
Addressing Blood–Brain Barrier Impairment in Alzheimer’s Disease



Abstract
:1. Introduction
2. Cellular and Noncellular Architecture of the BBB
2.1. Pathways Regulating BBB Integrity
2.2. BBB vs. Blood–CSF Barrier Alteration
3. Causes, Characteristics, and Consequences of BBB Breakdown
3.1. Aging: The Major Culprit for BBB Disruption
3.2. Peripheral Inflammation
4. AD
4.1. Clinical and Neuropathological Characteristics
4.2. Transgenic Mouse Models of AD
5. The Implication of BBB Dysfunction in AD
5.1. Neurovascular Unit Dysfunction
5.2. Accumulation of Neurotoxic Blood-Derived Products
5.3. Accumulation of Aβ in Cerebrovasculature
5.4. Accumulation of Phosphorylated Tau in the Cerebrovasculature
5.5. Alzheimer’s Disease and Vascular Dementia Co-Occur
5.6. Comparing BBB Dysfunction in AD vs. Other Neurodegenerative Diseases
6. BBB Dysfunction as a Tool for the Identification and Prediction of Early Cognitive Decline
7. Therapeutic Implications in BBB Impairment
8. Limitations of Current Studies and Future Challenges
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Engelhardt, B.; Coisne, C. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS 2011, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The Blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, B.; Sorokin, L. The blood–brain and the blood and dash; cerebrospinal fluid barriers: Function and dysfunction. Semin. Immunopathol. 2009, 31, 497–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redzic, Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and dysfunction of the blood-brain barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain endothelial cell-cell junctions: How to “Open” the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.; Campbell, M. Tight junction modulation of the blood brain barrier: CNS delivery of small molecules. Tissue Barriers 2016, 4, e1138017. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, function, and regulation of the blood-brain barrier tight junction in central nervous system disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, S.; Patkar, S.; Ogunshola, O. Cell-specific blood-brain barrier regulation in health and disease: A focus on hypoxia. Br. J. Pharmacol. 2014, 171, 1210–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.; Xiao, Z.J.; Yang, B.; Lan, Z.; Fang, F. Blood-brain barrier: More contributor to disruption of central nervous system homeostasis than victim in neurological disorders. Front. Neurosci. 2020, 14, 764. [Google Scholar] [CrossRef]
- Kaur, J.; Fahmy, L.M.; Davoodi-Bojd, E.; Zhang, L.; Ding, G.; Hu, J.; Zhang, Z.; Chopp, M.; Jiang, Q. Waste clearance in the Brain. Front. Neuroanat. 2021, 15. [Google Scholar] [CrossRef]
- Hladky, S.B.; Barrand, M.A. Mechanisms of fluid movement into, through and out of the brain: Evaluation of the evidence. Fluids Barriers CNS 2014, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Hladky, S.B.; Barrand, M.A. Elimination of substances from the brain parenchyma: Efflux via perivascular pathways and via the blood-brain barrier. Fluids Barriers CNS 2018, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, R.; Tao, C.; Xu, Z.; Chen, W.; Wang, C.; Song, L.; Zheng, J.; Gao, F. Blood-brain barrier disruption and perivascular beta-amyloid accumulation in the brain of aged rats with spontaneous hypertension: Evaluation with dynamic contrast-enhanced magnetic resonance imaging. Korean J. Radiol. 2018, 19, 498–507. [Google Scholar] [CrossRef] [Green Version]
- Shaltiel-Karyo, R.; Frenkel-Pinter, M.; Rockenstein, E.; Patrick, C.; Levy-Sakin, M.; Schiller, A.; Egoz-Matia, N.; Masliah, E.; Segal, D.; Gazit, E. A Blood-Brain Barrier (BBB) Disrupter Is Also a Potent α-Synuclein (α-syn) Aggregation Inhibitor: A novel dual mechanism of mannitol for the treatment of Parkinson disease (PD). J. Biol. Chem. 2013, 288, 17579–17588. [Google Scholar] [CrossRef] [Green Version]
- Dyrna, F.; Hanske, S.; Krueger, M.; Bechmann, I. The blood-brain barrier. J. Neuroimmune Pharmacol. 2013, 8, 763–773. [Google Scholar] [CrossRef]
- Patel, N.; Kirmi, O. Anatomy and imaging of the normal meninges. Semin. Ultrasound CT MRI 2009, 30, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.; Squier, W.; Eastman, J.T. Anatomy and development of the meninges: Implications for subdural collections and CSF circulation. Pediatr. Radiol. 2009, 39, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, F.; Creemers, J.; Lambrichts, I. Ultrastructure of the human spinal arachnoid mater and dura mater. J. Anat. 1996, 189, 417. [Google Scholar]
