
Mitochondrial Genetics Reinforces Multiple Layers of Interaction in Alzheimer’s Disease

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-seq Transcriptome Data from Brain Tissues
2.2. ADNI Cohort Data: mtDNA Sequence Data and Biomarkers
2.3. Differential Gene Expression Analysis in Brain Tissues
2.4. Gene–Gene Interaction Network Analysis
2.5. Epistasis between mtDNA Variants with an Impact on CSF Biomarker Levels
3. Results
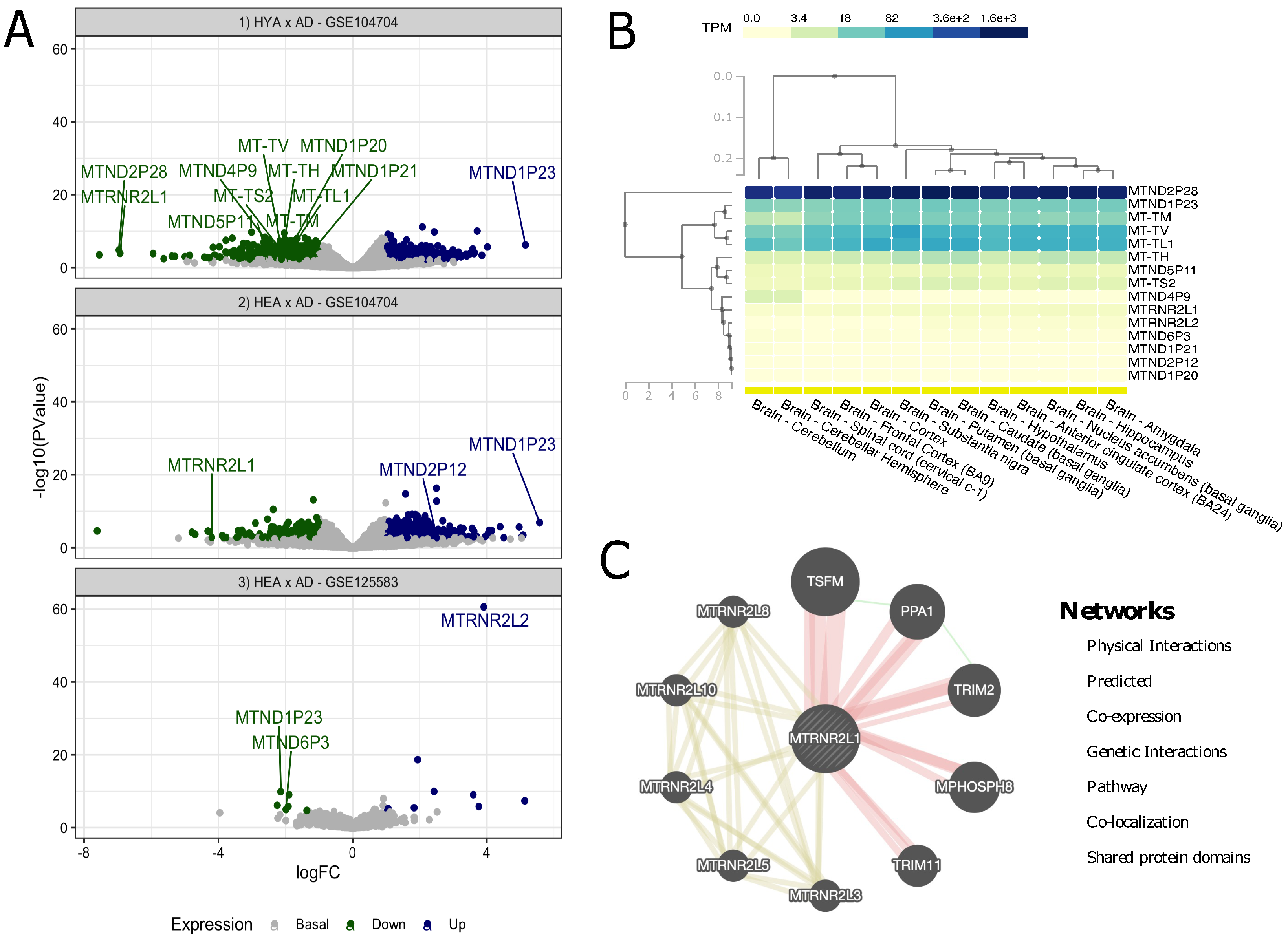
3.1. Screening for Differential Expression of mtDNA Genes and Pseudogenes
3.2. Epistasis between mtDNA and CSF PTAU and TAU Levels
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oliver, D.; Reddy, P.H. Molecular basis of Alzheimer’s disease: Focus on mitochondria. J. Alzheimer’s Dis. 2019, 72, S95–S116. [Google Scholar] [CrossRef] [PubMed]
- Bäckman, L.; Jones, S.; Berger, A.K.; Laukka, E.J.; Small, B. Multiple cognitive deficits during the transition to Alzheimer’s disease. J. Intern. Med. 2004, 256, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Bistaffa, E.; Tagliavini, F.; Matteini, P.; Moda, F. Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease. Brain Sci. 2020, 10, 815. [Google Scholar] [CrossRef] [PubMed]
- 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- Podlesniy, P.; Llorens, F.; Puigròs, M.; Serra, N.; Sepúlveda-Falla, D.; Schmidt, C.; Hermann, P.; Zerr, I.; Trullas, R. Cerebrospinal Fluid Mitochondrial DNA in Rapid and Slow Progressive Forms of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6298. [Google Scholar] [CrossRef]
- Tanaka, M.; Török, N.; Vécsei, L. Novel Pharmaceutical Approaches in Dementia. In NeuroPsychopharmacotherapy; Riederer, P., Laux, G., Nagatsu, T., Le, W., Riederer, C., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–18. [Google Scholar]
- Araújo, G.S.; Souza, M.R.; Oliveira, J.R.M.; Costa, I.G. Random Forest and Gene Networks for Association of SNPs to Alzheimer’s Disease. In Proceedings of the Brazilian Symposium on Bioinformatics, Recife, Brazil, 3–7 November 2013; Springer: Cham, Switzerland, 2013; pp. 104–115. [Google Scholar]
- Souza, M.B.R.; Araújo, G.S.; Costa, I.G.; Oliveira, J.R.M. Combined genome-wide CSF Aβ-42’s associations and simple network properties highlight new risk factors for Alzheimer’s disease. J. Mol. Neurosci. 2016, 58, 120–128. [Google Scholar] [CrossRef]
- Brito, L.M.; Ribeiro-dos Santos, Â.; Vidal, A.F.; de Araújo, G.S. Differential expression and mirna–gene interactions in early and late mild cognitive impairment. Biology 2020, 9, 251. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Mitochondria as potential targets in Alzheimer disease therapy: An update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Delbarba, A.; Abate, G.; Prandelli, C.; Marziano, M.; Buizza, L.; Arce Varas, N.; Novelli, A.; Cuetos, F.; Martínez, C.; Lanni, C.; et al. Mitochondrial alterations in peripheral mononuclear blood cells from Alzheimer’s disease and mild cognitive impairment patients. Oxid. Med. Cell. Longev. 2016, 2016, 5923938. [Google Scholar] [CrossRef] [Green Version]
