
Development of Alzheimer’s Disease Biomarkers: From CSF- to Blood-Based Biomarkers

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Discovery and Development of AD Biomarkers
2.1. History of CSF AD Biomarkers
2.2. Analytical Platforms of Core CSF Biomarkers
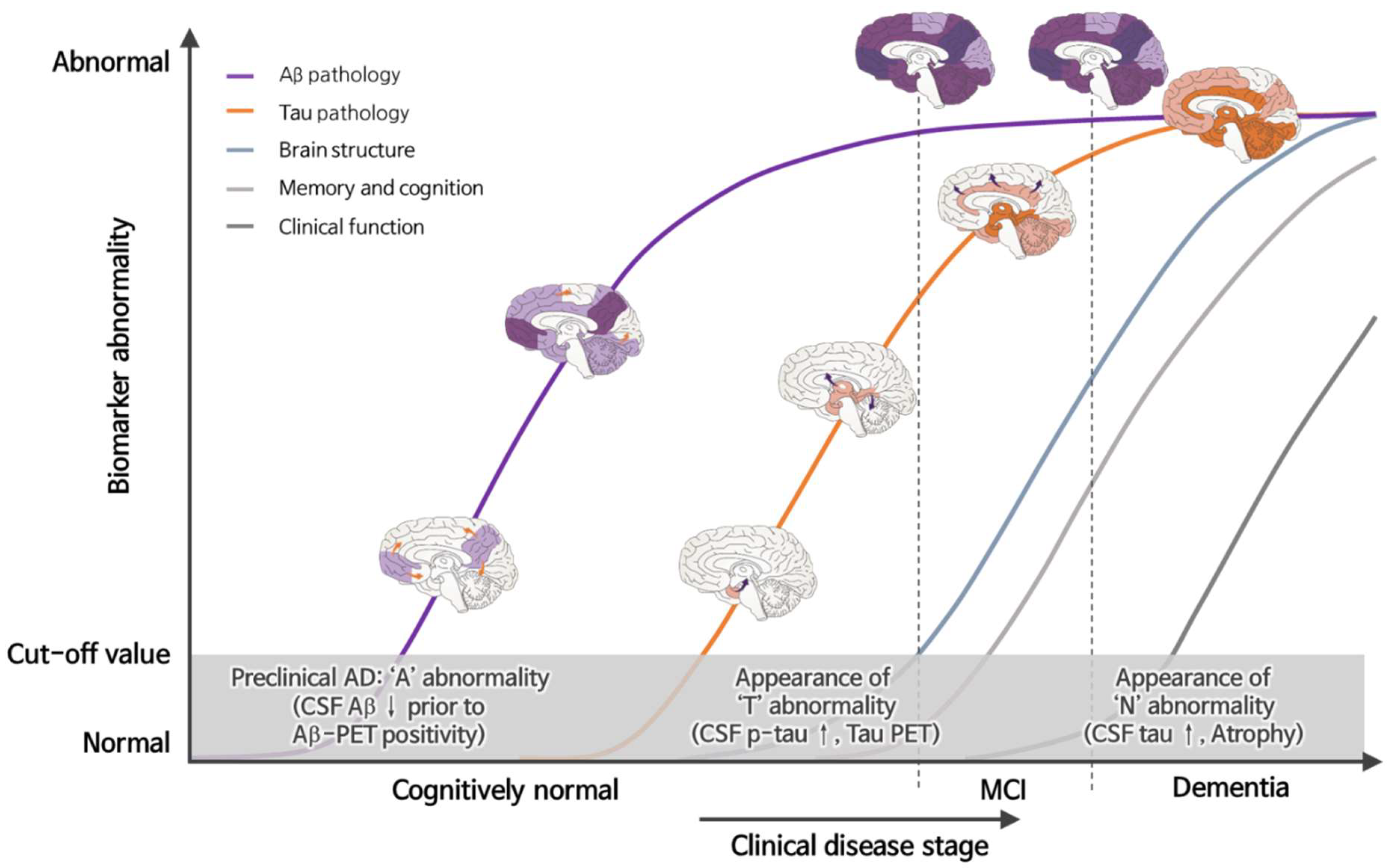
3. New Diagnostic Approach-Based A/T/N Biomarkers
4. Blood-Based Biomarkers for AD
4.1. Core AD Biomarkers (Aβ, p-Tau, and T-Tau)
4.2. Other AD-Associated Protein Biomarkers (Non-Aβ and Tau)
4.3. Exosome
4.4. MicroRNA
4.5. Lipids
4.6. Genetic Biomarkers
5. Perspective and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blennow, K.; Mattsson, N.; Schöll, M.; Hansson, O.; Zetterberg, H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol. Sci. 2015, 36, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H. Review: Tau in biofluids—Relation to pathology, imaging and clinical features. Neuropathol. Appl. Neurobiol. 2017, 43, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motter, R.; Vigo-Pelfrey, C.; Kholodenko, D.; Barbour, R.; Johnson-Wood, K.; Galasko, D.; Chang, L.; Miller, B.; Clark, C.; Green, R.; et al. Reduction of β-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 1995, 38, 643–648. [Google Scholar] [CrossRef]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergström, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging Brain Amyloid in Alzheimer’s Disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Weiner, M.W.; Veitch, D.P.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Cedarbaum, J.; Green, R.C.; Harvey, D.; Jack, C.R.; Jagust, W.; et al. 2014 Update of the Alzheimer’s Disease Neuroimaging Initiative: A review of papers published since its inception. Alzheimer’s Dement. 2015, 11, e1–e120. [Google Scholar] [CrossRef] [Green Version]
- Knopman, D.S.; Haeberlein, S.B.; Carrillo, M.C.; Hendrix, J.A.; Kerchner, G.; Margolin, R.; Maruff, P.; Miller, D.S.; Tong, G.; Tome, M.B.; et al. The National Institute on Aging and the Alzheimer’s Association Research Framework for Alzheimer’s disease: Perspectives from the Research Roundtable. Alzheimer’s Dement. 2018, 14, 563–575. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Verberk, I.M.W.; Thijssen, E.H.; Vermunt, L.; Hansson, O.; Zetterberg, H.; van der Flier, W.M.; Mielke, M.M.; del Campo, M. Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Albert, M.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carillo, M.; Thies, W.; Creighton, H. Phelps Introduction to Revised Criteria for the Diagnosis of Alzheimer’s Disease: National Institute on Aging and the Alzheimer Association Workgroups. Alzheimers Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 3, 137–152. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seubert, P.; Vigo-Pelfrey, C.; Esch, F.; Lee, M.; Dovey, H.; Davis, D.; Sinha, S.; Schiossmacher, M.; Whaley, J.; Swindlehurst, C.; et al. Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature 1992, 359, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Vandermeeren, M.; Mercken, M.; Vanmechelen, E.; Six, J.; Van de Voorde, A.; Martin, J.-J.; Cras, P. Detection of Proteins in Normal and Alzheimer’s Disease Cerebrospinal Fluid with a Sensitive Sandwich Enzyme-Linked Immunosorbent Assay. J. Neurochem. 1993, 61, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Vigo-Pelfrey, C.; Seubert, P.; Barbour, R.; Blomquist, C.; Lee, M.; Lee, D.; Coria, F.; Chang, L.; Miller, B.; Lieberburg, I.; et al. Elevation of microtubule-associated protein tau in the cerebrospinal fluid of patients with alzheimer’s disease. Neurology 1995, 45, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.F.; Shen, X.N.; Li, J.Q.; Suckling, J.; Tan, C.C.; Wang, Y.J.; Feng, L.; Zhang, C.; Tan, L.; Dong, Q.; et al. Clinical and biomarker trajectories in sporadic Alzheimer’s disease: A longitudinal study. Alzheimer’s Dement. 2020, 12, e12095. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.M.; Henson, R.L.; Li, Y.; Boerwinkle, A.H.; Xiong, C.; Bateman, R.J.; Goate, A.; Ances, B.M.; Doran, E.; Christian, B.T.; et al. Comparison of CSF biomarkers in Down syndrome and autosomal dominant Alzheimer’s disease: A cross-sectional study. Lancet Neurol. 2021, 20, 615–626. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiki, A.; Kamada, M.; Kawamura, Y.; Terao, C.; Shimoda, F.; Tomita, N.; Arai, H.; Furukawa, K. Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer’s disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. J. Neurochem. 