- Yasuda, K.; Cline, C.; Vogel, P.; Onciu, M.; Fatima, S.; Sorrentino, B.P.; Thirumaran, R.K.; Ekins, S.; Urade, Y.; Fujimori, K.; et al. Drug transporters on arachnoid barrier cells contribute to the blood-cerebrospinal fluid barrier. Drug Metab. Dispos. 2013, 41, 923–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchings, M.; Weller, R.O. Anatomical relationships of the pia mater to cerebral blood vessels in man. J. Neurosurg. 1986, 65, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Weller, R. Microscopic morphology and histology of the human meninges. Morphologie 2005, 89, 22–34. [Google Scholar] [CrossRef]
- Mastorakos, P.; McGavern, D. The anatomy and immunology of vasculature in the central nervous system. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates csf flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [Green Version]
- Howell, O.; Schulz-Trieglaff, E.K.; Carassiti, D.; Gentleman, S.M.; Nicholas, R.; Roncaroli, F.; Reynolds, R. Extensive grey matter pathology in the cerebellum in multiple sclerosis is linked to inflammation in the subarachnoid space. Neuropathol. Appl. Neurobiol. 2015, 41, 798–813. [Google Scholar] [CrossRef] [Green Version]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Chopp, M.; Chen, J. Multifaceted roles of pericytes in central nervous system homeostasis and disease. J. Cereb. Blood Flow Metab. 2020, 40, 1381–1401. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Sonar, S.A.; Lal, G. Blood-brain barrier and its function during inflammation and autoimmunity. J. Leukoc. Biol. 2018, 103, 839–853. [Google Scholar] [CrossRef]
- Haigh, J.; Morelli, P.I.; Gerhardt, H.; Haigh, K.; Tsien, J.; Damert, A.; Miquerol, L.; Muhlner, U.; Klein, R.; Ferrara, N.; et al. Cortical and retinal defects caused by dosage-dependent reductions in VEGF-A paracrine signaling. Dev. Biol. 2003, 262, 225–241. [Google Scholar] [CrossRef]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling? In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/β-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/β-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Tam, S.J.; Richmond, D.L.; Kaminker, J.S.; Modrusan, Z.; Martin-McNulty, B.; Cao, T.C.; Weimer, R.M.; Carano, R.A.; van Bruggen, N.; Watts, R.J. Death receptors DR6 and TROY regulate brain vascular development. Dev. Cell 2012, 22, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [Green Version]
- Ronaldson, P.T.; Davis, T.P. Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24. [Google Scholar] [CrossRef] [PubMed]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, M.J.; Damodarasamy, M.; Banks, W.A. The extracellular matrix of the blood–brain barrier: Structural and functional roles in health, aging, and Alzheimer’s disease. Tissue Barriers 2019, 7, 1651157. [Google Scholar] [CrossRef] [PubMed]
- Turtzo, L.C.; Jikaria, N.; Cota, M.R.; Williford, J.P.; Uche, V.; Davis, T.; MacLaren, J.; Moses, A.D.; Parikh, G.; Castro, M.A.; et al. Meningeal blood-brain barrier disruption in acute traumatic brain injury. Brain Commun. 2020, 2, fcaa143. [Google Scholar] [CrossRef]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Spector, R.; Snodgrass, S.R.; Johanson, C.E. A balanced view of the cerebrospinal fluid composition and functions: Focus on adult humans. Exp. Neurol. 2015, 273, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Verheggen, I.C.M.; de Jong, J.J.A.; van Boxtel, M.P.J.; Postma, A.A.; Jansen, J.F.A.; Verhey, F.R.J.; Backes, W.H. Imaging the role of blood-brain barrier disruption in normal cognitive ageing. GeroScience 2020, 42, 1751–1764. [Google Scholar] [CrossRef]
- Erdő, F.; Denes, L.; de Lange, E. Age-associated physiological and pathological changes at the blood–brain barrier: A review. J. Cereb. Blood Flow Metab. 2017, 37, 4–24. [Google Scholar] [CrossRef] [Green Version]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Tarumi, T.; Zhang, R. Cerebral blood flow in normal aging adults: Cardiovascular determinants, clinical implications, and aerobic fitness. J. Neurochem. 2018, 144, 595–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zeevi, N.; Pachter, J.; McCullough, L.D.; Wolfson, L.; Kuchel, G. The Blood-brain barrier: Geriatric relevance of a critical brain-body interface. J. Am. Geriatr. Soc. 2010, 58, 1749–1757. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Yang, Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg. Focus 2007, 22, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [Green Version]