- Zou, T.; Chen, W.; Zhou, X.; Duan, Y.; Ying, X.; Liu, G.; Zhu, M.; Pari, A.; Alimu, K.; Miao, H.; et al. Association of multiple candidate genes with mild cognitive impairment in an elderly Chinese Uygur population in Xinjiang. Psychogeriatrics 2019, 19, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Miton, C.M.; Buda, K.; Tokuriki, N. Epistasis and intramolecular networks in protein evolution. Curr. Opin. Struct. Biol. 2021, 69, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bennett, D.A.; De Jager, P.L.; Zhang, Q.Y.; Zhang, H.Y. Genome-wide epistasis analysis for Alzheimer’s disease and implications for genetic risk prediction. Alzheimer’s Res. Ther. 2021, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Escott-Price, V.; Schmidt, K.M. Probability of the Alzheimer’s disease based on common and rare genetic variants. Alzheimer’s Res. Ther. 2021, 13, 140. [Google Scholar] [CrossRef]
- Tan, M.S.; Yang, Y.X.; Xu, W.; Wang, H.F.; Tan, L.; Zuo, C.T.; Dong, Q.; Tan, L.; Suckling, J.; Yu, J.T. Associations of Alzheimer’s disease risk variants with gene expression, amyloidosis, tauopathy, and neurodegeneration. Alzheimer’s Res. Ther. 2021, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013, 45, 580. [Google Scholar] [CrossRef] [PubMed]
- Ridge, P.G.; Wadsworth, M.E.; Miller, J.B.; Saykin, A.J.; Green, R.C.; The Alzheimer’s Disease Neuroimaging Initiative; Kauwe, J.S. Assembly of 809 whole mitochondrial genomes with clinical, imaging, and fluid biomarker phenotyping. Alzheimer’s Dement. 2018, 14, 514–519. [Google Scholar] [CrossRef]
- Lakatos, A.; Derbeneva, O.; Younes, D.; Keator, D.; Bakken, T.; Lvova, M.; Brandon, M.; Guffanti, G.; Reglodi, D.; Saykin, A.; et al. Association between mitochondrial DNA variations and Alzheimer’s disease in the ADNI cohort. Neurobiol. Aging 2010, 31, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Hui, D.; Chalise, P.; Sharma, P.; Wang, X.; Andrews, S.J.; Pa, J.; Mahnken, J.D.; Morris, J.; Wilkins, H.M.; et al. Exploratory analysis of mtDNA haplogroups in two Alzheimer’s longitudinal cohorts. Alzheimer’s Dement. 2020, 16, 1164–1172. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Vandeerstichele, H.; Korecka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Update on the biomarker core of the Alzheimer’s Disease Neuroimaging Initiative subjects. Alzheimer’s Dement. 