2016, 136, 258–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, R.; Izumoto, T.; Fushiki, S. The cerebrospinal fluid level of glial fibrillary acidic protein is increased in cerebrospinal fluid from Alzheimer’s disease patients and correlates with severity of dementia. Eur. Neurol. 2001, 46, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Jesse, S.; Steinacker, P.; Cepek, L.; Arnim, C.V.; Tumani, H.; Lehnert, S.; Kretzschmar, H.A.; Baier, M.; Otto, M. Glial fibrillary acidic protein and protein S-100B: Different concentration pattern of glial proteins in cerebrospinal fluid of patients with alzheimer’s disease and creutzfeldt-jakob disease. J. Alzheimer’s Dis. 2009, 17, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Peskind, E.R.; Griffin, W.S.T.; Akama, K.T.; Raskind, M.A.; Van Eldik, L.J. Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer’s disease. Neurochem. Int. 2001, 39, 409–413. [Google Scholar] [CrossRef]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol. Psychiatry 2010, 68, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Antonell, A.; Mansilla, A.; Rami, L.; Lladó, A.; Iranzo, A.; Olives, J.; Balasa, M.; Sánchez-Valle, R.; Molinuevo, J.L. Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 901–908. [Google Scholar] [CrossRef]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 127, 459–472. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B.E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Waterloo, K.K.; Aarsland, D.; et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflammation 2019, 16, 46. [Google Scholar] [CrossRef]
- Ewers, M.; Franzmeier, N.; Suárez-Calvet, M.; Morenas-Rodriguez, E.; Caballero, M.A.A.; Kleinberger, G.; Piccio, L.; Cruchaga, C.; Deming, Y.; Dichgans, M.; et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6221. [Google Scholar] [CrossRef]
- Rauchmann, B.S.; Schneider-Axmann, T.; Alexopoulos, P.; Perneczky, R. CSF soluble TREM2 as a measure of immune response along the Alzheimer’s disease continuum. Neurobiol. Aging 2019, 74, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Ohara, T.; Hata, J.; Tanaka, M.; Honda, T.; Yamakage, H.; Yoshida, D.; Inoue, T.; Hirakawa, Y.; Kusakabe, T.; Shibata, M.; et al. Serum Soluble Triggering Receptor Expressed on Myeloid Cells 2 as a Biomarker for Incident Dementia: The Hisayama Study. Ann. Neurol. 2019, 85, 47–58. [Google Scholar] [CrossRef]
- Evers, B.M.; Rodriguez-Navas, C.; Tesla, R.J.; Prange-Kiel, J.; Wasser, C.R.; Yoo, K.S.; McDonald, J.; Cenik, B.; Ravenscroft, T.A.; Plattner, F.; et al. Lipidomic and Transcriptomic Basis of Lysosomal Dysfunction in Progranulin Deficiency. Cell Rep. 2017, 20, 2565–2574. [Google Scholar] [CrossRef] [Green Version]
- Batzu, L.; Westman, E.; Pereira, J.B. Cerebrospinal fluid progranulin is associated with increased cortical thickness in early stages of Alzheimer’s disease. Neurobiol. Aging 2020, 88, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Thientunyakit, T.; Sethanandha, C.; Muangpaisan, W.; Chawalparit, O.; Arunrungvichian, K.; Siriprapa, T.; Thongpraparn, T.; Chanachai, R.; Gelovani, J. Implementation of [ 18 F]-labeled amyloid brain PET imaging biomarker in the diagnosis of Alzheimer’s disease: First-hand experience in Thailand. Nucl. Med. Commun. 2018, 39, 186–192. [Google Scholar] [CrossRef]
- Li, Y.; Ng, Y.L.; Paranjpe, M.D.; Ge, Q.; Gu, F.; Li, P.; Yan, S.; Lu, J.; Wang, X.; Zhou, Y. Tracer-specific reference tissues selection improves detection of 18 F-FDG, 18 F -florbetapir, and 18 F -flortaucipir PET SUVR changes in Alzheimer’s disease. Hum. Brain Mapp. 2022. [Google Scholar] [CrossRef]
- Toledo, J.B.; Cairns, N.J.; Da, X.; Chen, K.; Carter, D.; Fleisher, A.; Householder, E.; Ayutyanont, N.; Roontiva, A.; Bauer, R.J.; et al. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol. Commun. 2013, 1, 65. [Google Scholar] [CrossRef] [Green Version]
- Toledo, J.B.; Korff, A.; Shaw, L.M.; Trojanowski, J.Q.; Zhang, J. CSF α-synuclein improves diagnostic and prognostic performance of CSF tau and Aβ in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 683–697. [Google Scholar] [CrossRef] [Green Version]
- Dorey, A.; Perret-Liaudet, A.; Tholance, Y.; Fourier, A.; Quadrio, I. Cerebrospinal fluid Aβ40 improves the interpretation of Aβ42 concentration for diagnosing Alzheimer’s disease. Front. Neurol. 2015, 6, 247. [Google Scholar] [CrossRef] [Green Version]
