
Detection of TERT Promoter Mutations as a Prognostic Biomarker in Gliomas: Methodology, Prospects, and Advances

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Classification of CNS Tumors
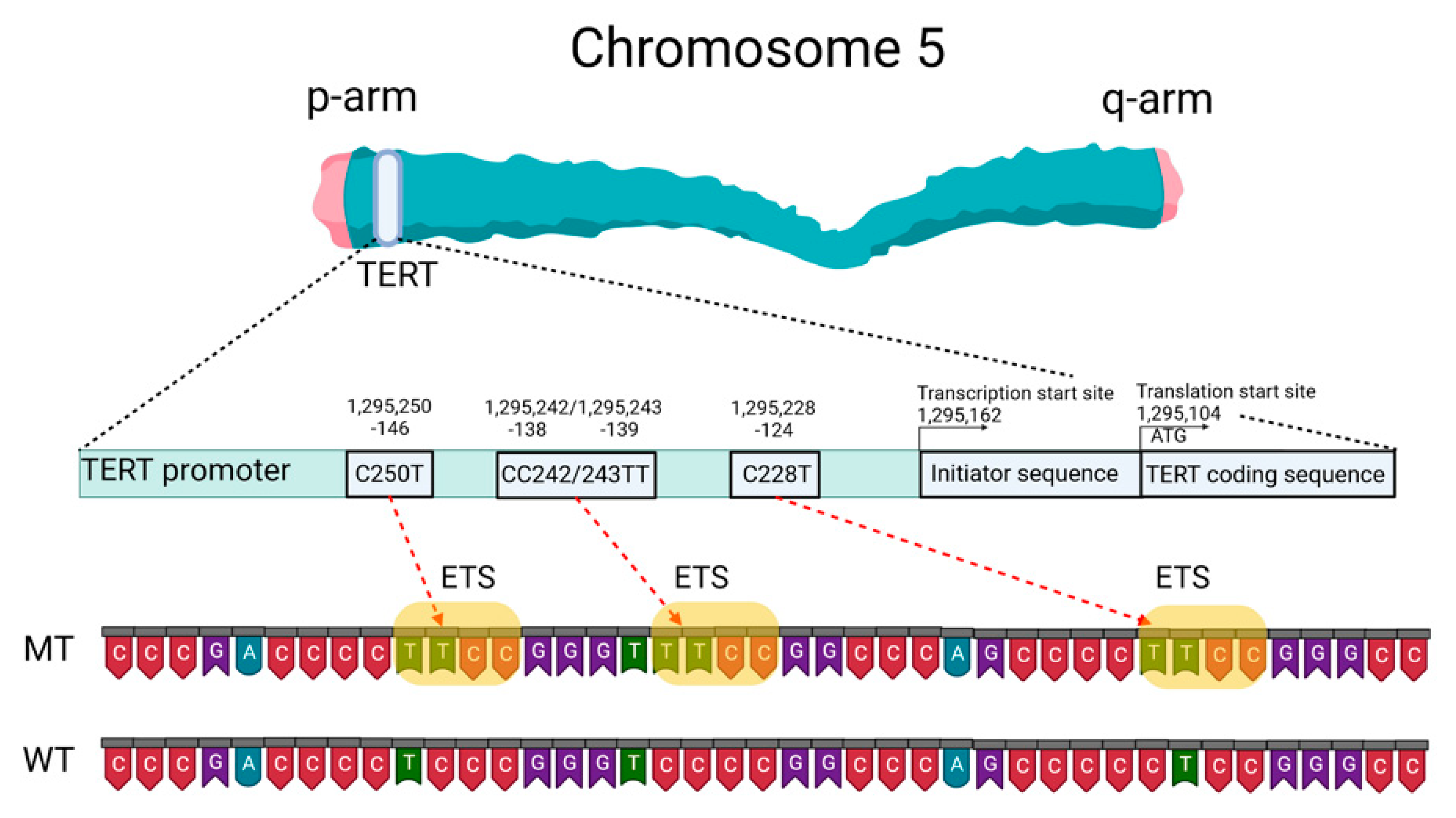
2.2. Telomeres, Telomerase, and the TERT Promoter
2.3. Telomere Length as a Prognostic Factor for Patients with CNS Tumors
2.4. Mutation Status of the TERT Promoter as a Prognostic Marker
2.5. Analysis of Methods to Detect TERT Promoter Mutations in CNS Tumors
2.5.1. Sanger Sequencing
2.5.2. Droplet Digital PCR (ddPCR)
2.5.3. Next-Generation Sequencing (NGS)
2.5.4. Nanopore Sequencing
2.5.5. Magnetic Resonance Imaging (MRI)
2.6. Specifics of Mutation Detection in the TERTp According to the DNA Source
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| [18F]-FET PET | O-(2-[18F]-Fluoroethyl)-l-tyrosine positron emission tomography |
| 1H-MRS | Short echo time |
| 5mC | 5-Methylcytosine |
| ADC | Apparent diffusion coefficient |
| ARMS-PCR | PCR based on the amplification-resistant mutation system |
| AUC | The area under the curve |
| BRAF | B-Raf murine sarcoma viral oncogene homolog |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| cfDNA | Circulating free DNA |
| CIC | Capicua transcriptional repressor |
| CNN | Convolutional neural network |
| CNS | Central nervous system |
| cT1WI | T1-weighted contrast-enhanced imaging |
| ctDNA | Circulating tumor DNA |
| DCE | Dynamic contrast enhancement |
| ddPCR | Droplet digital PCR |
| DNA | Deoxyribonucleic acid |
| dPCR | Digital PCR |
| DSC | Dynamic susceptibility contrast |
| EGFR | Epithelial growth factor receptor |
| FFPE | Formalin-fixed tumor samples embedded in paraffin |
| FUBP1 | Far upstream element-binding protein 1 |
| GABP | GA-binding protein |
| H3F3A | Histone family 3A |
| IDH1/2 | Isocitrate dehydrogenase 1 and/or 2 |
| IT-ddPCR | IDH1-TERT-mutation ddPCR |
| MAF | Mutant allele fraction |
| MALDI-TOF | Laser desorption ionization time-of-flight mass spectrometry platform |
| MassARRAY | Mass spectrometric assay |
| meth | Methylated |
| MGMT | O6-methylguanine-DNA methyltransferase |
| MGMTp | MGMT promotor |
| MR | Magnetic resonance |
| MRI | Magnetic resonance imaging |
| MRS | Magnetic resonance spectroscopy |
| mt | Mutant type |
| mut | Mutation |
| n.b. | Nucleotide bases |
| NGS | Next-generation sequencing |
| NOTCH1 | Notch receptor 1 |
| OS | Overall survival |
| PCR | Polymerase chain reaction |
| PFS | Progression-free survival |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-kinase catalytic subunit alpha |
| PTEN | Phosphatase and tensin homolog |
| ROIs | Regions of interest |
| RT-PCR | Real-time PCR |
| SEL1L | Suppressor/enhancer of Lin-12-like |
| TBR | Tumor-to-background ratio |
| TCIA | Cancer Imaging Archive |
| TERT | Telomerase reverse transcriptase |
| TERTp | TERT promoter |
| TTP | Time-to-peak |
| unmeth | Not methylated |
| VASARI | Visually Accessible Rembrandt Images |
| WHO | World Health Organization |
| wt | Wild-type |
References
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef]
- Hafezi, F.; Perez Bercoff, D. The Solo Play of TERT Promoter Mutations. Cells 2020, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Powter, B.; Jeffreys, S.A.; Sareen, H.; Cooper, A.; Brungs, D.; Po, J.; Roberts, T.; Koh, E.S.; Scott, K.F.; Sajinovic, M.; et al. Human TERT promoter mutations as a prognostic biomarker in glioma. J. Cancer Res. Clin. Oncol. 2021, 147, 1007–1017. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Packer, R.J. The 2021 WHO Classification of Tumors of the Central Nervous System: Clinical implications. Neuro-Oncology 2021, 23, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Śledzińska, P.; Bebyn, M.G.; Furtak, J.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Biomarkers in Gliomas. Int. J. Mol. Sci. 2021, 22, 10373. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22 (Suppl. 2), iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Reifenberger, G. Molecular neuropathology of gliomas. Int. J. Mol. Sci. 2009, 10, 184–212. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.A.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Q.L.; Sun, W.; Chandrasekharan, P.; Cheng, H.S.; Ying, Z.; Lakshmanan, M.; Raju, A.; Tenen, D.G.; Cheng, S.Y.; et al. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 2015, 10, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, Y.; Bharath, S.R.; Ozturk, M.B.; Bowler, M.W.; Loo, B.Z.L.; Tergaonkar, V.; Song, H. Structural basis for reactivating the mutant TERT promoter by cooperative binding of p52 and ETS1. Nat. Commun. 2018, 9, 3183. [Google Scholar] [CrossRef] [PubMed]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Lemma, R.B.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Pérez, N.M.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef]
