
Antiviral Cell Products against COVID-19: Learning Lessons from Previous Research in Anti-Infective Cell-Based Agents


 , ,
, , 
Abstract
1. Introduction
2. Promising BMCP in COVID-19 Prophylaxis and Treatment
2.1. DC Vaccines
2.2. CAR-Effector Cell Therapy

{kind=link}
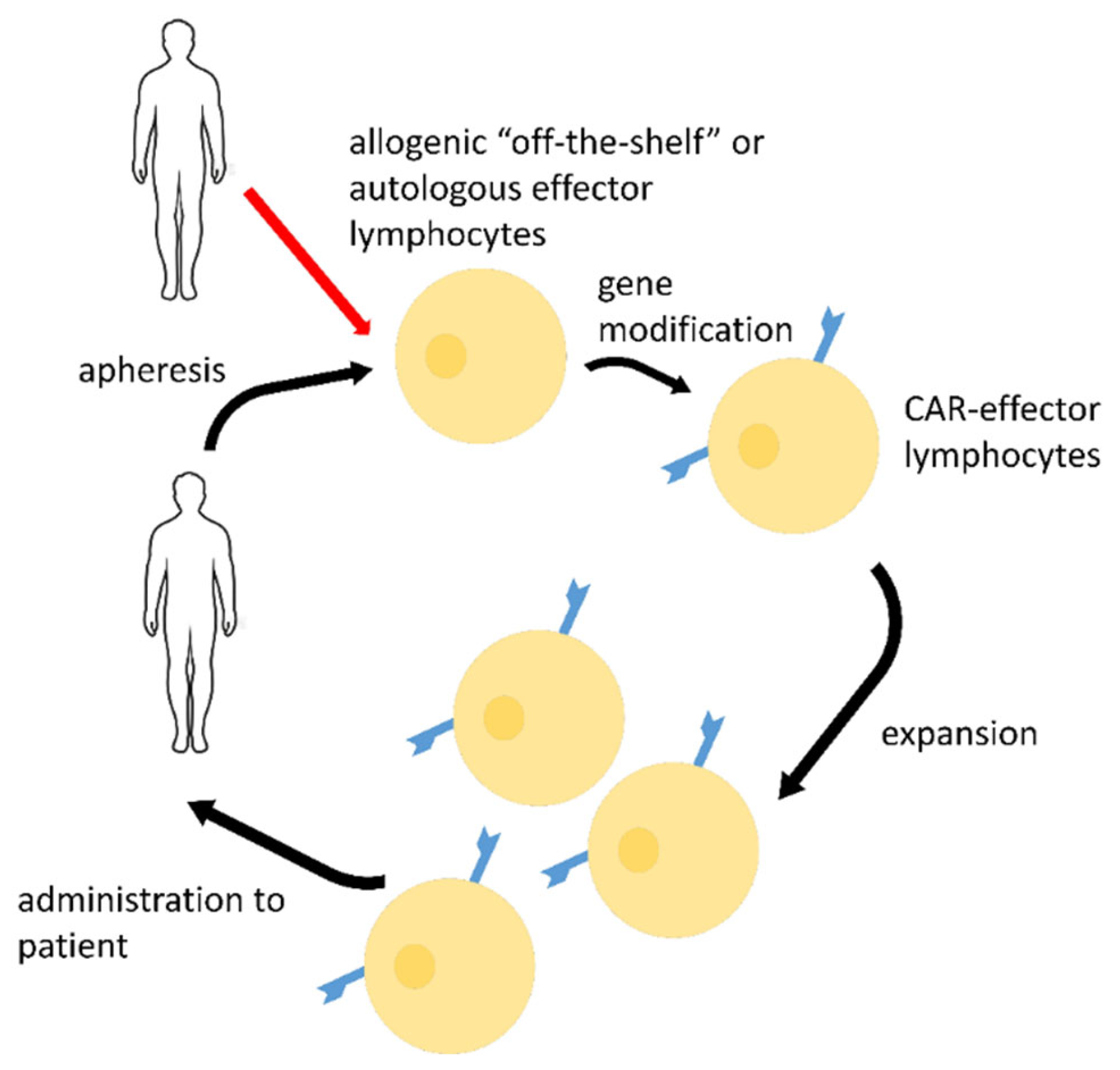{kind=link}
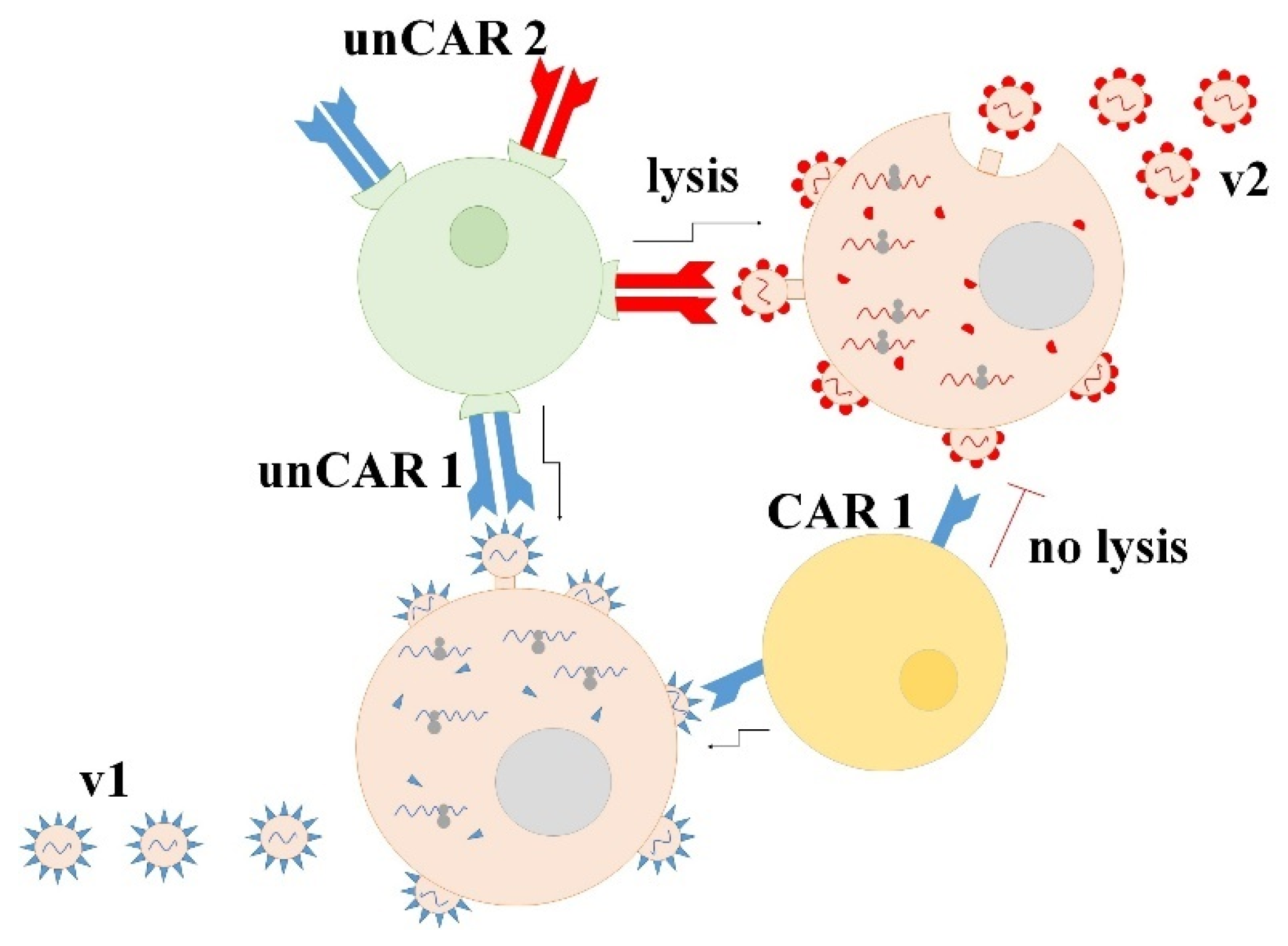{kind=link}
| CAR Details, Reference | Target Antigen | Effector Cells | Stage of the Study |
|---|---|---|---|
| scFv-based CR3022 2nd generation CAR, CD28, and CD3ζ activating domains [62] | RBD epitope | T-cells | Pre-clinical research |
| scFv-based CR3022, 3rd generation CAR, CD28, 4-1BB, and CD3ζ activating domains [71,72,73] | RBD epitope | NK-cells | Pre-clinical research |
| scFv-based S309, 3rd generation CAR, CD28, 4-1BB, and CD3ζ activating domains [72,73] | RBD epitope | NK-cells | Pre-clinical research |
| scFv-basedCR3022, MERTK, or MEGF10, or FcRγ(FCER1G), or CD3ζ activating domains [74] | RBD epitope | macrophages | Pre-clinical research |
| H84T-Banana Lectin (BanLec), 2nd generation CAR, 4-1BB, and CD3 ζ activating domains [75] | high mannose glycosites that decorate viral envelopes | NK-cells | Pre-clinical research |
| NKG2D-ACE2 CAR-NK cells for therapy of COVID-19, NCT04324996 [76] | S | NK-cells | Phase I/II clinical trial |
2.3. Potential Advantages of Universal CAR Technology in Anti-SARS-CoV-2 Treatment
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Li, C.X.; Noreen, S.; Zhang, L.X.; Saeed, M.; Wu, P.F.; Ijaz, M.; Dai, D.F.; Maqbool, I.; Madni, A.; Akram, F.; et al. A critical analysis of SARS-CoV-2 (COVID-19) complexities, emerging variants, and therapeutic interventions and vaccination strategies. Biomed. Pharmacother. 2022, 146, 112550. [Google Scholar] [CrossRef] [PubMed]
- Sarubbo, F.; El Haji, K.; Vidal-Balle, A.; Bargay Lleonart, J. Neurological consequences of COVID-19 and brain related pathogenic mechanisms: A new challenge for neuroscience. Brain Behav. Immun. Health 2022, 19, 100399. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.S.; Lee, P.I.; Hsueh, P.R. Treatment options for COVID-19: The reality and challenges. J. Microbiol. Immunol. Infect. 