
Targeting TKI-Activated NFKB2-MIF/CXCLs-CXCR2 Signaling Pathways in FLT3 Mutated Acute Myeloid Leukemia Reduced Blast Viability

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primary Leukemia Blasts
2.2. Cell Culture of AML Cells and Treatment of Blasts by TKIs
2.3. In Vitro & Ex Vivo Treatment of MIF and Small Molecular Inhibitors
2.4. Combination of GILT and Recombinant Human MIF Protein to Treat MV4-11 In Vitro
2.5. Cell Proliferation Assay
2.6. Transfection of siRNA-NFKB2 in MV4-11
2.7. Flow Cytometry (FC)
2.8. RNA Isolation and Real-Time Polymerase Chain Reaction (qPCR) Analysis
2.9. Proteomics and Blotting Analysis
2.10. Imaging Acquisition
2.11. Statistical Analysis
3. Results
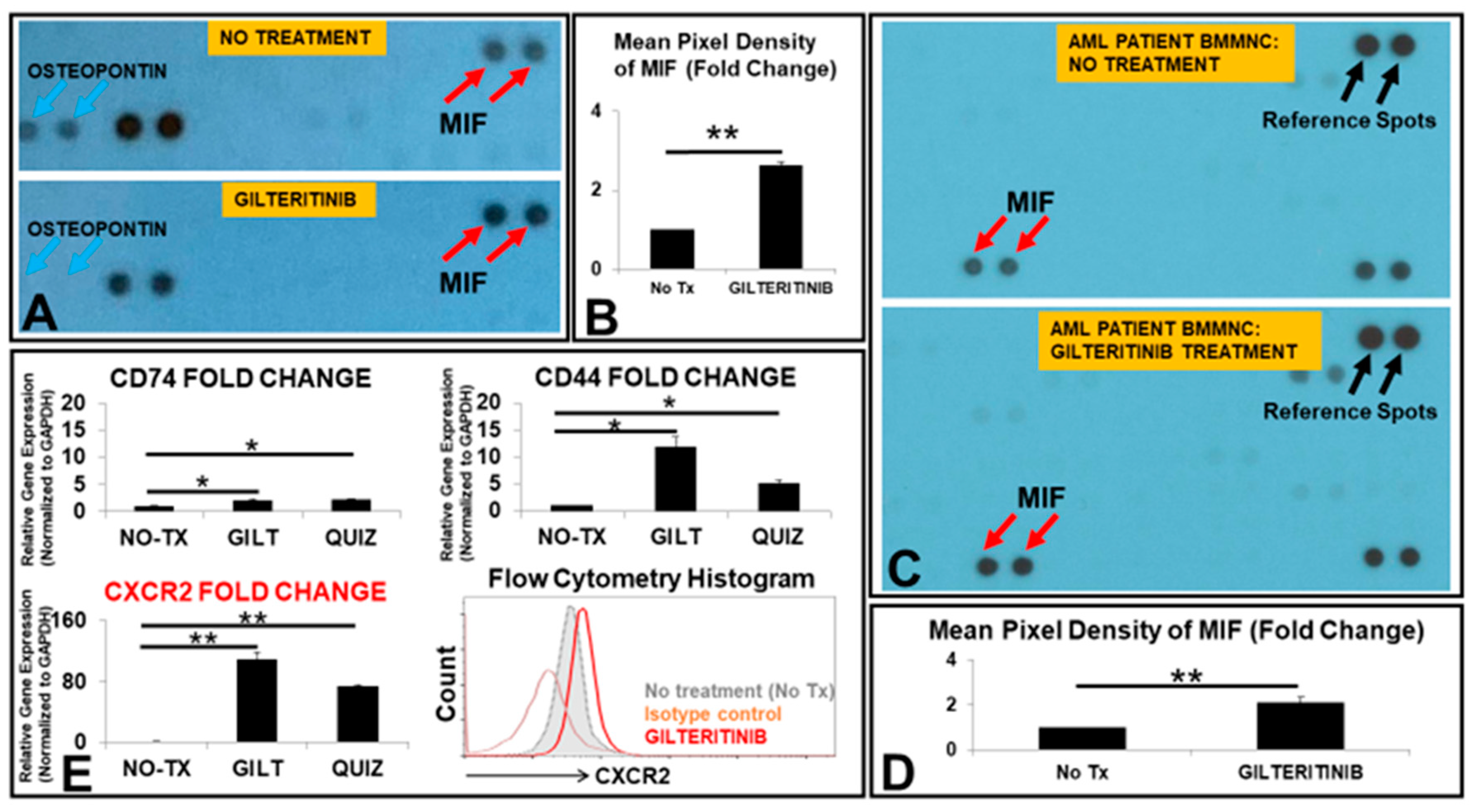
3.1. The Release of MIF Was Significantly Increased by GILT-Treated MV4-11 Cells In Vitro
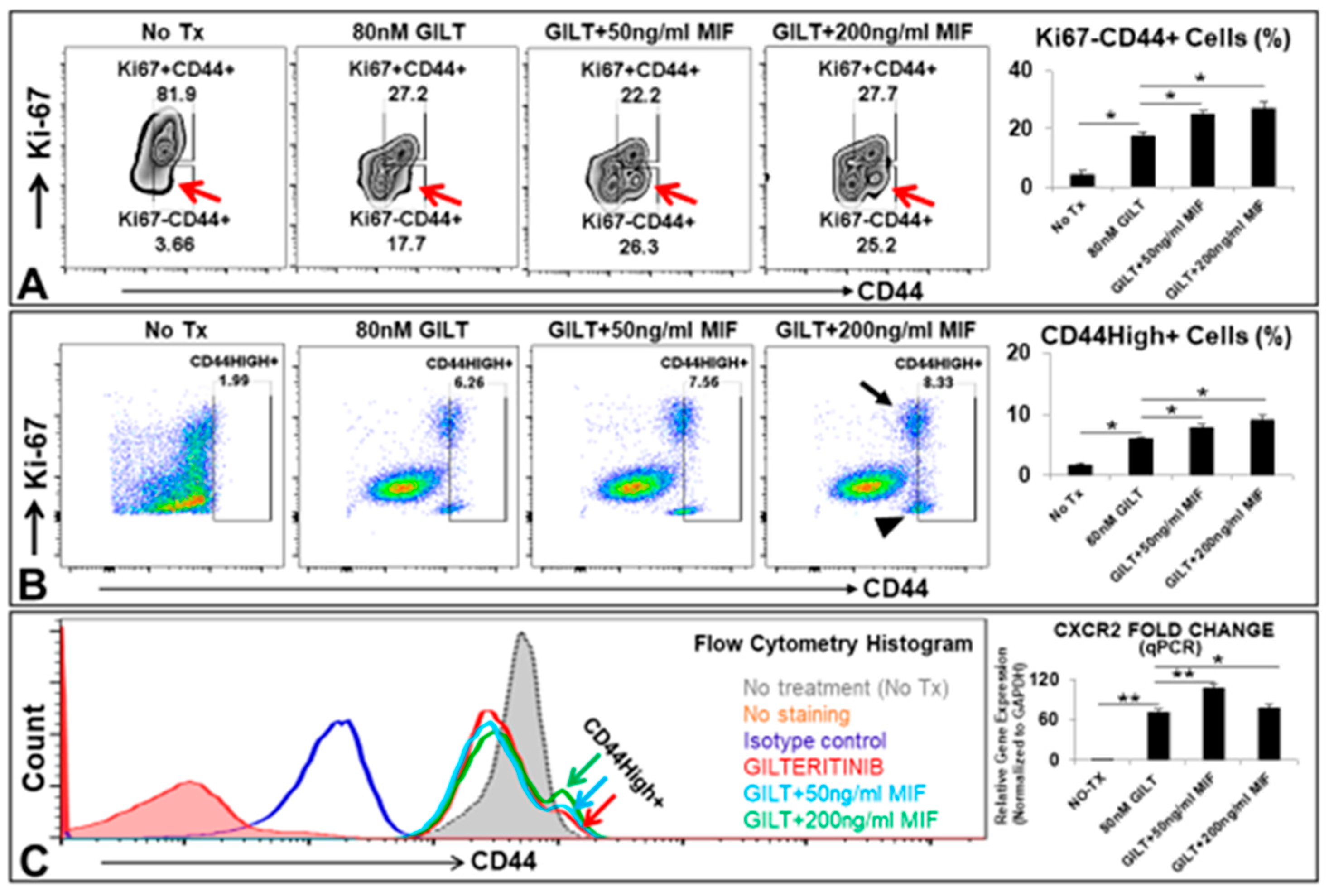
3.2. MIF Promoted the Proliferation of MV4-11 through the Activation of MIF/CXCLs-CXCR2 Signaling Pathways
3.3. MIF Promoted the Survival of A Group of CD44High+ Cells after the TKI Treatment In Vitro
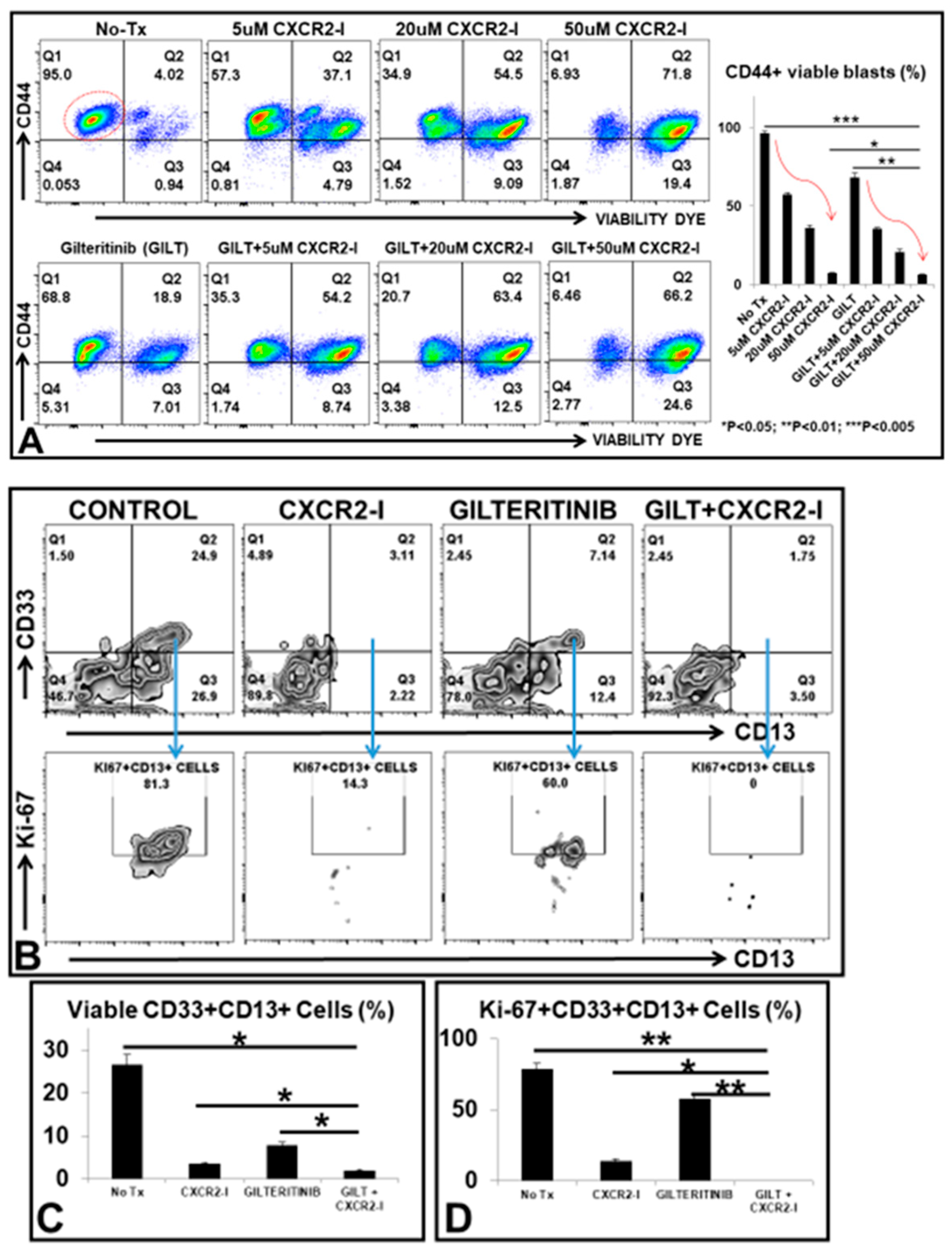
3.4. Targeting the MIF-CXCR2 Pathway in the Treatment of FLT3mut AML Blasts In Vitro
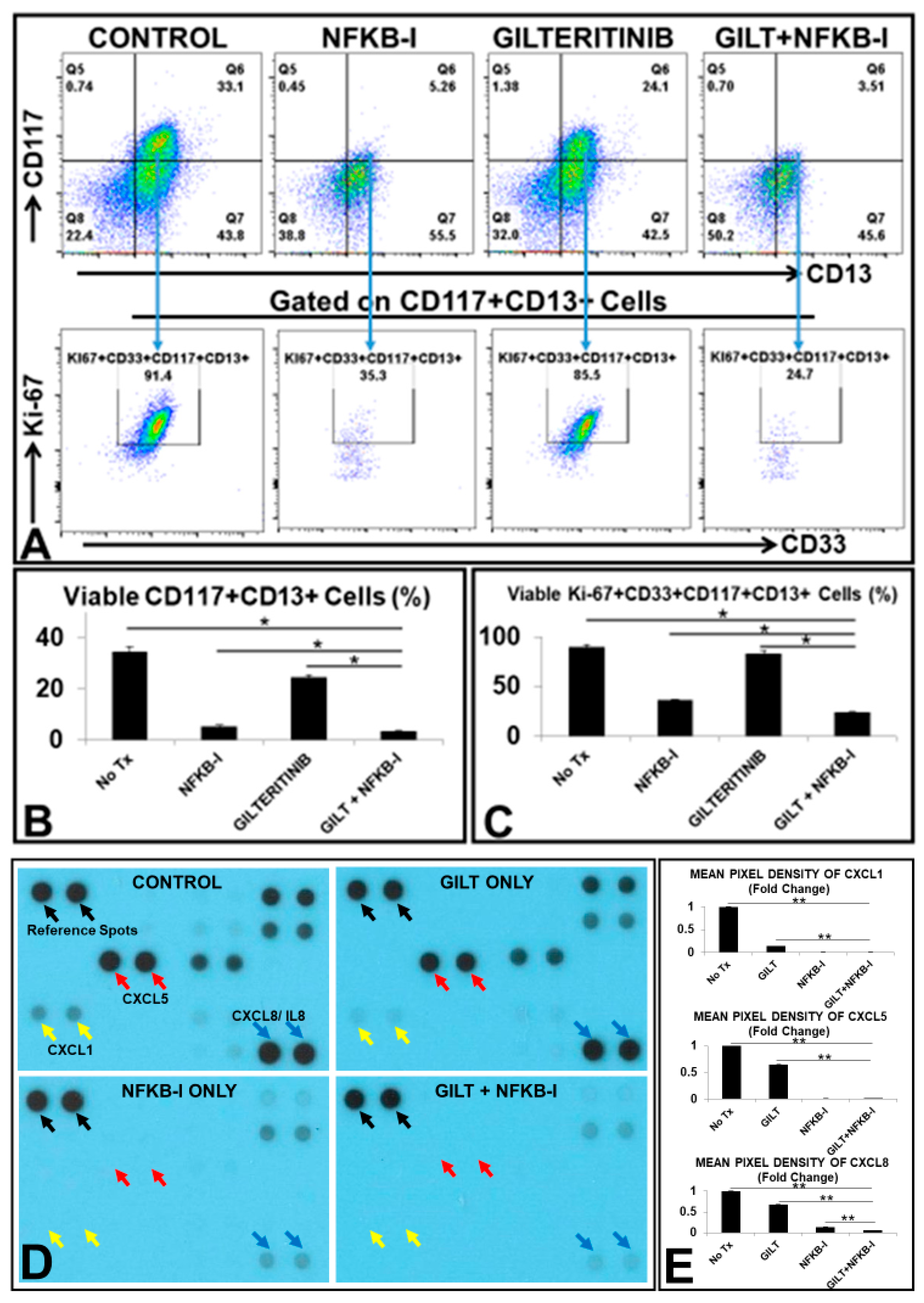
3.5. NFKB2 Could Activate MIF/CXCLs-CXCR2 Pathways in TKI-Treated Blasts In Vitro
3.6. The Combination of GILT and a NFKB Family Inhibitor (NFKB-I) Significantly Reduced the Viable Primary Blasts from FLT3mut AML Patients Ex Vivo
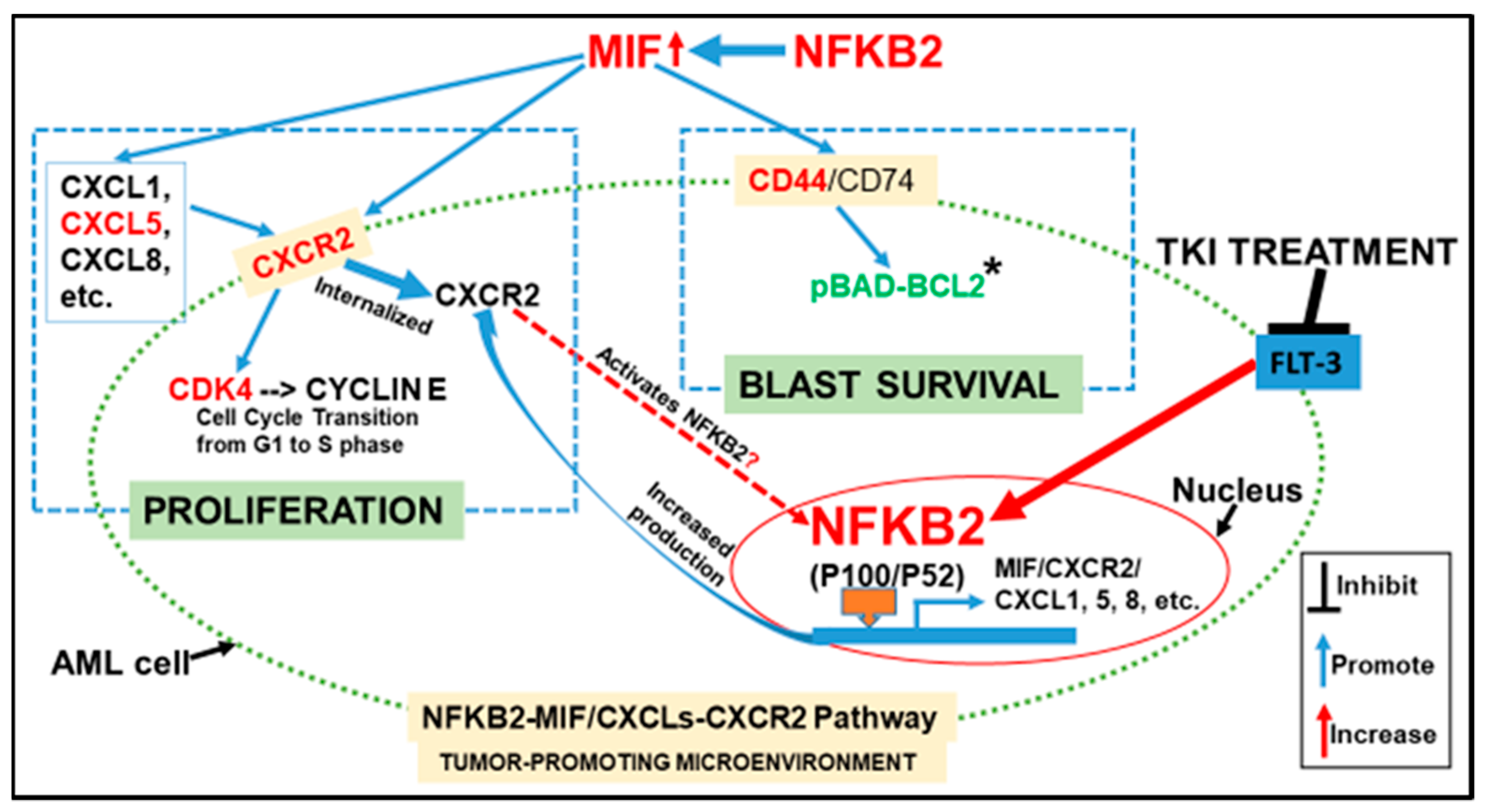
