Abstract
Immunologic and neuroinflammatory pathways have been found to play a major role in the pathogenesis of many neurological disorders such as epilepsy, proposing the use of novel therapeutic strategies. In the era of personalized medicine and in the face of the exhaustion of anti-seizure therapeutic resources, it is worth looking at the current or future possibilities that neuroimmunomodulator or anti-inflammatory therapy can offer us in the management of patients with epilepsy. For this reason, we performed a narrative review on the recent advances on the basic epileptogenic mechanisms related to the activation of immunity or neuroinflammation with special attention to current and future opportunities for novel treatments in epilepsy. Neuroinflammation can be considered a universal phenomenon and occurs in structural, infectious, post-traumatic, autoimmune, or even genetically based epilepsies. The emerging research developed in recent years has allowed us to identify the main molecular pathways involved in these processes. These molecular pathways could constitute future therapeutic targets for epilepsy. Different drugs current or in development have demonstrated their capacity to inhibit or modulate molecular pathways involved in the immunologic or neuroinflammatory mechanisms described in epilepsy. Some of them should be tested in the future as possible antiepileptic drugs.
1. Introduction
The immunologic and/or neuroinflammatory substrate has been found to play a role in the pathogenesis of many neurological disorders. In some conditions, this is considered to be a key element (vg demyelinating diseases of the central nervous system (CNS) [1]), while in others play a secondary role as an adjunct to other mechanisms (vg neurodegenerative diseases [2,3], or other diseases of a genetic or structural basis [4]). The evidence of this transversal involvement of neuro-inflammation and/or immunity in the pathogenic processes of neurological conditions is constantly increasing thanks to major advances in basic research, and epilepsy does not escape from this paradigm.
However, in the case of epilepsy, the classic debate has focused on elucidating whether the underlying immunitary or neuroinflammatory mechanisms are actually the cause or consequence of epileptic seizures [5]. It is incontrovertible that certain inflammatory-based pathological conditions are associated with an increased risk of developing epileptic seizures. Indeed, in most autoimmune diseases, there is a five-fold increased risk of epilepsy in children and a four-fold increased risk in non-elderly adults (aged < 65) [6]. Classic epidemiological studies labeled a large percentage of epilepsies as idiopathic or of unknown origin [7]. However, nowadays, a significant subgroup of these epilepsies is known to have an immune-mediated origin, to the point that the International League Against Epilepsy (ILAE) classification, in its latest update, recognizes immune-mediated epilepsy as one specific etiological category [8].
On the other hand, several studies have shown that epileptic seizures can generate neuroinflammatory activation that, in turn, could be involved in the progression of epileptogenesis [9]. A recent study showed that patients with epilepsy have abnormal levels of plasma inflammatory and neurotrophic markers independent of the underlying etiology, suggesting that such biomarkers may be rather a consequence than a cause of epilepsy [10]. Finally, different models of chemically and electrically induced seizures show upregulation of genes expressed in inflammatory cascades, as seen in patients [11].
Based on this bidirectional relationship between immunity and epilepsy, this review wants to focus on the available evidence of the possibility that biomarkers of inflammation have pathogenic value and, therefore, can become therapeutic targets for epilepsy [12].
2. Methods
We carried out a literature search through a Pubmed-Medline (https://pubmed.ncbi.nlm.nih.gov/; last access on 18 February 2022) using as keywords: “Epilepsy”, “Epileptogenesis”, “Brain innate immunity”, “Adaptive immunity”, “Neuroinflammation”, “Therapy” and “Antiepileptogenic drugs”. This search was extended with the bibliographic references found in the selected articles. Following the analysis of these articles, we carry out a narrative review of the state of the matter, including our most recent knowledge related to the following 3 items:
- Types of immunity and its relationship with the central nervous system (CNS);
- The mechanisms of immunomediated epileptogenesis;
- The epileptic disorders related to immunity;
- The immunomodulatory and anti-neuroinflammatory treatments of epilepsy.
3. Results
3.1. Types of Immunity and Its Relationship with the Central Nervous System (CNS)
Two main components of the immune system have been characterized, namely the innate (non-specific) immune system and the adaptive (specific) immune system. Their functions and mechanisms are closely linked, and both work synergically against germs or other harmful substances that occasionally may trigger a self-directed immune response against CNS antigens.
The innate immune system provides a general first-line defense in a non-specific manner, mainly through natural killer cells and phagocytes. Their equivalent in the CNS is the microglia [13].
Conversely, the adaptive immune system is considered a second-line defense, driving a targeted immune response mainly through antigen-specific antibodies.
In the last two decades, a better characterization of the role that immunity plays in the different morbid processes that affect CNS disorders has constituted one of the most exciting fields of research in neurology [14,15]. The major focus of research has been conditioned by the discovery of multiple autoantibodies directed against specific CNS antigens that have been proposed as diagnostic markers of these diseases. The target antigens of these antibodies basically include intracellular proteins (cytoplasmic or nuclear enzymes and proteins that act as RNA ligands) and membrane proteins (ion channels and neurotransmitter receptors) [15].
Therefore, it is well known that any immune pathology of the CNS requires the collaboration of a broad spectrum of immune and inflammatory systems [16].
Innate immunity can originate both at the peripheral level (outside the blood-brain barrier (BBB)) or at the central level (central innate immunity). On the other hand, adaptive immunity is always peripheral and requires a disruption of the BBB to exert its effect on the CNS.
3.1.1. Peripheral Immunity (Innate/Adaptive)
The peripheral innate immune response is responsible for the elimination of any challenge in a non-specific manner [17,18]. Its main effectors are neutrophils, monocytes/macrophages, dendritic cells, and natural killer (NK) cells. In addition, native CD4+ T cells are the starting point for the transition toward a specific immune response of the adaptive system, and depending on the inflammatory molecules found in their microenvironment, they can further polarize into various cell lines:
- T helper cells (Th cells): include three cell subtypes: Th1 cells, defined by the expression of lineage cytokine interferon (IFN)-γ, required for the control of intracellular viruses and bacteria; Th2 cells, defined by the expression of lineage cytokines interleukin (IL)-4/IL-5/IL-13 and the master transcription factor GATA3, which orchestrate the immune reaction against parasites; and Th17 cells, defined by the expression of lineage cytokines IL-17/IL-22, important for the immune response against certain extracellular bacteria and fungi., although they also play a major role in the development of autoimmunity.
- Regulatory T cells (Treg cells): Its main biological function consists of the suppression of self-reactive cells at the peripheral level.
Differentiation in either cell line is determined by molecular factors produced by antigen-presenting cells and B cells [18,19].
The adaptive immune response is specific to the pathogen presented by antigen-presenting cells, which leads to the clonal expansion of T and B lymphocytes. Each clone that originates from the original T or B lymphocyte has the same antigen receptor as the original and is targeted against the same pathogen or endogenous antigens in case of autoimmunity. In this regard, a field of research has emerged related to the existence of pathogenic autoantibodies against neuronal antigens that cause a broad spectrum of clinical phenotypes by affecting fundamental brain functions [20].
Most of these diseases are the result of an autoimmunization that occurs in the peripheral lymphoid tissue, and one of the most paradigmatic examples is paraneoplastic neurological syndromes (PNS) [21].
Traditionally, CNS has been considered an immuno-privileged organ due to the existence of a cellular barrier that surrounds the brain, mainly composed of endothelial cells, pericytes and astrocytes called the blood-brain barrier (BBB), which constitutes a defensive bastion against both cellular and molecular elements from the bloodstream [22].
For this reason, for all the cellular and molecular elements described in peripheral immunity to act in the CNS, it is necessary that there is a dysfunction of the BBB. This can happen in some circumstances, such as aging or metabolic or infectious insults [23] (Figure 1).
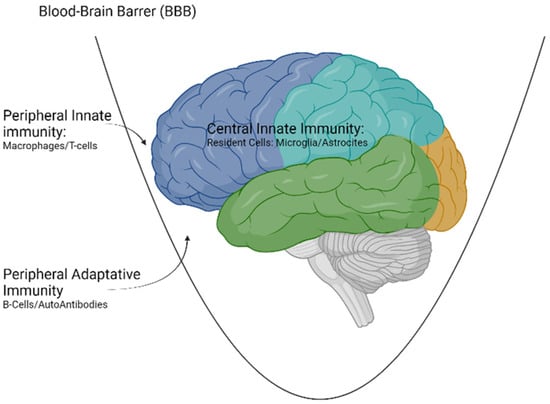
Figure 1.
Types of immunity acting at the level of the CNS. Created with www.Biorender.com (last access on 15 March 2022).
3.1.2. Central Innate Immunity of the Brain
In addition to the peripheral immunity described, an intrinsic immunity originated in the CNS itself has been identified, termed central innate immunity [23], that comprise the following CNS resident immune cells:
- Meningeal macrophages, located in the vicinity of the BBB. They play a main role in immunosurveillance and subsequent presentation of antigens to CD4+ T cells.
- Macrophages near the choroid plexus. Its basic function is also related to immunosurveillance tasks.
- Macrophages of the perivascular space. They are considered a part of the BBB and participate mostly in immunosurveillance and in the recruitment of circulating leukocytes.
- Parenchymal cells: Microglia and astrocytes. They are considered the key elements of central innate immunity but also participate in physiological processes regarding brain development and synaptic plasticity.