- Goodall, E.F.; Wang, C.; Simpson, J.E.; Baker, D.J.; Drew, D.R.; Heath, P.R.; Saffrey, M.J.; Romero, I.A.; Wharton, S.B. Age-associated changes in the blood-brain barrier: Comparative studies in human and mouse. Neuropathol. Appl. Neurobiol. 2018, 44, 747–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagare, A.P.; Sweeney, M.D.; Makshanoff, J.; Zlokovic, B.V. Shedding of soluble platelet-derived growth factor receptor-β from human brain pericytes. Neurosci. Lett. 2015, 607, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.J.; Duan, T.; Verkman, A.S. Aquaporin-4 reduces neuropathology in a mouse model of Alzheimer’s disease by remodeling peri-plaque astrocyte structure. Acta Neuropathol. Commun. 2019, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Duncombe, J.; Lennen, R.J.; Jansen, M.A.; Marshall, I.; Wardlaw, J.; Horsburgh, K. Ageing causes prominent neurovascular dysfunction associated with loss of astrocytic contacts and gliosis. Neuropathol. Appl. Neurobiol. 2017, 43, 477–491. [Google Scholar] [CrossRef] [Green Version]
- Ronaldson, P.T.; Davis, T. Blood-brain barrier integrity and glial support: Mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr. Pharm. Des. 2012, 18, 3624–3644. [Google Scholar] [CrossRef]
- Huber, J.D.; Campos, C.R.; Mark, K.S.; Davis, T.P. Alterations in blood-brain barrier ICAM-1 expression and brain microglial activation after λ -carrageenan-induced inflammatory pain. Am. J. Physiol. Circ. Physiol. 2006, 290, H732–H740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezowski, V.; Landry, C.; Dehouck, M.-P.; Cecchelli, R.; Fenart, L. Contribution of glial cells and pericytes to the mRNA profiles of P-glycoprotein and multidrug resistance-associated proteins in an in vitro model of the blood-brain barrier. Brain Res. 2004, 1018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Rafalski, V.A.; Meyer-Franke, A.; Adams, R.A.; Poda, S.B.; Coronado, P.E.R.; Pedersen, L.Ø.; Menon, V.; Baeten, K.M.; Sikorski, S.L.; et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 2018, 19, 1212–1223. [Google Scholar] [CrossRef]
- Merlini, M.; Rafalski, V.A.; Coronado, P.E.R.; Gill, T.M.; Ellisman, M.; Muthukumar, G.; Subramanian, K.S.; Ryu, J.K.; Syme, C.A.; Davalos, D.; et al. Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron 2019, 101, 1099–1108.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.J. Blood-brain barrier (BBB) and the complement landscape. Mol. Immunol. 2018, 102, 26–31. [Google Scholar] [CrossRef]
- Jacob, A.; Alexander, J.J. Complement and blood-brain barrier integrity. Mol. Immunol. 2014, 61, 149–152. [Google Scholar] [CrossRef]
- Orsini, F.; De Blasio, D.; Zangari, R.; Zanier, E.R.; de Simoni, M.G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell. Neurosci. 2014, 8, 380. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.-H.; Kim, J.-H.; Seo, J.-H.; Lee, J.; Choi, W.S.; Kim, Y.-S. Matrix metalloproteinase-3 causes dopaminergic neuronal death through nox1-regenerated oxidative stress. PLoS ONE 2014, 9, e115954. [Google Scholar] [CrossRef]
- Cai, Z.; Wan, C.-Q.; Liu, Z. Astrocyte and Alzheimer’s disease. J. Neurol. 2017, 264, 2068–2074. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Suidan, G.L.; Quan, J.; Pi, Y.Q.; O’Neill, S.M.; Ilardi, M.; Pozdnyakov, N.; Lanz, T.A.; Xi, H.; Bell, R.D.; et al. Inhibition of 2-AG hydrolysis differentially regulates blood brain barrier permeability after injury. J. Neuroinflammation 2018, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Labus, J.; Häckel, S.; Lucka, L.; Danker, K. Interleukin-1β induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain barrier model. J. Neurosci. Methods 2014, 228, 35–45. [Google Scholar] [CrossRef]
- Tan, S.; Shan, Y.; Lin, Y.; Liao, S.; Zhang, B.; Zeng, Q.; Wang, Y.; Deng, Z.; Chen, C.; Hu, X.; et al. Neutralization of interleukin-9 ameliorates experimental stroke by repairing the blood-brain barrierviadown-regulation of astrocyte-derived vascular endothelial growth factor-A. FASEB J. 2018, 33, 4376–4387. [Google Scholar] [CrossRef]
- Presta, I.; Vismara, M.F.M.; Novellino, F.; Donato, A.; Zaffino, P.; Scali, E.; Pirrone, K.C.; Spadea, M.F.; Malara, N.; Donato, G. Innate immunity cells and the neurovascular unit. Int. J. Mol. Sci. 2018, 19, 3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, F.L.; Kittel, Á.; Veszelka, S.; Palmela, I.; Tóth, A.; Brites, D.; Deli, M.A.; Brito, M.A. Exposure to lipopolysaccharide and/or unconjugated bilirubin impair the integrity and function of brain microvascular endothelial cells. PLoS ONE 2012, 7, e35919. [Google Scholar] [CrossRef] [PubMed]
- Mayerhofer, R.; Fröhlich, E.E.; Reichmann, F.; Farzi, A.; Kogelnik, N.; Fröhlich, E.; Sattler, W.; Holzer, P. Diverse action of lipoteichoic acid and lipopolysaccharide on neuroinflammation, blood-brain barrier disruption, and anxiety in mice. Brain Behav. Immun. 2017, 60, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [Green Version]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, J.; Duan, L.; Xiong, H.; Jiang, Y.; Liang, H. Microglia activation mediated by toll-like receptor-4 impairs brain white matter tracts in rats. J. Biomed. Res. 2017, 32, 136–144. [Google Scholar] [CrossRef]
