
Colorectal Cancer Heterogeneity and the Impact on Precision Medicine and Therapy Efficacy

 , ,
, ,
Abstract
:1. Introduction
2. Methods
3. Mutational Status of RAS
4. Mutational Status of BRAF
5. Microsatellite Instability and Immunotherapy
6. HER2 Inhibition
7. Targeting NTRK, ALK, and ROS1 Fusions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Koehler, A.; Bataille, F.; Schmid, C.; Ruemmele, P.; Waldeck, A.; Blaszyk, H.; Hartmann, A.; Hofstaedter, F.; Dietmaier, W. Gene expression profiling of colorectal cancer and metastases divides tumours according to their clinicopathological stage. J. Pathol. 2004, 204, 65–74. [Google Scholar] [CrossRef]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular biomarkers for the evaluation of colorectal cancer: Guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J. Clin. Oncol. 2017, 35, 1453–1486. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Parseghian, C.M.; Napolitano, S.; Loree, J.M.; Kopetz, S. Mechanisms of Innate and Acquired Resistance to Anti-EGFR Therapy: A Review of Current Knowledge with a Focus on Rechallenge Therapies. Clin. Cancer Res. 2019, 25, 6899–6908. [Google Scholar] [CrossRef]
- Bang, Y.H.; Hong, Y.S.; Lee, J.S.; Lee, K.W.; Han, H.S.; Kim, S.Y.; Kim, J.W.; Kim, H.K.; Kim, J.W.; Eun, C.K.; et al. Effectiveness of Combining Bevacizumab With First-Line Chemotherapy Regimens for Metastatic Colorectal Cancer in Real-World Practice. Clin. Colorectal Cancer 2021, 20, 101–112. [Google Scholar] [CrossRef]
- Saoudi Gonzalez, N.; Salva, F.; Ros, J.; Baraibar, I.; Marmolejo, D.; Valdivia, A.; Cuadra-Urteaga, J.L.; Mulet, N.; Tabernero, J.; Elez, E. Up-to-date role of aflibercept in the treatment of colorectal cancer. Expert Opin. Biol. Ther. 2021, 21, 1315–1324. [Google Scholar] [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Grothey, A.; Fakih, M.; Tabernero, J. Management of BRAF-mutant metastatic colorectal cancer: A review of treatment options and evidence-based guidelines. Ann. Oncol. 2021, 32, 959–967. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Kummar, S.; Lassen, U.N. TRK Inhibition: A New Tumor-Agnostic Treatment Strategy. Target Oncol. 2018, 13, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Weiss, J.; Yaeger, R.D.; Johnson, M.L.; Spira, A.; Klempner, S.J.; Barve, M.A.; Christensen, J.G.; Chi, A.; Der-Torossian, H.; Velastegui, K.; et al. LBA6 KRYSTAL-1: Adagrasib (MRTX849) as monotherapy or combined with cetuximab (Cetux) in patients (Pts) with colorectal cancer (CRC) harboring a KRASG12C mutation. Ann. Oncol. 2021, 32, S1294. [Google Scholar] [CrossRef]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Valtorta, E.; Martino, C.; Sartore-Bianchi, A.; Penaullt-Llorca, F.; Viale, G.; Risio, M.; Rugge, M.; Grigioni, W.; Bencardino, K.; Lonardi, S.; et al. Assessment of a HER2 scoring system for colorectal cancer: Results from a validation study. Mod. Pathol. 2015, 28, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Siena, S.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Fakih, M.; Elez, E.; et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2021, 22, 779–789. [Google Scholar] [CrossRef]
- Chou, A.; Fraser, T.; Ahadi, M.; Fuchs, T.; Sioson, L.; Clarkson, A.; Sheen, A.; Singh, N.; Corless, C.L.; Gill, A.J. NTRK gene rearrangements are highly enriched in MLH1/PMS2 deficient, BRAF wild-type colorectal carcinomas-a study of 4569 cases. Mod. Pathol. 2020, 33, 924–932. [Google Scholar] [CrossRef]