Microglia are considered the key activator of this type of immunity. Its function includes the monitoring and detection of disturbances in the brain microenvironment and contributes to restoring homeostasis. In addition to their function as the true “macrophages of the brain”, recent studies have shown that they are also critical for ensuring normal brain development and for tissue repair after a noxious stimulus [24]. Microglia recognizes this aggression and respond developing a phenomenon termed “microglial activation”, a common process in various pathologies including neuropathic pain, neurodegeneration, and traumatic brain injury [25].
The main recognition systems to these noxious stimuli used by the microglia are called innate immune receptors. Among them, the most important are the Toll-like receptor system (TLR), nod-like receptor system (NLR), and inflammasome-associated nucleotide-binding domain, leucine-rich repeat proteins.
All three are membrane-bound molecular chains that are activated after the recognition and binding with peripheral inflammation effectors, which are the “alarm” molecules, fundamentally: DAMPS (danger-associated molecular pattern) [26] as molecules generated by a heterogeneous group of insults both centrally (traumatic brain injury, cerebral ischemia) and peripherally (radiation, metabolic syndrome, aging [27]); and PAMPS (pathogens-associated molecular pattern) [28], small molecular fragments derived from different microorganisms, including HIV or SARS-CoV-2 [29].
After the binding of these innate immune receptors with their peripheral “alarm” ligand, the result will be a subsequent activation of microglia into two states:
- M1 state or proinflammatory state, which converts microglia into a secretory cell of elements such as cytokines (IL-1β, IL6, TNFα), chemokines (CCL2), and other products such as ROS (reactive oxygen species), NO (nitric oxide), or glutamate, responsible of cell destruction processes of inflammatory origin, also known as pyroptosis [30,31].
- M2 state or alternative activation, which has the opposite effect, resulting in secretion of anti-inflammatory or neurotrophic factors [32].
Peripheral tissue damage activators can, if they can pass through a dysfunctional BBB, induce conversion to M1 status. The outline of the processes described can be seen in Figure 2.
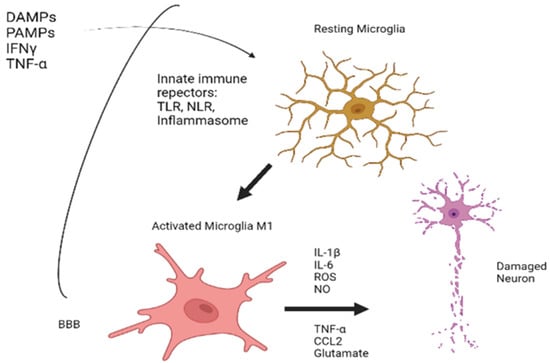
Figure 2.
An outline of the processes underlying the activation of microglia. Created with www.Biorender.com (last access on 15 March 2022).
Astrocytes also exert a key role in central innate immunity under tighter regulation of microglia [33]. It is mainly involved in the control and regulation of immune cells crossing through the BBB.
Under pathological conditions, activated microglia release factors that activate several astrocytic intracellular signaling pathways, such as:
- Mammalian target of rapamicin (mTOR) pathway;
- Nuclear factor k-B (NF-κB) pathway;
- Janus kinase (JAK)-signal transducer;
- Activator of transcription (STAT) pathway;
- Mitogen-activated protein kinase (MAPK) pathway [13,34]; and
- Purinergic signaling.
Figure 3 summarizes schematically some of the main intracellular molecular pathways leading to epileptogenesis.
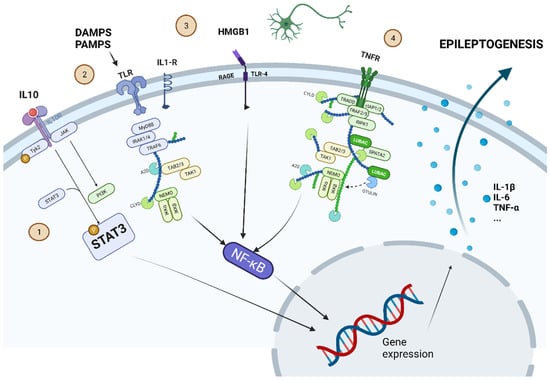
Figure 3.
Some of the most important intracellular molecular pathways leading to epileptogenesis are shown. 1. -JAK-STAT pathway: proinflammatory cytokines (such as IL-10) set this pathway in motion. The phosphorylated STAT3 protein can alter gene expression to induce secretion of proinflammatory cytokines (IL-1β, IL-6, or TNF-α). 2. -TLR pathway can be activated by DAMPS or PAMPS. 3. -HMGB1 is produced by cell damage and activates the pathway through its interaction on RAGE-TLR4. 4. -TNFR can be activated by the release of TNF-α. The pathways activated by TLR, RAGE-TLR4, and TNFR converge in the activation of NF-κβ, which also modifies gene expression to induce the secretion of proinflammatory cytokines. Created with www.Biorender.com (last access on 15 March 2022).
Subsequently, reactive astrocytes secrete factors that promote changes in BBB permeability, resulting in the recruitment of immune cells into the brain parenchyma, as well as several cytokines, amplifying the initial innate immune response and leading to neuroinflammation. Thus, constant communication (a dialogue) between microglia and astrocytes is a key element in the maintenance of the innate immune response [35].
3.2. Mechanisms of Immunomediated Epileptogenesis
3.2.1. Epileptogenesis Mediated by Peripheral Immunity
The epileptogenicity of the molecules arising from the activation of peripheral innate immunity remains a matter of debate [36].
Recent research studies have shown elevated plasmatic levels of these molecules in patients with epilepsy. Particularly, a recent systematic review showed increased levels of IL-1ra, IL-1, IL-6, and CXCL8/IL-8 in several different epilepsy syndromes independently of the underlying etiology [10,37].
In the case of post-traumatic epilepsy, many cytokines are released after severe brain injury for days, accompanied by an activation of ion channels and modifications of receptors associated with neuronal excitation and inhibition. Therefore, microglia and peripheral immunity are activated during hours or days, while plasmatic inflammation persists for weeks and coincides with neuronal loss. Subsequently, mossy fibers sprouting occurs in the hippocampus contributing to increase neuronal excitability [38].
Our understanding of the mechanisms of epileptogenesis related to peripheral adaptive immunity has undergone a radical change thanks to the description and characterization of a new group of autoimmune-based neurological pathologies that has led to the term autoimmune-based epilepsy [15].
Among epileptic disorders related to autoimmunity, it is important to distinguish between immune-mediated encephalitis (especially autoimmune limbic encephalitis (ALE)) and autoimmune-associated epilepsy (AAE), a chronic epileptic syndrome with the capacity for spontaneous recurrence of seizures in the long term [39]. The relationship between ALE and the risk of subsequently developing chronic epilepsy remains unclear and is considered to be mainly related to the nature of the associated neuronal antibody [40]:
- ALE with Autoantibodies against surface antigens: Infrequently associated with oncological pathology and with suitable response to immunomodulatory treatments [41]. Autoantibodies against surface antigens have an epileptogenic capacity per se, but the risk of developing AAE is lower due to the reversible effect of these antibodies after their removal.
- ALE with Autoantibodies against intracytoplasmic antigens: Includes classic paraneoplastic syndromes related to onconeural antibodies. Related AAE is characterized by its refractoriness to medical treatments [42,43]. Table 1 summarizes the characteristics of the main paraneoplastic syndromes with antibodies against intracellular antigens.
 Table 1. Antibodies directed against intracellular antigens.
Table 1. Antibodies directed against intracellular antigens.
AAE is increasingly recognized as a clinical entity with its own personality, thanks to the discovery of neural autoantibodies in patients with epilepsies of unknown origin. It is a field in evolution, and the percentage of patients with epilepsy of unknown origin in which an autoantibody can be identified is still modest [56].
Finally, although ALE is often associated with specific anti-neuronal antibodies, it is widely known that those targeting intracellular antigens might be merely biomarkers of the disease, sustained by their inability to reproduce the clinical phenotype in animal models and the prominent T cell-mediated nature of the immune response, which is more commonly associated with the development of AAE following ALE due to neuronal loss and gliosis [26].
3.2.2. Epileptogenesis and Brain Innate Immunity
Recent experimental studies and pathological analyses of brain tissue samples from patients with refractory epilepsy suggest that activation of microglia plays a key role in the etiopathology of epilepsy and seizure disorders [57].
Although the increase in biomarkers of neuroinflammation in patients with epilepsy has been known for a long time, some authors argued that they are a consequence of seizures, while others defended a primary role in epileptogenesis [36].
The biomarkers that have experimental evidence in animal models for modifying neuronal excitability are:
- IL-1 Receptor (IL-R1)/Toll-like Receptor (TLR) [58]: Preclinical investigations in experimental models using pharmacological and genetic tools have identified a significant contribution of interleukin-1 (IL-1) type 1 receptor/Toll-like receptor (IL-1R/TLR) signaling seizure activity. This signal can be activated by ligands associated with infections (PAMPS) or by endogenous molecules, such as proinflammatory cytokines (e.g., IL-1beta) or DAMPS (e.g., high-mobility group box 1 (HMGB1)) [59]. The activation of the IL-1β/IL-1β R axis is strictly linked to the secretion of the intracellular protein MyD88 after the activation of innate immune receptors (especially TLR) during pathogen recognition [31]. This activates an intracellular molecular cascade that, ultimately, can lead to an alteration of neuronal excitability.