- Moritz, K.E.; McCormack, N.M.; Abera, M.B.; Viollet, C.; Yauger, Y.J.; Sukumar, G.; Dalgard, C.L.; Burnett, B.G. The role of the immunoproteasome in interferon-γ-mediated microglial activation. Sci. Rep. 2017, 7, 9365. [Google Scholar] [CrossRef] [PubMed]
- Akundi, R.S.; Candelario-Jalil, E.; Hess, S.; Hüll, M.; Lieb, K.; Gebicke-Haerter, P.J.; Fiebich, B.L. Signal transduction pathways regulating cyclooxygenase-2 in lipopolysaccharide-activated primary rat microglia. Glia 2005, 51, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Moreno, B.; Jukes, J.-P.; Vergara-Irigaray, N.; Errea, O.; Villoslada, P.; Perry, V.H.; Newman, T.A. Systemic inflammation induces axon injury during brain inflammation. Ann. Neurol. 2011, 70, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Skelly, D.T.; Hennessy, E.; Dansereau, M.-A.; Cunningham, C. A Systematic Analysis of the Peripheral and CNS effects of systemic LPS, IL-1β, TNF-α and IL-6 Challenges in C57BL/6 Mice. PLoS ONE 2013, 8, e69123. [Google Scholar] [CrossRef] [Green Version]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Palmer, A.M. Neuroprotective therapeutics for Alzheimer’s disease: Progress and prospects. Trends Pharmacol. Sci. 2011, 32, 141–147. [Google Scholar] [CrossRef]
- Hardy, J. Has the amyloid cascade hypothesis for Alzheimer’s disease been proved? Curr. Alzheimer Res. 2006, 3, 71–73. [Google Scholar] [CrossRef]
- Morris, R.; Mucke, L. Alzheimer’s disease: A needle from the haystack. Nature 2006, 440, 284–285. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Deane, R.; Sallstrom, J.; Chow, N.; Miano, J. Neurovascular pathways and alzheimer amyloid β-peptide. Brain Pathol. 2006, 15, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglia and Alzheimer’s disease pathogenesis. J. Neurosci. Res. 2004, 77, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.; Robinson, S.; Verma, S.; Morley, J. Efflux of human and mouse amyloid β proteins 1–40 and 1–42 from brain: Impairment in a mouse model of Alzheimer’s disease. Neuroscience 2003, 121, 487–492. [Google Scholar] [CrossRef]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-brain Barrier pathology in Alzheimer’s and Parkinson’s disease: Implications for drug therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar] [CrossRef]
- Johnson, L.A.; Olsen, R.H.; Merkens, L.S.; DeBarber, A.; Steiner, R.D.; Sullivan, P.M.; Maeda, N.; Raber, J. Apolipoprotein E–low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol. Dis. 2014, 64, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramár, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C.; et al. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Sci. Data 2021, 8, 270. [Google Scholar] [CrossRef]
- Huang, H.; Nie, S.; Cao, M.; Marshall, C.; Gao, J.; Xiao, N.; Hu, G.; Xiao, M. Characterization of AD-like phenotype in aged APPSwe/PS1dE9 mice. AGE 2016, 38, 303–322. [Google Scholar] [CrossRef] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.K.; McLarnon, J.G. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 2009, 13, 2911–2925. [Google Scholar] [CrossRef] [Green Version]
- Raven, E.P.; Lu, P.H.; Tishler, T.A.; Heydari, P.; Bartzokis, G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J. Alzheimer’s Dis. 2013, 37, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Moon, G.J.; Kim, H.J.; Kim, D.; Kim, J.; Nam, Y.; Sharma, C.; Leem, E.; Lee, S.; Kim, K.; et al. Control of hippocampal prothrombin kringle-2 (pKr-2) expression reduces neurotoxic symptoms in five familial Alzheimer’s disease mice. J. Cereb. Blood Flow Metab. 2021. [Google Scholar] [CrossRef] [PubMed]
- Squire, L.R. Memory and the hippocampus: A synthesis from findings with rats, monkeys, and humans. Psychol. Rev. 1992, 99, 195–231. [Google Scholar] [CrossRef] [PubMed]
- Brkic, M.; Balusu, S.; Libert, C.; Vandenbroucke, R.E. Friends or Foes: Matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediat. Inflamm. 2015, 2015, 620581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brilha, S.; Ong, C.W.M.; Weksler, B.; Romero, N.; Couraud, P.-O.; Friedland, J.S. Matrix metalloproteinase-9 activity and a downregulated Hedgehog pathway impair blood-brain barrier function in an in vitro model of CNS tuberculosis. Sci. Rep. 2017, 7, 16031. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Shinohara, M.; Shinohara, M.; Yamazaki, A.; Murray, M.; Liesinger, A.M.; Heckman, M.G.; Lesser, E.R.; Parisi, J.E.; Petersen, R.C.; et al. Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain 2019, 142, 1077–1092. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, H.; Chow, N.; Sallstrom, J.; Bell, R.D.; Deane, R.; Brooks, A.I.; Kanagala, S.; Rubio, A.; Sagare, A.; et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 2005, 11, 959–965. [Google Scholar] [CrossRef]