- Afrăsânie, V.-A.; Marinca, M.V.; Alexa-Stratulat, T.; Gafton, B.; Păduraru, M.; Adavidoaiei, A.M.; Miron, L.; Rusu, C. KRAS, NRAS, BRAF, HER2 and microsatellite instability in metastatic colorectal cancer—Practical implications for the clinician. Radiol. Oncol. 2019, 53, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Cho, M.; Sy, M.; Salgia, R.; Fakih, M. Molecular profiling of metastatic colorectal tumors using next-generation sequencing: A single-institution experience. Oncotarget 2017, 8, 42198–42213. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, V.; Stintzing, S.; Kirchner, T.; Boeck, S.; Jung, A. Clinical relevance of EGFR- and KRAS-status in colorectal cancer patients treated with monoclonal antibodies directed against the EGFR. Cancer Treat. Rev. 2009, 35, 262–271. [Google Scholar] [CrossRef]
- Modest, D.P.; Ricard, I.; Heinemann, V.; Hegewisch-Becker, S.; Schmiegel, W.; Porschen, R.; Stintzing, S.; Graeven, U.; Arnold, D.; von Weikersthal, L.F.; et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: Pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann. Oncol. 2016, 27, 1746–1753. [Google Scholar] [CrossRef]
- Schirripa, M.; Cremolini, C.; Loupakis, F.; Morvillo, M.; Bergamo, F.; Zoratto, F.; Salvatore, L.; Antoniotti, C.; Marmorino, F.; Sensi, E.; et al. Role of NRAS mutations as prognostic and predictive markers in metastatic colorectal cancer. Int. J. Cancer 2015, 136, 83–90. [Google Scholar] [CrossRef]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D'Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Bokemeyer, C.; Bondarenko, I.; Hartmann, J.T.; de Braud, F.; Schuch, G.; Zubel, A.; Celik, I.; Schlichting, M.; Koralewski, P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: The OPUS study. Ann. Oncol. 2011, 22, 1535–1546. [Google Scholar] [CrossRef]
- Au, H.J.; Karapetis, C.S.; O’Callaghan, C.J.; Tu, D.; Moore, M.J.; Zalcberg, J.R.; Kennecke, H.; Shapiro, J.D.; Koski, S.; Pavlakis, N.; et al. Health-related quality of life in patients with advanced colorectal cancer treated with cetuximab: Overall and KRAS-specific results of the NCIC CTG and AGITG CO.17 Trial. J. Clin. Oncol. 2009, 27, 1822–1828. [Google Scholar] [CrossRef] [Green Version]
- Peeters, M.; Douillard, J.Y.; Van Cutsem, E.; Siena, S.; Zhang, K.; Williams, R.; Wiezorek, J. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: Assessment as prognostic and predictive biomarkers of response to panitumumab. J. Clin. Oncol. 2013, 31, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, F.; Lenz, H.-J.; Kohne, C.-H.; Heinemann, V.; Tejpar, S.; Esser, R.; Beier, F.; Stroh, C.; Duecker, K.; Van Cutsem, E. Effect of KRAS and NRAS mutational status on first-line treatment with FOLFIRI plus cetuximab in patients with metastatic colorectal cancer (mCRC): New results from the CRYSTAL trial. J. Clin. Oncol. 2014, 32, LBA443. [Google Scholar] [CrossRef]
- Allegra, C.J.; Rumble, R.B.; Hamilton, S.R.; Mangu, P.B.; Roach, N.; Hantel, A.; Schilsky, R.L. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J. Clin. Oncol. 2016, 34, 179–185. [Google Scholar] [CrossRef]
- Lakatos, G.; Kohne, C.H.; Bodoky, G. Current therapy of advanced colorectal cancer according to RAS/RAF mutational status. Cancer Metastasis Rev. 2020, 39, 1143–1157. [Google Scholar] [CrossRef]
- Stahler, A.; Heinemann, V.; Ricard, I.; von Einem, J.C.; Giessen-Jung, C.; Westphalen, C.B.; Michl, M.; Heinrich, K.; Miller-Phillips, L.; Jelas, I.; et al. Current treatment options in RAS mutant metastatic colorectal cancer patients: A meta-analysis of 14 randomized phase III trials. J. Cancer Res. Clin. Oncol. 2020, 146, 2077–2087. [Google Scholar] [CrossRef]
