
Immunosuppressive Signaling Pathways as Targeted Cancer Therapies

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Immune Evasion
2.1. Elimination
2.2. Equilibrium
2.3. Escape
2.3.1. Escaping the Antigen Presentation Pathway
2.3.2. The PD-1/PD-L1 Pathway as a Mechanism of Escape
3. The Tumor Microenvironment
4. Immunoregulatory Signaling Pathways
4.1. Myeloid-Derived Suppressor Cells
4.2. Tumor-Associated Macrophages
4.3. Regulatory T Cells
5. Personalized Precision Medicine and Combinatorial Therapies
 positive/negative response.
positive/negative response.6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, L.M.d.; Larsen, S.J.; Catin Kupper, B.E.; Begnami, M.D.F.d.S.; Scapulatempo-Neto, C.; Petersen, A.H.; Aagaard, M.M.; Baumbach, J.; Aguiar, S.; Rogatto, S.R. Increased Levels of Genomic Instability and Mutations in Homologous Recombination Genes in Locally Advanced Rectal Carcinomas. Front. Oncol. 2019, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.; Greer, S.U.; Hopmans, E.; Grimes, S.M.; Lee, H.; Zhao, L.; Miotke, L.; Suarez, C.; Almeda, A.F.; Haraldsdottir, S.; et al. Profiling diverse sequence tandem repeats in colorectal cancer reveals co-occurrence of microsatellite and chromosomal instability involving Chromosome 8. Genome Med. 2021, 13, 145. [Google Scholar] [CrossRef] [PubMed]
- Jardim, M.J.; Wang, Q.; Furumai, R.; Wakeman, T.; Goodman, B.K.; Wang, X.-F. Reduced ATR or Chk1 Expression Leads to Chromosome Instability and Chemosensitization of Mismatch Repair–deficient Colorectal Cancer Cells. Mol. Biol. Cell 2009, 20, 3801–3809. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Linstra, R.; van Vugt, M.A.T.M. Genomic instability, inflammatory signaling and response to cancer immunotherapy. Biochim. et Biophys. Acta (BBA) Rev. Cancer 2022, 1877, 188661. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K.; Arihiro, K. Tumor-Driven Evolution of Immunosuppressive Networks during Malignant Progression. Cancer Res. 2006, 66, 5527–5536. [Google Scholar] [CrossRef] [Green Version]
- Gu, D.; Ao, X.; Yang, Y.; Chen, Z.; Xu, X. Soluble immune checkpoints in cancer: Production, function and biological significance. J. Immunother Cancer 2018, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Tsai, C.-J.; Jang, H. A New View of Pathway-Driven Drug Resistance in Tumor Proliferation. Trends Pharmacol. Sci. 2017, 38, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, P. Ueber den jetzigen stand der karzinomforschung. vortrag gehalten vor den studenten der amsterdamer universitaet, vereinigung fuer wissenschaftliche arbeit 1 june 1908. printed in: P. ehrlich. Beitraege Exp. Pathol. Chemother. Akad. Verl. Leipz. 1909, 118–164. [Google Scholar]
