/Turabian StyleEbenezer, Oluwakemi, Michael Shapi, and Jack A. Tuszynski. 2022. "A Review of the Recent Development in the Synthesis and Biological Evaluations of Pyrazole Derivatives" Biomedicines 10, no. 5: 1124. https://doi.org/10.3390/biomedicines10051124
APA StyleEbenezer, O., Shapi, M., & Tuszynski, J. A. (2022). A Review of the Recent Development in the Synthesis and Biological Evaluations of Pyrazole Derivatives. Biomedicines, 10(5), 1124. https://doi.org/10.3390/biomedicines10051124