- Baldeiras, I.; Santana, I.; Leitão, M.J.; Gens, H.; Pascoal, R.; Tábuas-Pereira, M.; Beato-Coelho, J.; Duro, D.; Almeida, M.R.; Oliveira, C.R. Addition of the Aβ42/40 ratio to the cerebrospinal fluid biomarker profile increases the predictive value for underlying Alzheimer’s disease dementia in mild cognitive impairment. Alzheimer’s Res. Ther. 2018, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Korecka, M.; Toledo, J.B.; Trojanowski, J.Q.; Shaw, L.M. Clinical utility and analytical challenges in measurement of cerebrospinal fluid amyloid-β1-42 and τ proteins as alzheimer disease biomarkers. Clin. Chem. 2013, 59, 903–916. [Google Scholar] [CrossRef]
- Olsson, A.; Vanderstichele, H.; Andreasen, N.; De Meyer, G.; Wallin, A.; Holmberg, B.; Rosengren, L.; Vanmechelen, E.; Blennow, K. Simultaneous measurement of β-amyloid(1-42), total Tau, and phosphorylated Tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin. Chem. 2005, 51, 336–345. [Google Scholar] [CrossRef]
- Carrillo, M.C.; Blennow, K.; Soares, H.; Lewczuk, P.; Mattsson, N.; Oberoi, P.; Umek, R.; Vandijck, M.; Salamone, S.; Bittner, T.; et al. Global standardization measurement of cerebral spinal fluid for Alzheimer’s disease: An update from the Alzheimer’s Association Global Biomarkers Consortium. Alzheimer’s Dement. 2013, 9, 137–140. [Google Scholar] [CrossRef]
- Bittner, T.; Zetterberg, H.; Teunissen, C.E.; Ostlund, R.E.; Militello, M.; Andreasson, U.; Hubeek, I.; Gibson, D.; Chu, D.C.; Eichenlaub, U.; et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of β-amyloid (1-42) in human cerebrospinal fluid. Alzheimer’s Dement. 2016, 12, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Agnello, L.; Piccoli, T.; Vidali, M.; Cuffaro, L.; Lo Sasso, B.; Iacolino, G.; Giglio, V.R.; Lupo, F.; Alongi, P.; Bivona, G.; et al. Diagnostic accuracy of cerebrospinal fluid biomarkers measured by chemiluminescent enzyme immunoassay for Alzheimer disease diagnosis. Scand. J. Clin. Lab. Investig. 2020, 80, 313–317. [Google Scholar] [CrossRef]
- Delaby, C.; Teunissen, C.E.; Blennow, K.; Alcolea, D.; Arisi, I.; Amar, E.B.; Beaume, A.; Bedel, A.; Bellomo, G.; Bigot-Corbel, E.; et al. Clinical reporting following the quantification of cerebrospinal fluid biomarkers in Alzheimer’s disease: An international overview. Alzheimer’s Dement. 2021, 17, e057528. [Google Scholar] [CrossRef]
- Garrett, S.L.; McDaniel, D.; Obideen, M.; Trammell, A.R.; Shaw, L.M.; Goldstein, F.C.; Hajjar, I. Racial Disparity in Cerebrospinal Fluid Amyloid and Tau Biomarkers and Associated Cutoffs for Mild Cognitive Impairment. JAMA Netw. Open 2019, 2, e1917363. [Google Scholar] [CrossRef]
- Moon, S.; Kim, S.; Mankhong, S.; Choi, S.H.; Vandijck, M.; Kostanjevecki, V.; Jeong, J.H.; Yoon, S.J.; Park, K.W.; Kim, E.J.; et al. Alzheimer’s cerebrospinal biomarkers from Lumipulse fully automated immunoassay: Concordance with amyloid-beta PET and manual immunoassay in Koreans: CSF AD biomarkers measured by Lumipulse in Koreans. Alzheimer’s Res. Ther. 2021, 13, 22. [Google Scholar] [CrossRef]
- Lifke, V.; Kollmorgen, G.; Manuilova, E.; Oelschlaegel, T.; Hillringhaus, L.; Widmann, M.; von Arnim, C.A.F.; Otto, M.; Christenson, R.H.; Powers, J.L.; et al. Elecsys® Total-Tau and Phospho-Tau (181P) CSF assays: Analytical performance of the novel, fully automated immunoassays for quantification of tau proteins in human cerebrospinal fluid. Clin. Biochem. 2019, 72, 30–38. [Google Scholar] [CrossRef]
- Pannee, J.; Shaw, L.M.; Korecka, M.; Waligorska, T.; Teunissen, C.E.; Stoops, E.; Vanderstichele, H.M.J.; Mauroo, K.; Verberk, I.M.W.; Keshavan, A.; et al. The global Alzheimer’s Association round robin study on plasma amyloid β methods. Alzheimer’s Dement. 2021, 13, e12242. [Google Scholar] [CrossRef]
- Dakterzada, F.; López-Ortega, R.; Arias, A.; Riba-Llena, I.; Ruiz-Julián, M.; Huerto, R.; Tahan, N.; Piñol-Ripoll, G. Assessment of the Concordance and Diagnostic Accuracy Between Elecsys and Lumipulse Fully Automated Platforms and Innotest. Front. Aging Neurosci. 2021, 13, 91. [Google Scholar] [CrossRef]
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef]
- Landau, S.M.; Lu, M.; Joshi, A.D.; Pontecorvo, M.; Mintun, M.A.; Trojanowski, J.Q.; Shaw, L.M.; Jagust, W.J. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of β-amyloid. Ann. Neurol. 2013, 74, 826–836. [Google Scholar] [CrossRef]
- Blennow, K.; Shaw, L.M.; Stomrud, E.; Mattsson, N.; Toledo, J.B.; Buck, K.; Wahl, S.; Eichenlaub, U.; Lifke, V.; Simon, M.; et al. Predicting clinical decline and conversion to Alzheimer’s disease or dementia using novel Elecsys Aβ(1–42), pTau and tTau CSF immunoassays. Sci. Rep. 2019, 9, 19024. [Google Scholar] [CrossRef] [Green Version]