- Burnet, M. Cancer—A Biological Approach. III. Viruses Associated with Neoplastic Conditions. IV. Practical Applications. Br. Med. J. 1957, 1, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. On immunosurveillance in human cancer. Yale J. Biol. Med. 1982, 55, 329–333. [Google Scholar]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gicobi, J.K.; Barham, W.; Dong, H. Immune resilience in response to cancer therapy. Cancer Immunol. Immunother. 2020, 69, 2165–2167. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, M.G.; Li, Z. Heat-shock proteins in infection-mediated inflammation-induced tumorigenesis. J. Hematol. Oncol. 2009, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Bernardone, I.S. Role of NK cells and adaptive immunity in “immunoediting”: Recent developments. Inmunología 2008, 27, 141–146. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [PubMed]
- Kim, R. Chapter 2-Cancer Immunoediting: From Immune Surveillance to Immune Escape. In Cancer Immunotherapy; Prendergast, G.C., Jaffee, E.M., Eds.; Academic Press: Burlington, VT, USA, 2007; pp. 9–27. [Google Scholar]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological Aspects of Cancer Chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Zhang, J.; Shih, D.J.H.; Lin, S.-Y. Role of DNA repair defects in predicting immunotherapy response. Biomark. Res. 2020, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.; Blokzijl, F.; Kuijk, E.; Bertl, J.; Vougioukalaki, M.; Janssen, R.; Besselink, N.; Boymans, S.; de Ligt, J.; Pedersen, J.S. Deficiency of nucleotide excision repair is associated with mutational signature observed in cancer. Genome Res. 2019, 29, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Rondón, N.; Villegas, V.E.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, L.R.; Chen, H.; Collins, A.R.; Connell, M.; Damia, G.; Dasgupta, S.; Malhotra, M.; Meeker, A.K.; Amedei, A.; Amin, A.; et al. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin. Cancer Biol. 2015, 35, S5–S24. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1000165. [Google Scholar] [PubMed]
- Pieren, D.K.J.; Smits, N.A.M.; Imholz, S.; Nagarajah, B.; van Oostrom, C.T.; Brandt, R.M.C.; Vermeij, W.P.; Dollé, M.E.T.; Guichelaar, T. Compromised DNA Repair Promotes the Accumulation of Regulatory T Cells With an Aging-Related Phenotype and Responsiveness. Front. Aging 2021, 2, 13. [Google Scholar] [CrossRef]
- Nebot-Bral, L.; Coutzac, C.; Kannouche, P.L.; Chaput, N. Why is immunotherapy effective (or not) in patients with MSI/MMRD tumors? Bull. Cancer 2019, 106, 105–113. [Google Scholar] [CrossRef]
- Juvekar, A.; Hu, H.; Yadegarynia, S.; Lyssiotis, C.A.; Ullas, S.; Lien, E.C.; Bellinger, G.; Son, J.; Hok, R.C.; Seth, P.; et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc. Natl. Acad. Sci. USA 2016, 113, E4338–E4347. [Google Scholar] [CrossRef] [Green Version]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef] [PubMed]
- Wayne, J.; Brooks, T.; Landras, A.; Massey, A.J. Targeting DNA damage response pathways to activate the STING innate immune signaling pathway in human cancer cells. FEBS J. 2021, 288, 4507–4540. [Google Scholar] [CrossRef] [PubMed]