- Pavlova, A.V.; Kubareva, E.A.; Monakhova, M.V.; Zvereva, M.I.; Dolinnaya, N.G. Impact of G-Quadruplexes on the Regulation of Genome Integrity, DNA Damage and Repair. Biomolecules 2021, 11, 1284. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Arita, H.; Narita, Y.; Fukushima, S.; Tateishi, K.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Collins, V.P.; Kawahara, N.; et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013, 126, 267–276. [Google Scholar] [CrossRef]
- Eckel-Passow, J.; Lachance, D.; Molinaro, A.; Walsh, K.; Decker, P.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.; Smirnov, I.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef]
- Gao, K.; Li, G.; Qu, Y.; Wang, M.; Cui, B.; Ji, M.; Shi, B.; Hou, P. TERT promoter mutations and long telomere length predict poor survival and radiotherapy resistance in gliomas. Oncotarget 2016, 7, 8712–8725. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, B.; Rachakonda, S.; Hosen, I.; Volz, F.; Hemminki, K.; Weyerbrock, A.; Kumar, R. TERT promoter mutations and telomere length in adult malignant gliomas and recurrences. Oncotarget 2015, 6, 10617–10633. [Google Scholar] [CrossRef] [PubMed]
- Hosen, I.; Sheikh, M.; Zvereva, M.; Scelo, G.; Forey, N.; Durand, G.; Voegele, C.; Poustchi, H.; Khoshnia, M.; Roshandel, G.; et al. Urinary TERT promoter mutations are detectable up to 10 years prior to clinical diagnosis of bladder cancer: Evidence from the Golestan Cohort Study. EBioMedicine 2020, 53, 102643. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, T.; Sofiadis, A.; Juhlin, C.C.; Zedenius, J.; Höög, A.; Larsson, C.; Xu, D. TERT promoter mutation as an early genetic event activating telomerase in follicular thyroid adenoma (FTA) and atypical FTA. Cancer 2014, 120, 2965–2979. [Google Scholar] [CrossRef]
- Hysek, M.; Paulsson, J.O.; Jatta, K.; Shabo, I.; Stenman, A.; Höög, A.; Larsson, C.; Zedenius, J.; Juhlin, C.C. Clinical Routine TERT Promoter Mutational Screening of Follicular Thyroid Tumors of Uncertain Malignant Potential (FT-UMPs): A Useful Predictor of Metastatic Disease. Cancers 2019, 11, 1443. [Google Scholar] [CrossRef]
- Nault, J.C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.P.; Pincus, L.B.; Ruben, B.S.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Cheng, L.; Montironi, R.; Lopez-Beltran, A. TERT Promoter Mutations Occur Frequently in Urothelial Papilloma and Papillary Urothelial Neoplasm of Low Malignant Potential. Eur. Urol. 2017, 71, 497–498. [Google Scholar] [CrossRef]
- Mitchell, T.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018, 173, 611–623.e17. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.; Malta, T.; Sabedot, T.S.; Salama, S.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Abou-El-Ardat, K.; Seifert, M.; Becker, K.; Eisenreich, S.; Lehmann, M.; Hackmann, K.; Rump, A.; Meijer, G.; Carvalho, B.; Temme, A.; et al. Comprehensive molecular characterization of multifocal glioblastoma proves its monoclonal origin and reveals novel insights into clonal evolution and heterogeneity of glioblastomas. Neuro-Oncology 2017, 19, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Qi, C.; Maling, G.; Xiang, W.; Yanhui, L.; Ruofei, L.; Yunhe, M.; Jiewen, L.; Qing, M. TERT mutation in glioma: Frequency, prognosis and risk. J. Clin. Neurosci. 2016, 26, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Arita, H.; Yamasaki, K.; Matsushita, Y.; Nakamura, T.; Shimokawa, A.; Takami, H.; Tanaka, S.; Mukasa, A.; Shirahata, M.; Shimizu, S.; et al. A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol. Commun. 2016, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Pekmezci, M.; Rice, T.; Molinaro, A.M.; Walsh, K.; Decker, P.A.; Hansen, H.; Sicotte, H.; Kollmeyer, T.M.; McCoy, L.S.; Sarkar, G.; et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017, 133, 1001–1016. [Google Scholar] [CrossRef]
- Yang, P.; Cai, J.; Yan, W.; Yang, P.; Cai, J.; Yan, W.; Zhang, W.; Wang, Y.; Chen, B.; Li, G.; et al. Classification based on mutations of TERT promoter and IDH characterizes subtypes in grade II/III gliomas. Neuro-Oncology 2016, 18, 1099–1108. [Google Scholar] [CrossRef]