- Leitão, M.J.; Silva-Spínola, A.; Santana, I.; Olmedo, V.; Nadal, A.; Le Bastard, N.; Baldeiras, I. Clinical validation of the Lumipulse G cerebrospinal fluid assays for routine diagnosis of Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 91. [Google Scholar] [CrossRef] [Green Version]
- Hansson, O. Biomarkers for neurodegenerative diseases. Nat. Med. 2021, 27, 954–963. [Google Scholar] [CrossRef]
- Cousins, K.A.Q.; Phillips, J.S.; Irwin, D.J.; Lee, E.B.; Wolk, D.A.; Shaw, L.M.; Zetterberg, H.; Blennow, K.; Burke, S.E.; Kinney, N.G.; et al. ATN incorporating cerebrospinal fluid neurofilament light chain detects frontotemporal lobar degeneration. Alzheimer’s Dement. 2021, 17, 822–830. [Google Scholar] [CrossRef]
- Jack, C.R.; Knopman, D.S.; Chételat, G.; Dickson, D.; Fagan, A.M.; Frisoni, G.B.; Jagust, W.; Mormino, E.C.; Petersen, R.C.; Sperling, R.A.; et al. Suspected non-Alzheimer disease pathophysiology-concept and controversy. Nat. Rev. Neurol. 2016, 12, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Wisse, L.E.M.; de Flores, R.; Xie, L.; Das, S.R.; McMillan, C.T.; Trojanowski, J.Q.; Grossman, M.; Lee, E.B.; Irwin, D.; Yushkevich, P.A.; et al. Pathological drivers of neurodegeneration in suspected non-Alzheimer’s disease pathophysiology. Alzheimer’s Res. Ther. 2021, 13, 100. [Google Scholar] [CrossRef]
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer ’ s disease. Mol. Brain 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H. Blood-based biomarkers for Alzheimer’s disease—An update. J. Neurosci. Methods 2019, 319, 2–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetterberg, H.; Blennow, K. From Cerebrospinal Fluid to Blood: The Third Wave of Fluid Biomarkers for Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, S271–S279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Bolstad, N.; Warren, D.J.; Nustad, K. Heterophilic antibody interference in immunometric assays. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Figurski, M.J.; Waligórska, T.; Toledo, J.; Vanderstichele, H.; Korecka, M.; Lee, V.M.Y.; Trojanowski, J.Q.; Shaw, L.M. Improved protocol for measurement of plasma β-amyloid in longitudinal evaluation of Alzheimer’s Disease Neuroimaging Initiative study patients. Alzheimer’s Dement. 2012, 8, 250–260. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.C.; Yang, S.Y.; Chieh, J.J.; Horng, H.E.; Hong, C.Y.; Yang, H.C.; Chen, K.H.; Shih, B.Y.; Chen, T.F.; Chiu, M.J. Biofunctionalized magnetic nanoparticles for specifically detecting biomarkers of Alzheimer’s disease in vitro. ACS Chem. Neurosci. 2011, 2, 500–505. [Google Scholar] [CrossRef] [Green Version]
- Ashton, N.J.; Leuzy, A.; Karikari, T.K.; Mattsson-Carlgren, N.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Nordberg, A.; Ossenkoppele, R.; et al. The validation status of blood biomarkers of amyloid and phospho-tau assessed with the 5-phase development framework for AD biomarkers. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2140–2156. [Google Scholar] [CrossRef]
- Vergallo, A.; Mégret, L.; Lista, S.; Cavedo, E.; Zetterberg, H.; Blennow, K.; Vanmechelen, E.; De Vos, A.; Habert, M.O.; Potier, M.C.; et al. Plasma amyloid β 40/42 ratio predicts cerebral amyloidosis in cognitively normal individuals at risk for Alzheimer’s disease. Alzheimer’s Dement. 2019, 15, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.N.; Huang, Y.Y.; Chen, S.D.; Guo, Y.; Tan, L.; Dong, Q.; Yu, J.T. Plasma phosphorylated-tau181 as a predictive biomarker for Alzheimer’s amyloid, tau and FDG PET status. Transl. Psychiatry 2021, 11, 585. [Google Scholar] [CrossRef] [PubMed]
- Ovod, V.; Ramsey, K.N.; Mawuenyega, K.G.; Bollinger, J.G.; Hicks, T.; Schneider, T.; Sullivan, M.; Paumier, K.; Holtzman, D.M.; Morris, J.C.; et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimer’s Dement. 2017, 13, 841–849. [Google Scholar] [CrossRef]
- Kirmess, K.M.; Meyer, M.R.; Holubasch, M.S.; Knapik, S.S.; Hu, Y.; Jackson, E.N.; Harpstrite, S.E.; Verghese, P.B.; West, T.; Fogelman, I.; et al. The PrecivityADTM test: Accurate and reliable LC-MS/MS assays for quantifying plasma amyloid beta 40 and 42 and apolipoprotein E proteotype for the assessment of brain amyloidosis. Clin. Chim. Acta 2021, 519, 267–275. [Google Scholar] [CrossRef] [PubMed]