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H. Acute myeloid leukemia--major progress over four decades and glimpses into the future. Am. J. Hematol. 2016, 91, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, A.E. Availability of FLT3 inhibitors: How do we use them? Blood 2019, 134, 741–745. [Google Scholar] [CrossRef]
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Forman, S.J.; Rowe, J.M. The myth of the second remission of acute leukemia in the adult. Blood 2013, 121, 1077–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganzel, C.; Sun, Z.; Cripe, L.D.; Fernandez, H.F.; Douer, D.; Rowe, J.M.; Paietta, E.M.; Ketterling, R.; O’Connell, M.J.; Wiernik, P.H.; et al. Very poor long-term survival in past and more recent studies for relapsed AML patients: The ECOG-ACRIN experience. Am. J. Hematol. 2018, 93, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Lu, C.; Zhu, J.; Chen, X.; Hu, Y.; Xie, W.; Yao, J.; Huang, S. Risk Stratification in Acute Myeloid Leukemia Using CXCR Gene Signatures: A Bioinformatics Analysis. Front. Oncol. 2020, 10, 584766. [Google Scholar] [CrossRef]
- Bruserud, O.; Ryningen, A.; Olsnes, A.M.; Stordrange, L.; Oyan, A.M.; Kalland, K.H.; Gjertsen, B.T. Subclassification of patients with acute myelogenous leukemia based on chemokine responsiveness and constitutive chemokine release by their leukemic cells. Haematologica 2007, 92, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Riether, C.; Schurch, C.M.; Ochsenbein, A.F. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015, 22, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkaya, H.; Liu, S.; Wicha, M.S. Regulation of cancer stem cells by cytokine networks: Attacking cancer’s inflammatory roots. Clin. Cancer Res. 2011, 17, 6125–6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiehl, T.; Ho, A.D.; Marciniak-Czochra, A. Mathematical modeling of the impact of cytokine response of acute myeloid leukemia cells on patient prognosis. Sci. Rep. 2018, 8, 2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohl, S.R.; Bullinger, L.; Rucker, F.G. New Targeted Agents in Acute Myeloid Leukemia: New Hope on the Rise. Int. J. Mol. Sci. 2019, 20, 1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Baylink, D.J.; Cao, H.; Xiao, J.; Abdalla, M.I.; Wasnik, S.; Tang, X. Inflammation- and Gut-Homing Macrophages, Engineered to De Novo Overexpress Active Vitamin D, Promoted the Regenerative Function of Intestinal Stem Cells. Int. J. Mol. Sci. 2021, 22, 9516. [Google Scholar] [CrossRef]
- Rafat, A.; Dizaji Asl, K.; Mazloumi, Z.; Movassaghpour, A.A.; Talebi, M.; Shanehbandi, D.; Farahzadi, R.; Nejati, B.; Nozad Charoudeh, H. Telomerase inhibition on acute myeloid leukemia stem cell induced apoptosis with both intrinsic and extrinsic pathways. Life Sci. 2022, 295, 120402. [Google Scholar] [CrossRef]
- Cao, H.; Xiao, J.; Reeves, M.E.; Payne, K.; Chen, C.S.; Baylink, D.J.; Marcucci, G.; Xu, Y. Discovery of proangiogenic CD44+mesenchymal cancer stem cells in an acute myeloid leukemia patient’s bone marrow. J. Hematol. Oncol. 2020, 13, 63. [Google Scholar] [CrossRef]
- Xu, Y.; Payne, K.; Pham, L.H.G.; Eunwoo, P.; Xiao, J.; Chi, D.; Lyu, J.; Campion, R.; Wasnik, S.; Jeong, I.S.; et al. A novel vitamin D gene therapy for acute myeloid leukemia. Transl. Oncol. 2020, 13, 100869. [Google Scholar] [CrossRef]