- TNF-α: TNF-α affects seizure susceptibility in animal models in a dual pattern. The mechanism of action seems to be exerted through receptors TNFR1 (p55) or TNFR2 (p75) receptor signaling [60]. In general, TNFR1 has been reported to mediate the ictogenic effects of TNF-α, whereas TNFR2 mediates the neuroprotective actions of this cytokine.
- High-Mobility Proteins (HMGB1): These molecules are components of chromatin and are passively released from necrotic cells and actively released by cells that are exposed to deep stress [61]. Recent studies have described models of epilepsy induced by bicuculline and kainic acid that highlight the nature of HMGB1-TLR4 interactions [62], as well as its role in epileptic recurrence, emphasizing the role of immune-related molecules in epileptogenesis [63].
- Cyclooxygenase-2 (COX-2): COX-2 is an enzyme synthesizing prostaglandin (PGs) that has also received attention due to its possible involvement in seizure generation [64]. It seems that the presence of COX-2 facilitates the recurrence of seizures in the hippocampus and may upregulate P-Glycoprotein at the BBB, causing AED resistance [65].
In addition to these molecular pathways that have direct evidence, we must not forget that all the astrocytic intracellular signaling pathways outlined in the section dedicated to the basic mechanisms of central immunity are also indirectly related to mechanisms of epileptogenesis.
Finally, both in status epilepticus and in acute symptomatic seizures, there is activation of microglia and astrocytes. Interestingly, experimental evidence found a release of intraparenchymal inflammatory markers such as IL-1β, IL-6, and TNF-α in the hippocampus or neocortex [66].
Table 2 summarizes the main molecular mechanisms related to immune-mediated epileptogenesis at both the peripheral and central immunity levels.
Table 2.
Molecular mechanisms underlying immune-mediated epileptogenesis.
3.3. The Epileptic Disorders Related to Immunity
In the previous sections, we have described the main immunological and neuroinflammatory substrates that can be identified in epileptic disorders. It is important to recognize that these substrates have two features:
- Universality. Most epileptic disorders, regardless of their etiology (genetic, structural, autoimmune, infectious, traumatic, etc.), involve a greater or lesser extent the processes described. This universality suggests that these substrates may become valid therapeutic targets regardless of the etiology.
- Not compartmentalized. We will hardly find a pathology that exclusively affects a type of immunity, and a synergic collaboration between the different immune systems is presumed.
Despite these intrinsic features, and mainly considering the therapeutic orientation, it is interesting to establish a classification of epileptic disorders according to the predominant associated type of immunity.
In this section, we intend to establish a classification with a clinical orientation of the main epileptic disorders in which immune mechanisms play a relevant pathogenic role.
3.3.1. Epileptic Disorders Secondary to Systemic Autoimmune Disease
Primary Antiphospholipid syndrome (APS) or Hughes syndrome: APS is an immune-mediated disorder characterized by pregnancy morbidity and arterial or venous thrombotic events associated with persistent antiphospholipid antibodies (aPL), including lupus anticoagulant (LA), anticardiolipin (aCL) anti-β2-glycoprotein I antibodies (aB2GPI). Epilepsy has a prevalence of around 6% to 9% in patients with APS [67]. Different mechanisms of epileptogenesis have been proposed in this disease, including aPL-mediated inhibition of GABA receptors and immune-mediated neuronal damage [68].
Epilepsy-associated Systemic Lupus Erythematosus (SLE): SLE is an autoimmune disease in which several organs, tissues, and cells are damaged by adherence to autoantibodies and immune complexes. Anti-DNA antibodies are a subgroup of antinuclear antibodies that can bind to single-stranded DNA, double-stranded DNA, or both and are usually IgM or IgG antibodies. Several recent studies have shown the predominant role of central innate immunity in the pathogenesis of these encephalopathies [69].
Hashimoto’s encephalitis: A classical entity included in the steroid-responsive encephalopathy whose clinical expression is an epileptic encephalopathy with focal seizures and frequent secondary generalization, associated with anti-thyroid autoantibodies directed against alpha-enolase, dimethyl-argininase-I, and aldehyde-reductase-I. However, the pathogenic value of these autoantibodies in epileptogenesis is discussed [70].
Encephalopathy associated with gluten-related disease: Although the neurological manifestations of celiac disease usually affect other areas of the brain, it is true that people with celiac disease have an elevated risk of chronic focal epilepsy [71]. Neuropathology usually shows predominant T cell infiltration [72] and activation of humoral immunity with the synthesis of antigliadin antibodies.
Encephalopathy associated with vasculitis and Anti-Neutrophilic Cytoplasm antibodies (ANCA): It is a type of vasculitis that rarely produces epileptic seizures in the context of a steroid-responsive encephalopathy [70].
3.3.2. Autoimmune Diseases Primarily Involving CNS
ALE: Discussed in the section dedicated to adaptive immunity.
AAE: The most frequently detected neuronal antibody has been anti-GAD65, defining a refractory epileptic syndrome with its own personality [73,74]. The neurological anti-GAD65 syndrome is a heterogeneous disease, including stiff person spectrum disorders, cerebellar ataxia, AAE, and ALE in isolation or as part of an overlap syndrome. In general, it is a picture with poor response to immunotherapies, although, in a recent review, epileptic seizures were the manifestation with the best response to long-term immunomodulatory therapy [75].
Encephalitis with Anti-myelin oligodendrocyte glycoprotein (MOG) antibodies: Anti-MOG-associated disease is a recently identified autoimmune disorder that occurs in both adults and children as CNS demyelination. However, its clinical presentation includes, in more than 20% of cases, epileptic seizures, which is why it should be mentioned in this section [76].
Rasmussen encephalitis (RE): RE is the prototype of an epileptic disease related to the activation of cerebral innate immunity. Neuropathological studies have provided evidence of a progressive immune-mediated process of neuronal damage with prominent inflammation dominated by T cells, microglial activation, microglial nodules, and astrogliosis [77]. The natural history of RE is well known, with progressive cognitive impairment, brain hemi-atrophy, and continuous partial epilepsy [78].
ALE due to pharmacological inhibition of immune checkpoints: The development of oncology immunotherapy has led to the emergence of a new category of neurological immune-related adverse events secondary to the pharmacological blockade of immune checkpoints. The clinical expression is very heterogeneous, but a relevant percentage of these patients develop LE. The pathological substrate of these patients is infiltration of T cells as well as activation of microglia, consistent with a predominance of central innate immunity [79].
In a recent retrospective series that included 63 patients with immune checkpoint inhibitors-related neurologic autoimmunity, 21 (33%) patients whose clinical manifestations included epileptic seizures were described, in most cases, related to LE [80]. A total of 77% of these patients had detectable neural-specific autoantibodies, which implies synergic collaboration between the different immune systems. The drugs most frequently implicated in the literature are nivolumab [81,82,83] and pembrolizumab [84,85].
3.3.3. Epilepsies Secondary to Structural Etiologies with a Predominant Role of Autoimmunity
Hippocampal Sclerosis (HS): In human epilepsy surgical resections as well as in animal models, involvement of the adaptive immune system was observed in this type of epilepsy. T cell numbers (CD3+ as well as CD8+) are significantly elevated in HS compared to healthy controls [86]. Secondly, T cell numbers in HS correlated with the degree of neuronal loss.
mTORpathies (structural pathologies secondary to alteration of the mTOR pathway):
- Tuberous Sclerosis Complex (TSC): It is a genetic disease with a predisposition to the development of structural alterations of the cerebral cortex called tubers, as well as neoplasms such as subependymal giant cell astrocytoma (SEGA). The pathogenic mechanism is related to central innate immunity abnormalities, mainly mutations leading to a hyperactivation of the mTOR pathway. However, inhibition of this system can be achieved pharmacologically through mTOR inhibitor drugs such as everolimus. This is the first of the genetically based etiologies of which, today, we have a personalized therapeutic strategy that acts directly on the basic mechanisms of epileptogenesis [87].
- Focal Cortical Dysplasia (FCD): Some FCD produces an activation of cerebral innate immunity, mainly microglial activation. Although their exact pathogenesis remains poorly understood, somatic mutations in the mTOR pathway have been found in FCD type IIb [88].
3.4. The Immunomodulatory and/or Anti-Inflammatory Treatments in Epilepsy
All the aforementioned evidence opens the door to the emergence of new antiepileptic therapeutic strategies that surpass the classic paradigm of anti-seizure drugs and inaugurates the field of anti-epileptogenic treatment. Some of them have already been used in the history of epilepsy thanks to the serendipity, such as steroids or adrenocorticotropic hormone (ACTH) for certain catastrophic childhood epilepsies such as West syndrome [89].
Table 3 summarizes the main therapeutic lines according to the degree of development in clinical research.
Table 3.
Main therapeutic lines according to the degree of development in clinical research. Those drugs that have some study related to epilepsy are marked with an asterisk (*).
3.4.1. Established Treatments Used in Clinical Practice
Corticotherapy: Steroid therapy has a powerful anti-neuroinflammatory effect due to its genomic effects at transcriptional and post-transcriptional levels, primary exerted on the molecular pathways that converge into the nuclear factor-κβ (NF-κβ) [92], but also prevents microglial activation. It is currently the first-line therapeutic resource in cases of acute onset in the etiologies encompassed within what has been called steroid-responsive encephalopathies (SREAT) such as Hashimoto’s encephalitis [90], paraneoplastic LE [91], lupic encephalopathy, and vasculitis-associated encephalopathy with anti-neutrophilic cytoplasm (ANCA) antibodies [93].