- West, T.; Kirmess, K.M.; Meyer, M.R.; Holubasch, M.S.; Knapik, S.S.; Hu, Y.; Contois, J.H.; Jackson, E.N.; Harpstrite, S.E.; Bateman, R.J.; et al. A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: Findings from a multi cohort validity analysis. Mol. Neurodegener. 2021, 16, 30. [Google Scholar] [CrossRef]
- Pérez-Grijalba, V.; Arbizu, J.; Romero, J.; Prieto, E.; Pesini, P.; Sarasa, L.; Guillen, F.; Monleón, I.; San-José, I.; Martínez-Lage, P.; et al. Plasma Aβ42/40 ratio alone or combined with FDG-PET can accurately predict amyloid-PET positivity: A cross-sectional analysis from the AB255 Study. Alzheimer’s Res. Ther. 2019, 11, 1–9. [Google Scholar] [CrossRef]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.S.; Xiong, C.; et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, E1647–E1659. [Google Scholar] [CrossRef]
- Babapour Mofrad, R.; Scheltens, P.; Kim, S.Y.; Kang, S.; Youn, Y.C.; An, S.S.A.; Tomassen, J.; van Berckel, B.N.M.; Visser, P.J.; van der Flier, W.M.; et al. Plasma amyloid-β oligomerization assay as a pre-screening test for amyloid status. Alzheimer’s Res. Ther. 2021, 13, 133. [Google Scholar] [CrossRef]
- Fossati, S.; Ramos Cejudo, J.; Debure, L.; Pirraglia, E.; Sone, J.Y.; Li, Y.; Chen, J.; Butler, T.; Zetterberg, H.; Blennow, K.; et al. Plasma tau complements CSF tau and P-tau in the diagnosis of Alzheimer’s disease. Alzheimers Dement (Amst) 2019, 11, 483–492. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A. The role of protein misfolding and tau oligomers (TauOs) in Alzheimer’s disease (AD). Int. J. Mol. Sci. 2019, 20, 4661. [Google Scholar] [CrossRef] [Green Version]
- Pereira, J.B.; Janelidze, S.; Stomrud, E.; Palmqvist, S.; Van Westen, D.; Dage, J.L.; Mattsson-Carlgren, N.; Hansson, O. Plasma markers predict changes in amyloid, tau, atrophy and cognition in non-demented subjects. Brain 2021, 144, 2826–2836. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, H.; Kasai, T.; Ohmichi, T.; Kishi, Y.; Kakeya, T.; Waragai, M.; Kondo, M.; Allsop, D.; Tokuda, T. Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: Pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol. Neurodegener. 2017, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brickman, A.M.; Manly, J.J.; Honig, L.S.; Sanchez, D.; Reyes-Dumeyer, D.; Lantigua, R.A.; Lao, P.J.; Stern, Y.; Vonsattel, J.P.; Teich, A.F.; et al. Plasma p-tau181, p-tau217, and other blood-based Alzheimer’s disease biomarkers in a multi-ethnic, community study. Alzheimer’s Dement. 2021, 17, 1353–1364. [Google Scholar] [CrossRef]
- Mattsson-Carlgren, N.; Janelidze, S.; Palmqvist, S.; Cullen, N.; Svenningsson, A.L.; Strandberg, O.; Mengel, D.; Walsh, D.M.; Stomrud, E.; Dage, J.L.; et al. Longitudinal plasma p-tau217 is increased in early stages of Alzheimer’s disease. Brain 2021, 143, 3234–3241. [Google Scholar] [CrossRef]
- Doecke, J.D.; Pérez-Grijalba, V.; Fandos, N.; Fowler, C.; Villemagne, V.L.; Masters, C.L.; Pesini, P.; Sarasa, M. Total Aβ42/Aβ40 ratio in plasma predicts amyloid-PET status, independent of clinical AD diagnosis. Neurology 2020, 94, E1580–E1591. [Google Scholar] [CrossRef] [Green Version]
- Janelidze, S.; Stomrud, E.; Palmqvist, S.; Zetterberg, H.; Van Westen, D.; Jeromin, A.; Song, L.; Hanlon, D.; Tan Hehir, C.A.; Baker, D.; et al. Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci. Rep. 2016, 6, 26801. [Google Scholar] [CrossRef]
- Li, Y.; Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Weiner, M.W.; Leslie, S.M.; Masters, C.L.; Fowler, C.J.; Trojanowski, J.Q.; et al. Validation of Plasma Amyloid-β 42/40 for Detecting Alzheimer Disease Amyloid Plaques. Neurology 2022, 98, e688–e699. [Google Scholar] [CrossRef]
- Mielke, M.M.; Hagen, C.E.; Xu, J.; Chai, X.; Vemuri, P.; Lowe, V.J.; Airey, D.C.; Knopman, D.S.; Roberts, R.O.; Machulda, M.M.; et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimer’s Dement. 2018, 14, 989–997. [Google Scholar] [CrossRef]
- Lussier, F.Z.; Benedet, A.L.; Therriault, J.; Pascoal, T.A.; Tissot, C.; Chamoun, M.; Mathotaarachchi, S.; Savard, M.; Ashton, N.J.; Karikari, T.K.; et al. Plasma levels of phosphorylated tau 181 are associated with cerebral metabolic dysfunction in cognitively impaired and amyloid-positive individuals. Brain Commun. 2021, 3, fcab073. [Google Scholar] [CrossRef]
- Thijssen, E.H.; La Joie, R.; Strom, A.; Fonseca, C.; Iaccarino, L.; Wolf, A.; Spina, S.; Allen, I.E.; Cobigo, Y.; Heuer, H.; et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: A retrospective diagnostic performance study. Lancet Neurol. 2021, 20, 739–752. [Google Scholar] [CrossRef]