2020, 53, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Zinatizadeh, M.R.; Zarandi, P.K.; Zinatizadeh, M.; Yousefi, M.H.; Amani, J.; Rezaei, N. Efficacy of mRNA, adenoviral vector, and perfusion protein COVID-19 vaccines. Biomed. Pharm. 2022, 146, 112527. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Gam-COVID-Vac Vaccine Trial Group. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Galmiche, S.; Luong Nguyen, L.B.; Tartour, E.; de Lamballerie, X.; Wittkop, L.; Loubet, P.; Launay, O. Immunological and clinical efficacy of COVID-19 vaccines in immunocompromised populations: A systematic review. Clin. Microbiol. Infect. 2022, 28, 163–177. [Google Scholar] [CrossRef]
- Zhou, R.; To, K.K.; Wong, Y.C.; Liu, L.; Zhou, B.; Li, X.; Huang, H.; Mo, Y.; Luk, T.Y.; Lau, T.T.; et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity 2020, 53, 864–877. [Google Scholar] [CrossRef]
- Filin, I.Y.; Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Recent Advances in Experimental Dendritic Cell Vaccines for Cancer. Front. Oncol. 2021, 11, 730824. [Google Scholar] [CrossRef]
- Zhang, X.; Gordon, J.R.; Xiang, J. Advances in dendritic cell-based vaccine of cancer. Cancer Biother. Radiopharm. 2002, 6, 601–619. [Google Scholar] [CrossRef]
- Sadeghzadeh, M.; Bornehdeli, S.; Mohahammadrezakhani, H.; Abolghasemi, M.; Poursaei, E.; Asadi, M.; Zafari, V.; Aghebati-Maleki, L.; Shanehbandi, D. Dendritic cell therapy in cancer treatment; the state-of-the-art. Life Sci. 2020, 254, 117580. [Google Scholar] [CrossRef]
- Saadeldin, M.K.; Abdel-Aziz, A.K.; Abdellatif, A. Dendritic cell vaccine immunotherapy; the beginning of the end of cancer and COVID-19. A hypothesis. Med. Hypotheses 2021, 146, 110365. [Google Scholar] [CrossRef] [PubMed]
- Brusko, M.A.; Stewart, J.M.; Posgai, A.L.; Wasserfall, C.H.; Atkinson, M.A.; Brusko, T.M.; Keselowsky, B.G. Immunomodulatory Dual-Sized Microparticle System Conditions Human Antigen Presenting Cells into a Tolerogenic Phenotype In Vitro and Inhibits Type 1 Diabetes-Specific Autoreactive T Cell Responses. Front. Immunol. 2020, 11, 574447. [Google Scholar] [CrossRef] [PubMed]
- Adorini, L.; Penna, G.; Giarratana, N.; Uskokovic, M. Tolerogenic dendritic cells induced by vitamin D receptor ligands enhance regulatory T cells inhibiting allograft rejection and autoimmune diseases. J. Cell. Biochem. 2003, 88, 227–233. [Google Scholar] [CrossRef]