Immunoglobulins IV (IVIg)/Plasmapheresis (PLEX): IgIV and PLEX are generally used as a supplement to steroids in the event of acute exposure, but also as maintenance therapy in the event of relapse or chronic conditions. In both types of treatments, the mechanism of action consists in their ability to block or remove pathogenic elements from the circulation (vg autoantibodies or immunocomplexes), but also modulating the proliferation of B cells and plasma cells and production of cytokines [95].
Immunosuppressors:
- Rituximab: Anti-CD20 monoclonal antibody used in cases of involvement of adaptive humoral immunity. It is widely used due to its suitable tolerability and safety. Rituximab can be used as both a second-line agent for acute immunosuppression and as a long-term immunosuppressant for recurrent cases. Rituximab, however, does not deplete antibody-secreting cells, which are typically CD20-negative. Therefore, rituximab may work by deleting the antigen-specific memory B-cell populations that secrete the pathogenic antibodies [43].
- Azathioprine: It is an antagonist of the synthesis of purines and, consequently, of the production of DNA/RNA for the proliferation of white blood cells. Azathioprine usually takes 6 to 8 months to be effective, so it is often needed along with the progressive and concomitant reduction in oral steroids [15].
- Cyclophosphamide: It induces cell apoptosis through irreversible alterations of DNA. It is generally reserved for severe cases refractory to other immunotherapies due to the strong immunosuppressive effect and increased risk of adverse events including nausea and vomiting, alopecia, hemorrhagic cystitis, agranulocytosis, infertility, and increased risk of tumors [96].
- Mycophenolate mofetil: This drug depletes guanosine nucleotides preferentially in T and B lymphocytes, and inhibits their proliferation, thereby suppressing cell-mediated immune responses and antibody synthesis [96].
- mTOR pathway-modulating drugs: This line of research has produced the first drugs approved for the treatment of some epileptic disorders of neuroinflammatory basis, such as those included within the tuberous sclerosis complex. Everolimus has shown to be effective in the treatment of epilepsy related to tuberous sclerosis with level of evidence I [97]. Additionally, there is an ongoing clinical trial of everolimus in type II FCD (ClinicalTrials.gov Identifier: NCT03198949).
3.4.2. Future Therapeutic Strategies
Novel immunomodulatory strategies will be developed over the next few years due to a better understanding of the basic mechanism underlying the immunopathogenesis of epilepsy. Therefore, most of the following therapies could be proposed.
Among the drugs that modulate innate central immunity, we shall emphasize:
- TLR pathway inhibitor drugs: Research on selective TLR pathway blockade strategy (IL-1β/IL-1βRaxis) has generated some molecules that have shown antiepileptic efficacy in animal models. Among them, perhaps the most promising is resveratrol [115].
- HMGB1 inhibitor drugs: Emerging evidence suggest that HMGB1 may contribute to the pathogenesis of epilepsy [62] since glycyrrhizin, an HMGB1 inhibitor, exhibits neuroprotective and antiepileptic effects in different animal models of epilepsy [116,118]. However, this drug has not been assessed in clinical trials for epilepsy.
- TNF-α inhibitor drugs: There are four TNF-α inhibitors approved as treatments for ulcerative colitis and/or Crohn’s disease: infliximab, adalimumab, golimumab, and certolizumab pegol [98]. The mechanism of action is based on both the neutralization of TNF-α bioactivity and the induction of apoptosis of TNF-expressing mononuclear cells [99]. Adalimumab is a fully human IgG1 monoclonal antibody that specifically binds to TNF-α, which showed to be reduced seizures and functional impairment in patients with RE [100]. Indeed, there is an ongoing clinical trial trying to evaluate the benefit of adalimumab in patients with RE. (ClinicalTrials.gov Identifier: NCT04003922). In addition, infliximab was effective in a patient with relapsing polychondritis and LE [101]. Conversely, golimumab and certolizumab have not been tested for epilepsy.
- Drugs modulating T lymphocytes: A large experience has been gathered about these drugs regarding efficacy and safety due to their use as disease-modifying therapies in multiple sclerosis.
- Natalizumab is a humanized monoclonal anti-α4-integrin antibody approved for the treatment of multiple sclerosis. Integrins are heterodimeric proteins expressed on the cell surface of leukocytes that participate in a wide variety of functions, such as survival, growth, differentiation, migration, inflammatory responses, and tumor invasion, among others [126]. Natalizumab interferes with leukocyte migration across the BBB, which is mediated by interaction between α4-integrin and vascular cell adhesion molecule-1, resulting in a selective CNS immunosuppression due to lower recruitment of immune cells in the cerebral parenchyma [127]. The administration of anti–α4-integrin antibodies showed to reduce seizures in a mouse model of epilepsy [128]. However, a recent phase II clinical trial in refractory epilepsy did not meet the primary endpoint of decreasing seizures, although no adverse events were reported [102]. Further exploration of possible anti-inflammatory therapies for drug-resistant epilepsy is warranted [103]. Interestingly, some cases of RE have been successfully treated with natalizumab [104].
- Inebilizumab: this drug is a promising therapeutic monoclonal antibody against the B-cell surface antigen CD19 that has recently been shown to be safe and efficacious in the treatment of neuromyelitis optica spectrum disorder, another antibody-mediated disorder of the CNS [105]. Compared to rituximab, inebilizumab not only depletes CD20+ B cells but also CD20- plasmablasts and plasma cells, resulting in robust and sustained suppression of humoral immunity. There is an ongoing clinical trial phase II with inebilizumab for the treatment of limbic encephalitis with anti-NMDAR antibodies (ClinicalTrials.gov Identifier: NCT04372615).
- Cytokine-targeted therapies: Given the central role of cytokines in many epileptic disorders, these therapies are promising for patients with epilepsy [109]. Among them, we mainly distinguish anti-interleukin drugs (anakinra and tocilizumab) and anti-interferon-γ drugs (situximab and emapalumab).
- Anakinra, an anti-IL1 receptor, has shown antiepileptic properties in patients with super refractory epileptic status [110], RE [111], and especially in cases of febrile infection-related epilepsy syndrome (FIRES), for which it has been proposed as a first-line therapeutic alternative [112,113]. Additionally, anakinra has been shown to reduce seizures in animal models of anti-NMDAR encephalitis and lithium-pilocarpine-induced epilepsy.
- Tocilizumab, an anti-IL6 receptor, has been reported to be effective in FIRES refractory to anakinra [129]. Currently, there are no research reports with situximab or emapalumab for epilepsy.
- Anti-neonatal Fc receptor (FcRn) therapies: A novel treatment approach targets the neonatal Fc receptor (FcRn) of several immune cells. The primary function of FcRn is to prevent IgG and albumin from lysosomal degradation through the recycling and transcytosis of IgG, therefore, prolonging its half-life. Antagonism of this receptor causes IgG catabolism, resulting in reduced overall IgG and pathogenic autoantibody levels [106,107]. There is currently an ongoing phase II clinical trial with rozanolixizumab (ClinicalTrials.gov Identifier: NCT04875975), a high-affinity human neonatal FC receptor (IgG4P) monoclonal antibody (IgG4P), developed to reduce pathogenic IgG in autoimmune and alloimmune diseases, such as encephalitis with anti-LGI1 antibodies.
- Proteasome inhibitors: Drugs targeting long-lived plasma cells (LLPCs) are a promising treatment for antibody-mediated neurological conditions. Specifically, bortezomid is a selective inhibitor of the S26 protasome for which a possible efficacy has been suggested for the treatment of limbic encephalitis with anti-NMDA antibodies. A recent systematic review shows that more than 50% of the patients reported in the literature treated in this way showed improvement. In any case, specifically designed studies are necessary to evaluate this topic [114].
- Janus Kinase/Signal Transducer and Activator of Transcription (JAK-STAT) Inhibitors: Inhibitors of the JAK-STAT pathway are proposed as a future therapeutic target in neuroinflammatory-based processes since they modulate the intracellular signaling pathways of multiple cytokine receptors [120]. Among these molecules, we find AZD1480, tofacitinib, baricitinib, SAR317461, momelotinib, filgotinib, baricitinib, ruxolitinib, lestaurtinib, or pacritinib [130], although lestaurtinib has any promising studies on animal models of epilepsy [119].
- NF-κβ inhibitors: As mentioned before, steroids preferentially and indirectly act on the NF-κβ transcription complex, although other novel drugs may selectively block this relevant pathway of neuroinflammation. Among them, dimethyl-fumarate, fingolimod, or teriflunamide have been widely used in patients with multiple sclerosis [131]. Interestingly, promising results have been reported on animal models of epilepsy treated with dimethyl-fumarate and fingolimod [122,123]. Emerging experimental findings suggest that fingolimod exerts disease-modifying antiepileptic effects based on its anti-neuroinflammatory properties, potent neuroprotection, anti-gliotic effects, myelin protection, reduction in the mTOR signaling pathway, and activation of microglia and astrocytes. Thus, the antiepileptic efficacy of this drug seems to be supported by various mechanisms of action that converge on the modulation of innate brain immunity [117,122].