- Xu, J.; Liu, T.; Tang, W.; Chang, W.; Feng, Q.; Wei, Y.; Ren, L.; Ye, Q.; Cui, Y.; He, G.; et al. Bevacizumab plus chemotherapy versus chemotherapy alone as first-line treatment for patients with RAS mutant unresectable colorectal liver-limited metastases: A single center randomized control trial. Ann. Oncol. 2019, 30, v867. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Cremolini, C.; Antoniotti, C.; Rossini, D.; Lonardi, S.; Loupakis, F.; Pietrantonio, F.; Bordonaro, R.; Latiano, T.P.; Tamburini, E.; Santini, D.; et al. Upfront FOLFOXIRI plus bevacizumab and reintroduction after progression versus mFOLFOX6 plus bevacizumab followed by FOLFIRI plus bevacizumab in the treatment of patients with metastatic colorectal cancer (TRIBE2): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2020, 21, 497–507. [Google Scholar]
- Rosati, G.; Aprile, G.; Basile, D.; Avallone, A. Perspectives in the Treatment of RAS or BRAF Mutated Metastatic Colorectal Cancer Patients. Front. Oncol. 2021, 11, 602596. [Google Scholar] [CrossRef]
- Romero, D. Two new agents target KRAS G12C. Nat. Rev. Clin. Oncol. 2020, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, J.F.; Fisher, D.J.; Brown, L.C.; Adams, R.A.; Graham, J.; Quirke, P.; Richman, S.D.; Butler, R.; Domingo, E.; Blake, A.; et al. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) With Active Monitoring. J. Clin. Oncol. 2021, 39, 3705–3715. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Kim, T.W.; Lee, C.B.; Goh, B.C.; Miller, W.H., Jr.; Oh, D.Y.; Jamal, R.; Chee, C.E.; Chow, L.Q.M.; Gainor, J.F.; et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol. 2019, 30, 1134–1142. [Google Scholar] [CrossRef]
- Eng, C.; Kim, T.W.; Bendell, J.; Argiles, G.; Tebbutt, N.C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.H.; et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019, 20, 849–861. [Google Scholar] [CrossRef]
- Antoniotti, C.; Borelli, B.; Rossini, D.; Pietrantonio, F.; Morano, F.; Salvatore, L.; Lonardi, S.; Marmorino, F.; Tamberi, S.; Corallo, S.; et al. AtezoTRIBE: A randomised phase II study of FOLFOXIRI plus bevacizumab alone or in combination with atezolizumab as initial therapy for patients with unresectable metastatic colorectal cancer. BMC Cancer 2020, 20, 683. [Google Scholar] [CrossRef]
- Cremolini, C.; Rossini, D.; Antoniotti, C.; Pietrantonio, F.; Lonardi, S.; Salvatore, L.; Marmorino, F.; Borelli, B.; Ambrosini, M.; Barsotti, G. FOLFOXIRI plus bevacizumab (bev) plus atezolizumab (atezo) versus FOLFOXIRI plus bev as first-line treatment of unresectable metastatic colorectal cancer (mCRC) patients: Results of the phase II randomized AtezoTRIBE study by GONO. Ann. Oncol. 2021, 32, S1294–S1295. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Yoshino, T.; Arnold, D.; Taniguchi, H.; Pentheroudakis, G.; Yamazaki, K.; Xu, R.H.; Kim, T.W.; Ismail, F.; Tan, I.B.; Yeh, K.H.; et al. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: A JSMO-ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann. Oncol. 2018, 29, 44–70. [Google Scholar] [CrossRef]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Cederquist, L.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; et al. Rectal Cancer, Version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 874–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrun, H.; Turpin, A.; Zerbib, P. Therapeutic implications of B-RAF mutations in colorectal cancer. J. Visc. Surg. 2021, 158, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Moretto, R.; Aprile, G.; Muntoni, M.; Cremolini, C.; Iacono, D.; Casagrande, M.; Ferrari, L.; Salvatore, L.; Schirripa, M.; et al. Clinico-pathological nomogram for predicting BRAF mutational status of metastatic colorectal cancer. Br. J. Cancer 2016, 114, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br. J. Cancer 2015, 112, 1888–1894. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. (Non-V600) BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef]