- Shen, C.; Wang, Z.; Zhao, F.; Yang, Y.; Li, J.; Yuan, J.; Wang, F.; Li, D.; Yang, M.; Xing, L.; et al. Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA 2020, 323, 1582–1589. [Google Scholar] [CrossRef]
- Tosta, E. The protective immunity induced by SARS-CoV-2 infection and vaccination: A critical appraisal. Explor. Immunol. 2021, 1, 199–225. [Google Scholar] [CrossRef]
- Zohar, T.; Loos, C.; Fischinger, S.; Atyeo, C.; Wang, C.; Slein, M.D.; Burke, J.; Yu, J.; Feldman, J.; Hauser, B.M.; et al. Compromised Humoral Functional Evolution Tracks with SARS-CoV-2 Mortality. Cell 2020, 183, 1508–1519.e12. [Google Scholar] [CrossRef]
- Wu, Y.; Huang, Z.; Harrison, R.; Liu, L.; Zhu, L.; Situ, Y.; Wang, Y. Engineering CAR T cells for enhanced efficacy and safety. APL Bioeng. 2022, 6, 011502. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Grupp, S.A.; Porter, D.L.; June, C.H. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Loskog, A.; Giandomenico, V.; Rossig, C.; Pule, M.; Dotti, G.; Brenner, M.K. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia 2006, 20, 1819–1828. [Google Scholar] [CrossRef]
- Hombach, A.A.; Abken, H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28–OX40 signalling. Int. J. Cancer 2011, 129, 2935–2944. [Google Scholar] [CrossRef] [PubMed]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D. Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCRζ chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, P.; Fang, H.; Wang, G.; Zeng, X. Paving the Way Towards Universal Chimeric Antigen Receptor Therapy in Cancer Treatment: Current Landscape and Progress. Front. Immunol. 2020, 11, 604915. [Google Scholar] [CrossRef]
- Wu, J.; Mishra, H.K.; Walcheck, B. Role of ADAM17 as a regulatory checkpoint of CD16A in NK cells and as a potential target for cancer immunotherapy. J. Leukoc. Biol. 2019, 105, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Walcheck, B.; Wu, J. iNK-CD64/16A cells: A promising approach for ADCC? Expert Opin. Biol. Ther. 2019, 19, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Phase I–II Trial of Dendritic Cell Vaccine to Prevent COVID-19 in Adults. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04386252 (accessed on 12 February 2022).