- Lau, S.-F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef]
- Taylor, X.; Cisternas, P.; You, Y.; You, Y.; Xiang, S.; Marambio, Y.; Zhang, J.; Vidal, R.; Lasagna-Reeves, C.A. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J. Neuroinflammation 2020, 17, 223. [Google Scholar] [CrossRef]
- Ding, R.; Hase, Y.; Ameen-Ali, K.E.; Ndung’U, M.; Stevenson, W.; Barsby, J.; Gourlay, R.; Akinyemi, T.; Akinyemi, R.; Uemura, M.T.; et al. Loss of capillary pericytes and the blood–brain barrier in white matter in poststroke and vascular dementias and Alzheimer’s disease. Brain Pathol. 2020, 30, 1087–1101. [Google Scholar] [CrossRef]
- Halliday, M.R.; Rege, S.V.; Ma, Q.; Zhao, Z.; Miller, C.A.; Winkler, E.A.; Zlokovic, B.V. Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2016, 36, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, K.; Hosono, T.; Nakamura, T.; Bu, G.; Michikawa, M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem. 2011, 286, 17536–17542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alafuzoff, I.; Adolfsson, R.; Bucht, G.; Winblad, B. Albumin and immunoglobulin in plasma and cerebrospinal fluid, and blood-cerebrospinal fluid barrier function in patients with dementia of alzheimer type and multi-infarct dementia. J. Neurol. Sci. 1983, 60, 465–472. [Google Scholar] [CrossRef]
- Tibbling, G.; Link, H.; Öhman, S. Principles of albumin and IgG analyses in neurological disorders. I. Establishment of reference values. Scand. J. Clin. Lab. Investig. 1977, 37, 385–390. [Google Scholar] [CrossRef]
- Hampel, H.; Kötter, H.U.; Möller, H.-J. Blood-cerebrospinal fluid barrier dysfunction for high molecular weight proteins in alzheimer disease and major depression. Alzheimer Dis. Assoc. Disord. 1997, 11, 78–87. [Google Scholar] [CrossRef]
- Skoog, I.; Wallin, A.; Fredman, P.; Hesse, C.; Aevarsson, O.; Karlsson, I.; Gottfries, C.G.; Blennow, K. A population study on blood-brain barrier function in 85-year-olds: Relation to Alzheimer’s disease and vascular dementia. Neurology 1998, 50, 966–971. [Google Scholar] [CrossRef]
- Slemmon, J.R.; Hughes, C.M.; Campbell, G.A.; Flood, D.G. Increased levels of hemoglobin-derived and other peptides in Alzheimer’s disease cerebellum. J. Neurosci. 1994, 14, 2225–2235. [Google Scholar] [CrossRef] [Green Version]
- Paul, J.; Strickland, S.; Melchor, J.P. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J. Exp. Med. 2007, 204, 1999–2008. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and β-amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron 2010, 66, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Zipser, B.; Johanson, C.; Gonzalez, L.; Berzin, T.; Tavares, R.; Hulette, C.; Vitek, M.; Hovanesian, V.; Stopa, E. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef]
- Davie, E.W.; Fujikawa, K.; Kisiel, W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 2002, 30, 10363–10370. [Google Scholar] [CrossRef]
- Choi, S.-H.; Lee, D.Y.; Kim, S.U.; Jin, B.K. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: Role of microglial NADPH oxidase. J. Neurosci. 2005, 25, 4082–4090. [Google Scholar] [CrossRef] [PubMed]
- Freeze, W.M.; Bacskai, B.J.; Frosch, M.P.; Jacobs, H.I.; Backes, W.H.; Greenberg, S.M.; van Veluw, S.J. Blood-brain barrier leakage and microvascular lesions in cerebral amyloid angiopathy. Stroke 2019, 50, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, P.A.; Desmond, P.M.; Phal, P.M.; Steward, C.; Szoeke, C.; Salvado, O.; Ellis, K.A.; Martins, R.N.; Masters, C.L.; Ames, D.; et al. Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology 2014, 82, 1266–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Shi, J.; Smallman, R.; Iwatsubo, T.; Mann, D.M.A. Relationships in Alzheimer’s disease between the extent of Abeta deposition in cerebral blood vessel walls, as cerebral amyloid angiopathy, and the amount of cerebrovascular smooth muscle cells and collagen. Neuropathol. Appl. Neurobiol. 2006, 32, 332–340. [Google Scholar] [CrossRef]
- Wu, S.; Liu, H.; Zhao, H.; Wang, X.; Chen, J.; Xia, D.; Xiao, C.; Cheng, J.; Zhao, Z.; He, Y. Environmental lead exposure aggravates the progression of Alzheimer’s disease in mice by targeting on blood brain barrier. Toxicol. Lett. 2020, 319, 138–147. [Google Scholar] [CrossRef]
- He, J.-T.; Zhao, X.; Xu, L.; Mao, C.-Y. Vascular Risk Factors and Alzheimer’s Disease: Blood-brain barrier disruption, metabolic syndromes, and molecular links. J. Alzheimer’s Dis. 2020, 73, 39–58. [Google Scholar] [CrossRef]