- Yaeger, R.; Kotani, D.; Mondaca, S.; Parikh, A.R.; Bando, H.; Van Seventer, E.E.; Taniguchi, H.; Zhao, H.; Thant, C.N.; de Stanchina, E.; et al. Response to Anti-EGFR Therapy in Patients with BRAF non-V600-Mutant Metastatic Colorectal Cancer. Clin. Cancer Res. 2019, 25, 7089–7097. [Google Scholar] [CrossRef] [Green Version]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Cederquist, L.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; et al. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Loupakis, F.; Cremolini, C.; Masi, G.; Lonardi, S.; Zagonel, V.; Salvatore, L.; Cortesi, E.; Tomasello, G.; Ronzoni, M.; Spadi, R.; et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N. Engl. J. Med. 2014, 371, 1609–1618. [Google Scholar] [CrossRef] [Green Version]
- Cremolini, C.; Antoniotti, C.; Stein, A.; Bendell, J.; Gruenberger, T.; Rossini, D.; Masi, G.; Ongaro, E.; Hurwitz, H.; Falcone, A.; et al. Individual Patient Data Meta-Analysis of FOLFOXIRI Plus Bevacizumab Versus Doublets Plus Bevacizumab as Initial Therapy of Unresectable Metastatic Colorectal Cancer. J. Clin. Oncol. 2020, 38, 3314–3324. [Google Scholar] [CrossRef]
- Wirapati, P.; Pomella, V.; Vandenbosch, B.; Kerr, P.; Maiello, E.; Jeffery, G.M.; Curca, R.-O.D.; Karthaus, M.; Bridgewater, J.A.; Mihailov, A.C.; et al. Velour trial biomarkers update: Impact of RAS, BRAF, and sidedness on aflibercept activity. J. Clin. Oncol. 2017, 35, 3538. [Google Scholar] [CrossRef]
- Yoshino, T.; Portnoy, D.C.; Obermannova, R.; Bodoky, G.; Prausova, J.; Garcia-Carbonero, R.; Ciuleanu, T.; Garcia-Alfonso, P.; Cohn, A.L.; Van Cutsem, E.; et al. Biomarker analysis beyond angiogenesis: RAS/RAF mutation status, tumour sidedness, and second-line ramucirumab efficacy in patients with metastatic colorectal carcinoma from RAISE-a global phase III study. Ann. Oncol. 2019, 30, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- van Geel, R.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF-Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Cercek, A.; O’Reilly, E.M.; Reidy, D.L.; Kemeny, N.; Wolinsky, T.; Capanu, M.; Gollub, M.J.; Rosen, N.; Berger, M.F.; et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin. Cancer Res. 2015, 21, 1313–1320. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [Green Version]
- Huijberts, S.; Boelens, M.C.; Bernards, R.; Opdam, F.L. Mutational profiles associated with resistance in patients with BRAFV600E mutant colorectal cancer treated with cetuximab and encorafenib +/− binimetinib or alpelisib. Br. J. Cancer 2021, 124, 176–182. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Tabernero, J. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. Reply. N. Engl. J. Med. 2020, 382, 877–878. [Google Scholar]