- Immunity and Safety of COVID-19 Synthetic Minigene Vaccine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04276896?term=DC&cond=SARS+CoV+2+Infection&draw=2&rank=1 (accessed on 12 February 2022).
- Zhou, Q.; Gu, H.; Sun, S.; Zhang, Y.; Hou, Y.; Li, C.; Zhao, Y.; Ma, P.; Lv, L.; Aji, S.; et al. Large-Sized Graphene Oxide Nanosheets Increase DC-T-Cell Synaptic Contact and the Efficacy of DC Vaccines against SARS-CoV-2. Adv. Mater. 2021, 33, e2102528. [Google Scholar] [CrossRef]
- Reuter, T.; Heldmann, M.; Schimmer, S.; Schepers, K.; Dittmer, U. Protection of mice against Friend retrovirus infection by vaccination with antigen-loaded, spleen-derived dendritic cells. Vaccine 2004, 22, 2686–2689. [Google Scholar] [CrossRef]
- Norton, T.D.; Miller, E.A. Recent Advances in Lentiviral Vaccines for HIV-1 Infection. Front. Immunol. 2016, 7, 243. [Google Scholar]
- Mohamed, H.; Miller, V.; Jennings, S.R.; Wigdahl, B.; Krebs, F.C. The Evolution of Dendritic Cell Immunotherapy against HIV-1 Infection: Improvements and Outlook. J. Immunol. Res. 2020, 2020, 9470102. [Google Scholar] [CrossRef]
- Norton, T.D.; Zhen, A.; Tada, T.; Kim, J.; Kitchen, S.; Landau, N.R. Lentiviral Vector-Based Dendritic Cell Vaccine Suppresses HIV Replication in Humanized Mice. Mol. Ther. 2019, 27, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Spadaccia, M.; Sabado, R.; Chertova, E.; Bess, J.; Trubey, C.M.; Holman, R.M.; Salazar, A.; Lifson, J.; Bhardwaj, N. Autologous aldrithiol-2-inactivated HIV-1 combined with polyinosinic-polycytidylic acid-poly-L-lysine carboxymethylcellulose as a vaccine platform for therapeutic dendritic cell immunotherapy. Vaccine 2015, 33, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Lee, S.H.; Song, X.T.; Jones, L.; Machida, K.; Huang, X.F.; Chen, S.Y. A super TLR agonist to improve efficacy of dendritic cell vaccine in induction of anti-HCV immunity. PLoS ONE 2012, 7, e48614. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, F.; Chen, L.; Ma, L.; Wang, Y.; He, Y.; Ma, Z.; Liu, H.; Guo, Y.; Zhang, Y.; et al. Development of a dendritic cell vaccine encoding multiple cytotoxic T lymphocyte epitopes targeting hepatitis C virus. Int. J. Mol. Med. 2013, 32, 901–909. [Google Scholar] [CrossRef][Green Version]
- Mekonnen, Z.A.; Masavuli, M.G.; Yu, W.; Gummow, J.; Whelan, D.M.; Al-Delfi, Z.; Torresi, J.; Gowans, E.J.; Grubor-Bauk, B. Enhanced T Cell Responses Induced by a Necrotic Dendritic Cell Vaccine, Expressing HCV NS3. Front. Microbiol. 2020, 11, 559105. [Google Scholar] [CrossRef]
- Ostanin, A.A.; Chernykh, E.R. Autologous Dendritic Cell Vaccine for Treatment of Patients with Chronic HCV-Infection. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03119025?term=DC+vaccine&cond=Hepatitis+C&draw=2&rank=1 (accessed on 15 February 2022).
- Phase I–II Vaccination of Autologous Dendritic Cells Transduced with Adenoviral Vector Encoding NS3 in Hepatitis C Encoding NS3 in Hepatitis C. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02309086?term=DC+vaccine&cond=Hepatitis+C&draw=2&rank=2 (accessed on 15 February 2022).
- Chen, M.; Li, Y.G.; Zhang, D.Z.; Wang, Z.Y.; Zeng, W.Q.; Shi, X.F.; Guo, Y.; Guo, S.H.; Ren, H. Therapeutic effect of autologous dendritic cell vaccine on patients with chronic hepatitis B: A clinical study. World J. Gastroenterol. 2005, 11, 1806–1808. [Google Scholar] [CrossRef]
- A Clinical Trial on Hepatitis B Vaccine Activated-Dendritic Cells Combined with Anti-HBV Drugs in CHB (CTHBVACADCHB). ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02615639?term=DC+vaccine&cond=Hepatitis+B&draw=2&rank=1 (accessed on 17 February 2022).