- MAPK pathway inhibitors: Several drugs perform a selective blockade of this pathway, such as SB203580 [121], macranthoin G, and PD-0325901 (a derivative of CI-1040). The inhibition of p38-MAPK by SB203580 may regulate epileptic activity by decreasing expression levels of adenosine A1 receptor (A1R) and the type 1 equilibrative nucleoside transporter (ENT1) in animal models [121,124].
- COX-2 inhibitor drugs: COX-2 inhibitors have sown anti-seizure properties in various acute and chronic models of epilepsy. Aspirin, naproxen, rofecoxib, or nimesulide protected mice from mortality caused by pentylenetetrazol-induced seizure (PTZ) [132].
- Purinergic signaling modulating drugs: Increasing evidence suggests purinergic signaling via extracellularly released ATP as shared pathological mechanisms across numerous brain diseases, including epilepsy. Once released, ATP activates specific purinergic receptors, such as the ionotropic P2X7 receptor (P2X7R). Suggesting the therapeutic potential of drugs targeting the P2X7R for epilepsy, P2X7R expression increases following status epilepticus and during epilepsy, and P2X7R antagonism modulates seizure severity and epilepsy development. JNJ-47965567, a selective P2X7R antagonist, has some evidence in animal models of focal epilepsy [125].
4. Conclusions
The avalanche of new evidence revealing the pathogenesis of neuroinflammatory and/or neuroimmunological mechanisms at the origin of many epileptic disorders has broadened the possibility of testing new undiscovered therapies. Several drugs have shown that they can inhibit or modulate the molecular pathways of these immune mechanisms. However, while some therapies such as everolimus have already demonstrated antiepileptic activity in humans, most novel therapies have only suggested their ability to improve seizures in in vitro or animal models, and further studies should assess their potential as antiepileptic drugs.
We find it particularly interesting to point out that these neuroinflammatory mechanisms play a transversal role in the pathogenesis of multiple neurological pathologies. (such as Alzheimer’s disease [2,133] or Parkinson’s disease [134]) and even in psychiatric diseases [135,136,137]. In this sense, a two-way relationship exists between epilepsy and depression and anxiety disorders [138], suggesting a common basis. From this perspective, it is not excluded that the search for new therapeutic strategies that act on the basic mechanisms of epilepsy also influence these comorbidities.
Future studies assessing the efficacy and safety of these therapies in patients with epilepsy are warranted.
Author Contributions
Conceptualization, M.J.A.-C., P.J.S.-C., G.E.-T. and N.L.C.-P.; methodology, M.J.A.-C., P.C.-G., G.G.-M. and M.M.-G.; writing—original draft preparation, M.J.A.-C., P.J.S.-C. and N.L.C.-P.; writing—review and editing, M.J.A.-C., N.L.C.-P. and P.J.S.-C.; supervision, P.J.S.-C.; funding acquisition, P.J.S.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Andalusian Network of Clinical and Translational Research in Neurology (Neuro-RECA) of the Consejería de Salud y Familias de la Junta de Andalucía (Code: RIC-0111-2019).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
References
- Nylander, A.; Hafler, D.A. Multiple sclerosis. J. Clin. Invest. 2012, 122. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Wesselingh, R.; Butzkueven, H.; Buzzard, K.; Tarlinton, D.; O’Brien, T.; Monif, M. Innate Immunity in the Central Nervous System: A Missing Piece of the Autoimmune Encephalitis Puzzle? Front. Immunol. 2019, 10, 2066. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, X.; Zhang, Y.; Liu, H.; Liao, J.; Shao, K.; Chu, Y.; Liu, G. Modulation of TSC-m, TOR signaling on immune cells in immunity and autoimmunity. J. Cell. Physiol. 2014, 229, 17–26. [Google Scholar] [PubMed]
- Pracucci, E.; Pillai, V.; Lamers, D.; Parra, R.; Landi, S. Neuroinflammation: A Signature or a Cause of Epilepsy? Int. J. Mol. Sci. 2021, 22, 6981. [Google Scholar] [CrossRef] [PubMed]
- Ong, M.-S.; Kohane, I.S.; Cai, T.; Gorman, M.P.; Mandl, K.D. Population-Level Evidence for an Autoimmune Etiology of Epilepsy. JAMA Neurol. 2014, 71, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.W.; Shorvon, S.D. Epidemiology of the epilepsies. J. Neurol. Neurosurg. Psychiatry 1996, 61, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshe, S.; Peltola, J.; Perez, E.R.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Yao, L. The role of inflammation in epileptogenesis. Acta Epileptol. 2020, 2, 15. [Google Scholar] [CrossRef]
- Alvim, M.K.M.; Morita-Sherman, M.E.; Yasuda, C.L.; Rocha, N.P.; Vieira Érica, L.; Pimentel-Silva, L.R.; Nogueira, M.H.; Barbosa, R.; Watanabe, N.; Coan, A.C.; et al. Inflammatory and neurotrophic factor plasma levels are related to epilepsy independently of etiology. Epilepsia 2021, 62, 2385–2394. [Google Scholar] [CrossRef]
- Kim, I.; Mlsna, L.M.; Yoon, S.; Le, B.; Yu, S.; Xu, D.; Koh, S. A postnatal peak in microglial development in the mouse hippocampus is correlated with heightened sensitivity to seizure triggers. Brain Behav. 2015, 5, e00403. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. The holy grail of epilepsy prevention: Preclinical approaches to antiepileptogenic treatments. Neuropharmacol. 2020, 167, 107605. [Google Scholar] [CrossRef] [PubMed]
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Liblau, R.; Becher, B. Innate and adaptive immune responses in the CNS. Lancet Neurol. 2015, 14, 945–955. [Google Scholar] [CrossRef]
- Sechi, E.; Flanagan, E.P. Antibody-Mediated Autoimmune Diseases of the CNS: Challenges and Approaches to Diagnosis and Management. Front. Neurol. 2021, 12. [Google Scholar] [CrossRef]
- Gendelman, H.E. Neural Immunity: Friend or Foe? J. Neuro. Virol. 2002, 8, 474–479. [Google Scholar] [CrossRef]
- Le Page, A.; Dupuis, G.; Frost, E.H.; Larbi, A.; Pawelec, G.; Witkowski, J.M.; Fulop, T. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp. Gerontol. 2018, 107, 59–66. [Google Scholar] [CrossRef]
- Huang, X.; Hussain, B.; Chang, J. Peripheral inflammation and blood–brain barrier disruption: Effects and mechanisms. CNS Neurosci. Ther. 2021, 27, 36–47. [Google Scholar] [CrossRef]
- Van Hamburg, J.P.; de Bruijn, M.J.W.; Ribeiro de Almeida, C.; van Zwam, M.; van Meurs, M.; de Haas, E.; Boon, L.; Samsom, J.N.; Hendrinks, R.W. Enforced expression of GATA3 allows differentiation of IL-17-producing cells, but constrains Th17-mediated pathology. Eur. J. Immunol. 2008, 38, 2573–2586. [Google Scholar] [CrossRef]
- Alexopoulos, H.; Dalakas, M.C. The immunobiology of autoimmune encephalitides. J. Autoimmun. 2019, 104, 102339. [Google Scholar] [CrossRef]
- Iorio, R.; Lennon, V.A. Neural antigen-specific autoimmune disorders. Immunol Rev. 2012, 248, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Melzer, N.; Meuth, S.G.; Wiendl, H. Neuron-directed autoimmunity in the central nervous system: Entities, mechanisms, diagnostic clues, and therapeutic options. Curr. Opin. Neurol. 2012, 25, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.M.; Yaqubi, M.; Ludwin, S.; Antel, J.P. Species differences in immune-mediated CNS tissue injury and repair: A (neuro)inflammatory topic. Glia 2020, 68, 811–829. [Google Scholar] [CrossRef] [PubMed]
- Michell-Robinson, M.; Touil, H.; Healy, L.; Owen, D.; Durafourt, B.; Bar-Or, A.; Antel, J.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef] [Green Version]
- Chasaide, C.N.; Lynch, M.A. The role of the immune system in driving neuroinflammation. Brain Neurosci. Adv. 2020, 4, 239821281990108. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.H.; Perucca, P.; O’brien, T.J.; Kwan, P.; Monif, M. Inflammation, ictogenesis, and epileptogenesis: An exploration through human disease. Epilepsia 2020, 62, 303–324. [Google Scholar] [CrossRef] [PubMed]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Aguilera, G.; Colín-González, A.L.; Rangel-Lopez, E.; Chavarria, A.; Santamaría, A. Redox Signaling, Neuroinflammation, and Neurodegeneration. Antioxid. Redox Signal. 2018, 28, 1626–1651. [Google Scholar] [CrossRef]
- Serrano-Castro, P.J.; Estivill-Torrús, G.; Cabezudo-García, P.; Reyes-Bueno, J.A.; Petersen, N.C.; Aguilar-Castillo, M.J.; Suarez-Perez, J.; Jimenez-Hernandez, M.D.; Moya-Molina, M.A.; Oliver-Martos, B.; et al. Influencia de la infección SARS-Cov2 sobre Enfermedades Neurodegenerativas y Neuropsiquiátricas: ¿Una pandemia demorada? Neurología. 2020. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0213485320300670 (accessed on 20 April 2020).