- Reisländer, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef]
- Yaghmour, G.; Pandey, M.; Ireland, C.; Patel, K.; Nunnery, S.; Powell, D.; Baum, S.; Wiedower, E.; Schwartzberg, L.S.; Martin, M.G. Role of Genomic Instability in Immunotherapy with Checkpoint Inhibitors. Anticancer Res. 2016, 36, 4033–4038. [Google Scholar] [CrossRef]
- Burster, T.; Gärtner, F.; Bulach, C.; Zhanapiya, A.; Gihring, A.; Knippschild, U. Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells. Pharmaceuticals 2021, 14, 236. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Wang, Z.; Arienti, F.; Rivoltini, L.; Parmiani, G.; Ferrone, S. beta2-Microglobulin mutations, HLA class I antigen loss, and tumor progression in melanoma. J. Clin. Investig. 1998, 101, 2720–2729. [Google Scholar] [CrossRef]
- Chang, C.-C.; Campoli, M.; Restifo, N.P.; Wang, X.; Ferrone, S. Immune Selection of Hot-Spot β2 Microglobulin Gene Mutations, HLA-A2 Allospecificity Loss, and Antigen-Processing Machinery Component Down-Regulation in Melanoma Cells Derived from Recurrent Metastases following Immunotherapy. J. Immunol. 2005, 174, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.Q.; Zhang, J.Q.; Xia, M.; Miao, F.Q.; Shan, X.N.; Xie, W. Low-molecular-weight protein (LMP)2/LMP7 abnormality underlies the downregulation of human leukocyte antigen class I antigen in a hepatocellular carcinoma cell line. J. Gastroenterol. Hepatol. 2007, 22, 1155–1161. [Google Scholar] [CrossRef]
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.F.; DuPage, M.J.P.; Dong, H.K.; Chen, J.; Jacks, T. Regulated Expression of a Tumor-Associated Antigen Reveals Multiple Levels of T-Cell Tolerance in a Mouse Model of Lung Cancer. Cancer Res. 2008, 68, 9459–9468. [Google Scholar] [CrossRef] [Green Version]
- Nefedova, Y.; Cheng, P.; Gilkes, D.; Blaskovich, M.; Beg, A.A.; Sebti, S.M.; Gabrilovich, D.I. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J. Immunol. 2005, 175, 4338–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.C.; Varner, J.A. Myeloid Cells in the Tumor Microenvironment: Modulation of Tumor Angiogenesis and Tumor Inflammation. J. Oncol. 2010, 2010, 201026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monu, N.R.; Frey, A.B. Myeloid-derived suppressor cells and anti-tumor T cells: A complex relationship. Immunol. Investg. 2012, 41, 595–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Wang, H.; Li, C.; Fang, J.-Y.; Xu, J. Cancer Cell-Intrinsic PD-1 and Implications in Combinatorial Immunotherapy. Front. Immunol. 2018, 9, 1774. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Haining, W.N.; Freeman, G.J.; Sharpe, A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef]
- Filippone, A.; Lanza, M.; Mannino, D.; Raciti, G.; Colarossi, C.; Sciacca, D.; Cuzzocrea, S.; Paterniti, I. PD1/PD-L1 immune checkpoint as a potential target for preventing brain tumor progression. Cancer Immunol. Immunother. 2022. [Google Scholar] [CrossRef]