- Paris, D.; Patel, N.; DelleDonne, A.; Quadros, A.; Smeed, R.; Mullan, M. Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci. Lett. 2004, 366, 80–85. [Google Scholar] [CrossRef]
- Yin, K.-J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.-F.; Turk, J.; et al. Matrix Metalloproteinases Expressed by Astrocytes Mediate Extracellular Amyloid-beta Peptide Catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef]
- Melchor, J.P.; Pawlak, R.; Strickland, S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates Amyloid-β (Aβ) degradation and inhibits Aβ-Induced neurodegeneration. J. Neurosci. 2003, 23, 8867–8871. [Google Scholar] [CrossRef] [Green Version]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid -protein, and the -amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.-J.; Saido, T.C. Metabolic regulation of brain abeta by neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Kook, S.-Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Aβ₁₋₄₂-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca2+-Calcineurin Signaling. J. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; Rozemuller, A.J.; van Horssen, J.; de Vries, H.E. Amyloid beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef]
- Bell, R.D.; Deane, R.; Chow, N.; Long, X.; Sagare, A.; Singh, I.; Streb, J.W.; Guo, H.; Rubio, A.; Van Nostrand, W.; et al. SRF and myocardin regulate LRP-mediated amyloid-β clearance in brain vascular cells. Nat. Cell Biol. 2009, 11, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Weller, R.O.; Subash, M.; Preston, S.; Mazanti, I.; Carare, R.O. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2007, 18, 253–266. [Google Scholar] [CrossRef]
- Ujiie, M.; Dickstein, D.L.; Carlow, D.A.; Jefferies, W.A. Blood-brain barrier permeability precedes senile plaque formation in an alzheimer disease model. Microcirculation 2003, 10, 463–470. [Google Scholar] [CrossRef]
- Stunff, H.L.; Véret, J.; Kassis, N.; Denom, J.; Meneyrol, K.; Paul, J.L.; Cruciani-Guglielmacci, C.; Magnan, C.; Janel, N. Deciphering the link between hyperhomocysteinemia and ceramide metabolism in Alzheimer-type neurodegeneration. Front Neurol. 2019, 10, 807. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [Green Version]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, N.; Brännström, K.; Byman, E.; Moussaud, S.; Nielsen, H.M.; Olofsson, A.; Wennström, M.; Bank, T.N.B. Amyloid-beta 1–40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 2018, 17, e12728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [Green Version]
- Kimbrough, I.F.; Robel, S.; Roberson, E.D.; Sontheimer, H. Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 2015, 138, 3716–3733. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Carranza, D.L.; Nilson, A.N.; Van Skike, C.E.; Jahrling, J.B.; Patel, K.; Garach, P.; Gerson, J.E.; Sengupta, U.; Abisambra, J.; Nelson, P.; et al. Cerebral microvascular accumulation of tau oligomers in Alzheimer’s disease and related tauopathies. Aging Dis. 2017, 8, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Canepa, E.; Fossati, S. Impact of tau on neurovascular pathology in Alzheimer’s disease. Front. Neurol. 2021, 11. [Google Scholar] [CrossRef]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef] [Green Version]
- Michalicova, A.; Majerova, P.; Kovac, A. Tau protein and its role in blood–brain barrier dysfunction. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef]
- Albrecht, D.; Isenberg, A.L.; Stradford, J.; Monreal, T.; Sagare, A.; Pachicano, M.; Sweeney, M.; Toga, A.; Zlokovic, B.; Chui, H.; et al. Associations between vascular function and tau PET are associated with global cognition and amyloid. J. Neurosci. 2020, 40, 8573–8586. [Google Scholar] [CrossRef]
- Liu, C.; Yamazaki, Y.; Heckman, M.G.; Martens, Y.A.; Jia, L.; Yamazaki, A.; Diehl, N.N.; Zhao, J.; Zhao, N.; DeTure, M.; et al. Tau and apolipoprotein E modulate cerebrovascular tight junction integrity independent of cerebral amyloid angiopathy in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Emrani, S.; Lamar, M.; Price, C.C.; Wasserman, V.; Matusz, E.; Au, R.; Swenson, R.; Nagele, R.; Heilman, K.M.; Libon, D.J. Alzheimer’s/Vascular spectrum dementia: Classification in addition to diagnosis. J. Alzheimer’s Dis. 2020, 73, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.A.; Yu, L.; Wilson, R.S.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann. Neurol. 2018, 83, 74–83. [Google Scholar] [CrossRef]