- Luo, J.; Li, J.; Chen, R.L.; Nie, L.; Huang, J.; Liu, Z.W.; Luo, L.; Yan, X.J. Autologus dendritic cell vaccine for chronic hepatitis B carriers: A pilot, open label, clinical trial in human volunteers. Vaccine 2010, 28, 2497–2504. [Google Scholar] [CrossRef]
- Akbar, S.M.; Furukawa, S.; Horiike, N.; Abe, M.; Hiasa, Y.; Onji, M. Safety and immunogenicity of hepatitis B surface antigen-pulsed dendritic cells in patients with chronic hepatitis B. J. Viral Hepat. 2011, 18, 408–414. [Google Scholar] [CrossRef]
- Wei, M.J.; Pan, X.N.; Wei, K.P.; Li, X.H.; Liu, X.L.; Zhang, X.M.; Jiang, Y.L.; Zhang, C.Y.; Shen, J.K. Efficacy of HBV-pulsed DCs in combination with entecavir in patients with chronic hepatitis B infection. Int. Immunopharmacol. 2015, 27, 238–243. [Google Scholar] [CrossRef]
- Yang, J.Y.; Cao, D.Y.; Liu, W.C.; Zhang, H.M.; Teng, Z.H.; Ren, J. Dendritic cell generated from CD34+ hematopoietic progenitors can be transfected with adenovirus containing gene of HBsAg and induce antigen-specific cytotoxic T cell responses. Cell Immunol. 2006, 240, 14–21. [Google Scholar] [CrossRef]
- Long, J.; Zhou, B.; Li, H.; Dai, Q.; Zhang, B.; Xing, S.; Zeng, Z.; Chen, W.; Yang, J. Improvement of HBsAg gene-modified dendritic cell-based vaccine efficacy by optimizing immunization method or the application of β-glucosylceramide. Immunol. Investig. 2013, 42, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Chemaly, R.F.; Ullmann, A.J.; Stoelben, S.; Richard, M.P.; Bornhäuser, M.; Groth, C.; Einsele, H.; Silverman, M.; Mullane, K.M.; Brown, J.; et al. Letermovir for cytomegalovirus prophylaxis in hematopoietic-cell transplantation. N. Engl. J. Med. 2014, 370, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Van Craenenbroeck, A.H.; Smits, E.L.; Anguille, S.; Van de Velde, A.; Stein, B.; Braeckman, T.; Van Camp, K.; Nijs, G.; Ieven, M.; Goossens, H.; et al. Induction of cytomegalovirus-specific T cell responses in healthy volunteers and allogeneic stem cell recipients using vaccination with messenger RNA-transfected dendritic cells. Transplantation 2015, 99, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.K.K.; Clancy, L.; Simms, R.; Burgess, J.; Deo, S.; Blyth, E.; Micklethwaite, K.P.; Gottlieb, D.J. Adjuvant Peptide Pulsed. Adjuvant Peptide Pulsed Dendritic Cell Vaccination in Addition to T Cell Adoptive Immunotherapy for Cytomegalovirus Infection in Allogeneic Hematopoietic Stem Cell Transplantation Recipients. Biol. Blood Marrow Transplant. 2018, 24, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Cytomegalovirus (CMV) RNA-Pulsed Dendritic Cells for Pediatric Patients and Young Adults with WHO Grade IV Glioma, Recurrent Malignant Glioma, or Recurrent Medulloblastoma (ATTAC-P). ClinicalTryals.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03615404?term=DC&cond=CMV&draw=2&rank=1 (accessed on 16 February 2022).
- Ueno, K.; Kinjo, Y.; Okubo, Y.; Aki, K.; Urai, M.; Kaneko, Y.; Shimizu, K.; Wang, D.N.; Okawara, A.; Nara, T.; et al. Dendritic cell-based immunization ameliorates pulmonary infection with highly virulent Cryptococcus gattii. Infect. Immun. 2015, 83, 1577–1586. [Google Scholar] [CrossRef]
- Ueno, K.; Urai, M.; Ohkouchi, K.; Miyazaki, Y.; Kinjo, Y. Dendritic Cell-Based Vaccine Against Fungal Infection. Methods Mol. Biol. 2016, 1403, 537–549. [Google Scholar]
- Ueno, K.; Urai, M.; Takatsuka, S.; Abe, M.; Miyazaki, Y.; Kinjo, Y. Immunization with Antigen-Pulsed Dendritic Cells Against Highly Virulent Cryptococcus gattii Infection: Analysis of Cytokine-Producing T Cells. Methods Mol. Biol. 2017, 1625, 327–339. [Google Scholar]
- Silva, L.B.R.; Dias, L.S.; Rittner, G.M.G.; Muñoz, J.E.; Souza, A.C.O.; Nosanchuk, J.D.; Travassos, L.R.; Taborda, C.P. Dendritic Cells Primed with Paracoccidioides brasiliensis Peptide P10 Are Therapeutic in Immunosuppressed Mice with Paracoccidioidomycosis. Front. Microbiol. 2017, 8, 1057. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Premkumar, L.; Segovia-Chumbez, B.; Jadi, R.; Martinez, D.R.; Raut, R.; Markmann, A.; Cornaby, C.; Bartelt, L.; Weiss, S.; Park, Y.; et al. The receptor binding domain of the viral spike protein is an immunodominant and highly specific target of antibodies in SARS-CoV-2 patients. Sci. Immunol. 2020, 5, eabc8413. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Su, B.; Guo, X.; Sun, W.; Deng, Y.; Bao, L.; Zhu, Q.; Zhang, X.; Zheng, Y.; Geng, C.; et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell 2020, 182, 73–84.e16. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Kazanova, A.; Thurmond, S.; Saragovi, H.U.; Rudd, C.E. Effective chimeric antigen receptor T cells against SARS-CoV-2. iScience 2021, 24, 103295. [Google Scholar] [CrossRef]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef]