- Mészáros, Á.; Molnár, K.; Nógrádi, B.; Hernádi, Z.; Nyúl-Tóth, Á.; Wilhelm, I.; Krizbai, I.A. Neurovascular Inflammaging in Health and Disease. Cells 2020, 9, 1614. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Godbout, J.P. Basic Aspects of the Immunology of Neuroinflammation. Mod. Trends Pharm. 2013, 28, 1–19. [Google Scholar] [CrossRef]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jo, M.; Kim, J.-H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscience 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Sinha, S.; Patil, S.; Jayalekshmy, V.; Satishchandra, P. Do cytokines have any role in epilepsy? Epilepsy Res. 2008, 82, 171–176. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.E.; van den Munckhof, B.; Braun, K.P.J.; van Royen-Kerkhof, A.; de Jager, W.; Jansen, F.E. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2016, 63, 177–190. [Google Scholar] [CrossRef]
- Webster, K.M.; Sun, M.; Crack, P.; O’brien, T.J.; Shultz, S.R.; Semple, B.D. Inflammation in epileptogenesis after traumatic brain injury. J. Neuroinflamm. 2017, 14, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Steriade, C.; Britton, J.; Dale, R.C.; Gadoth, A.; Irani, S.R.; Linnoila, J.; McKeon, A.; Shao, X.; Venegas, V.; Bien, C.G. Acute symptomatic seizures secondary to autoimmune encephalitis and autoimmune-associated epilepsy: Conceptual definitions. Epilepsia 2020, 61, 1341–1351. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, L.; Lu, D.; Dai, S.; Han, Y.; Wu, Z.; Xu, P.; Chang, L.; Wu, Q. Association between autoimmune encephalitis and epilepsy: Systematic review and meta-analysis. Seizure 2021, 91, 346–359. [Google Scholar] [CrossRef]
- Romoli, M.; Krashia, P.; Sen, A.; Franciotta, D.; Gastaldi, M.; Nobili, A.; Mancini, A.; Cesarini, E.N.; Nigro, P.; Tambasco, N.; et al. Hippocampal epileptogenesis in autoimmune encephalitis. Ann. Clin. Transl. Neurol. 2019, 6, 2261–2269. [Google Scholar] [CrossRef] [Green Version]
- Bien, C.G.; Scheffer, I. Autoantibodies and epilepsy. Epilepsia 2011, 52, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Abboud, H.; Probasco, J.C.; Irani, S.; Ances, B.; Benavides, D.R.; Bradshaw, M.; Christo, P.P.; Dale, R.C.; Fernandez-Fournier, M.; Flanagan, E.P.; et al. Autoimmune encephalitis: Proposed best practice recommendations for diagnosis and acute management. J. Neurol. Neurosurg. Psychiatry 2021, 92, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Eichler, T.W.; Totland, C.; Haugen, M.; Qvale, T.H.; Mazengia, K.; Storstein, A.; Haukanes, B.I.; Vedeler, C.A. CDR2L Antibodies: A New Player in Paraneoplastic Cerebellar Degeneration. PLoS ONE 2013, 8, e66002. [Google Scholar] [CrossRef]
- Greenlee, J.E.; Carlson, N.G.; Abbatemarco, J.R.; Herdlevær, I.; Clardy, S.L.; Vedeler, C.A. Paraneoplastic and Other Autoimmune Encephalitides: Antineuronal Antibodies, T Lymphocytes, and Questions of Pathogenesis. Front. Neurol. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Binks, S.; Uy, C.; Honnorat, J.; Irani, S.R. Paraneoplastic neurological syndromes: A practical approach to diagnosis and management. Pr. Neurol. 2021, 22, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kawamura, M.; Sakimura, K.; Kato, N. Significance of Autoantibodies in Autoimmune Encephalitis in Relation to Antigen Localization: An Outline of Frequently Reported Autoantibodies with a Non-Systematic Review. Int. J. Mol. Sci. 2020, 21, 4941. [Google Scholar] [CrossRef] [PubMed]
- Takkar, A.; Mehta, S.; Gupta, N.; Bansal, S.; Lal, V. Anti- RI antibody associated progressive supranuclear palsy like presentation in a patient with breast carcinoma. J. Neuroimmunol. 2020, 347. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Ortega Suero, G.; Sola-Valls, N.; Escudero, D.; Saiz, A.; Graus, F. Síndromes neurológicos paraneoplásicos asociados a anticuerpos anti-Ma y anti-Ma. Neurologia 2018, 33, 35–46. [Google Scholar] [CrossRef]
- Shah, S.; Vazquez Do Campo, R.; Kumar, N.; McKeon, A.; Flanagan, E.P.; Klein, C.; Pittock, S.J.; Dubey, D. Paraneoplastic Myeloneuropathies: Clinical, Oncologic, and Serologic Accompaniments. Neurology 2021, 96, e632–e639. [Google Scholar] [CrossRef]
- Bernardo, F.; Rebordão, L.; Rêgo, A.; Machado, S.; Passos, J.; Costa, C.; Cruz, S.; Pinto, A.N.; Santos, M. Stiff person spectrum disorders: An illustrative case series of their phenotypic and antibody diversity. J. Neuroimmunol. 2020, 341, 577192. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Tan, J.; Sun, H.; Liu, Y.; Guan, W.; Jia, J.; Wang, Z. Anti-SOX1 Antibodies in Paraneoplastic Neurological Syndrome. J. Clin. Neurol. 2020, 16, 530–546. [Google Scholar] [CrossRef] [PubMed]
- Heimfarth, L.; Passos, F.R.S.; Monteiro, B.S.; Araújo, A.A.D.S.; Júnior, L.J.Q.; Quintans, J.D.S.S. Serum glial fibrillary acidic protein is a body fluid biomarker: A valuable prognostic for neurological disease—A systematic review. Int. Immunopharmacol. 2022, 107, 108624. [Google Scholar] [CrossRef] [PubMed]
- Loehrer, P.A.; Timmermann, L.; Pehl, A.; Bien, C.I.; Pfestroff, A.; Pedrosa, D.J. Rhombencephalitis associated with isolated Zic4-antibodies in Paraneoplastic cerebellar degeneration: A case report. BMC Neurol. 2020, 20, 208. [Google Scholar] [CrossRef] [PubMed]
- Cabezudo-García, P.; Mena-Vázquez, N.; Ciano-Petersen, N.; García-Martín, G.; Estivill-Torrús, G.; Serrano-Castro, P. Prevalence of Neural Autoantibodies in Epilepsy of Unknown Etiology: Systematic Review and Meta-Analysis. Brain Sci. 2021, 11, 392. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhu, C.; Huang, D. Microglial activation: An important process in the onset of epilepsy. Am. J. Transl. Res. 2018, 10, 2877–2889. [Google Scholar]
- Vezzani, A.; Maroso, M.; Balosso, S.; Sanchez, M.-A.; Bartfai, T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav. Immun. 2011, 25, 1281–1289. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Bianchi, M.E.; Vezzani, A. Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: The importance of IL-1beta and high-mobility group box. J. Intern. Med. 2011, 270, 319–326. [Google Scholar] [CrossRef]
- Balosso, S.; Ravizza, T.; Aronica, E.; Vezzani, A. The dual role of TNF-α and its receptors in seizures. Exp. Neurol. 2013, 247, 267–271. [Google Scholar] [CrossRef]
- Guan, Y.; Li, T. The Potential Therapeutic Role of the HMGB1-TLR Pathway in Epilepsy. Curr. Drug Targets 2021, 22, 171–182. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Semple, B.D.; Jones, N.C.; Othman, I.; Shaikh, M.F. High mobility group box 1 (HMGB1) as a novel frontier in epileptogenesis: From pathogenesis to therapeutic approaches. J. Neurochem. 2019, 151, 542–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matin, N.; Tabatabaie, O.; Falsaperla, R.; Lubrano, R.; Pavone, P.; Mahmood, F.; Gullotta, M.; Serra, A.; Di Mauro, P.; Cocuzza, S.; et al. Epilepsy and innate immune system: A possible immunogenic predisposition and related therapeutic implications. Hum. Vaccines Immunother. 2015, 11, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Rawat, C.; Kukal, S.; Dahiya, U.R.; Kukreti, R. Cyclooxygenase-2 (COX-2) inhibitors: Future therapeutic strategies for epilepsy management. J. Neuroinflamm. 2019, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łukawski, K.; Czuczwar, S.J. Understanding mechanisms of drug resistance in epilepsy and strategies for overcoming it. Expert Opin. Drug Metab. Toxicol. 2021, 17, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Kuruba, R. Experimental Models of Status Epilepticus and Neuronal Injury for Evaluation of Therapeutic Interventions. Int. J. Mol. Sci. 2013, 14, 18284–18318. [Google Scholar] [CrossRef] [PubMed]
- Rato, M.L.; Bandeira, M.; Romão, V.C.; de Sousa, D.A. Neurologic Manifestations of the Antiphospholipid Syndrome—An Update. Curr. Neurol. Neurosci. Rep. 2021, 21, 1–13. [Google Scholar] [CrossRef]