2010, 6, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, A.; Hannon, G. Fastx-Toolkit. FASTQ/A Short-Reads Pre-Processing Tools. 2017. Available online: http://hannonlab.cshl.edu/fastx_toolkit (accessed on 4 August 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Cavalcante, G.C.; Magalhães, L.; Ribeiro-dos Santos, Â.; Vidal, A.F. Mitochondrial Epigenetics: Non-Coding RNAs as a Novel Layer of Complexity. Int. J. Mol. Sci. 2020, 21, 1838. [Google Scholar] [CrossRef] [Green Version]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef] [Green Version]
- Streck, E.L.; Gonçalves, C.L.; Furlanetto, C.B.; Scaini, G.; Dal-Pizzol, F.; Quevedo, J. Mitochondria and the central nervous system: Searching for a pathophysiological basis of psychiatric disorders. Braz. J. Psychiatry 2014, 36, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral-Costa, J.; Kowaltowski, A. Neurological disorders and mitochondria. Mol. Asp. Med. 2020, 71, 100826. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis. 2010, 20, S265–S279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridge, P.G.; Kauwe, J.S. Mitochondria and Alzheimer’s disease: The role of mitochondrial genetic variation. Curr. Genet. Med. Rep. 2018, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagnoni, G.M.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G. Pathology and hippocampal atrophy in Alzheimer’s disease. Lancet Neurol. 2017, 16, 862–864. [Google Scholar] [CrossRef]
- Pierrick, C.; Manjón, J.V.; Enrique, L.; Gwenaelle, C. Lifespan Changes of the Human Brain In Alzheimer’s Disease. Sci. Rep. 2019, 9, 3998. [Google Scholar]
- Whitwell, J.L. Progression of atrophy in Alzheimer’s disease and related disorders. Neurotox. Res. 2010, 18, 339–346. [Google Scholar] [CrossRef]
- Sintini, I.; Graff-Radford, J.; Senjem, M.L.; Schwarz, C.G.; Machulda, M.M.; Martin, P.R.; Jones, D.T.; Boeve, B.F.; Knopman, D.S.; Kantarci, K.; et al. Longitudinal neuroimaging biomarkers differ across Alzheimer’s disease phenotypes. Brain 2020, 143, 2281–2294. [Google Scholar] [CrossRef]
- Kiernan, J. Anatomy of the temporal lobe. Epilepsy Res. Treat. 2012, 2012, 176157. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.S.; Erickson, D.J.; Sleeper, L.A.; Haynes, R.L.; Kinney, H.C. The lateral temporal lobe in early human life. J. Neuropathol. Exp. Neurol. 2017, 76, 424–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palejwala, A.H.; O’Connor, K.P.; Milton, C.K.; Anderson, C.; Pelargos, P.; Briggs, R.G.; Conner, A.K.; O’Donoghue, D.L.; Glenn, C.A.; Sughrue, M.E. Anatomy and white matter connections of the fusiform gyrus. Sci. Rep. 2020, 10, 13489. [Google Scholar] [CrossRef] [PubMed]