- Silbert, L.C.; Dodge, H.H.; Perkins, L.G.; Sherbakov, L.; Lahna, D.; Erten-Lyons, D.; Woltjer, R.; Shinto, L.; Kaye, J.A. Trajectory of white matter hyperintensity burden preceding mild cognitive impairment. Neurology 2012, 79, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Malek, N.; Lawton, M.A.; Swallow, D.M.A.; Grosset, K.A.; Marrinan, S.L.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Hardy, J.; Morris, H.R.; et al. Vascular disease and vascular risk factors in relation to motor features and cognition in early Parkinson’s disease. Mov. Disord. 2016, 31, 1518–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drouin-Ouellet, J.; Sawiak, S.; Cisbani, G.; Lagacé, M.; Kuan, W.-L.; Saint-Pierre, M.; Dury, R.J.; Alata, W.; St-Amour, I.; Mason, S.L.; et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol. 2015, 78, 160–177. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Hsu, Y.-H.; Lin, M.-H.; Yang, T.-H.; Chen, H.-M.; Chen, Y.-C.; Hsiao, H.-Y.; Chen, C.-C.; Chern, Y.; Chang, C. Neurovascular abnormalities in humans and mice with Huntington’s disease. Exp. Neurol. 2013, 250, 20–30. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood–spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 125, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Gaitán, M.I.; Shea, C.D.; Evangelou, I.E.; Bs, R.D.S.; Ms, K.M.F.; Bielekova, B.; Massacesi, L.; Reich, D.S. Evolution of the blood-brain barrier in newly forming multiple sclerosis lesions. Ann. Neurol. 2011, 70, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’S disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-brain stimulation associates with improved microvascular integrity in the subthalamic nucleus in Parkinson’s disease. Neurobiol. Dis. 2015, 74, 392–405. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.; Sanberg, P.R. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Bowser, R.; Appel, S.H. decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology 2009, 72, 1614–1616. [Google Scholar] [CrossRef]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef]
- Kirk, J.; Plumb, J.; Mirakhur, M.; McQuaid, S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J. Pathol. 2003, 201, 319–327. [Google Scholar] [CrossRef]
- Gold, B.T.; Shao, X.; Sudduth, T.L.; Jicha, G.A.; Wilcock, D.M.; Seago, E.R.; Wang, D.J. Water exchange rate across the blood-brain barrier is associated with CSF amyloid-&β 42 in healthy older adults. Alzheimer’s Dement. 2021, 17, 2020–2029. [Google Scholar] [CrossRef]
- Toornvliet, R.; Vanberckel, B.; Luurtsema, G.; Lubberink, M.; Geldof, A.A.; Bosch, T.M.; Oerlemans, R.; Lammertsma, A.A.; Franssen, E.J.; Berckel, B.N. Effect of age on functional P-glycoprotein in the blood-brain barrier measured by use of (R)-[11C] verapamil and positron emission tomography. Clin. Pharmacol. Ther. 2006, 79, 540–548. [Google Scholar] [CrossRef]
- Cao, C.; Cirrito, J.R.; Lin, X.; Wang, L.; Verges, D.K.; Dickson, A.; Mamcarz, M.; Zhang, C.; Mori, T.; Aredash, G.W.; et al. Caffeine Suppresses Amyloid-β levels in plasma and brain of alzheimer’s disease transgenic mice. J. Alzheimer’s Dis. 2009, 17, 681–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, R.; Johanson, C.E. Review: Vitamin transport and homeostasis in mammalian brain: Focus on Vitamins B and E. J. Neurochem. 2007, 103, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Salvador, E.; Shityakov, S.; Förster, C. Glucocorticoids and endothelial cell barrier function. Cell Tissue Res. 2014, 355, 597–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-C.; Sonninen, T.-M.; Peltonen, S.; Koistinaho, J.; Lehtonen, Š. Blood-brain barrier and neurodegenerative diseases—Modeling with iPSC-derived brain cells. Int. J. Mol. Sci. 2021, 22, 7710. [Google Scholar] [CrossRef]
- Hue, C.D.; Cho, F.S.; Cao, S.; Dale Bass, C.R.; Meaney, D.F.; Morrison, I.B., 3rd. Dexamethasone potentiates in vitro blood-brain barrier recovery after primary blast injury by glucocorticoid receptor-mediated upregulation of ZO-1 tight junction protein. J. Cereb. Blood Flow Metab. 2015, 35, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y. Angiogenesis and blood-brain barrier permeability in vascular remodeling after stroke. Curr. Neuropharmacol. 2020, 18, 1250–1265. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [Green Version]
- Van Skike, C.E.; Jahrling, J.B.; Olson, A.B.; Sayre, N.L.; Hussong, S.A.; Ungvari, Z.; Lechleiter, J.D.; Galvan, V. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer’s disease and vascular cognitive impairment. Am. J. Physiol. Circ. Physiol. 2018, 314, H693–H703. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Garrett, M.R.; Men, P.; Zhu, X.; Perry, G.; Smith, M.A. Nanoparticle and other metal chelation therapeutics in Alzheimer disease. Biochim. Biophys. Acta 2005, 1741, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Bana, L.; Minniti, S.; Salvati, E.; Sesana, S.; Zambelli, V.; Cagnotto, A.; Orlando, A.; Cazzaniga, E.; Zwart, R.; Scheper, W.; et al. Liposomes bi-functionalized with phosphatidic acid and an ApoE-derived peptide affect Aβ aggregation features and cross the blood-brain barrier: Implications for therapy of Alzheimer disease. Nanomedicine 2014, 10, 1583–1590. [Google Scholar] [CrossRef]