- Van Cutsem, E.; Taieb, J.; Yaeger, R.; Yoshino, T.; Maiello, E.; Elez, E.; Dekervel, J.; Ross, P.; Ruiz Casado, A.; Graham, J.; et al. O-10 ANCHOR CRC: Results from a single-arm, phase 2 study of encorafenib, binimetinib plus cetuximab in previously untreated BRAF V600E–mutant metastatic colorectal cancer. Ann. Oncol. 2021, 32, S222. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J. Mol. Diagn. 2008, 10, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.C.; Li, A.F.; Lin, P.C.; Lin, C.C.; Lin, H.H.; Huang, S.C.; Lin, C.H.; Liang, W.Y.; Chen, W.S.; Jiang, J.K.; et al. Clinicopathological and Molecular Profiles of Sporadic Microsatellite Unstable Colorectal Cancer with or without the CpG Island Methylator Phenotype (CIMP). Cancer 2020, 12, 3487. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Roy, H.K.; Lynch, H.T. Lynch syndrome in the 21st century: Clinical perspectives. QJM Int. J. Med. 2016, 109, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Latham, A.; Srinivasan, P.; Kemel, Y. Microsatellite instability is associated with the presence of lynch syndrome pan-cancer. J. Clin. Oncol. 2018, 37, 286–297. [Google Scholar] [CrossRef]
- Ashktorab, H.; Ahuja, S.; Kannan, L.; Llor, X.; Ellis, N.A.; Xicola, R.M.; Laiyemo, A.O.; Carethers, J.M.; Brim, H.; Nouraie, M. A meta-analysis of MSI frequency and race in colorectal cancer. Oncotarget 2016, 7, 34546–34557. [Google Scholar] [CrossRef]
- Koopman, M.; Kortman, G.A.; Mekenkamp, L.; Ligtenberg, M.J.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.; van Krieken, J.H. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Sinicrope, F.A.; Mahoney, M.R.; Smyrk, T.C.; Thibodeau, S.N.; Warren, R.S.; Bertagnolli, M.M.; Nelson, G.D.; Goldberg, R.M.; Sargent, D.J.; Alberts, S.R. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J. Clin. Oncol. 2013, 31, 3664–3672. [Google Scholar] [CrossRef]
- Hutchins, G.; Southward, K.; Handley, K.; Magill, L.; Beaumont, C.; Stahlschmidt, J.; Richman, S.; Chambers, P.; Seymour, M.; Kerr, D.; et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J. Clin. Oncol. 2011, 29, 1261–1270. [Google Scholar] [CrossRef]
- Cohen, R.; Taieb, J.; Fiskum, J.; Yothers, G.; Goldberg, R.; Yoshino, T.; Alberts, S.; Allegra, C.; de Gramont, A.; Seitz, J.F.; et al. Microsatellite Instability in Patients With Stage III Colon Cancer Receiving Fluoropyrimidine With or Without Oxaliplatin: An ACCENT Pooled Analysis of 12 Adjuvant Trials. J. Clin. Oncol. 2021, 39, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Andre, T.; Amonkar, M.; Norquist, J.M.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.; Garcia-Carbonero, R.; et al. Health-related quality of life in patients with microsatellite instability-high or mismatch repair deficient metastatic colorectal cancer treated with first-line pembrolizumab versus chemotherapy (KEYNOTE-177): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 665–677. [Google Scholar] [CrossRef]
- Fanotto, V.; Ongaro, E.; Rihawi, K.; Avallone, A.; Silvestris, N.; Fornaro, L.; Vasile, E.; Antonuzzo, L.; Leone, F.; Rosati, G.; et al. HER-2 inhibition in gastric and colorectal cancers: Tangible achievements, novel acquisitions and future perspectives. Oncotarget 2016, 7, 69060–69074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, D.M.; Piha-Paul, S.A.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697, Erratum in: Lancet 2010, 376, 1302. [Google Scholar] [CrossRef]