- Ansell, S.M.; Vonderheide, R.H. Cellular composition of the tumor microenvironment. Am. Soc. Clin. Oncol. Educ. Book 2013, e91–e97. [Google Scholar] [CrossRef]
- Schiavoni, G.; Gabriele, L.; Mattei, F. The tumor microenvironment: A pitch for multiple players. Front. Oncol. 2013, 3, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forget, M.-A.; Haymaker, C.; Hess, K.R.; Meng, Y.J.; Creasy, C.; Karpinets, T.; Fulbright, O.J.; Roszik, J.; Woodman, S.E.; Kim, Y.U.; et al. Prospective Analysis of Adoptive TIL Therapy in Patients with Metastatic Melanoma: Response, Impact of Anti-CTLA4, and Biomarkers to Predict Clinical Outcome. Clin. Cancer Res. 2018, 24, 4416–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Dou, B.; Tan, H.; Feng, Y.; Wang, N.; Wang, D. Tumor microenvironment-driven non-cell-autonomous resistance to antineoplastic treatment. Mol. Cancer 2019, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Laitala, A.; Erler, J.T. Hypoxic Signalling in Tumour Stroma. Front. Oncol. 2018, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef] [Green Version]
- Setlai, B.P.; Hull, R.; Reis, R.M.; Agbor, C.; Ambele, M.A.; Mulaudzi, T.V.; Dlamini, Z. MicroRNA Interrelated Epithelial Mesenchymal Transition (EMT) in Glioblastoma. Genes 2022, 13, 244. [Google Scholar] [CrossRef]
- Sun, I.-H.; Oh, M.-H.; Zhao, L.; Patel, C.H.; Arwood, M.L.; Xu, W.; Tam, A.J.; Blosser, R.L.; Wen, J.; Powell, J.D. mTOR Complex 1 Signaling Regulates the Generation and Function of Central and Effector Foxp3(+) Regulatory T Cells. J. Immunol. 2018, 201, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Mafi, S.; Mansoori, B.; Taeb, S.; Sadeghi, H.; Abbasi, R.; Cho, W.C.; Rostamzadeh, D. mTOR-Mediated Regulation of Immune Responses in Cancer and Tumor Microenvironment. Front. Immunol. 2022, 12, 774103. [Google Scholar] [CrossRef]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef] [Green Version]
- Dutcher, J.P. Mammalian Target of Rapamycin Inhibition. Clin. Cancer Res. 2004, 10, 6382S–6387S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017, 8, 3649–3665. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef]
- Chevalier, M.F.; Trabanelli, S.; Racle, J.; Salomé, B.; Cesson, V.; Gharbi, D.; Bohner, P.; Domingos-Pereira, S.; Dartiguenave, F.; Fritschi, A.S.; et al. ILC2-modulated T cell-to-MDSC balance is associated with bladder cancer recurrence. J. Clin. Investig. 2017, 127, 2916–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.-F.; Song, S.-Y.; Wang, T.-J.; Ji, W.-J.; Li, S.-W.; Liu, N.; Yan, C.-X. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology 2018, 7, e1494113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.; Iyer, S.; Kerr, W.G. Role of SHIP1 in cancer and mucosal inflammation. Ann. N. Y. Acad. Sci. 2013, 1280, 6–10. [Google Scholar] [CrossRef]