- Janelidze, S.; Berron, D.; Smith, R.; Strandberg, O.; Proctor, N.K.; Dage, J.L.; Stomrud, E.; Palmqvist, S.; Mattsson-Carlgren, N.; Hansson, O. Associations of Plasma Phospho-Tau217 Levels with Tau Positron Emission Tomography in Early Alzheimer Disease. JAMA Neurol. 2021, 78, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Alawode, D.O.T.; Heslegrave, A.J.; Ashton, N.J.; Karikari, T.K.; Simrén, J.; Montoliu-Gaya, L.; Pannee, J.; O’Connor, A.; Weston, P.S.J.; Lantero-Rodriguez, J.; et al. Transitioning from cerebrospinal fluid to blood tests to facilitate diagnosis and disease monitoring in Alzheimer’s disease. J. Intern. Med. 2021, 290, 583–601. [Google Scholar] [CrossRef] [PubMed]
- Lewczuk, P.; Ermann, N.; Andreasson, U.; Schultheis, C.; Podhorna, J.; Spitzer, P.; Maler, J.M.; Kornhuber, J.; Blennow, K.; Zetterberg, H. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 71. [Google Scholar] [CrossRef]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Gräber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 2019, 25, 277–283. [Google Scholar] [CrossRef]
- Mattsson, N.; Cullen, N.C.; Andreasson, U.; Zetterberg, H.; Blennow, K. Association between Longitudinal Plasma Neurofilament Light and Neurodegeneration in Patients with Alzheimer Disease. JAMA Neurol. 2019, 76, 791–799. [Google Scholar] [CrossRef]
- Gafson, A.R.; Barthélemy, N.R.; Bomont, P.; Carare, R.O.; Durham, H.D.; Julien, J.P.; Kuhle, J.; Leppert, D.; Nixon, R.A.; Weller, R.O.; et al. Neurofilaments: Neurobiological foundations for biomarker applications. Brain 2020, 143, 1975–1998. [Google Scholar] [CrossRef]
- Chatterjee, P.; Pedrini, S.; Stoops, E.; Goozee, K.; Villemagne, V.L.; Asih, P.R.; Verberk, I.M.W.; Dave, P.; Taddei, K.; Sohrabi, H.R.; et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Transl. Psychiatry 2021, 11, 27. [Google Scholar] [CrossRef]
- Verberk, I.M.W.; Thijssen, E.; Koelewijn, J.; Mauroo, K.; Vanbrabant, J.; De Wilde, A.; Zwan, M.D.; Verfaillie, S.C.J.; Ossenkoppele, R.; Barkhof, F.; et al. Combination of plasma amyloid beta(1-42/1-40)and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimer’s Res. Ther. 2020, 12, 1–14. [Google Scholar] [CrossRef]
- Oeckl, P.; Halbgebauer, S.; Anderl-Straub, S.; Steinacker, P.; Huss, A.M.; Neugebauer, H.; Von Arnim, C.A.F.; Diehl-Schmid, J.; Grimmer, T.; Kornhuber, J.; et al. Glial fibrillary acidic protein in serum is increased in Alzheimer’s disease and correlates with cognitive impairment. J. Alzheimer’s Dis. 2019, 67, 481–488. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Smith, R.; Mattsson-Carlgren, N.; Palmqvist, S.; Teunissen, C.E.; Zetterberg, H.; Stomrud, E.; Ashton, N.J.; Blennow, K.; et al. Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer’s disease. Brain 2021, 144, 3505–3516. [Google Scholar] [CrossRef] [PubMed]
- Kester, M.I.; Teunissen, C.E.; Crimmins, D.L.; Herries, E.M.; Ladenson, J.K.H.; Scheltens, P.; Van Der Flier, W.M.; Morris, J.C.; Holtzman, D.M.; Fagan, A.M. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol. 2015, 72, 1275–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saman, S.; Kim, W.H.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.Y.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Mankhong, S.; Kang, J.H. Extracellular vesicle as a source of alzheimer’s biomarkers: Opportunities and challenges. Int. J. Mol. Sci. 2019, 20, 1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A.F. Inhibition of γ-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008, 22, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimer’s Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Gu, H.; Guo, Q.; Liang, S.; Xue, J.; Yao, F.; Liu, X.; Li, F.; Liu, H.; Sun, L.; et al. Profiling of Serum Exosome MiRNA Reveals the Potential of a MiRNA Panel as Diagnostic Biomarker for Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3084–3094. [Google Scholar] [CrossRef]
- Xing, W.; Gao, W.; Lv, X.; Xu, X.; Zhang, Z.; Yan, J.; Mao, G.; Bu, Z. The Diagnostic Value of Exosome-Derived Biomarkers in Alzheimer’s Disease and Mild Cognitive Impairment: A Meta-Analysis. Front. Aging Neurosci. 2021, 13, 637218. [Google Scholar] [CrossRef]
- Zhao, Y.; Jaber, V.; Alexandrov, P.N.; Vergallo, A.; Lista, S.; Hampel, H.; Lukiw, W.J. microRNA-Based Biomarkers in Alzheimer’s Disease (AD). Front. Neurosci. 2020, 14, 1028. [Google Scholar] [CrossRef]