- Mehrabadi, A.Z.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Hamidinejad, M.A.; Ghaleh, H.E.G. Therapeutic potential of CAR T cell in malignancies: A scoping review. Biomed. Pharm. 2022, 146, 112512. [Google Scholar] [CrossRef]
- Björkström, N.K.; Strunz, B.; Ljunggren, H.G. Natural killer cells in antiviral immunity. Nat. Rev. Immunol. 2022, 22, 112–123. [Google Scholar] [CrossRef]
- Carlsten, M.; Childs, R.W. Genetic manipulations of NK cells for cancer immunotherapy. Front. Immunol. 2015, 6, 266. [Google Scholar] [CrossRef]
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front. Immunol. 2017, 8, 465. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Li, L.; McCarty, J.; Kaur, I.; Yvon, E.; Shaim, H.; Muftuoglu, M.; Liu, E.; Orlowski, R.Z.; Cooper, L.; et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br. J. Haematol. 2017, 177, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Mamonkin, M.; Brenner, M.K.; Heslop, H.E. Taking T-Cell Oncotherapy Off-the-Shelf. Trends Immunol. 2021, 42, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Badeti, S.; Geng, K.; Liu, D. Efficacy of Targeting SARS-CoV-2 by CAR-NK Cells. BioRxiv 2020, 247320. [Google Scholar] [CrossRef]
- Ma, M.; Badeti, S.; Chen, C.H.; Pinter, A.; Jiang, Q.; Shi, L.; Zhou, R.; Xu, H.; Li, Q.; Gause, W.; et al. CAR-NK Cells Effectively Target the D614 and G614 SARS-CoV-2-infected Cells. BioRxiv 2021, 426742. [Google Scholar] [CrossRef]
- Ma, M.T.; Badeti, S.; Chen, C.H.; Kim, J.; Choudhary, A.; Honnen, B.; Reichman, C.; Calianese, D.; Pinter, A.; Jiang, Q.; et al. CAR-NK Cells Effectively Target SARS-CoV-2-Spike-Expressing Cell Lines In Vitro. Front. Immunol. 2021, 12, 652223. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Ma, Z.; Qian, K.; Li, T.; Zhao, J.; Hu, S. CAR Macrophages for SARS-CoV-2 Immunotherapy. Front. Immunol. 2021, 12, 669103. [Google Scholar] [CrossRef]
- Christodoulou, I.; Rahnama, R.; Ravich, J.W.; Seo, J.; Zolov, S.N.; Marple, A.N.; Markovitz, D.M.; Bonifant, C.L. Glycoprotein Targeted CAR-NK Cells for the Treatment of SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 763460. [Google Scholar] [CrossRef]
- A Phase I/II Study of Universal Off-the-Shelf NKG2D-ACE2 CAR-NK Cells for Therapy of COVID-19. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04324996?term=CAR&cond=COVID-19&draw=2&rank=1 (accessed on 19 February 2022).
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef]
- Sohail, A.; Yu, Z.; Arif, R.; Nutini, A.; Nofal, T.A. Piecewise differentiation of the fractional order CAR-T cells-SARS-2 virus model. Results Phys. 2022, 33, 105046. [Google Scholar] [CrossRef] [PubMed]
- Al-Utaibi, K.A.; Nutini, A.; Sohail, A.; Arif, R.; Tunc, S.; Sait, S.M. Forecasting the action of CAR-T cells against SARS-corona virus-II infection with branching process. Model. Earth Syst. Environ. 2021, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Xiao, Y.; Meng, X.; Tang, L.; Li, B.; Zhao, Z.; Tan, Q.; Shan, H.; Liu, L.; Huang, X. Nanovesicles derived from bispecific CAR-T cells targeting the spike protein of SARS-CoV-2 for treating COVID-19. J. Nanobiotechnology 2021, 19, 391. [Google Scholar] [CrossRef] [PubMed]
- Bednar, C.; Ensser, A. CARs-A New Perspective to HCMV Treatment. Viruses 2021, 13, 1563. [Google Scholar] [CrossRef] [PubMed]
- Seif, M.; Einsele, H.; Löffler, J. CAR T Cells Beyond Cancer: Hope for Immunomodulatory Therapy of Infectious Diseases. Front. Immunol. 2019, 10, 2711. [Google Scholar] [CrossRef]
- Slabik, C.; Kalbarczyk, M.; Danisch, S.; Zeidler, R.; Klawonn, F.; Volk, V.; Krönke, N.; Feuerhake, F.; Ferreira de Figueiredo, C.; Blasczyk, R.; et al. CAR-T Cells Targeting Epstein-Barr Virus gp350 Validated in a Humanized Mouse Model of EBV Infection and Lymphoproliferative Disease. Mol. Ther. Oncolytics 2020, 18, 504–524. [Google Scholar] [CrossRef]
- Tang, X.; Zhou, Y.; Li, W.; Tang, Q.; Chen, R.; Zhu, J.; Feng, Z. T cells expressing a LMP1-specific chimeric antigen receptor mediate antitumor effects against LMP1-positive nasopharyngeal carcinoma cells in vitro and in vivo. J. Biomed. Res. 2014, 28, 468–475. [Google Scholar]
- Kieser, A.; Sterz, K.R. The Latent Membrane Protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar]
- LMP1 Positive Infectious Diseases and Hematological Malignancies. ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04657965?term=CAR&cond=Infections&draw=2&rank=3 (accessed on 23 February 2022).