- Liersch, R.; Gerss, J.; Schliemann, C.; Bayer, M.; Schwoppe, C.; Biermann, C.; Appelmann, I.; Kessler, T.; Lowenberg, B.; Buchner, T.; et al. Osteopontin is a prognostic factor for survival of acute myeloid leukemia patients. Blood 2012, 119, 5215–5220. [Google Scholar] [CrossRef] [Green Version]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Li, Z.; Li, X.; Huo, Z. High CXCR2 expression predicts poor prognosis in adult patients with acute myeloid leukemia. Ther. Adv. Hematol. 2020, 11, 2040620720958586. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, X.; Shen, J.; Yao, J. Macrophage migration inhibitory factor in the pathogenesis of leukemia (Review). Int. J. Oncol. 2021, 59, 62. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ma, X.L.; Wei, Y.Q.; Wei, X.W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Fillmore, C.M.; Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Schinke, C.; Giricz, O.; Li, W.; Shastri, A.; Gordon, S.; Barreyro, L.; Bhagat, T.; Bhattacharyya, S.; Ramachandra, N.; Bartenstein, M.; et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 2015, 125, 3144–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matityahu, E.; Feniger-Barish, R.; Meshel, T.; Zaslaver, A.; Ben-Baruch, A. Intracellular trafficking of human CXCR1 and CXCR2: Regulation by receptor domains and actin-related kinases. Eur. J. Immunol. 2002, 32, 3525–3535. [Google Scholar] [CrossRef]