- Brooks, R.; Fuhler, G.M.; Iyer, S.; Smith, M.J.; Park, M.-Y.; Paraiso, K.H.T.; Engelman, R.W.; Kerr, W.G. SHIP1 Inhibition Increases Immunoregulatory Capacity and Triggers Apoptosis of Hematopoietic Cancer Cells. J. Immunol. 2010, 184, 3582–3589. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sasaki, T.; Kozieradzki, I.; Wakeham, A.; Itie, A.; Dumont, D.J.; Penninger, J.M. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999, 13, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Gilby, D.C.; Goodeve, A.C.; Winship, P.R.; Valk, P.J.; Delwel, R.; Reilly, J.T. Gene structure, expression profiling and mutation analysis of the tumour suppressor SHIP1 in Caucasian acute myeloid leukaemia. Leukemia 2007, 21, 2390–2393. [Google Scholar] [CrossRef] [Green Version]
- Houghton, A.M.; Rzymkiewicz, D.M.; Ji, H.; Gregory, A.D.; Egea, E.E.; Metz, H.E.; Stolz, D.B.; Land, S.R.; Marconcini, L.A.; Kliment, C.R.; et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 2010, 16, 219–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauls, S.D.; Marshall, A.J. Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur. J. Immunol. 2017, 47, 932–945. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chapuis, N.; Tamburini, J.; Bardet, V.; Cornillet-Lefebvre, P.; Willems, L.; Green, A.; Mayeux, P.; Lacombe, C.; Bouscary, D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010, 95, 819–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darici, S.; Alkhaldi, H.; Horne, G.; Jørgensen, H.G.; Marmiroli, S.; Huang, X. Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. J. Clin. Med. 2020, 9, 2934. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Yu, J.; Li, H.; Zhang, Z.; Lin, W.; Wei, X.; Shao, B. Targeting the MDSCs of Tumors In Situ With Inhibitors of the MAPK Signaling Pathway to Promote Tumor Regression. Front. Oncol. 2021, 11, 647312. [Google Scholar] [CrossRef]
- Lee, E.R.; Kim, J.Y.; Kang, Y.J.; Ahn, J.Y.; Kim, J.H.; Kim, B.W.; Choi, H.Y.; Jeong, M.Y.; Cho, S.G. Interplay between PI3K/Akt and MAPK signaling pathways in DNA-damaging drug-induced apoptosis. Biochim. Biophys. Acta 2006, 1763, 958–968. [Google Scholar] [CrossRef] [Green Version]
- Sasidharan Nair, V.; Saleh, R.; Toor, S.M.; Alajez, N.M.; Elkord, E. Transcriptomic Analyses of Myeloid-Derived Suppressor Cell Subsets in the Circulation of Colorectal Cancer Patients. Front. Oncol 2020, 10, 1530. [Google Scholar] [CrossRef]
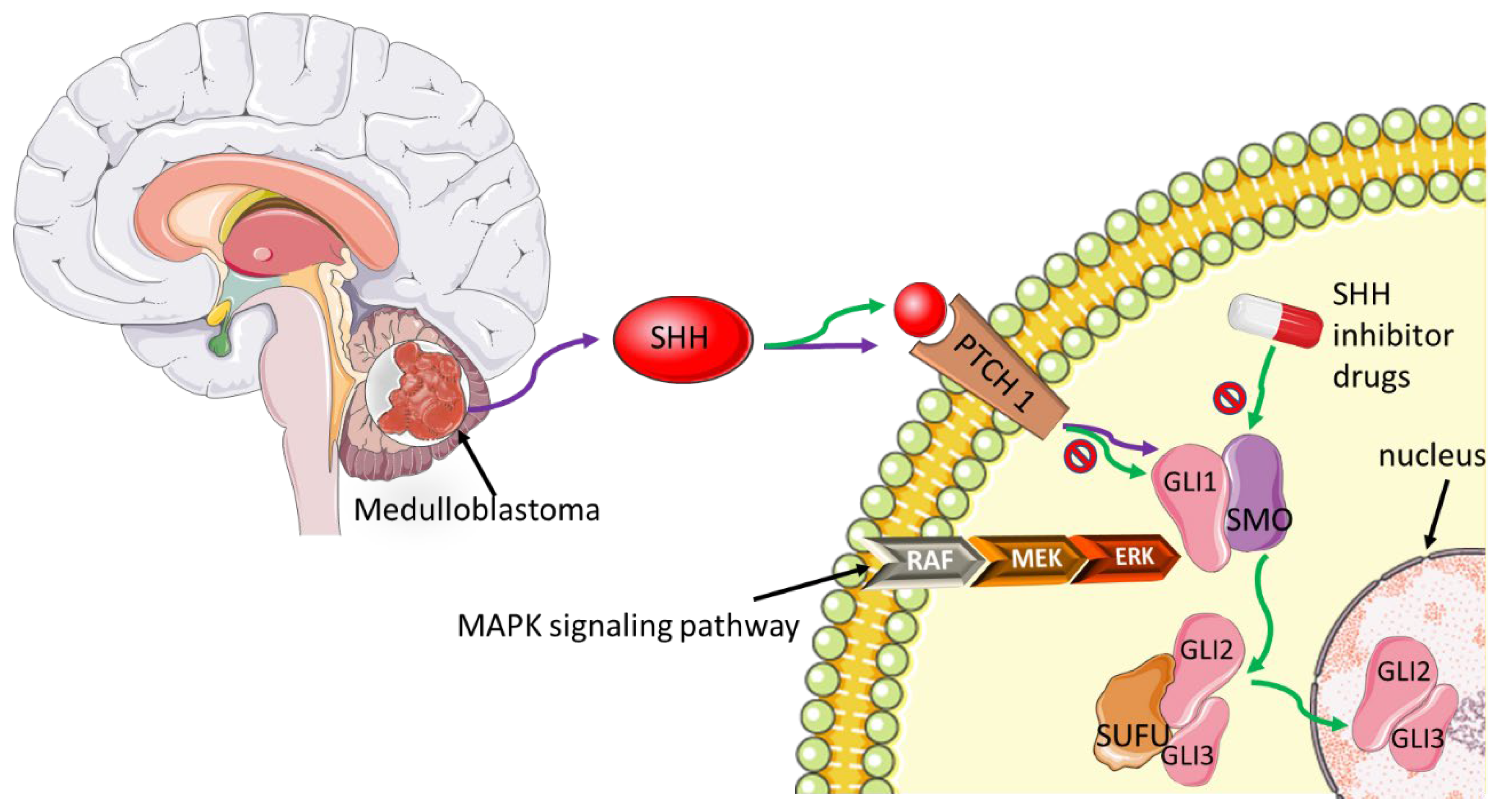
- Rovida, E.; Stecca, B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin Cancer Biol. 2015, 35, 154–167. [Google Scholar] [CrossRef]
- Lemm, E.A.; Valle-Argos, B.; Smith, L.D.; Richter, J.; Gebreselassie, Y.; Carter, M.J.; Karolova, J.; Svaton, M.; Helman, K.; Weston-Bell, N.J.; et al. Preclinical Evaluation of a Novel SHIP1 Phosphatase Activator for Inhibition of PI3K Signaling in Malignant B Cells. Clin. Cancer Res. 2020, 26, 1700–1711. [Google Scholar] [CrossRef]