- Javle, M.; Borad, M.J.; Azad, N.S.; Kurzrock, R.; Abou-Alfa, G.K.; George, B.; Hainsworth, J.; Meric-Bernstam, F.; Swanton, C.; Sweeney, C.J.; et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): A multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021, 22, 1290–1300. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Roviello, G.; Catalano, M.; Iannone, L.F.; Marano, L.; Brugia, M.; Rossi, G.; Aprile, G.; Antonuzzo, L. Current status and future perspectives in HER2 positive advanced gastric cancer. Clin. Transl. Oncol. 2022, in press. [Google Scholar] [CrossRef]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Amatu, A.; Porcu, L.; Ghezzi, S.; Lonardi, S.; Leone, F.; Bergamo, F.; Fenocchio, E.; Martinelli, E.; Borelli, B.; et al. HER2 Positivity Predicts Unresponsiveness to EGFR-Targeted Treatment in Metastatic Colorectal Cancer. Oncologist 2019, 24, 1395–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosi, F.; Sartore-Bianchi, A.; Lonardi, S.; Amatu, A.; Leone, F.; Ghezzi, S.; Martino, C.; Bencardino, K.; Bonazzina, E.; Bergamo, F.; et al. Long-term Clinical Outcome of Trastuzumab and Lapatinib for HER2-positive Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2020, 19, 256–262.e2. [Google Scholar] [CrossRef] [PubMed]
- Aprile, G.; De Maglio, G.; Menis, J.; Casagrande, M.; Tuniz, F.; Pisa, E.F.; Fontanella, C.; Skrap, M.; Beltrami, A.C.; Fasola, G.; et al. HER-2 Expression in Brain Metastases from Colorectal Cancer and Corresponding Primary Tumors: A Case Cohort Series. Int. J. Mol. Sci. 2013, 14, 2370–2387. [Google Scholar] [CrossRef] [Green Version]
- Siena, S.; Sartore-Bianchi, A.; Marsoni, S.; Hurwitz, H.I.; McCall, S.J.; Penault-Llorca, F.; Srock, S.; Bardelli, A.; Trusolino, L. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann. Oncol. 2018, 29, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Vernieri, C.; Milano, M.; Brambilla, M.; Mennitto, A.; Maggi, C.; Cona, M.S.; Prisciandaro, M.; Fabbroni, C.; Celio, L.; Mariani, G.; et al. Resistance mechanisms to anti-HER2 therapies in HER2-positive breast cancer: Current knowledge, new research directions and therapeutic perspectives. Crit. Rev. Oncol. Hematol. 2019, 139, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Piro, G.; Carbone, C.; Cataldo, I.; Di Nicolantonio, F.; Giacopuzzi, S.; Aprile, G.; Simionato, F.; Boschi, F.; Zanotto, M.; Mina, M.M.; et al. An FGFR3 Autocrine Loop Sustains Acquired Resistance to Trastuzumab in Gastric Cancer Patients. Clin. Cancer Res. 2016, 22, 6164–6175. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Fucà, G.; Morano, F.; Gloghini, A.; Corso, S.; Aprile, G.; Perrone, F.; De Vita, F.; Tamborini, E.; Tomasello, G.; et al. Biomarkers of Primary Resistance to Trastuzumab in HER2-Positive Metastatic Gastric Cancer Patients: The AMNESIA Case-Control Study. Clin. Cancer Res. 2018, 24, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Mangiapane, L.R.; Nicotra, A.; Turdo, A.; Gaggianesi, M.; Bianca, P.; Di Franco, S.; Sardina, D.S.; Veschi, V.; Signore, M.; Beyes, S.; et al. PI3K-driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut 2022, 71, 119–128. [Google Scholar] [CrossRef]
- Guarini, C.; Grassi, T.; Pezzicoli, G.; Porta, C. Beyond RAS and BRAF: HER2, a New Actionable Oncotarget in Advanced Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 6813. [Google Scholar] [CrossRef]