- Maldini, C.R.; Ellis, G.I.; Riley, J.L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018, 18, 605–616. [Google Scholar] [CrossRef]
- Liu, L.; Patel, B.; Ghanem, M.H.; Bundoc, V.; Zheng, Z.; Morgan, R.A.; Rosenberg, S.A.; Dey, B.; Berger, E.A. Novel CD4-Based Bispecific Chimeric Antigen Receptor Designed for Enhanced Anti-HIV Potency and Absence of HIV Entry Receptor Activity. J. Virol. 2015, 89, 6685–6694. [Google Scholar] [CrossRef]
- Zhen, A.; Peterson, C.W.; Carrillo, M.A.; Reddy, S.S.; Youn, C.S.; Lam, B.B.; Chang, N.Y.; Martin, H.A.; Rick, J.W.; Kim, J.; et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog. 2017, 13, e1006753. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Liang, H.; Pan, H.; Liang, Y.; Wang, H.; Yang, X.; Lu, P.; Zhang, X.; Yang, J.; Zhang, D.; et al. HIV-1-Specific CAR-T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade Enhance Anti-HIV Efficacy in vivo. Front. Microbiol. 2021, 12, 684016. [Google Scholar] [CrossRef] [PubMed]
- Pampusch, M.S.; Abdelaal, H.M.; Cartwright, E.K.; Molden, J.S.; Davey, B.C.; Sauve, J.D.; Sevcik, E.N.; Rendahl, A.K.; Rakasz, E.G.; Connick, E.; et al. CAR/CXCR5-T cell immunotherapy is safe and potentially efficacious in promoting sustained remission of SIV infection. PLoS Pathog. 2022, 18, e1009831. [Google Scholar] [CrossRef] [PubMed]
- Leslie, G.J.; Wang, J.; Richardson, M.W.; Haggarty, B.S.; Hua, K.L.; Duong, J.; Secreto, A.J.; Jordon, A.P.; Romano, J.; Kumar, K.E.; et al. Potent and Broad Inhibition of HIV-1 by a Peptide from the gp41 Heptad Repeat-2 Domain Conjugated to the CXCR4 Amino Terminus. PLoS Pathog. 2016, 12, e1005983. [Google Scholar] [CrossRef]
- Maldini, C.R.; Gayout, K.; Leibman, R.S.; Dopkin, D.L.; Mills, J.P.; Shan, X.; Glover, J.A.; Riley, J.L. HIV-Resistant and HIV-Specific CAR-Modified CD4+ T Cells Mitigate HIV Disease Progression and Confer CD4+ T Cell Help In Vivo. Mol. Ther. 2020, 28, 1585–1599. [Google Scholar] [CrossRef]
- Kim, G.B.; Hege, K.; Riley, J.L. CAR Talk: How Cancer-Specific CAR T Cells Can Instruct How to Build CAR T Cells to Cure HIV. Front. Immunol. 2019, 10, 2310. [Google Scholar] [CrossRef]
- CD4 CAR+ ZFN-Modified T Cells in HIV Therapy. ClinicalTryal.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03617198 (accessed on 23 February 2022).
- CAR-T Cells for HIV Infection. ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04648046?term=CAR&cond=Infections&draw=2&rank=1 (accessed on 23 February 2022).
- Third-Generation CAR-T-Cell Therapy in Individuals with HIV-1 Infection (TCTIWHI). ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04863066?term=CAR&cond=Infections&draw=2&rank=2 (accessed on 23 February 2022).
- The Effect of Chimeric Antigen Receptor (CAR)-T Cell Therapy on the Reconstitution of HIV-specific Immune Function. ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03240328?term=CAR&cond=Infections&draw=2&rank=10 (accessed on 23 February 2022).