- Nasser, M.W.; Raghuwanshi, S.K.; Malloy, K.M.; Gangavarapu, P.; Shim, J.Y.; Rajarathnam, K.; Richardson, R.M. CXCR1 and CXCR2 activation and regulation. Role of aspartate 199 of the second extracellular loop of CXCR2 in CXCL8-mediated rapid receptor internalization. J. Biol. Chem. 2007, 282, 6906–6915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naugler, W.E.; Karin, M. NF-kappaB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankauskas, S.S.; Wong, D.W.L.; Bucala, R.; Djudjaj, S.; Boor, P. Evolving complexity of MIF signaling. Cell. Signal. 2019, 57, 76–88. [Google Scholar] [CrossRef]
- Balaji, S.; Ahmed, M.; Lorence, E.; Yan, F.; Nomie, K.; Wang, M. NF-kappaB signaling and its relevance to the treatment of mantle cell lymphoma. J. Hematol. Oncol. 2018, 11, 83. [Google Scholar] [CrossRef]
- Farr, L.; Ghosh, S.; Moonah, S. Role of MIF Cytokine/CD74 Receptor Pathway in Protecting Against Injury and Promoting Repair. Front. Immunol. 2020, 11, 1273. [Google Scholar] [CrossRef]
- Bach, J.P.; Rinn, B.; Meyer, B.; Dodel, R.; Bacher, M. Role of MIF in inflammation and tumorigenesis. Oncology 2008, 75, 127–133. [Google Scholar] [CrossRef]
- Simpson, K.D.; Templeton, D.J.; Cross, J.V. Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J. Immunol. 2012, 189, 5533–5540. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ye, Y.L.; Li, M.X.; Ye, S.B.; Huang, W.R.; Cai, T.T.; He, J.; Peng, J.Y.; Duan, T.H.; Cui, J.; et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene 2017, 36, 2095–2104. [Google Scholar] [CrossRef]