- Chamberlain, T.C.; Cheung, S.T.; Yoon, J.S.J.; Ming-Lum, A.; Gardill, B.R.; Shakibakho, S.; Dzananovic, E.; Ban, F.; Samiea, A.; Jawanda, K.; et al. Interleukin-10 and Small Molecule SHIP1 Allosteric Regulators Trigger Anti-inflammatory Effects through SHIP1/STAT3 Complexes. iScience 2020, 23, 101433. [Google Scholar] [CrossRef] [PubMed]
- So, Y.W. The Role of SHIP1 in IL-10 Signalling Pathway in Primary Macrophages. Text; University of British Columbia: Vancouver, BC, Canada, 2014. [Google Scholar]
- Shevach, E.M. Foxp3+ T Regulatory Cells: Still Many Unanswered Questions-A Perspective after 20 Years of Study. Front. Immunol. 2018, 9, 1048. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Tumor-associated macrophages. Curr. Biol. 2020, 30, R246–R248. [Google Scholar] [CrossRef] [PubMed]
- Valeta-Magara, A.; Gadi, A.; Volta, V.; Walters, B.; Arju, R.; Giashuddin, S.; Zhong, H.; Schneider, R.J. Inflammatory Breast Cancer Promotes Development of M2 Tumor-Associated Macrophages and Cancer Mesenchymal Cells through a Complex Chemokine Network. Cancer Res. 2019, 79, 3360–3371. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, W.; Huo, M.; Wang, P.; Liu, X.; Wang, Y.; Li, Y.; Zhou, Z.; Xu, N.; Zhu, H. XBP1 regulates the protumoral function of tumor-associated macrophages in human colorectal cancer. Signal Transduct. Target. Ther. 2021, 6, 357. [Google Scholar] [CrossRef]
- Kemp, S.B.; Steele, N.G.; Carpenter, E.S.; Donahue, K.L.; Bushnell, G.G.; Morris, A.H.; The, S.; Orbach, S.M.; Sirihorachai, V.R.; Nwosu, Z.C.; et al. Pancreatic cancer is marked by complement-high blood monocytes and tumor-associated macrophages. Life Sci. Alliance 2021, 4, e202000935. [Google Scholar] [CrossRef]
- Yuri, P.; Shigemura, K.; Kitagawa, K.; Hadibrata, E.; Risan, M.; Zulfiqqar, A.; Soeroharjo, I.; Hendri, A.Z.; Danarto, R.; Ishii, A.; et al. Increased tumor-associated macrophages in the prostate cancer microenvironment predicted patients′ survival and responses to androgen deprivation therapies in Indonesian patients cohort. Prostate Int. 2020, 8, 62–69. [Google Scholar] [CrossRef]
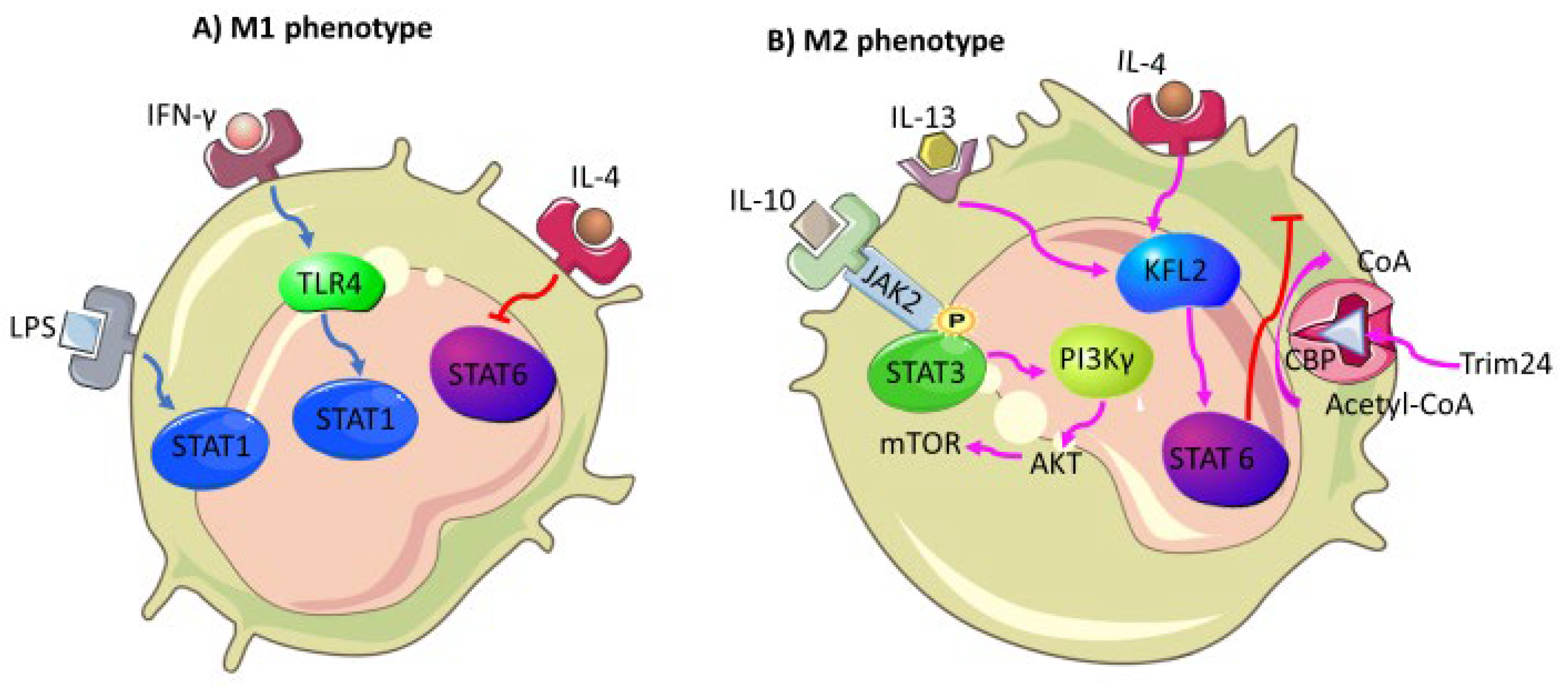
- Almatroodi, S.A.; McDonald, C.F.; Darby, I.A.; Pouniotis, D.S. Characterization of M1/M2 Tumour-Associated Macrophages (TAMs) and Th1/Th2 Cytokine Profiles in Patients with NSCLC. Cancer Microenviron 2016, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.; Jiang, L.; Hao, S.; Liu, Z.; Ding, S.; Zhang, W.; Yang, X.; Li, S. Activation of the IL-4/STAT6 Signaling Pathway Promotes Lung Cancer Progression by Increasing M2 Myeloid Cells. Front. Immunol. 2019, 10, 2638. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Dong, Y.; Peng, L.; Yang, M.; Niu, L.; Liu, Z.; Xie, J. Tumor-associated macrophages affect the biological behavior of lung adenocarcinoma A549 cells through the PI3K/AKT signaling pathway. Oncol. Lett. 2019, 18, 1840–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, M.T.; Castoldi, A.; Andrade-Oliveira, V.; Latancia, M.T.; Terra, F.F.; Correa-Costa, M.; Breda, C.N.S.; Felizardo, R.J.F.; Pereira, W.O.; da Silva, M.B.; et al. The lack of PI3Kγ favors M1 macrophage polarization and does not prevent kidney diseases progression. Int. Immunopharmacol. 