- Elfakhri, K.H.; Abdallah, I.M.; Brannen, A.D.; Kaddoumi, A. Multi-faceted therapeutic strategy for treatment of Alzheimer’s disease by concurrent administration of etodolac and α-tocopherol. Neurobiol. Dis. 2019, 125, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Ongnok, B.; Khuanjing, T.; Chunchai, T.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, N.; Chattipakorn, S.C. Donepezil provides neuroprotective effects against brain injury and Alzheimer’s pathology under conditions of cardiac ischemia/reperfusion injury. Biochim. Biophys. Acta 2020, 1867, 165975. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid- deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, B.; Hartz, A.M.S.; Miller, D.S. Tumor necrosis factor α and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol. Pharmacol. 2007, 71, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakki, S.L.; Senthil, V.; Chandrasekar, M. The blood brain barrier and its role in Alzheimer’s therapy: An overview. Curr. Drug Targets 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.E.; Greig, N.H.; Giacobini, E. Why Do So Many Drugs for Alzheimer’s Disease Fail in Development? Time for New Methods and New Practices? J. Alzheimer’s Dis. 2008, 15, 303–325. [Google Scholar] [CrossRef] [Green Version]

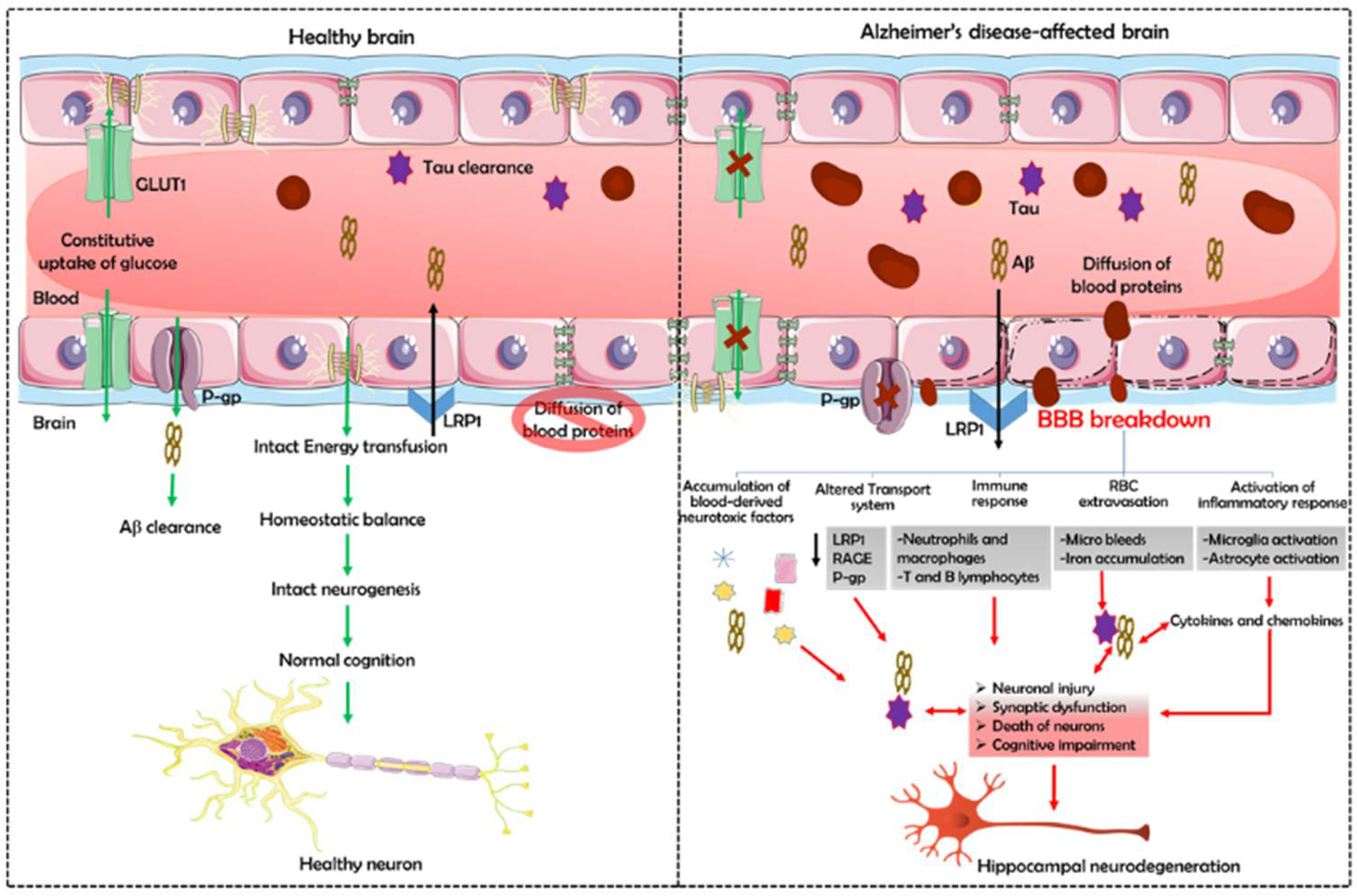

{kind=link}
{kind=link}
| Transgenic Mouse Strain | Genetic Background | Disease Status | References |
|---|---|---|---|
| 5XFAD | (C57BL/6 × SJL)F1 and C57BL/6 J | Amyloid plaques, accompanied by gliosis; Neuron loss occurs in multiple brain regions, beginning at approximately 6 months in the areas with the most pronounced amyloidosis; Mice display a range of cognitive and motor deficits. | [97] |
| APP/PS1 | C57BL/6J | Amyloid plaque formation in the neocortex begins around 6 weeks of age. At 3–4 months, deposits are observed in the hippocampus, and at 4–5 months, deposits appear in the striatum, thalamus, and brainstem; All congophilic amyloid deposits have phosphorylated tau-positive neuritic processes. | [98] |
| Tg2576 | C7BL/6;129 × 1/SvJ;129S1/Sv | Exhibit age-associated cognitive deficits such as impaired spatial learning and deficits in working memory as well as contextual fear conditioning at <6 months of age. | [99] |
| Neurodegenerative Diseases | Representative BBB Changes | References |
|---|---|---|
| Alzheimer’s disease | Accumulation of fibrinogen, thrombin, albumin, IgG, and hemosiderin in the cortex and hippocampus; loss of pericyte capillary coverage and numbers in the cortex and hippocampus; reduced tight junction proteins; RBC extravasation and peripheral macrophage infiltration; neutophil infiltration; and increased levels of angiogenic proteins | [100,111,119,120] |
| Parkinson’s disease | Accumulation of fibrinogen in the striatum as well as IgG and hemosiderin in the globus pallidus; microvascular degeneration, reduced and disrupted tight junctions, altered capillary basement membrane in the subthalamic nucleus; extravasation of RBCs in the striatum; increased endothelial cell numbers in the substantia nigra | [163,164] |
| Huntington’s disease | Leakage of fibrin in the putamen; decreased and disrupted tight junction protein expression in the putamen; accumulation of Huntingtin protein aggregates in the endothelial cells | [158,159] |
| Amyotrophic lateral sclerosis | Leakage of fibrinogen, thrombin, IgG, collagen type IV, and iron-containing proteins; loss of pericytes in the medulla; microvascular degeneration, intracellular vacuolization; reduced and disrupted tight junctions; and enlarged perivascular spaces | [160,165,166,167] |
| Multiple sclerosis | Fibrinogen leakage; decreased and disrupted tight junctions; and leukocyte infiltration | [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, C.; Woo, H.; Kim, S.R. Addressing Blood–Brain Barrier Impairment in Alzheimer’s Disease. Biomedicines 2022, 10, 742. https://doi.org/10.3390/biomedicines10040742
Sharma C, Woo H, Kim SR. Addressing Blood–Brain Barrier Impairment in Alzheimer’s Disease. Biomedicines. 2022; 10(4):742. https://doi.org/10.3390/biomedicines10040742
Chicago/Turabian StyleSharma, Chanchal, Hanwoong Woo, and Sang Ryong Kim. 2022. "Addressing Blood–Brain Barrier Impairment in Alzheimer’s Disease" Biomedicines 10, no. 4: 742. https://doi.org/10.3390/biomedicines10040742
APA StyleSharma, C., Woo, H., & Kim, S. R. (2022). Addressing Blood–Brain Barrier Impairment in Alzheimer’s Disease. Biomedicines, 10(4), 742. https://doi.org/10.3390/biomedicines10040742