- Wen, Y.; Cai, W.; Yang, J.; Fu, X.; Putha, L.; Xia, Q.; Windsor, J.A.; Phillips, A.R.; Tyndall, J.D.A.; Du, D.; et al. Targeting Macrophage Migration Inhibitory Factor in Acute Pancreatitis and Pancreatic Cancer. Front. Pharmacol. 2021, 12, 638950. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, A.M.; Shafat, M.S.; Mehta, T.K.; Di Palma, F.; Lawes, M.J.; Rushworth, S.A.; Bowles, K.M. MIF-Induced Stromal PKCbeta/IL8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 2017, 77, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deynoux, M.; Sunter, N.; Herault, O.; Mazurier, F. Hypoxia and Hypoxia-Inducible Factors in Leukemias. Front. Oncol. 2016, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; Trzyna, W.C.; McClintock, D.S.; Schumacker, P.T. Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J. Immunol. 2000, 165, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar] [CrossRef]
- Kitano, H. Biological robustness. Nat. Rev. Genet. 2004, 5, 826–837. [Google Scholar] [CrossRef]
- Lavi, O. Redundancy: A critical obstacle to improving cancer therapy. Cancer Res. 2015, 75, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, R.; Gade, P.; Wilson-Weekes, A.; Sayar, H.; Suvannasankha, A.; Goswami, C.; Li, L.; Gupta, S.; Cardoso, A.A.; Baghdadi, T.A.; et al. A noncanonical Flt3ITD/NF-kappaB signaling pathway represses DAPK1 in acute myeloid leukemia. Clin. Cancer Res. 2012, 18, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Olsnes, A.M.; Ersvaer, E.; Ryningen, A.; Paulsen, K.; Hampson, P.; Lord, J.M.; Gjertsen, B.T.; Kristoffersen, E.K.; Bruserud, O. The protein kinase C agonist PEP005 increases NF-kappaB expression, induces differentiation and increases constitutive chemokine release by primary acute myeloid leukaemia cells. Br. J. Haematol. 