2018, 64, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, B.; Tang, K.; Dong, X.; Xue, L.; Su, G.; Jin, Y. Aquaporin 1 alleviates acute kidney injury via PI3K-mediated macrophage M2 polarization. Inflamm. Res. 2020, 69, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chen, J.; Wang, P.; Fan, H.; Hou, S.; Gong, Y. Major signaling pathways and key mediators of macrophages in acute kidney injury (Review). Mol. Med. Rep. 2021, 23, 455. [Google Scholar] [CrossRef]
- di Magliano, M.P.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Petty, A.J.; Li, A.; Wang, X.; Dai, R.; Heyman, B.; Hsu, D.; Huang, X.; Yang, Y. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J. Clin. Investig. 2019, 129, 5151–5162. [Google Scholar] [CrossRef] [Green Version]
- Raleigh, D.R.; Reiter, J.F. Misactivation of Hedgehog signaling causes inherited and sporadic cancers. J. Clin. Investig. 2019, 129, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Pereira, V.; Torrejon, J.; Kariyawasam, D.; Berlanga, P.; Guerrini-Rousseau, L.; Ayrault, O.; Varlet, P.; Tauziède-Espariat, A.; Puget, S.; Bolle, S.; et al. Clinical and molecular analysis of smoothened inhibitors in Sonic Hedgehog medulloblastoma. Neurooncol. Adv. 2021, 3, vdab097. [Google Scholar] [CrossRef] [PubMed]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T.C. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef] [PubMed]
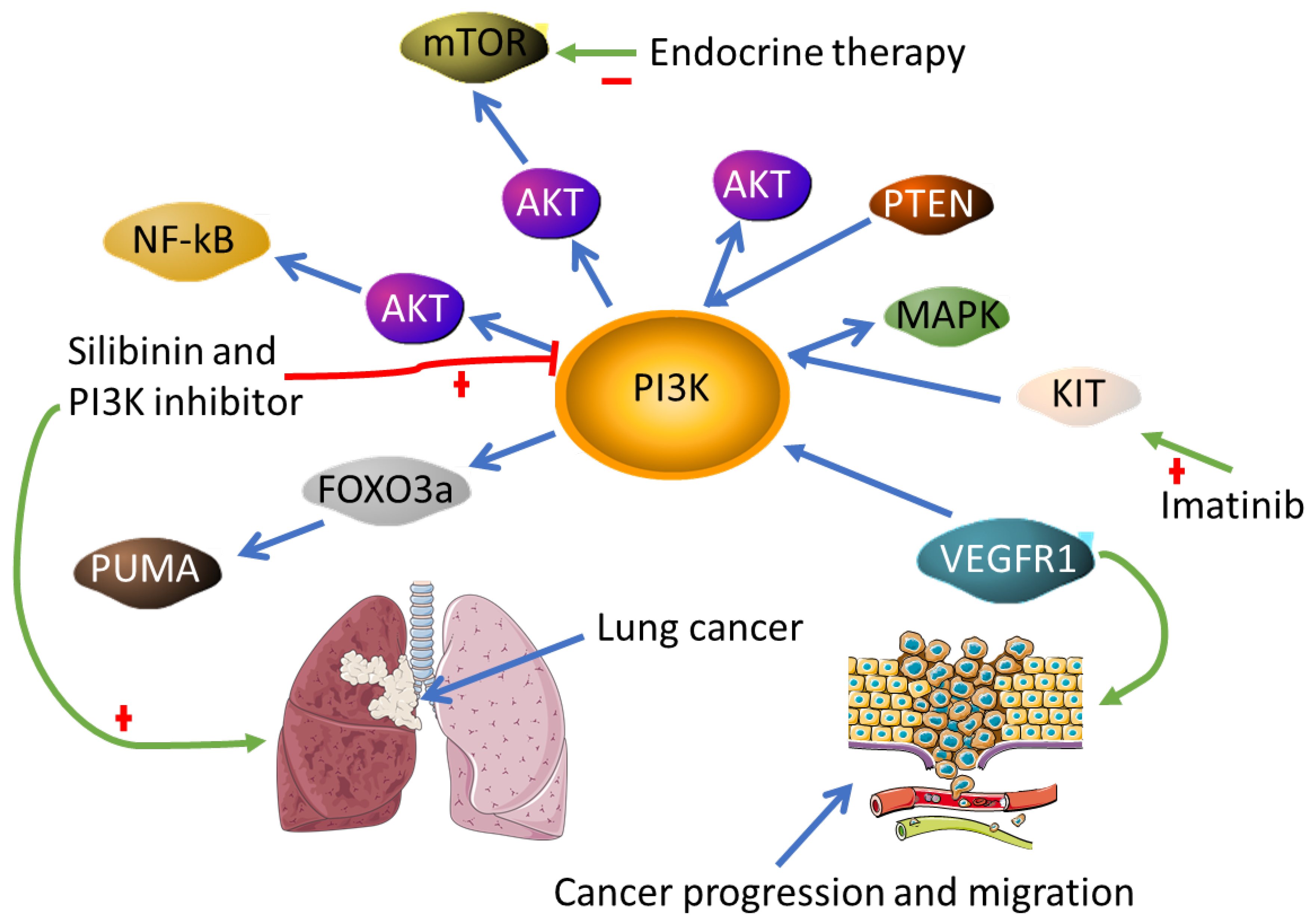
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Yang, W.Y.; Saaoud, F.; Drummer, C.T.; Sun, Y.; Xu, K.; Lu, Y.; Shan, H.; Shevach, E.M.; Jiang, X.; et al. IL-35 promotes CD4+Foxp3+ Tregs and inhibits atherosclerosis via maintaining CCR5-amplified Treg-suppressive mechanisms. JCI Insight 2021, 6, e152511. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Lu, W.; Sindhava, V.J.; Huang, Y.; Burkhardt, J.K.; Yang, E.; Riese, M.J.; Maltzman, J.S.; Jordan, M.S.; Kambayashi, T. Regulatory T cells require TCR signaling for their suppressive function. J. Immunol. 2015, 194, 4362–4370. [Google Scholar] [CrossRef] [Green Version]
- Abu-Eid, R.; Samara, R.N.; Ozbun, L.; Abdalla, M.Y.; Berzofsky, J.A.; Friedman, K.M.; Mkrtichyan, M.; Khleif, S.N. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol. Res. 2014, 2, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
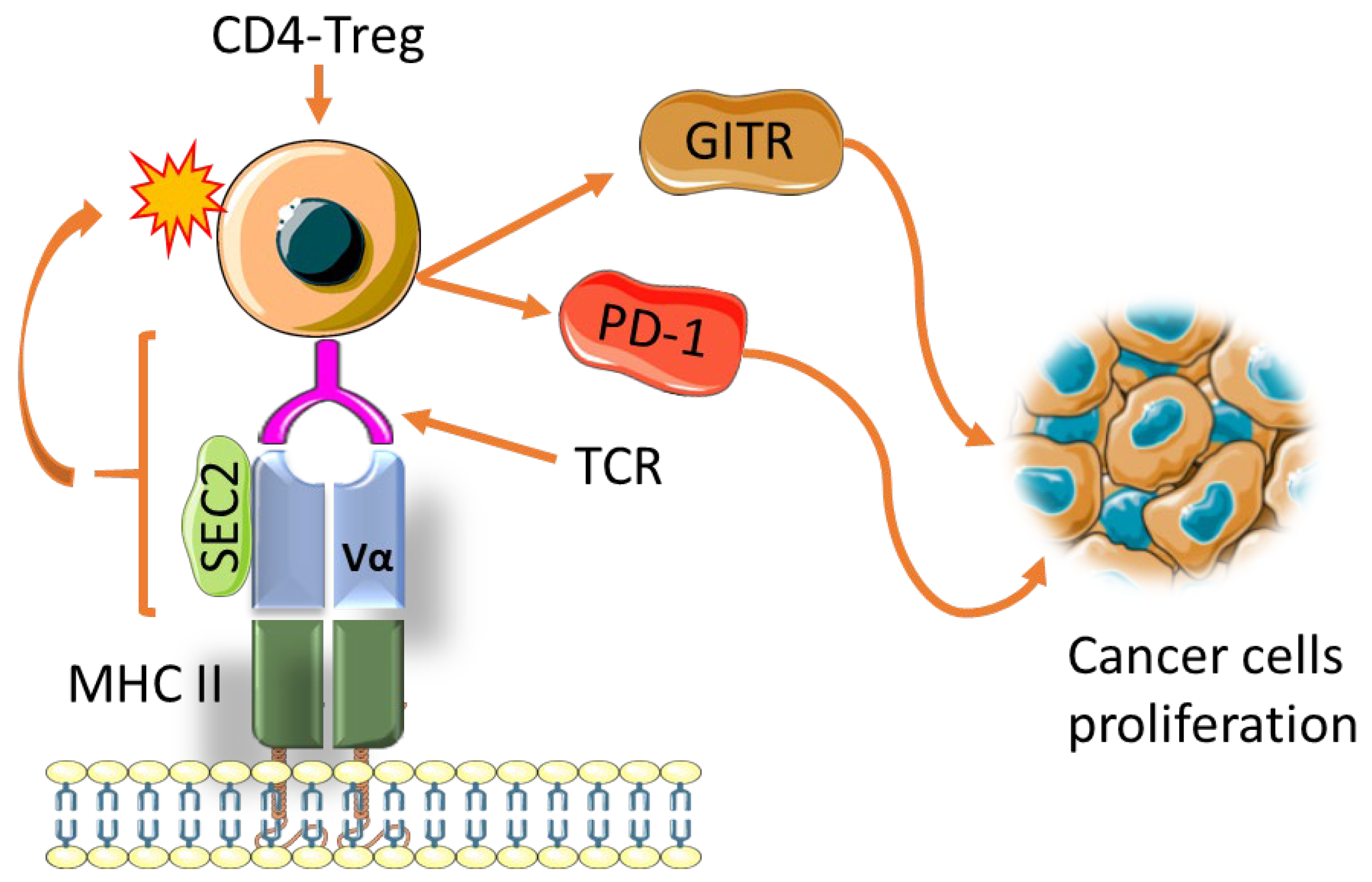
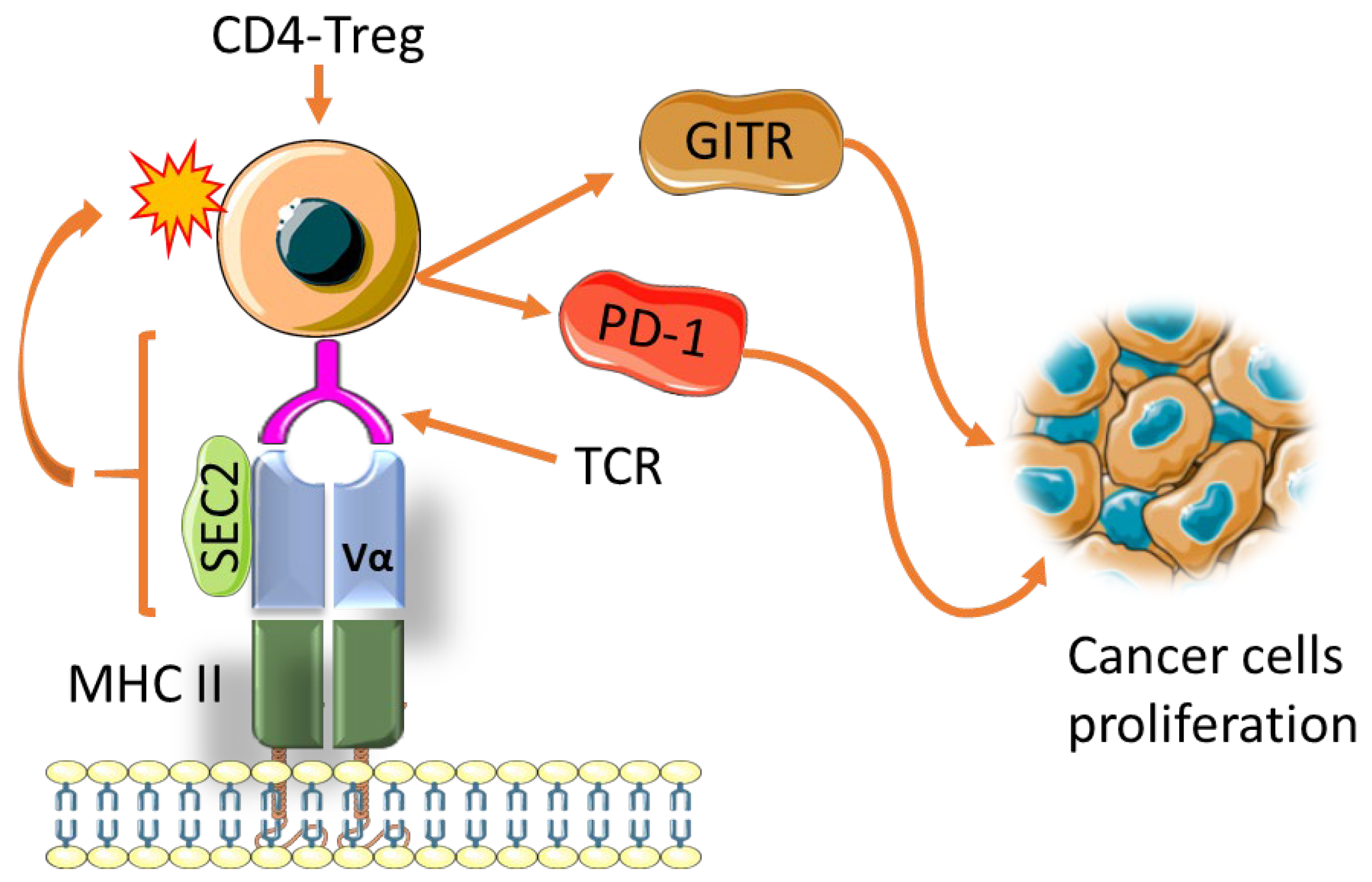
- Li, Y.; Xu, M.; Li, Y.; Zhang, Z.; Gu, W.; Halimu, G.; Li, Y.; Zhang, H.; Zhang, C. Induction of CD4+ regulatory T cells by stimulation with Staphylococcal Enterotoxin C2 through different signaling pathways. Biomed. Pharm. 2021, 143, 112204. [Google Scholar] [CrossRef]