- Noureldine, M.H.; Harifi, G.; Berjawi, A.; Haydar, A.; Nader, M.; Elnawar, R.; Sweid, A.; Al Saleh, J.; A Khamashta, M.; Uthman, I. Hughes syndrome and epilepsy: When to test for antiphospholipid antibodies? Lupus 2016, 25, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Xia, Y.; Yang, Q.; Zhao, Y. The contribution of macrophages to systemic lupus erythematosus. Clin. Immunol. 2019, 207, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lin, A.; Wang, X. Seizures in steroid-responsive encephalopathy. Neurol. Sci. 2021, 42, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Julian, T.; Hadjivassiliou, M.; Zis, P. Gluten sensitivity and epilepsy: A systematic review. J. Neurol. 2019, 266, 1557–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouvroye, M.D.; Zis, P.; Van Dam, A.-M.; Rozemuller, A.J.M.; Bouma, G.; Hadjivassiliou, M. The Neuropathology of Gluten-Related Neurological Disorders: A Systematic Review. Nutrients 2020, 12, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daif, A.; Lukas, R.V.; Issa, N.P.; Javed, A.; Van Haerents, S.; Reder, A.T.; Tao, J.X.; Warnke, P.; Rose, S.; Towle, V.L.; et al. Antiglutamic acid decarboxylase 65 (GAD65) antibody-associated epilepsy. Epilepsy Behav. 2018, 80, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Fredriksen, J.R.; Carr, C.M.; Koeller, K.K.; Verdoorn, J.T.; Gadoth, A.; Pittock, S.J.; Kotsenas, A.L. MRI findings in glutamic acid decarboxylase associated autoimmune epilepsy. Neuroradiology 2018, 60, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Budhram, A.; Sechi, E.; Flanagan, E.P.; Dubey, D.; Zekeridou, A.; Shah, S.S.; Gadoth, A.; Naddaf, E.; McKeon, A.; Pittock, S.J.; et al. Clinical spectrum of high-titre GAD65 antibodies. J. Neurol. Neurosurg. Psychiatry 2021, 92, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-H.; Zheng, Y.; Cai, M.-T.; Yang, F.; Fang, W.; Zhang, Y.-X.; Ding, M.-P. Seizure occurrence in myelin oligodendrocyte glycoprotein antibody-associated disease: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2020, 42, 102057. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Mühlebner, A. Neuropathology of epilepsy. Handb. Clin. Neurol. 2018, 145, 193–216. [Google Scholar]
- Bien, C.G.; Widman, G.; Urbach, H.; Sassen, R.; Kuczaty, S.; Wiestler, O.D.; Schramm, J.; Elger, C.E. The natural history of Rasmussen’s encephalitis. Brain 2002, 125, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Villagrán, M.; Jové, M.; Simó, M.; Vilariño, N.; Alemany, M.; Palmero, R.; Martinez-Villacamps, M.M.; Nadal, E.; Bruna, J. Encephalitis Induced by Immune Checkpoint Inhibitors: A Systematic Review. JAMA Neurol. 2021, 78, 864–873. [Google Scholar] [CrossRef]
- Sechi, E.; Markovic, S.N.; McKeon, A.; Dubey, D.; Liewluck, T.; Lennon, V.A.; Lopez-Chiriboga, A.S.; Klein, C.J.; Mauremann, M.; Pittock, S.J.; et al. Neurologic autoimmunity and immune checkpoint inhibitors: Autoantibody profiles and outcomes. Neurology 2020, 95, e2442–e2452. [Google Scholar] [CrossRef]
- Maniscalco, G.T.; Zekeridou, A.; Allegorico, L.; Ranieri, A.; Napolitano, M.; Pezzella, M.; Della Gatta, L.; Manzo, V.; Ferrari, S.; Mariotto, S. GAD65 autoimmunity after treatment with nivolumab: A multifocal presentation. Neurol. Sci. 2021, 42, 4289–4291. [Google Scholar] [CrossRef]
- Erol-Yıldız, R.; Kızılay, T.; Tüzün, E.; Mısırlı, H.; Türkoğlu, R. Nivolumab-induced autoantibody negative limbic encephalitis in a patient with Hodgkin lymphoma. Leuk. Lymphoma 2020, 61, 1519–1521. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Jaffer, M.; Verma, N.; Mokhtari, S.; Ramsakal, A.; Peguero, E. Immune checkpoint inhibitor induced anti-glutamic acid decarboxylase 65 (Anti-GAD 65) limbic encephalitis responsive to intravenous immunoglobulin and plasma exchange. J. Neurol. 2020, 267, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.P.; Hissaria, P.; Hsieh, A.H.; Kneebone, C.; Vallat, W. Autoimmune limbic encephalitis with anti-contactin-associated protein-like 2 antibody secondary to pembrolizumab therapy. J. Neuroimmunol. 2017, 305, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Salam, S.; Lavin, T.; Turan, A. Limbic encephalitis following immunotherapy against metastatic malignant melanoma. BMJ Case Rep. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tröscher, A.R.; Sakaraki, E.; Mair, K.M.; Köck, U.; Racz, A.; Borger, V.; Cloppenborg, T.; Becker, A.J.; Bien, C.G.; Bauer, J. T cell numbers correlate with neuronal loss rather than with seizure activity in medial temporal lobe epilepsy. Epilepsia 2021, 62, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Mcdaniel, S.S.; Wong, M. Therapeutic role of mamalian target of rapamycin (m, TOR) inhibition in preventing epileptogenesis. Neurosci. Lett. 2011, 497, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, T.S.; Broekaart, D.W.M.; Luinenburg, M.; Mijnsbergen, C.; Anink, J.J.; Sim, N.S.; Michailidou, I.; Jansen, F.E.; van Rijen, P.C.; Lee, J.H.; et al. Balloon cells promote immune system activation in focal cortical dysplasia type 2b. Neuropathol. Appl. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Sorel, L.; Dusaucy-Bauloye, A. A propos de 21 cas d’hypsarhythmia de Gibbs; son traitement spectaculaire par l’ACTH. Acta Neurol. Psychiatr. Belg. 1958, 58, 130–141. [Google Scholar]
- Laurent, C.; Capron, J.; Quillerou, B.; Thomas, G.; Alamowitch, S.; Fain, O.; Mekinian, A. Steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT): Characteristics, treatment and outcome in 251 cases from the literature. Autoimmun. Rev. 2016, 15, 1129–1133. [Google Scholar] [CrossRef]
- Kyritsis, A.P.; Markoula, S.; Alexiou, G.; Asimakopoulos, A.; Jabbour, P.; Fotopoulos, A.; Sioka, C. Diagnosis and treatment of limbic encephalitis in the cancer patient. Future Oncol. 2020, 16, 1649–1657. [Google Scholar] [CrossRef]
- Hwang, C.J.; Choi, D.; Park, M.; Hong, J. NF-κB as a Key Mediator of Brain Inflammation in Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2019, 18, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Lapides, D.A.; McDonald, M.M. Inflammatory Manifestations of Systemic Diseases in the Central Nervous System. Curr. Treat. Options Neurol. 2020, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, X. Immunotherapy for Refractory Autoimmune Encephalitis. Front. Immunol. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Reeves, H.M.; Winters, J.L. The mechanisms of action of plasma exchange. Br. J. Haematol. 2014, 164, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Alamilla-Sanchez, M.E.; Alcala-Salgado, M.A.; Alonso-Bello, C.D.; Fonseca-Gonzalez, G.T. Mechanism of Action and Efficacy of Immunosupressors in Lupus Nephritis. Int. J. Nephrol. Renov. Dis. 2021, 14, 441–458. [Google Scholar] [CrossRef]
- Krueger, D.A.; Wilfong, A.A.; Holland-Bouley, K.; Anderson, A.E.; Agricola, K.; Tudor, C.; Mays, M.; Lopez, C.M.; Kim, M.-O.; Franz, D.N. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann. Neurol. 2013, 74, 679–687. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Sandborn, W.J.; Panaccione, R.; Domènech, E.; Pouillon, L.; Siegmund, B.; Danese, S.; Ghosh, S. Tumour necrosis factor inhibitors in inflammatory bowel disease: The story continues. Ther. Adv. Gastroenterol. 2021, 14. [Google Scholar] [CrossRef]
- Vena, G.A.; Cassano, N. Drug focus: Adalimumab in the treatment of moderate to severe psoriasis. Biol. Targets Ther. 2007, 1, 93–103. [Google Scholar]
- Lagarde, S.; Villeneuve, N.; Trébuchon, A.; Kaphan, E.; Lepine, A.; McGonigal, A.; Roubertie, A.; Barthez, M.-A.J.; Trommsdorff, V.; Lefranc, J.; et al. Anti-tumor necrosis factor alpha therapy (adalimumab) in Rasmussen’s encephalitis: An open pilot study. Epilepsia 2016, 57, 956–966. [Google Scholar] [CrossRef]
- Kondo, T.; Fukuta, M.; Takemoto, A.; Takami, Y.; Sato, M.; Takahashi, N.; Suzuki, T.; Sato, J.; Atsuta, N.; Sobue, G.; et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: A case report and review of the literature. Nagoya J. Med. Sci. 2014, 76, 361–368. [Google Scholar]
- French, J.A.; Cole, A.J.; Faught, E.; Theodore, W.H.; Vezzani, A.; Liow, K.; Halford, J.J.; Armstrong, R.; Szaflarski, J.P.; Hubbard, S.; et al. Safety and Efficacy of Natalizumab as Adjunctive Therapy for People with Drug-Resistant Epilepsy. Neurology 2021, 97, e1757–e1767. [Google Scholar] [CrossRef] [PubMed]