- Kim, H.S.; Kwon, M.J.; Song, J.H.; Kim, E.S.; Kim, H.Y.; Min, K.W. Clinical implications of TERT promoter mutation on IDH mutation and MGMT promoter methylation in diffuse gliomas. Pathol. Res. Pract. 2018, 214, 881–888. [Google Scholar] [CrossRef]
- You, H.; Wu, Y.; Chang, K.; Shi, X.; Chen, X.; Yan, W.; Li, R. Paradoxical prognostic impact of TERT promoter mutations in gliomas depends on different histological and genetic backgrounds. CNS Neurosci. Ther. 2017, 23, 790–797. [Google Scholar] [CrossRef]
- Huang, D.-S.; Wang, Z.; He, X.-J.; Diplas, B.H.; Yang, R.; Killela, P.J.; Meng, Q.; Ye, Z.-Y.; Wang, W.; Jiang, X.-T.; et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer 2015, 51, 969–976. [Google Scholar] [CrossRef]
- Koelsche, C.; Sahm, F.; Capper, D.; Reuss, D.; Sturm, D.; Jones, D.T.W.; Kool, M.; Northcott, P.A.; Wiestler, B.; Böhmer, K.; et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013, 126, 907–915. [Google Scholar] [CrossRef]
- Nonoguchi, N.; Ohta, T.; Oh, J.E.; Kim, Y.H.; Kleihues, P.; Ohgaki, H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol. 2013, 126, 931–937. [Google Scholar] [CrossRef]
- Akyerli, C.B.; Yüksel, Ş.; Can, Ö.; Erson-Omay, E.Z.; Oktay, Y.; Coşgun, E.; Ülgen, E.; Erdemgil, Y.; Sav, A.; von Deimling, A.; et al. Use of telomerase promoter mutations to mark specific molecular subsets with reciprocal clinical behavior in IDH mutant and IDH wild-type diffuse gliomas. J. Neurosurg. 2018, 128, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Hewer, E.; Phour, J.; Gutt-Will, M.; Schucht, P.; Dettmer, M.S.; Vassella, E. TERT Promoter Mutation Analysis to Distinguish Glioma from Gliosis. J. Neuropathol. Exp. Neurol. 2020, 79, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Mosrati, M.A.; Malmström, A.; Lysiak, M.; Krysztofiak, A.; Hallbeck, M.; Milos, P.; Hallbeck, A.L.; Bratthäll, C.; Strandéus, M.; Stenmark-Askmalm, M.; et al. TERT promoter mutations and polymorphisms as prognostic factors in primary glioblastoma. Oncotarget 2015, 6, 16663–16673. [Google Scholar] [CrossRef]
- Olympios, N.; Gilard, V.; Marguet, F.; Clatot, F.; Di Fiore, F.; Fontanilles, M. TERT Promoter Alterations in Glioblastoma: A Systematic Review. Cancers 2021, 13, 1147. [Google Scholar] [CrossRef]
- Shu, C.; Wang, Q.; Yan, X.; Wang, J. The TERT promoter mutation status and MGMT promoter methylation status, combined with dichotomized MRI-derived and clinical features, predict adult primary glioblastoma survival. Cancer Med. 2018, 7, 3704–3712. [Google Scholar] [CrossRef]
- Sun, Z.-L.; Chan, A.K.-Y.; Chen, L.-C.; Tang, C.; Zhang, Z.-Y.; Ding, X.-J.; Wang, Y.; Sun, C.-R.; Ng, H.-K.; Yao, Y.; et al. TERT promoter mutated WHO grades II and III gliomas are located preferentially in the frontal lobe and avoid the midline. Int. J. Clin. Exp. Pathol. 2015, 8, 11485–11494. [Google Scholar]
- Zhang, Z.Y.; Chan, A.K.; Ding, X.J.; Qin, Z.Y.; Hong, C.S.; Chen, L.C.; Zhang, X.; Zhao, F.P.; Wang, Y.; Wang, Y.; et al. TERT promoter mutations contribute to IDH mutations in predicting differential responses to adjuvant therapies in WHO grade II and III diffuse gliomas. Oncotarget 2015, 6, 24871–24883. [Google Scholar] [CrossRef]
- Adachi, J.-I.; Shirahata, M.; Suzuki, T.; Mishima, K.; Uchida, E.; Sasaki, A.; Nishikawa, R. Droplet digital PCR assay for detecting TERT promoter mutations in patients with glioma. Brain Tumor Pathol. 2021, 38, 201–209. [Google Scholar] [CrossRef]
- Dubbink, H.J.; Atmodimedjo, P.N.; Kros, J.M.; French, P.; Sanson, M.; Idbaih, A.; Wesseling, P.; Enting, R.; Spliet, W.; Tijssen, C.; et al. Molecular classification of anaplastic oligodendroglioma using next-generation sequencing: A report of the prospective randomized EORTC Brain Tumor Group 26,951 phase III trial. Neuro-Oncology 2016, 18, 388–400. [Google Scholar] [CrossRef]
- Brandner, S. Molecular Diagnostics of Adult Gliomas in Neuropathological Practice. Acta Med. Acad. 2021, 50, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Bai, S.; Li, X.; Zhang, Y.; Li, Y.; He, F.; Cheng, W. Establishment and Validation of the Detection of TERT Promoter Mutations by Human Gliomas U251 Cell Lines. Biomed. Res. Int. 2021, 2021, 3271395. [Google Scholar] [CrossRef] [PubMed]
- Labussière, M.; Di Stefano, A.L.; Gleize, V.; Boisselier, B.; Giry, M.; Mangesius, S.; Bruno, A.; Paterra, R.; Marie, Y.; Rahimian, A.; et al. TERT promoter mutations in gliomas, genetic associations and clinico-pathological correlations. Br. J. Cancer 2014, 111, 2024–2032. [Google Scholar] [CrossRef]