- Weiner, K.S.; Zilles, K. The anatomical and functional specialization of the fusiform gyrus. Neuropsychologia 2016, 83, 48–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, S.H.; Hong, S.H. Face-name memory in Alzheimer’s disease. Geriatr. Nurs. 2014, 35, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Giegé, T.; Giegé, R.; Giegé, P. tRNA biology in mitochondria. Int. J. Mol. Sci. 2015, 16, 4518–4559. [Google Scholar] [CrossRef] [Green Version]
- Garone, C.; Minczuk, M.; D’Souza, A.R. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Bik-Multanowski, M.; Pietrzyk, J.J.; Midro, A. MTRNR2L12: A candidate blood marker of early Alzheimer’s disease-like dementia in adults with down syndrome. J. Alzheimer’s Dis. 2015, 46, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Aβ. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Tas, E.; Muzumdar, R. Humanin and age-related diseases: A new link? Front. Endocrinol. 2014, 5, 210. [Google Scholar] [CrossRef] [Green Version]
- Bodzioch, M.; Lapicka-Bodzioch, K.; Zapala, B.; Kamysz, W.; Kiec-Wilk, B.; Dembinska-Kiec, A. Evidence for potential functionality of nuclearly-encoded humanin isoforms. Genomics 2009, 94, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, D.J.H.; Pardiñas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; Coleman, A.; Santos, R.D.; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Hazafa, A.; Batool, A.; Ahmad, S.; Amjad, M.; Chaudhry, S.N.; Asad, J.; Ghuman, H.F.; Khan, H.M.; Naeem, M.; Ghani, U. Humanin: A mitochondrial-derived peptide in the treatment of apoptosis-related diseases. Life Sci. 2021, 264, 118679. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Guterman, P.; Albert, A.Y.; Barha, C.K.; Galea, L.A.M.; on behalf of the Alzheimer’s Disease Neuroimaging Initiative. Sex influences the effects of APOE genotype and Alzheimer’s diagnosis on neuropathology and memory. Psychoneuroendocrinology 2021, 129, 105248. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Fulton-Howard, B.; Patterson, C.; McFall, G.P.; Gross, A.; Michaelis, E.K.; Goate, A.; Swerdlow, R.H.; Pa, J.; Initiative, A.D.N.; et al. Mitonuclear interactions influence Alzheimer’s disease risk. Neurobiol. Aging 2020, 87, 138.e7–138.e14. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| GEO Accession | Brain Tissue | Group/Samples/Age | |
|---|---|---|---|
| GSE104704 | Lateral temporal lobe | Healthy young adults | 8/(42–59) |
| Healthy elderly adults | 10/(61–77) | ||
| AD cases | 12/(61–79) | ||
| GSE125583 | Fusiform gyrus | Healthy elderly adults | 70/(71–103) |
| AD cases | 219/(60–103) | ||
| (1) Healthy Young Adults vs. AD Cases—GSE104704 | |||||
|---|---|---|---|---|---|
| HGNC Symbol | Gene | logFC | FDR | Expression | Biotype |
| MT-TL1 | ENSG00000209082.1 | −1.84 | 0.0014 | Down | tRNA |