2009, 145, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Labbozzetta, M.; Notarbartolo, M.; Poma, P. Can NF-kappaB Be Considered a Valid Drug Target in Neoplastic Diseases? Our Point of View. Int. J. Mol. Sci. 2020, 21, 3070. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: Mechanism and clinical study. Signal. Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Diagnosis | Age | Sex | Disease Status | Gene Mutations |
|---|---|---|---|---|---|
| #1 | AML | 55 | M | Newly diagnosed | 1. FLT3: 40% allele frequency (c.2503G>T; p.D835Y) 2. NPM1: SR 0.72 (c.863_864insCTTG; p.W288Cfs*12) |
| #2 | AML | 41 | M | Relapsed/Refractory | FLT3 Internal Tandem Duplication (ITD): Allele Frequency: (2 separate mutations) 0.25% & 0.52% |
| #3 | AML | 69 | M | Relapsed/Refractory, On treatment | 1. FLT 3 ITD: SR 0.89 (c.1789delins25; p.Y597delins9) (23% allele frequency) 2.RUNX1: 94% allele frequency (c.602G>A; p.R201Q) 3.IDH2: 65% allele frequency (c.419G>A; p.R140Q) 4. DNMT3A: 48% allele frequency (c.2645G>A; p.R882H) 5.SRSF2: 46% allele frequency (c.284_307del24; p.P95_R102del) |
| #4 | AML | 53 | F | Newly Diagnosed | 1. CBFB-MYH11: Allele Frequency 102.3453% 2. FLT3 (c.1793_1794ins21;p.Y597_E598insDDPSLID): Allele Frequency 0.65% 3. KIT (c.1251_1257dekubsGGCA;p.Y418_D419delinsA): Allele Frequency 2% |
| #5 | AML | 62 | M | Newly diagnosed | 1. FLT3-ITD: Level = 0.98 |