- Nagaraj, S.; Laskowska-Kaszub, K.; Debski, K.J.; Wojsiat, J.; Dabrowski, M.; Gabryelewicz, T.; Kuznicki, J.; Wojda, U. Profile of 6 microRNA in blood plasma distinguish early stage Alzheimer’s disease patients from non-demented subjects. Oncotarget 2017, 8, 16122–16143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, B.; Zhou, H.; Zhang, R.; Song, M.; Yu, L.; Wang, L.; Liu, Z.; Zhang, Q.; Cui, D.; Wang, X.; et al. Serum miR-206 and miR-132 as Potential Circulating Biomarkers for Mild Cognitive Impairment. J. Alzheimer’s Dis. 2015, 45, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yu, J.T.; Liu, Q.Y.; Tan, M.S.; Zhang, W.; Hu, N.; Wang, Y.L.; Sun, L.; Jiang, T.; Tan, L. Circulating miR-125b as a biomarker of Alzheimer’s disease. J. Neurol. Sci. 2014, 336, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Leidinger, P.; Backes, C.; Deutscher, S.; Schmitt, K.; Mueller, S.C.; Frese, K.; Haas, J.; Ruprecht, K.; Paul, F.; Stähler, C.; et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013, 14, R78. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Vijayan, M.; Reddy, P.H. MicroRNA-455-3p as a potential peripheral biomarker for Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 3808–3822. [Google Scholar] [CrossRef]
- Hara, N.; Kikuchi, M.; Miyashita, A.; Hatsuta, H.; Saito, Y.; Kasuga, K.; Murayama, S.; Ikeuchi, T.; Kuwano, R. Serum microRNA miR-501-3p as a potential biomarker related to the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.H.; Bai, S.F.; Yan, J.Q. Blood circulating miRNAs as biomarkers of Alzheimer’s disease: A systematic review and meta-analysis. Biomark. Med. 2019, 13, 1045–1054. [Google Scholar] [CrossRef]
- Takousis, P.; Sadlon, A.; Schulz, J.; Wohlers, I.; Dobricic, V.; Middleton, L.; Lill, C.M.; Perneczky, R.; Bertram, L. Differential expression of microRNAs in Alzheimer’s disease brain, blood, and cerebrospinal fluid. Alzheimer’s Dement. 2019, 15, 1468–1477. [Google Scholar] [CrossRef]
- Lee, H.M.; Wong, W.K.K.; Fan, B.; Lau, E.S.; Hou, Y.; Luk, A.O.Y.; Chow, E.Y.K.; Ma, R.C.W.; Chan, J.C.N.; Kong, A.P.S. Detection of increased serum miR-122-5p and miR-455-3p levels before the clinical diagnosis of liver cancer in people with type 2 diabetes. Sci. Rep. 2021, 11, 23756. [Google Scholar] [CrossRef]
- Sun, L.; Xu, M.; Zhang, G.; Dong, L.; Wu, J.; Wei, C.; Xu, K.; Zhang, L. Identification of Circulating Exosomal miR-101 and miR-125b Panel Act as a Potential Biomarker for Hepatocellular Carcinoma. Int. J. Genomics 2021, 2021, 1326463. [Google Scholar] [CrossRef]
- Siedlecki-Wullich, D.; Català-Solsona, J.; Fábregas, C.; Hernández, I.; Clarimon, J.; Lleó, A.; Boada, M.; Saura, C.A.; Rodríguez-Álvarez, J.; Miñano-Molina, A.J. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Heinicke, F.; Zhong, X.; Zucknick, M.; Breidenbach, J.; Sundaram, A.Y.M.; Flåm, S.; Leithaug, M.; Dalland, M.; Farmer, A.; Henderson, J.M.; et al. Systematic assessment of commercially available low-input miRNA library preparation kits. RNA Biol. 2020, 17, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, M.; Khan, S. Plasma Lipids as Biomarkers for Alzheimer’s Disease: A Systematic Review. Cureus 2020, 4, e12008. [Google Scholar] [CrossRef]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; Macarthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef]
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of blood lipids with Alzheimer’s disease: A comprehensive lipidomics analysis. Alzheimer’s Dement. 2017, 13, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Olazarán, J.; Gil-De-Gómez, L.; Rodríguez-Martín, A.; Valentí-Soler, M.; Frades-Payo, B.; Marín-Muñoz, J.; Antúnez, C.; Frank-García, A.; Acedo-Jiménez, C.; Morlán-Gracia, L.; et al. A blood-based, 7-metabolite signature for the early diagnosis of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 1157–1173. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, S.; Risacher, S.L.; Yoder, K.K.; West, J.D.; Shen, L.; Kim, S.; Inlow, M.; Foroud, T.; Jagust, W.J.; Koeppe, R.A.; et al. Association of plasma and cortical amyloid beta is modulated by APOE ε4 status. Alzheimer’s Dement. 2014, 10, e9–e18. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Korecka, M.; Figurski, M.J.; Toledo, J.B.; Blennow, K.; Zetterberg, H.; Waligorska, T.; Brylska, M.; Fields, L.; Shah, N.; et al. The Alzheimer’s Disease Neuroimaging Initiative 2 Biomarker Core: A review of progress and plans. Alzheimer’s Dement. 2015, 11, 772–791. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.Á.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM 2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Aβ Biomarkers (A) | Analysis Method | Sample Size | Correlation ith Reference Standard | References |