| MT-TV | ENSG00000210077.1 | −2.16 | 6.38 | Down | tRNA |
| MT-TM | ENSG00000210112.1 | −1.93 | 0.0002 | Down | tRNA |
| MT-TH | ENSG00000210176.1 | −2.07 | 0.0025 | Down | tRNA |
| MT-TS2 | ENSG00000210184.1 | −2.12 | 0.0018 | Down | tRNA |
| MTND2P28 | ENSG00000225630.1 | −6.96 | 0.0014 | Down | pseudogene |
| MTND1P23 | ENSG00000225972.1 | 5.14 | 0.0002 | Up | pseudogene |
| MTND1P20 | ENSG00000226794.1 | −2.04 | 0.0481 | Down | pseudogene |
| MTND1P21 | ENSG00000235940.1 | −1.32 | 0.0151 | Down | pseudogene |
| MTND5P11 | ENSG00000248923.1 | −2.52 | 0.0419 | Down | pseudogene |
| MTND4P9 | ENSG00000250050.1 | −2.18 | 0.0302 | Down | pseudogene |
| MTRNR2L1 | ENSG00000256618.1 | −6.92 | 0.0050 | Down | protein coding |
| (2) Health Elderly Adults vs. AD Cases—GSE104704 | |||||
| HGNC Symbol | Gene | logFC | FDR | Expression | Biotype |
| MTND1P23 | ENSG00000225972.1 | 5.57 | 0.0001 | Up | pseudogene |
| MTND2P12 | ENSG00000228725.3 | 2.49 | 0.0158 | Up | pseudogene |
| MTRNR2L1 | ENSG00000256618.1 | −4.19 | 0.0430 | Down | protein coding |
| (3) Health Elderly Adults vs. AD Cases—GSE125583 | |||||
| HGNC Symbol | Gene | logFC | FDR | Expression | Biotype |
| MTND1P23 | ENSG00000225972.1 | −2.13 | 1.36 | Down | pseudogene |
| MTND6P3 | ENSG00000254132.1 | −1.98 | 0.02653 | Down | pseudogene |
| MTRNR2L2 | ENSG00000271043.1 | 3.90 | 1.13 | Up | protein coding |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavalcante, G.C.; Brito, L.M.; Schaan, A.P.; Ribeiro-dos-Santos, Â.; de Araújo, G.S.; on behalf of Alzheimer’s Disease Neuroimaging Initiative. Mitochondrial Genetics Reinforces Multiple Layers of Interaction in Alzheimer’s Disease. Biomedicines 2022, 10, 880. https://doi.org/10.3390/biomedicines10040880
Cavalcante GC, Brito LM, Schaan AP, Ribeiro-dos-Santos Â, de Araújo GS, on behalf of Alzheimer’s Disease Neuroimaging Initiative. Mitochondrial Genetics Reinforces Multiple Layers of Interaction in Alzheimer’s Disease. Biomedicines. 2022; 10(4):880. https://doi.org/10.3390/biomedicines10040880
Chicago/Turabian StyleCavalcante, Giovanna Chaves, Leonardo Miranda Brito, Ana Paula Schaan, Ândrea Ribeiro-dos-Santos, Gilderlanio Santana de Araújo, and on behalf of Alzheimer’s Disease Neuroimaging Initiative. 2022. "Mitochondrial Genetics Reinforces Multiple Layers of Interaction in Alzheimer’s Disease" Biomedicines 10, no. 4: 880. https://doi.org/10.3390/biomedicines10040880
APA StyleCavalcante, G. C., Brito, L. M., Schaan, A. P., Ribeiro-dos-Santos, Â., de Araújo, G. S., & on behalf of Alzheimer’s Disease Neuroimaging Initiative. (2022). Mitochondrial Genetics Reinforces Multiple Layers of Interaction in Alzheimer’s Disease. Biomedicines, 10(4), 880. https://doi.org/10.3390/biomedicines10040880