| #6 | AML | 65 | F | Refractory | 1. FLT 3: ITD SR 0.57 2. PHF6: 41% allele frequency (c.821G>A; p.R274Q) 3. RUNX1: 34% allele frequency (c.494_497dup; p.G168Kfs*46) 4. WT1: 82% allele frequency (c.1140dup; p.S381Vfs*4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, H.; Tadros, V.; Hiramoto, B.; Leeper, K.; Hino, C.; Xiao, J.; Pham, B.; Kim, D.H.; Reeves, M.E.; Chen, C.-S.; et al. Targeting TKI-Activated NFKB2-MIF/CXCLs-CXCR2 Signaling Pathways in FLT3 Mutated Acute Myeloid Leukemia Reduced Blast Viability. Biomedicines 2022, 10, 1038. https://doi.org/10.3390/biomedicines10051038
Cao H, Tadros V, Hiramoto B, Leeper K, Hino C, Xiao J, Pham B, Kim DH, Reeves ME, Chen C-S, et al. Targeting TKI-Activated NFKB2-MIF/CXCLs-CXCR2 Signaling Pathways in FLT3 Mutated Acute Myeloid Leukemia Reduced Blast Viability. Biomedicines. 2022; 10(5):1038. https://doi.org/10.3390/biomedicines10051038
Chicago/Turabian StyleCao, Huynh, Verena Tadros, Benjamin Hiramoto, Kevin Leeper, Christopher Hino, Jeffrey Xiao, Bryan Pham, Do Hyun Kim, Mark E. Reeves, Chien-Shing Chen, and et al. 2022. "Targeting TKI-Activated NFKB2-MIF/CXCLs-CXCR2 Signaling Pathways in FLT3 Mutated Acute Myeloid Leukemia Reduced Blast Viability" Biomedicines 10, no. 5: 1038. https://doi.org/10.3390/biomedicines10051038
APA StyleCao, H., Tadros, V., Hiramoto, B., Leeper, K., Hino, C., Xiao, J., Pham, B., Kim, D. H., Reeves, M. E., Chen, C.-S., Zhong, J. F., Zhang, K. K., Xie, L., Wasnik, S., Baylink, D. J., & Xu, Y. (2022). Targeting TKI-Activated NFKB2-MIF/CXCLs-CXCR2 Signaling Pathways in FLT3 Mutated Acute Myeloid Leukemia Reduced Blast Viability. Biomedicines, 10(5), 1038. https://doi.org/10.3390/biomedicines10051038