- Meng, Z.; Chen, Y.; Lu, M. Advances in Targeting the Innate and Adaptive Immune Systems to Cure Chronic Hepatitis B Virus Infection. Front. Immunol. 2020, 10, 3127. [Google Scholar] [CrossRef]
- Bohne, F.; Chmielewski, M.; Ebert, G.; Wiegmann, K.; Kürschner, T.; Schulze, A.; Urban, S.; Krönke, M.; Abken, H.; Protzer, U. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology 2008, 134, 239–247. [Google Scholar] [CrossRef]
- Krebs, K.; Böttinger, N.; Huang, L.R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef]
- Kruse, R.L.; Shum, T.; Tashiro, H.; Barzi, M.; Yi, Z.; Whitten-Bauer, C.; Legras, X.; Bissig-Choisat, B.; Garaigorta, U.; Gottschalk, S.; et al. HBsAg-redirected T cells exhibit antiviral activity in HBV-infected human liver chimeric mice. Cytotherapy 2018, 20, 697–705. [Google Scholar] [CrossRef]
- Klopp, A.; Schreiber, S.; Kosinska, A.D.; Pulé, M.; Protzer, U.; Wisskirchen, K. Depletion of T cells via Inducible Caspase 9 Increases Safety of Adoptive T-Cell Therapy Against Chronic Hepatitis B. Front. Immunol. 2021, 12, 734246. [Google Scholar] [CrossRef] [PubMed]
- Festag, M.M.; Festag, J.; Fräßle, S.P.; Asen, T.; Sacherl, J.; Schreiber, S.; Mück-Häusl, M.A.; Busch, D.H.; Wisskirchen, K.; Protzer, U. Evaluation of a Fully Human, Hepatitis B Virus-Specific Chimeric Antigen Receptor in an Immunocompetent Mouse Model. Mol. Ther. 2019, 27, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Sautto, G.A.; Wisskirchen, K.; Clementi, N.; Castelli, M.; Diotti, R.A.; Graf, J.; Clementi, M.; Burioni, R.; Protzer, U.; Mancini, N. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut 2016, 65, 512–523. [Google Scholar] [CrossRef]
- Kudo, K.; Imai, C.; Lorenzini, P.; Kamiya, T.; Kono, K.; Davidoff, A.M.; Chng, W.J.; Campana, D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014, 74, 93–103. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, Z.; Li, G.; Liu, G.; Lin, S.; Chen, W.; Xiong, S. An NK cell line (NK92-41BB) expressing high levels of granzyme is engineered to express the high affinity chimeric genes CD16/CAR. Cytotechnology 2021, 73, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Li, H.K.; Hsiao, C.W.; Yang, S.H.; Yang, H.P.; Wu, T.S.; Lee, C.Y.; Lin, Y.L.; Pan, J.; Cheng, Z.F.; Lai, Y.D.; et al. A Novel off-the-Shelf Trastuzumab-Armed NK Cell Therapy (ACE1702) Using Antibody-Cell-Conjugation Technology. Cancers 2021, 13, 2724. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; You, F.; Jiang, L.; Li, J.; Zhu, X.; Bao, Y.; Sun, X.; Tang, X.; Meng, H.; An, G.; et al. Gene-modified NK-92MI cells expressing a chimeric CD16-BB-ζ or CD64-BB-ζ receptor exhibit enhanced cancer-killing ability in combination with therapeutic antibody. Oncotarget 2017, 8, 37128–37139. [Google Scholar] [CrossRef] [PubMed]
- Snyder, K.M.; Hullsiek, R.; Mishra, H.K.; Mendez, D.C.; Li, Y.; Rogich, A.; Kaufman, D.S.; Wu, J.; Walcheck, B. Expression of a Recombinant High Affinity IgG Fc Receptor by Engineered NK Cells as a Docking Platform for Therapeutic mAbs to Target Cancer Cells. Front. Immunol. 2018, 9, 2873. [Google Scholar] [CrossRef]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J., Jr. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef]
- Lim, R.M.; Rong, L.; Zhen, A.; Xie, J. A Universal CAR-NK Cell Targeting Various Epitopes of HIV-1 gp160. ACS Chem. Biol. 2020, 15, 2299–2310. [Google Scholar] [CrossRef] [PubMed]



| Name of the Vaccine, ClinicalTrials.gov Identifier or Authors if Applicable, Reference | Type of the Antigen and Adjuvant | Stage of the Study |
|---|---|---|
| AV-COVID-19, NCT04386252 [27] | S-protein with GM-CSF or without an adjuvant | Phase I–II clinical trial, not yet recruiting |
| LV-SMENP DC, NCT04276896 [28] | lentivirus vectors expressing COVID-19 minigene | Phase I–II clinical trial, recruiting |
| Zhou et al. [29] | S-protein with graphene oxide (GO) nanosheets | pre-clinical research |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chikileva, I.; Shubina, I.; Burtseva, A.-M.; Kirgizov, K.; Stepanyan, N.; Varfolomeeva, S.; Kiselevskiy, M. Antiviral Cell Products against COVID-19: Learning Lessons from Previous Research in Anti-Infective Cell-Based Agents. Biomedicines 2022, 10, 868. https://doi.org/10.3390/biomedicines10040868
Chikileva I, Shubina I, Burtseva A-M, Kirgizov K, Stepanyan N, Varfolomeeva S, Kiselevskiy M. Antiviral Cell Products against COVID-19: Learning Lessons from Previous Research in Anti-Infective Cell-Based Agents. Biomedicines. 2022; 10(4):868. https://doi.org/10.3390/biomedicines10040868
Chicago/Turabian StyleChikileva, Irina, Irina Shubina, Anzhelika-Mariia Burtseva, Kirill Kirgizov, Nara Stepanyan, Svetlana Varfolomeeva, and Mikhail Kiselevskiy. 2022. "Antiviral Cell Products against COVID-19: Learning Lessons from Previous Research in Anti-Infective Cell-Based Agents" Biomedicines 10, no. 4: 868. https://doi.org/10.3390/biomedicines10040868
APA StyleChikileva, I., Shubina, I., Burtseva, A.-M., Kirgizov, K., Stepanyan, N., Varfolomeeva, S., & Kiselevskiy, M. (2022). Antiviral Cell Products against COVID-19: Learning Lessons from Previous Research in Anti-Infective Cell-Based Agents. Biomedicines, 10(4), 868. https://doi.org/10.3390/biomedicines10040868