- Chan, A.K.-Y.; Yao, Y.; Zhang, Z.; Chung, N.Y.-F.; Liu, J.S.-M.; Li, K.K.-W.; Shi, Z.; Chan, D.T.-M.; Poon, W.S.; Zhou, L.; et al. TERT promoter mutations contribute to subset prognostication of lower-grade gliomas. Mod. Pathol. 2015, 28, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, Z.; Sun, M.; Chen, K.; Yuan, W.; Jiang, G. The Sensitive Detection of Telomerase Reverse Transcriptase Promoter Mutation by Amplification Refractory Mutation System-PCR. Genet. Test Mol. Biomark. 2016, 20, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Komori, T.; Kato, Y.; Masutomi, K.; Ichimura, K.; Ogasawara, S.; Kaneko, M.K.; Oki, H.; Suzuki, H.; Nitta, M.; et al. Elevated TERT Expression in TERT-Wildtype Adult Diffuse Gliomas: Histological Evaluation with a Novel TERT-Specific Antibody. Biomed. Res. Int. 2018, 2018, 7945845. [Google Scholar] [CrossRef]
- Diplas, B.H.; Liu, H.; Yang, R.; Hansen, L.J.; Zachem, A.L.; Zhao, F.; Bigner, D.D.; McLendon, R.E.; Jiao, Y.; He, Y.; et al. Sensitive and rapid detection of TERT promoter and IDH mutations in diffuse gliomas. Neuro-Oncology 2019, 21, 440–450. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, X.; Yan, X.; Sun, M.; Fan, Y.; Huang, Y. Significance of TERT and ATRX mutations in glioma. Oncol. Lett. 2019, 17, 95–102. [Google Scholar] [CrossRef]
- Qu, C.X.; Ji, H.M.; Shi, X.C.; Bi, H.; Zhai, L.Q.; Han, D.W. Characteristics of the isocitrate dehydrogenase gene and telomerase reverse transcriptase promoter mutations in gliomas in Chinese patients. Brain Behav. 2020, 10, e01583. [Google Scholar] [CrossRef]
- Pierini, T.; Nardelli, C.; Fernandez, A.G.L.; Pierini, V.; Pellanera, F.; Nofrini, V.; Gorello, P.; Moretti, M.; Arniani, S.; Roti, G.; et al. New somatic TERT promoter variants enhance the Telomerase activity in Glioblastoma. Acta Neuropathol. Commun. 2020, 8, 145. [Google Scholar] [CrossRef]
- Wong, Q.H.; Li, K.K.; Wang, W.W.; Malta, T.M.; Noushmehr, H.; Grabovska, Y.; Jones, C.; Chan, A.K.; Kwan, J.S.; Huang, Q.J.; et al. Molecular landscape of IDH-mutant primary astrocytoma Grade IV/glioblastomas. Mod. Pathol. 2021, 34, 1245–1260. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Hata, N.; Akagi, Y.; Kuga, D.; Hatae, R.; Sangatsuda, Y.; Michiwaki, Y.; Amemiya, T.; Takigawa, K.; Funakoshi, Y.; et al. Molecular diagnosis of diffuse glioma using a chip-based digital PCR system to analyze IDH, TERT, and H3 mutations in the cerebrospinal fluid. J. Neurooncol. 2021, 152, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Koh, J.; Kim, S.-I.; Won, J.K.; Park, C.-K.; Choi, S.H.; Park, S.-H. The frequency and prognostic effect of TERT promoter mutation in diffuse gliomas. Acta Neuropathol. Commun. 2017, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.; Azevedo, A.; Esteves, S.; Marques, A.R.; Martins, C.; Costa, I.; Mafra, M.; Marques, J.M.B.; Roque, L.; Pojo, M. Clinical insights gained by refining the 2016 WHO classification of diffuse gliomas with: EGFR amplification, TERT mutations, PTEN deletion and MGMT methylation. BMC Cancer 2019, 19, 968. [Google Scholar] [CrossRef] [PubMed]
- Bertero, L.; Anfossi, V.; Osella-Abate, S.; Disanto, M.G.; Mantovani, C.; Zenga, F.; Rudà, R.; Garbossa, D.; Soffietti, R.; Ricardi, U.; et al. Pathological prognostic markers in central nervous system solitary fibrous tumour/hemangiopericytoma: Evidence from a small series. PLoS ONE 2018, 13, e0203570. [Google Scholar] [CrossRef]
- Miki, S.; Satomi, K.; Ohno, M.; Matsushita, Y.; Kitahara, M.; Miyakita, Y.; Takahashi, M.; Matsuda, M.; Ishikawa, E.; Matsumura, A.; et al. Highly sensitive detection of TERT promoter mutations in recurrent glioblastomas using digital PCR. Brain Tumor Pathol. 2020, 37, 154–158. [Google Scholar] [CrossRef]
- Pesenti, C.; Paganini, L.; Fontana, L.; Veniani, E.; Runza, L.; Ferrero, S.; Bosari, S.; Menghi, M.; Marfia, G.; Caroli, M.; et al. Mass spectrometry-based assay for the molecular diagnosis of glioma: Concomitant detection of chromosome 1p/19q co-deletion, and IDH1, IDH2, and TERT mutation status. Oncotarget 2017, 8, 57134–57148. [Google Scholar] [CrossRef][Green Version]
- Muralidharan, K.; Yekula, A.; Small, J.L.; Rosh, Z.S.; Kang, K.M.; Wang, L.; Lau, S.; Zhang, H.; Lee, H.; Bettegowda, C.; et al. TERT Promoter Mutation Analysis for Blood-Based Diagnosis and Monitoring of Gliomas. Clin. Cancer Res. 2021, 27, 169–178. [Google Scholar] [CrossRef]
- Ge, J.; Liu, M.Y.; Li, L.; Deng, Q.; Liu, F.; Luo, Y.; Wang, L.; Yao, G.; Zhu, D.; Lu, H.; et al. Detection of IDH1 and TERT promoter mutations with droplet digital PCR in diffuse gliomas. Int. J. Clin. Exp. Pathol. 2020, 13, 230–238. [Google Scholar]