|---|---|---|---|---|
| Plasma Aβ42/Aβ40 | SIMOA | n = 719 | Negative correlation Aβ PET SUVR Positive correlation CSF Aβ42 | [87] |
| Plasma Aβ42/40 | High-precision immunoprecipitation mass spectrometry (IPMS) | n = 158 (Cognitively normal) | Negative correlation Aβ PET positivity (AUC 0.88, 95% CI 0.82–0.93) CSF p-tau181/Aβ42 (AUC 0.85, 95% CI 0.79–0.92) | [77] |
| Plasma Aβ42/Aβ40 | ELISA (Aβtest, TP42/40) | n = 135 (18-month) n = 169 (36-month) n = 135 (54-month) | Negative correlation Aβ PET SUVR (r = −0.63, p < 0.0001) | [86] |
| Plasma Aβ42/Aβ40 | Liquid chromatography-tandem mass spectrometry (LC-MS/MS) | n = 414 | Negative correlation Aβ PET positivity (AUC 0.81, 95% CI 0.77–0.85) | [75] |
| Plasma Aβ Oligomer | Multimer detection system (MDS) | n = 399 (MCI n = 42) (AD dementia n = 164) (non-AD dementia n = 58) (Other disease n = 61) (Normal control n = 60) (Subjective cognitive decline n = 14) | Positive correlation CSF T-tau (r = 0.20, p = 0.01) Negative correlation CSF Aβ (r = −0.20, p = 0.035) No correlation p-tau (r = 0.12, p > 0.05) | [78] |
| Plasma Aβ42/Aβ40 | High-precision immunoprecipitation mass spectrometry (IPMS) | n = 465 | Negative correlation Aβ PET positivity (AUC 0.84, 95% CI 0.80–0.87) Positive correlation CSF Aβ42/Aβ40 (AUC 0.85, 95% CI 0.78–0.91) | [88] |
| p-tau Biomarkers (T) | Analysis Method | Sample Size | Correlation | References |
| Plasma p-tau181 | SIMOA (* substituting the detection antibody for a p-tau 181-specific monoclonal antibody) | Cognitively unimpaired; n = 172, MCI; n = 57, AD dementia; n = 40 | Positive correlation Tau PET SUVR (p-tau181; r = 0.580, p < 0.001) | [89] |
| Plasma p-tau217 Plasma p-tau181 | Electrochemiluminescence-based assays (different in the biotinylated antibody epitope) | n = 593 | Positive correlation Aβ PET positivity (p-tau217: AUC = 0.91, 95% CI = 0.88–0.94, p-tau181 AUC = 0.89, 95% CI = 0.86–0.93) | [91] |
| Plasma p-tau217 Plasma p-tau181 | SIMOA | Subgroup of 40 subjects with Aβ PET (n = 300) Autopsied sample n = 113 | Positive correlation Aβ PET positivity (p-tau217: AUC = 0.84, 95% CI = 0.68–0.99, p-tau181: AUC = 0.82, 95% CI = 0.65–0.99) Presence of AD pathology (p < 0.001) | [84] |
| Plasma p-tau181 | SIMOA | n = 1189 | Positive correlation Aβ PET SUVR (r = 0.45, p < 0.0001) Tau PET SUVR (r = 0.25 p = 0.0003) Negative correlation FGD PET uptake (r = −0.37, p < 0.0001) | [72] |
| Plasma p-tau217 | Meso Scale Discovery-based immunoassays | n = 490 Cognitively health control n = 225; Subjective cognitive decline n = 89 MCI n = 176 | Positive correlation CSF p-tau217 (r = 0.709 in Aβ-PET positive Control; r = 0.543 in Aβ-PET positive MCI) Entorhinal Tau PET (87% agreement) | [92] |
| Neuronal Injury Biomarkers (N) | Analysis Method | Sample Size | Correlation | References |
| Plasma T-tau | SIMOA | n = 97 (Normal control n = 68) (AD n = 29) | Poor correlation CSF T-tau (r = 0.26, p = 0.09) Positive correlation CSF p-tau 181 (r = 0.29, p = 0.003) | [79] |
| Plasma T-tau | SIMOA | Cognitively unimpaired; n = 172, MCI; n = 57, AD dementia; n = 40 | Positive correlation Tau PET SUVR (T-tau; r = 0.194, p = 0.022) | [89] |
| NfL * | SIMOA | Autopsied samples (n = 113) | Positive correlation Presence of AD pathology (p = 0.07) | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mankhong, S.; Kim, S.; Lee, S.; Kwak, H.-B.; Park, D.-H.; Joa, K.-L.; Kang, J.-H. Development of Alzheimer’s Disease Biomarkers: From CSF- to Blood-Based Biomarkers. Biomedicines 2022, 10, 850. https://doi.org/10.3390/biomedicines10040850
Mankhong S, Kim S, Lee S, Kwak H-B, Park D-H, Joa K-L, Kang J-H. Development of Alzheimer’s Disease Biomarkers: From CSF- to Blood-Based Biomarkers. Biomedicines. 2022; 10(4):850. https://doi.org/10.3390/biomedicines10040850
Chicago/Turabian StyleMankhong, Sakulrat, Sujin Kim, Seongju Lee, Hyo-Bum Kwak, Dong-Ho Park, Kyung-Lim Joa, and Ju-Hee Kang. 2022. "Development of Alzheimer’s Disease Biomarkers: From CSF- to Blood-Based Biomarkers" Biomedicines 10, no. 4: 850. https://doi.org/10.3390/biomedicines10040850
APA StyleMankhong, S., Kim, S., Lee, S., Kwak, H.-B., Park, D.-H., Joa, K.-L., & Kang, J.-H. (2022). Development of Alzheimer’s Disease Biomarkers: From CSF- to Blood-Based Biomarkers. Biomedicines, 10(4), 850. https://doi.org/10.3390/biomedicines10040850