- Gall, V.A.; Philips, A.V.; Qiao, N.; Clise-Dwyer, K.; Perakis, A.A.; Zhang, M.; Clifton, G.T.; Sukhumalchandra, P.; Ma, Q.; Reddy, S.M.; et al. Trastuzumab Increases HER2 Uptake and Cross-Presentation by Dendritic Cells. Cancer Res. 2017, 77, 5374–5383. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Jiang, Z.; Mortenson, E.D.; Deng, L.; Radkevich-Brown, O.; Yang, X.; Sattar, H.; Wang, Y.; Brown, N.K.; Greene, M.; et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010, 18, 160–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantino, P.; Modi, S.; Tolaney, S.M.; Cortés, J.; Hamilton, E.P.; Kim, S.B.; Toi, M.; Andrè, F.; Curigliano, G. Interstitial Lung Disease Induced by Anti-ERBB2 Antibody-Drug Conjugates: A Review. JAMA Oncol. 2021, 7, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Giaccone, G.; Im, S.A.; Oh, D.Y.; Bauer, T.M.; Nordstrom, J.L.; Li, H.; Chichili, G.R.; Moore, P.A.; Hong, S.; et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann. Oncol. 2017, 28, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Lonardi, S.; Martino, C.; Fenocchio, E.; Tosi, F.; Ghezzi, S.; Leone, F.; Bergamo, F.; Zagonel, V.; Ciardiello, F.; et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: The phase II HERACLES-B trial. ESMO Open 2020, 5, e000911. [Google Scholar] [CrossRef]
- Nakamura, Y.; Okamoto, W.; Kato, T.; Esaki, T.; Kato, K.; Komatsu, Y.; Yuki, S.; Masuishi, T.; Nishina, T.; Ebi, H.; et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: A phase 2 trial. Nat. Med. 2021, 27, 1899–1903. [Google Scholar] [CrossRef]
- Gupta, R.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; D’Andre, S.D.; Meiri, E.; Shrestha, S.; Warren, S.L.; Ranasinghe, S.; Schilsky, R.L. Pertuzumab plus trastuzumab (P+T) in patients (Pts) with colorectal cancer (CRC) with ERBB2 amplification or overexpression: Results from the TAPUR Study. J. Clin. Oncol. 2020, 38 (suppl. 4), 133. [Google Scholar] [CrossRef]
- Siena, S.; Elez, E.; Peeters, M.; André, T.; Van den Eynde, M.; Ng, K.; Cercek, A.; Fountzilas, C.; Sanchez, F.; Hubbard, J.; et al. PD-1 MOUNTAINEER: Open-label, phase 2 study of tucatinib combined with trastuzumab for HER2-positive metastatic colorectal cancer (SGNTUC-017, trial in progress). Ann. Oncol. 2021, 32, S199. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Beeram, M.; Mayordomo, J.I.; Hanna, D.L.; Ajani, J.A.; Blum Murphy, M.A.; Murthy, R.K.; Piha-Paul, S.A.; Bauer, T.M.; Bendell, J.C.; et al. Single agent activity of ZW25, a HER2-targeted bispecific antibody, in heavily pretreated HER2-expressing cancers. J. Clin. Oncol. 2018, 36 (suppl. 15), 2500. [Google Scholar] [CrossRef]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563–575.e11. [Google Scholar] [CrossRef]
- Siravegna, G.; Lazzari, L.; Crisafulli, G.; Sartore-Bianchi, A.; Mussolin, B.; Cassingena, A.; Martino, C.; Lanman, R.B.; Nagy, R.J.; Fairclough, S.; et al. Radiologic and Genomic Evolution of Individual Metastases during HER2 Blockade in Colorectal Cancer. Cancer Cell 2018, 34, 148–162.e7. [Google Scholar] [CrossRef] [Green Version]
- Raghav, K.P. Sequencing strategies in the management of metastatic colorectal cancer with HER2 amplification. Clin. Adv. Hematol. Oncol. 2022, 20, 86–88. [Google Scholar] [PubMed]
- Manca, P.; Corallo, S.; Busico, A.; Lonardi, S.; Corti, F.; Antoniotti, C.; Procaccio, L.; Clavarezza, M.; Smiroldo, V.; Tomasello, G.; et al. The Added Value of Baseline Circulating Tumor DNA Profiling in Patients with Molecularly Hyperselected, Left-sided Metastatic Colorectal Cancer. Clin. Cancer Res. 2021, 27, 2505–2514. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Meng, Q.; Sun, H.; Zhang, X.; Yun, J.; Li, B.; Wu, S.; Li, X.; Yang, H.; Zhu, H.; et al. HER2-specific chimeric antigen receptor-T cells for targeted therapy of metastatic colorectal cancer. Cell Death Dis. 2021, 12, 1109. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.A.; Malley, R.; Weinberg, B.A. Molecular Profiling in Metastatic Colorectal Cancer. Oncology 2020, 34, 352–355. [Google Scholar] [PubMed]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Tejpar, S.; Sartore-Bianchi, A.; Hechtman, J.F.; Christiansen, J.; Novara, L.; Tebbutt, N.; et al. ALK, ROS1, and NTRK Rearrangements in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djx089. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Barlesi, F.; Siena, S.; Ahn, M.J.; Drilon, A.; Conley, A.; Rolfo, C.; Wolf, J.; Seto, T.; Doebele, R.; et al. Patient-reported outcomes from STARTRK-2: A global phase II basket study of entrectinib for ROS1 fusion-positive non-small-cell lung cancer and NTRK fusion-positive solid tumours. ESMO Open 2021, 6, 100113. [Google Scholar] [CrossRef]
- He, X.; Jiao, X.D.; Liu, K.; Qin, B.D.; Wu, Y.; Ling, Y.; Liu, J.; Xu, A.Q.; Song, K.; Zang, Y.S. Clinical Responses to Crizotinib, Alectinib, and Lorlatinib in a Metastatic Colorectal Carcinoma Patient With ALK Gene Rearrangement: A Case Report. JCO Precis. Oncol. 2021, 5, PO.20.00534. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, G.; Liu, Y.; Feng, L.; Wang, M.; Liu, J.; Chen, Y.; Ouyang, L. Development of small-molecule tropomyosin receptor kinase (TRK) inhibitors for NTRK fusion cancers. Acta Pharm. Sin. B 2021, 11, 355–372. [Google Scholar] [CrossRef]
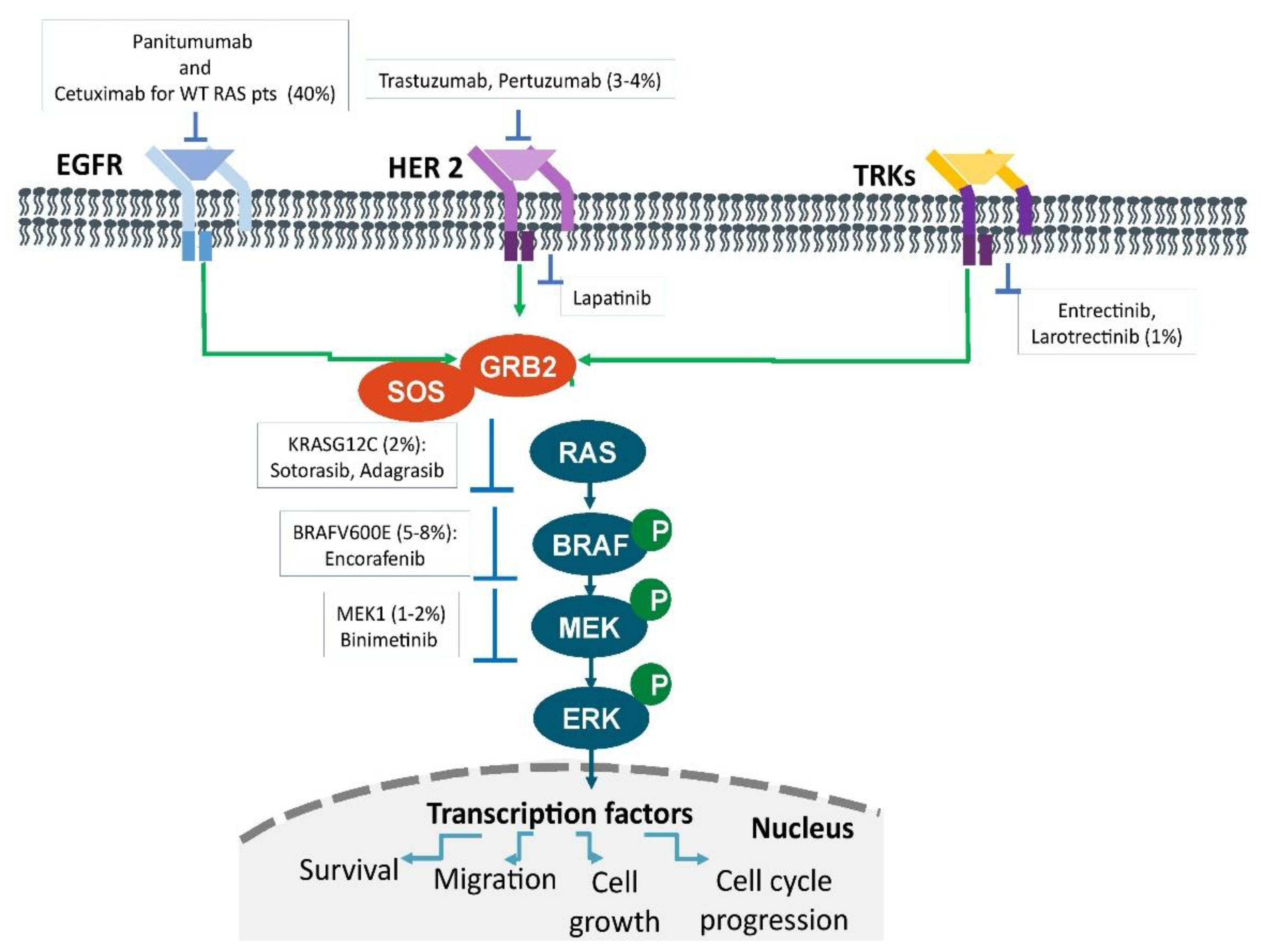

{kind=link}
| Biomarker | References | No. of Patients | Treatment | Key Findings |
|---|---|---|---|---|
| KRASG12C | [16] | 42 | Sotorasib | Median PFS 4.0 months; PR 7.1%; SD 66.7% |
| KRYSTAL-1 [17] | 28 | Adagrasib + cetuximab | ORR 43%; DCR 100% | |
| BRAF | BEACON [18] | 665 | Encorafenib + Cmab vs. Encorafenib + Cmab + binimetinib vs. Cmab + CT | Median OS (9.0 and 8.4 months vs. 5.4 months, p < 0.001); ORR (26% and 20% vs. 2%, p < 0.001) |
| MSI | CheckMate-142 [19] | 119 | Nivolumab + ipilimumab | ORR 55%; DCR > 12-week of 80% |
| Keynote 177 [13] | 307 | Pembrolizumab vs. CT | PFS 16.5 vs. 8.2 months, p = 0.0002; ORR 43.8% vs. 33.1% | |
| HER-2 | HERACLES [14,20] | 32 | Trastuzumab + lapatinib | ORR 28%; DCR 69%; median PFS 4.7 months |
| Destiny-CRC01 [21] | 53 | Trastuzumab deruxtecan | ORR 45%; median PFS 7 months | |
| NTRK | NAVIGATE [22] | 40 | Larotrectinib | ORR 50%; median OS 30 months |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosati, G.; Aprile, G.; Colombo, A.; Cordio, S.; Giampaglia, M.; Cappetta, A.; Porretto, C.M.; De Stefano, A.; Bilancia, D.; Avallone, A. Colorectal Cancer Heterogeneity and the Impact on Precision Medicine and Therapy Efficacy. Biomedicines 2022, 10, 1035. https://doi.org/10.3390/biomedicines10051035
Rosati G, Aprile G, Colombo A, Cordio S, Giampaglia M, Cappetta A, Porretto CM, De Stefano A, Bilancia D, Avallone A. Colorectal Cancer Heterogeneity and the Impact on Precision Medicine and Therapy Efficacy. Biomedicines. 2022; 10(5):1035. https://doi.org/10.3390/biomedicines10051035
Chicago/Turabian StyleRosati, Gerardo, Giuseppe Aprile, Alfredo Colombo, Stefano Cordio, Marianna Giampaglia, Alessandro Cappetta, Concetta Maria Porretto, Alfonso De Stefano, Domenico Bilancia, and Antonio Avallone. 2022. "Colorectal Cancer Heterogeneity and the Impact on Precision Medicine and Therapy Efficacy" Biomedicines 10, no. 5: 1035. https://doi.org/10.3390/biomedicines10051035
APA StyleRosati, G., Aprile, G., Colombo, A., Cordio, S., Giampaglia, M., Cappetta, A., Porretto, C. M., De Stefano, A., Bilancia, D., & Avallone, A. (2022). Colorectal Cancer Heterogeneity and the Impact on Precision Medicine and Therapy Efficacy. Biomedicines, 10(5), 1035. https://doi.org/10.3390/biomedicines10051035