- Higa, N.; Akahane, T.; Yokoyama, S.; Yonezawa, H.; Uchida, H.; Takajo, T.; Kirishima, M.; Hamada, T.; Matsuo, K.; Fujio, S.; et al. A tailored next-generation sequencing panel identified distinct subtypes of wild-type IDH and TERT promoter glioblastomas. Cancer Sci. 2020, 111, 3902–3911. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Wald, A.I.; Melan, M.A.; Roy, S.; Zhong, S.; Hamilton, R.L.; Lieberman, F.S.; Drappatz, J.; Amankulor, N.M.; Pollack, I.F.; et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro-Oncology 2016, 18, 379–387. [Google Scholar] [CrossRef]
- Sahm, F.; Schrimpf, D.; Jones, D.T.; Meyer, J.; Kratz, A.; Reuss, D.; Capper, D.; Koelsche, C.; Korshunov, A.; Wiestler, B.; et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 2016, 131, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Synhaeve, N.E.; Bent, M.J.V.D.; French, P.J.; Dinjens, W.N.M.; Atmodimedjo, P.N.; Kros, J.M.; Verdijk, R.; Dirven, C.M.F.; Dubbink, H.J. Clinical evaluation of a dedicated next generation sequencing panel for routine glioma diagnostics. Acta Neuropathol. Commun. 2018, 6, 126. [Google Scholar] [CrossRef]
- E Piccioni, D.; Achrol, A.S.; A Kiedrowski, L.; Banks, K.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef] [PubMed]
- Euskirchen, P.; Bielle, F.; Labreche, K.; Kloosterman, W.P.; Rosenberg, S.; Daniau, M.; Schmitt, C.; Masliah-Planchon, J.; Bourdeaut, F.; Dehais, C.; et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol. 2017, 134, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Ozturk-Isik, E.; Cengiz, S.; Ozcan, A.; Yakicier, C.; Danyeli, A.E.; Pamir, M.N.; Özduman, K.; Dincer, A. Identification of IDH and TERTp mutation status using 1 H-MRS in 112 hemispheric diffuse gliomas. J. Magn. Reson. Imaging 2020, 51, 1799–1809. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, J.; Li, M.; Wang, H.; Lou, F.; Cao, S.; Lu, C. Comprehensive Molecular Characterization of Chinese Patients with Glioma by Extensive Next-Generation Sequencing Panel Analysis. Cancer Manag. Res. 2021, 13, 3573–3588. [Google Scholar] [CrossRef] [PubMed]
- Zacher, A.; Kaulich, K.; Stepanow, S.; Wolter, M.; Köhrer, K.; Felsberg, J.; Malzkorn, B.; Reifenberger, G. Molecular Diagnostics of Gliomas Using Next Generation Sequencing of a Glioma-Tailored Gene Panel. Brain Pathol. 2017, 27, 146–159. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Z.; Li, J.; Huang, T.; Wang, Y.; Chang, L.; Zheng, W.; Ma, Y.; Chen, F.; Gong, X.; et al. Genomic analysis of primary and recurrent gliomas reveals clinical outcome related molecular features. Sci. Rep. 2019, 9, 16058. [Google Scholar] [CrossRef]
- Petersen, J.K.; Boldt, H.B.; Sørensen, M.D.; Blach, S.; Dahlrot, R.H.; Hansen, S.; Burton, M.; Thomassen, M.; Kruse, T.; Poulsen, F.R.; et al. Targeted next-generation sequencing of adult gliomas for retrospective prognostic evaluation and up-front diagnostics. Neuropathol. Appl. Neurobiol. 2021, 47, 108–126. [Google Scholar] [CrossRef]
- Tian, H.; Wu, H.; Wu, G.; Xu, G. Noninvasive Prediction of TERT Promoter Mutations in High-Grade Glioma by Radiomics Analysis Based on Multiparameter MRI. Biomed. Res. Int. 2020, 2020, 3872314. [Google Scholar] [CrossRef]
- Jiang, C.; Kong, Z.; Zhang, Y.; Liu, S.; Liu, Z.; Chen, W.; Liu, P.; Liu, D.; Wang, Y.; Lyu, Y.; et al. Conventional magnetic resonance imaging-based radiomic signature predicts telomerase reverse transcriptase promoter mutation status in grade II and III gliomas. Neuroradiology 2020, 62, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kaiser, L.; Holzgreve, A.; Ruf, V.C.; Suchorska, B.; Wenter, V.; Quach, S.; Herms, J.; Bartenstein, P.; Tonn, J.-C.; et al. Prediction of TERTp-mutation status in IDH-wildtype high-grade gliomas using pre-treatment dynamic [18F] FET PET radiomics. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 4415–4425. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, B.; Zhang, S.; Cheng, J.; Liu, X.; Wang, W.; Dong, Y.; Zhang, L.; Mo, X.; Chen, Q.; et al. Quantitative MRI-based radiomics for noninvasively predicting molecular subtypes and survival in glioma patients. NPJ Precis. Oncol. 2021, 5, 72. [Google Scholar] [CrossRef]
- Zhang, H.-W.; Lyu, G.-W.; He, W.-J.; Lei, Y.; Lin, F.; Wang, M.-Z.; Zhang, H.; Liang, L.-H.; Feng, Y.-N.; Yang, J.-H. DSC and DCE Histogram Analyses of Glioma Biomarkers, Including IDH, MGMT, and TERT, on Differentiation and Survival. Acad. Radiol. 2020, 27, e263–e271. [Google Scholar] [CrossRef] [PubMed]