- Melvin, J.J.; Hardison, H.H. Immunomodulatory Treatments in Epilepsy. Semin. Pediatr. Neurol. 2014, 21, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Simon, O.J.; Göbel, K.; Bien, C.G.; Meuth, S.G.; Wiendl, H. Rasmussen encephalitis treated with natalizumab. Neurology 2013, 81, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Rensel, M.; Zabeti, A.; Mealy, M.A.; Cimbora, D.; She, D.; Drappa, J.; Katz, E. Long-term efficacy and safety of inebilizumab in neuromyelitis optica spectrum disorder: Analysis of aquaporin-4-immunoglobulin G-seropositive participants taking inebilizumab for ≥4 years in the N-MOmentum trial. Mult. Scler. [CrossRef]
- Gable, K.L.; Guptill, J.T. Antagonism of the Neonatal Fc Receptor as an Emerging Treatment for Myasthenia Gravis. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Smets, I.; Titulaer, M.J. Antibody Therapies in Autoimmune Encephalitis. Neurotherapeutics 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fabene, P.F.; Bramanti, P.; Constantin, G. The emerging role for chemokines in epilepsy. J. Neuroimmunol. 2010, 224, 22–27. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Choi, C.-H.; Ma, S.-H.; Ma, K.K.; Leung, H.; Mok, V.C. Super-refractory status epilepticus in autoimmune encephalitis treated with interleukin-1 receptor antagonist, anakinra. Epileptic Disord. 2021, 23, 500–505. [Google Scholar] [CrossRef]
- Mochol, M.; Taubøll, E.; Sveberg, L.; Tennøe, B.; Olsen, K.B.; Heuser, K.; Svalheim, S. Seizure control after late introduction of anakinra in a patient with adult onset Rasmussen’ encephalitis. Epilepsy Behav. Rep. 2021, 16, 100462. [Google Scholar] [CrossRef]
- Yamanaka, G.; Ishida, Y.; Kanou, K.; Suzuki, S.; Watanabe, Y.; Takamatsu, T.; Morichi, S.; Go, S.; Oana, S.; Yamazaki, T.; et al. Towards a Treatment for Neuroinflammation in Epilepsy: Interleukin-1 Receptor Antagonist, Anakinra, as a Potential Treatment in Intractable Epilepsy. Int. J. Mol. Sci. 2021, 22, 6282. [Google Scholar] [CrossRef]
- Koh, S.; Wirrell, E.; Vezzani, A.; Nabbout, R.; Muscal, E.; Kaliakatsos, M.; Wickström, R.; Riviello, J.J.; Brunklaus, A.; Payne, E.; et al. Proposal to optimize evaluation and treatment of Febrile infection-related epilepsy syndrome (FIRES): A Report from FIRES workshop. Epilepsia Open 2021, 6, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Dinoto, A.; Cheli, M.; Bratina, A.; Sartori, A.; Manganotti, P. Bortezomib in anti-N-Methyl-d-Aspartate-Receptor (NMDA-R) encephalitis: A systematic review. J. Neuroimmunol. 2021, 356, 577586. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Akhter, J.; Aarzoo; Bashir, D.J.; Manzoor, S.; Rastogi, S.; Arora, I.; Aggarwal, N.B.; Samim, M. Resveratrol loaded nanoparticles attenuate cognitive impairment and inflammatory markers in PTZ-induced kindled mice. Int. Immunopharmacol. 2021, 101, 108287. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Khan, S.U.; Othman, I.; Shaikh, M.F. Naturally Occurring HMGB1 Inhibitor, Glycyrrhizin, Modulates Chronic Seizures-Induced Memory Dysfunction in Zebrafish Model. ACS Chem. Neurosci. 2021, 12, 3288–3302. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Gnatkovsky, V.; Othman, I.; Shaikh, M.F. From the Molecular Mechanism to Pre-clinical Results: Anti-epileptic Effects of Fingolimod. Curr. Neuropharmacol. 2020, 18, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Wang, L.; Zhang, B.; Gao, F.; Yang, C.M. Glycyrrhizin, an HMGB1 inhibitor, exhibits neuroprotective effects in rats after lithium-pilocarpine-induced status epilepticus. J. Pharm. Pharmacol. 2019, 71, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Medlej, Y.; Salah, H.; Wadi, L.; Saad, S.; Bashir, B.; Allam, J.; Atoui, Z.; Darwish, N.; Karnib, N.; Darwish, H.; et al. Lestaurtinib (CEP-701) modulates the effects of early life hypoxic seizures on cognitive and emotional behaviors in immature rats. Epilepsy Behav. 2019, 92, 332–340. [Google Scholar] [CrossRef]
- Jamilloux, Y.; El Jammal, T.; Vuitton, L.; Gerfaud-Valentin, M.; Kerever, S.; Sève, P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun. Rev. 2019, 18, 102390. [Google Scholar] [CrossRef] [PubMed]
- Tai, T.Y.; Warner, L.N.; Jones, T.D.; Jung, S.; Concepcion, F.A.; Skyrud, D.W.; Fender, J.; Liu, Y.; Williams, A.D.; Neumaier, J.F.; et al. Antiepileptic action of c-Jun N-terminal kinase (JNK) inhibition in an animal model of temporal lobe epilepsy. Neuroscience 2017, 349, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Bascuñana, P.; Möhle, L.; Brackhan, M.; Pahnke, J. Fingolimod as a Treatment in Neurologic Disorders Beyond Multiple Sclerosis. Drugs R&D 2020, 20, 197–207. [Google Scholar] [CrossRef]
- Singh, N.; Saha, L.; Kumari, P.; Singh, J.; Bhatia, A.; Banerjee, D.; Chakrabarti, A. Effect of dimethyl fumarate on neuroinflammation and apoptosis in pentylenetetrazol kindling model in rats. Brain Res. Bull. 2019, 144, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, Q.; Huang, H.; Zhang, J.; Wang, J.; Chen, Y.; Peng, Y.; Zhang, H.; Zeng, J.; Feng, Z.; et al. Inhibition of p38 MAPK regulates epileptic severity by decreasing expression levels of A1R and ENT. Mol. Med. Rep. 2020, 22, 5348–5357. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Pacheco, A.; Diaz-Hernandez, M.; Arribas-Blázquez, M.; Sanz-Rodriguez, A.; Olivos-Oré, L.A.; Artalejo, A.R.; Alves, M.; Letavic, M.; Miras-Portugal, M.T.; Conroy, R.M.; et al. Transient P2X7 Receptor Antagonism Produces Lasting Reductions in Spontaneous Seizures and Gliosis in Experimental Temporal Lobe Epilepsy. J. Neurosci. 2016, 36, 5920–5932. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.F.; Silva, L.B.; Cruz, J.V.; Araújo, P.H.; Kimani, N.M.; Leite, F.H.; Campos, J.M.; Santos, C.B. An Overview of the α4β1 Integrin and the Potential Therapeutic Role of its Antagonists. Curr. Med. Chem. 2021, 28, 5884–5895. [Google Scholar] [CrossRef]
- Miller, J.W. Inflammation as a Target for Epilepsy Therapy. Neurology 2021, 97, 845–846. [Google Scholar] [CrossRef]
- Fabene, P.F.; Mora, G.N.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef]
- Stredny, C.M.; Case, S.; Sansevere, A.J.; Son, M.; Henderson, L.; Gorman, M.P. Interleukin-6 Blockade with Tocilizumab in Anakinra-Refractory Febrile Infection-Related Epilepsy Syndrome (FIRES). Child Neurol. Open 2020, 7. [Google Scholar] [CrossRef]
- Nailwal, N.P.; Doshi, G.M. Role of intracellular signaling pathways and their inhibitors in the treatment of inflammation. Inflammopharmacology 2021, 29, 617–640. [Google Scholar] [CrossRef]
- Ambrosius, B.; Gold, R.; Chan, A.; Faissner, S. Antineuroinflammatory drugs in HIV-associated neurocognitive disorders as potential therapy. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e551. [Google Scholar] [CrossRef] [Green Version]
- Dhir, A. An update of cyclooxygenase (COX)-inhibitors in epilepsy disorders. Expert Opin. Investig. Drugs 2019, 28, 191–205. [Google Scholar] [CrossRef]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Gelders, G.; Baekelandt, V.; Van Der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte-Silva, E.; Macedo, D.; Maes, M.; Peixoto, C.A. Novel insights into the mechanisms underlying depression-associated experimental autoimmune encephalomyelitis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 93, 4784268. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Berk, M.; Klein, H.; Walder, K.; Galecki, P.; Maes, M. Nitrosative Stress, Hypernitrosylation, and Autoimmune Responses to Nitrosylated Proteins: New Pathways in Neuroprogressive Disorders Including Depression and Chronic Fatigue Syndrome. Mol. Neurobiol. 2017, 54, 4271–4291. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Yirmyia, R.; Noraberg, J.; Brene, S.; Hibbeln, J.; Perini, G.; Kubera, M.; Bob, P.; Lerer, B.; Maj, M. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: Leads for future research and new drug developments in depression. Metab. Brain Dis. 2009, 24, 27–53. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.E.; Garcia-Morales, I.; Gil-Nagel, A. Prevalence of depressive symptoms and their impact on quality of life in patients with drug-resistant focal epilepsy (IMDYVA study). Epilepsy Res. 2015, 110, 157–165. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).