- Fukuma, R.; Yanagisawa, T.; Kinoshita, M.; Shinozaki, T.; Arita, H.; Kawaguchi, A.; Takahashi, M.; Narita, Y.; Terakawa, Y.; Tsuyuguchi, N.; et al. Prediction of IDH and TERT promoter mutations in low-grade glioma from magnetic resonance images using a convolutional neural network. Sci. Rep. 2019, 9, 20311. [Google Scholar] [CrossRef] [PubMed]
- Ivanidze, J.; Lum, M.; Pisapia, D.; Magge, R.; Ramakrishna, R.; Kovanlikaya, I.; Fine, H.A.; Chiang, G.C. MRI Features Associated with TERT Promoter Mutation Status in Glioblastoma. J. Neuroimaging 2019, 29, 357–363. [Google Scholar] [CrossRef]
- Wei, R.L.; Wei, X.T. Advanced Diagnosis of Glioma by Using Emerging Magnetic Resonance Sequences. Front. Oncol. 2021, 11, 694498. [Google Scholar] [CrossRef]
- Williams, C.; Pontén, F.; Moberg, C.; Söderkvist, P.; Uhlén, M.; Pontén, J.; Sitbon, G.; Lundeberg, J. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am. J. Pathol. 1999, 155, 1467–1471. [Google Scholar] [CrossRef]
- Fontanilles, M.; Marguet, F.; Beaussire, L.; Magne, N.; Pépin, L.-F.; Alexandru, C.; Tennevet, I.; Hanzen, C.; Langlois, O.; Jardin, F.; et al. Cell-free DNA and circulating TERT promoter mutation for disease monitoring in newly-diagnosed glioblastoma. Acta Neuropathol. Commun. 2020, 8, 179. [Google Scholar] [CrossRef]
- Juratli, T.A.; Stasik, S.; Zolal, A.; Schuster, C.; Richter, S.; Daubner, D.; Juratli, M.A.; Thowe, R.T.; Hennig, S.; Makina, M.; et al. TERT Promoter Mutation Detection in Cell-Free Tumor-Derived DNA in Patients with IDH Wild-Type Glioblastomas: A Pilot Prospective Study. Clin. Cancer Res. 2018, 24, 5282–5291. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Shah, R.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Avogbe, P.H.; Manel, A.; Vian, E.; Durand, G.; Forey, N.; Voegele, C.; Zvereva, M.; Hosen, I.; Meziani, S.; De Tilly, B.; et al. Urinary TERT promoter mutations as noninvasive biomarkers for the comprehensive detection of urothelial cancer. EBioMedicine 2019, 44, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Hosen, I.; Forey, N.; Durand, G.; Voegele, C.; Bilici, S.; Avogbe, P.H.; Delhomme, T.M.; Foll, M.; Manel, A.; Vian, E.; et al. Development of Sensitive Droplet Digital PCR Assays for Detecting Urinary TERT Promoter Mutations as Noninvasive Biomarkers for Detection of Urothelial Cancer. Cancers 2020, 12, 3541. [Google Scholar] [CrossRef]


{kind=link}
| Authors: | Arita et al. [19] | Heidenreich et al. [22] | Yuan et al. [32] | Arita et al. [33] | Pekmezci et al. [34] | Yang et al. [35] | Kim et al. [36] | You et al. [37] | Huang et al. [38] | |
|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis: | ||||||||||
| Diffuse astrocytoma | 19% | 29% | 33% | 20% | ||||||
| Anaplastic astrocytoma | 25% | 33% | 33% | 30% | 32% | 33% | ||||
| Astrocytoma | 39% | 24% | 22% | 7% | 11% | |||||
| Glioblastoma | 70% | 80% | 69% | 58% | 66% | 64% | 42% | 84% | ||
| Oligoastrocytoma | 36% | 38% | 49% | 54% | 54% | |||||
| Anaplastic oligoastrocytoma | 40% | 44% | 42% | 41% | ||||||
| Oligodendroglioma | 74% | 70% | 72% | 83% | 96% | 74% | 76% | 70% | ||
| Anaplastic oligodendroglioma | 74% | 74% | 67% | 100% | 53% | |||||
| Number of patients in the research | 546 | 303 | 3477 | 758 | 1208 | 377 | 67 | 684 | 204 | |
| Method, Patient Group | Type of Tumor, Number of Patients | Material of Tumor DNA | MAF, % | Primers on TERTp Sequence (forward and Reverse) | PCR Product Length, n.b. (If Specified) | Reference |
|---|---|---|---|---|---|---|
| Group 1: Pyrosequencing group 2: PCR and Sanger sequencing | Glioma (1) 242/304 (2) 127 | Frozen tissue samples | dnp * | Primers to amplify the region including both sites: 5′-TCCCTCGGGTTACCCCACAG-3′ and 5′-AAAGGAAGGGGAGGGGCTG-3′ (biotinylated at the 5′-end) The primer for pyrosequencing (5′-ACCCCGCCCCGTCCCGACCCC-3′) was constructed above C250T, so the two hotspots are analyzed in the same assay with the dispensation order 5′-GTCGTCCGCATGCCTC-3′ to pyrosequence 5′-TT/CCCGGGTCCCCGGCCCAGCCCCT/CTCCG-3′ (AQ PyroMark Q96 (version 2.5.7) analysis, applied to underlined positions) 5’-TCCCTCGGGTTACCCCACAG-3′ and 5′-AAAGGAAGGGGAGGGGCTG-3′ | 356 ** | [19] |
| PCR and Sanger sequencing | Gliomas, 325 | Frozen tissue | dnp | 5′-CCCACGTGCGCAGCAGGAC-3′ and 5′-CTCCCAGTGGATTCGCGGGC-3′ | 260 | [22] |
| Nested PCR and Sanger sequencing | Glioblastoma, 358/32 | FFPE samples | 20% | 1st PCR: 5′-GTCCTGCCCCTTCACCTT-3′ and 5′-GCACCTCGCGGTAGTGG-3′ Nested PCR: 5′-CCGTCCTGCCCCTTCACC-3′ and 5′-GGGCCGCGGAAAGGAAG-3′ For samples which were not amplified, used set 3: 5′-TTCCAGCTCCGCCTCCT-3′ and 5′-GCGCTGCCTGAAACTCG-3′ | 273 128 145 | [40] |
| Reverse transcription PCR (“RT-PCR”) and Sanger sequencing | Gliomas class II, III and IV (1) group 235 (2) group 897 Total control group 1090 | (1) Blood (2) Frozen tissue and FFPE samples | dnp | TERTp was amplified using 5′-GGCCGATTCGACCTCTCT-3′ (5′-GTCCTGCCCCTTCACCTT-3′ for FFPE samples) and 5′-AGCACCTCGCGGTAGTGG-3′ | 489 ** | [52] |
| PCR and Sanger sequencing | Gliomas of low malignancy, 237 | FFPE samples | dnp | 5′-GTCCTGCCCCTTCACCTT-3′ 5′-CAGCGCTGCCTGAAACTC-3′ | 163 | [53] |
| Nested PCR and Sanger sequencing | Grade III gliomas, 377 | FFPE samples | dnp | 1st PCR: 5′-GTCCTGCCCCTTCACCTT-3′ and 5′-GCACCTCGCGGTAGTGG-3′ Nested PCR: 5′-CCGTCCTGCCCCTTCACC-3′ 5′-GGGCCGCGGAAAGGAAG-3′ | 273 128 | [35] |
| Nested PCR and Sanger sequencing | Glioma, 887 | Tissue frozen in liquid nitrogen (80% of tumor cells) | dnp | 1st PCR: 5′-GTCCTGCCCCTTCACCTT-3′ 5′-GCACCTCGCGGTAGTGG-3′ Nested PCR: 5′-CCGTCCTGCCCCTTCACC-3′ 5′-GGGCCGCGGAAAGGAAG-3′ | 273 128 | [37] |
| Sanger sequencing ddPCR | Glioma, 9 | Frozen tissue and FFPE samples | C228T C250T 1.0% | 5′–TCCCTCGGGTTACCCCACAG–3′ and 5′–AAAGGAAGGGGAGGGGCTG–3′ | 356 ** | [48] |
| PCR and Sanger sequencing | Glioma, 147 | Frozen tissue and FFPE | C228T 10% | M13F: 5′-AGTGGATTCGCGGGCACAGA-3′ and M13R: 5′-CAGCGCTGCCTGAAACTC-3′ Primer M13: 5′-TGTAAAACGACGGCCAGT-3′ and 5′-CAGGAAACAGCTATGACC-3′ | 235 | [51] |
| PCR and Sanger sequencing | Glioma, 41/4 | FFPE samples | dnp | 5′-TCCCTCGGGTTACCCCACAG-3′ and 5′-AAAGGAAGGGGAGGGGCTG-3′ | 356 ** | [55] |
| Allele-specific quantitative PCR assay “GliomaDx” | 39 diffuse glioma | Frozen tissue | 0,1% MAF, or 0.2% tumor cells | 5′-CAGCGCTGCCTGAAACTC3′ and 5′-GTCCTGCCCCTTCACCTTC-3′ | 163 ** | [56] |
| Pyrosequencing | Glioma, 179 | Tumor tissue (>80% tumor cells) | dnp | 5′-GTCCTGCCCCTTCACCTT-3′ and 5′-GCACCTCGCGGTAGTGG-3′ (both biotinylated at the 5′ end) Primer for pyrosequencing: 5′-TGTAAAACGACGGCCAGT-3′ 5′-CAGGAAACAGCTATGACC-3′ (both biotinylated at the 5′ end) | 273 ** | [57] |
| PCR and Sanger sequencing | Glioma, 124 | FFPE samples (>50% of tumor cells) | dnp | 5′-AGCACCTCGCGGTAGTGG-3′ | dnp | [58] |
| PCR and Sanger sequencing | Primary CNS tumors, 301 | FFPE samples and blood (ctDNA and cfDNA) | dnp | 5′-GTCCTGCCCCTTCACCTTC-3′ and 5′-AGCACCTCGCGGTAGTGG-3′ | 274 | [59] |
| PCR and Sanger sequencing | Primary glioblastoma, 67 | FFPE samples | dnp | 5′-GTCCTGCCCCTTCACCTT-3′ and 5′-CAGCGCTGCCTGAAACTC-3′ | 163 ** | [60] |
| Chip-based dPCR system | Diffuse glioma, 34 | Samples of cerebrospinal fluid | dnp | dnp | dnp | [61] |
| Sanger sequencing | Glioma, 168 | FFPE samples | dnp | 5′-M13-GTAAAACGACGGCCAGTCACCCGTCCTGCCCCTTCACCTT-3′ (M13: 5′-GTAAAACGACGGCCAGT-3′ and 5′-GCACCTCGCGGTAGTGG-3′) | 300–310 | [62] |
| Sanger sequencing | Glioma, 200 | FFPE samples | dnp | 5′-CACCCGTCCTGCCCCTTCACCTT-3′ and 5′-GGCTTCCCACGTGCGCAGCAGGA-3′. | 193 ** | [42] |
| Sanger sequencing | Glioma, 444 | FFPE samples | dnp | 5′-GCACAGACGCCCAGGACCGCGCT-3′ and 5′-TTCCCACGTGCGCAGCAGGACGCA-3′ | 196 | [63] |
| RT-PCR | Glioma, 1208 | FFPE samples | dnp | 5′-AGTGGATTCGCGGGCACAGA-3′ and 5′-AGCACCTCGCGGTAGTGG-3′ | 346 | [36] |
| Sanger sequencing | Glioma, 15 | FFPE samples | dnp | 5′-CAGCGCTGCCTGAAACTC-3′ and 5′-GTCCTGCCCCTTCACCTT-3′ | 163 ** | [64] |
| Method | Detectable Markers | Material of Tumor DNA | Type of Tumor, Number of Patients | Limit of Detection | Sensitivity | Reference |
|---|---|---|---|---|---|---|
| ddPCR | TERTp mutations | Fresh-frozen samples and FFPE samples | 9 gliomas | 1% mutant DNA | dnp | [48] |
| MassARRAY Mass spectrometry | 1p/19q co-deletion mutations TERTp and IDH | FFPE samples (tumor cell content in all samples was at least 70%) | 50 gliomas | dnp * | dnp | [66] |
| ddPCR | TERTp mutations | Plasma cfDNA | 157 gliomas | 0.27% (C250T) and 0.42% (C228T). | 62.5% | [67] |
| IDH1-TERT-mutation ddPCR (IT-ddPCR) | Mutations TERTp and IDH1 | FFPE samples | 80 gliomas | 0.1% mutant DNA | dnp | [68] |
| NGS analysis | Analyzes 48 genes including TERT | FFPE samples | 106 gliomas | dnp | dnp | [69] |
| NGS analysis (GlioSeq) | Analyzes 68 genes including TERT | Frozen tissue and FFPE samples from surgically removed CNS tumors | 54 tumors of CNS | 3–5% mutant alleles for SNV and 1–5% for gene fusions. | 100% | [70] |
| NGS analysis | Analyzes 130 genes including TERT | FFPE samples | 150 CNS tumors | dnp | 99.0% | [71] |
| NGS analysis | ATRX, CIC, EGFR, FUBP1, NOTCH1, PTEN, H3F3A, IDH1/2, PIK3CA, and BRAF, amplification of EGFR, MDM, chromosome copy number changes 1p, 7, 10 and 19q | FFPE samples | 433 diffuse gliomas | dnp | dnp | [72] |
| NGS analysis Guardant360 test | 54, 68, 70 and 73 genes including TERT | ctDNA | 419 primary brain tumors | dnp | dnp | [73] |
| Sequencing 3rd Generation (Nanopore) | IDH1, IDH2, TP53, H3F3A, and the TERT | Fresh-frozen tumor tissue | 28 CNS tumors | dnp | dnp | [74] |
| Magnetic resonance imaging | TERTp and IDH mutations | Not applicable | 112 diffuse gliomas | dnp | 83.33% | [75] |
| Multigene (NGS) panel | TP53, TERT, IDH1, PTEN, ATRX, EGFR, and others | FFPE samples | 81 gliomas | dnp | dnp | [76] |
| Multigene (NGS) panel | ATRX, BRAF, CDKN2A, CDKN2B, CDKN2C), CIC, EGFR, FUBP1, H3F3A, IDH1, IDH2, NF1, NF2, NRAS, PIK3CA, PIK3R1, PTEN, RB1, TERTp,TP53 | 58 fresh-frozen samples and 80 FFPE samples | 121 diffuse gliomas | dnp | 100% | [77] |
| Multigene (NGS) panel | TERT, IDH1, TP53, PTEN, NOTCH1, EGFR, and others | Fresh-frozen samples | 81 gliomas, 303 glioblastomas, 509 lower-grade gliomas | dnp | dnp | [78] |
| Multigene (NGS) panel | ATRX, BRAF, CDKN2A, CDKN2B, CDKN2C), CIC, EGFR, FUBP1, H3F3A, IDH1, IDH2, NF1, NF2, NRAS, PIK3CA, PIK3R1, PTEN, RB1, TERTp, TP53 | FFPE samples | 345 diffuse gliomas | dnp | dnp | [79] |
| The Method | The Amount of Material Required | Number of Mutations Determined Simultaneously | Sensitivity of the Method | Method Specificity | Cost of Analysis | Technical Complexity | Time of Analysis | Pros, Cons and Limitations |
|---|---|---|---|---|---|---|---|---|
| ddPCR | Low (1–5 ng of DNA) | One | 1% | High | High | High | During the working day | +: Low amount of the DNA template; absolute quantification; resistance to PCR inhibitors −: High cost of assays; need for well-trained staff; higher contamination risk; limited and defined targets |
| Multiplexed dPCR | Low (1–5 ng of DNA) | Several | 1–2% | High | Medium | Medium | During the working day | +: Low amount of the DNA template; feasibility; absolute quantification; resistance to PCR inhibitors; user-friendly system −: Considerable cost of analyses; high contamination risk; limited and defined targets |
| dPCR | Low (1–5 ng of DNA) | One | 1% | High | Medium | Medium | During the working day | +: Low amount of the DNA template; feasibility; absolute quantification; resistance to PCR inhibitors; user-friendly system −: Considerable cost of analyses; single-target method; high contamination risk; limited and defined targets |
| NGS mutation panel | Moderate (5–10 ng of DNA) | A lot of | 1 to 5% | High | High | Very high | In a few days | +: Satisfactory estimate of MAF; availability of diverse commercial tests; possibility of detecting large diversity of targets including unpredictable mutations and allelic forms −: High cost of assays; time-consuming method |
| Nanopore sequencing | Very low (1 ng of DNA) | A lot of | 1% | High | High | High | Within 6 h | +: Possibility of detecting large diversity of targets including unpredictable mutations and allelic forms −: High error rates; high cost |
| Sanger sequencing | High (10 ng of DNA) | Several | 10 to 20% | Medium | Low | Low | Within 4–6 h | +: Low cost −: High error level; high Limit of Detection; quantitative tests are problematic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasanau, T.; Pisarev, E.; Kisil, O.; Nonoguchi, N.; Le Calvez-Kelm, F.; Zvereva, M. Detection of TERT Promoter Mutations as a Prognostic Biomarker in Gliomas: Methodology, Prospects, and Advances. Biomedicines 2022, 10, 728. https://doi.org/10.3390/biomedicines10030728
Hasanau T, Pisarev E, Kisil O, Nonoguchi N, Le Calvez-Kelm F, Zvereva M. Detection of TERT Promoter Mutations as a Prognostic Biomarker in Gliomas: Methodology, Prospects, and Advances. Biomedicines. 2022; 10(3):728. https://doi.org/10.3390/biomedicines10030728
Chicago/Turabian StyleHasanau, Tsimur, Eduard Pisarev, Olga Kisil, Naosuke Nonoguchi, Florence Le Calvez-Kelm, and Maria Zvereva. 2022. "Detection of TERT Promoter Mutations as a Prognostic Biomarker in Gliomas: Methodology, Prospects, and Advances" Biomedicines 10, no. 3: 728. https://doi.org/10.3390/biomedicines10030728
APA StyleHasanau, T., Pisarev, E., Kisil, O., Nonoguchi, N., Le Calvez-Kelm, F., & Zvereva, M. (2022). Detection of TERT Promoter Mutations as a Prognostic Biomarker in Gliomas: Methodology, Prospects, and Advances. Biomedicines, 10(3), 728. https://doi.org/10.3390/biomedicines10030728