- Hui, J.; Cao, Y.; Xiao, F.; Zhang, J.; Li, H.; Hu, F. Staphylococcus aureus enterotoxin C2 mutants: Biological activity assay in vitro. J. Ind. Microbiol. Biotechnol. 2008, 35, 975–980. [Google Scholar] [CrossRef]
- Zhu, M.M.T.; Burugu, S.; Gao, D.; Yu, J.; Kos, Z.; Leung, S.; Horst, B.A.; Nielsen, T.O. Evaluation of glucocorticoid-induced TNF receptor (GITR) expression in breast cancer and across multiple tumor types. Mod. Pathol. 2020, 33, 1753–1763. [Google Scholar] [CrossRef]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.A.; Manlove, L.S.; Farrar, M.A. Interleukin-2 and STAT5 in regulatory T cell development and function. JAKSTAT 2013, 2, e23154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Giantonio, B.J.; Alpaugh, R.K.; Schultz, J.; McAleer, C.; Newton, D.W.; Shannon, B.; Guedez, Y.; Kotb, M.; Vitek, L.; Persson, R.; et al. Superantigen-based immunotherapy: A phase I trial of PNU-214565, a monoclonal antibody-staphylococcal enterotoxin A recombinant fusion protein, in advanced pancreatic and colorectal cancer. J. Clin. Oncol 1997, 15, 1994–2007. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Liang, P.; Dong, B.; Yu, X.; Han, X.; Wang, Y.; Han, Z. Long-term results of a phase II clinical trial of superantigen therapy with staphylococcal enterotoxin C after microwave ablation in hepatocellular carcinoma. Int. J. Hyperth. 2011, 27, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.M.; Connolly, N.B.; Patel, P.M.; Kilany, S.; Hedlund, G.; Nordle, O.; Forsberg, G.; Zweit, J.; Stern, P.L.; Hawkins, R.E. A phase II study of a 5T4 oncofoetal antigen tumour-targeted superantigen (ABR-214936) therapy in patients with advanced renal cell carcinoma. Br. J. Cancer 2007, 96, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, L.; Yin, L.; Yu, H.; Jing, H.; Liu, Y.; Kong, C.; Xu, M. Superantigen staphylococcal enterotoxin C1 inhibits the growth of bladder cancer. Biosci. Biotechnol. Biochem. 2017, 81, 1741–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.; Feng, X.H. TGF-β signaling in cancer. Acta Biochim. Biophys Sin. 2018, 50, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Zhang, P.; Liangfang, Y.; Wenshe, S.; Wang, H.; Lin, X.; Dai, Y.; Feng, X.H.; Moses, R.; Wang, D.; et al. KLF17 empowers TGF-β/Smad signaling by targeting Smad3-dependent pathway to suppress tumor growth and metastasis during cancer progression. Cell Death Dis. 2015, 6, e1681. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhang, Y.Y.; Chen, Y.; Wang, J.; Wang, Q.; Lu, H. TGF-β Signaling and Resistance to Cancer Therapy. Front. Cell Dev. Biol. 2021, 9, 786728. [Google Scholar] [CrossRef]
- Wan, Y.Y.; Flavell, R.A. ′Yin-Yang′ functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol. Rev. 2007, 220, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.M.; Kasahara, Y. Chapter 19—Molecular Diagnostics in the Evaluation of Cancer: Modern Concepts and Overview. In Molecular Diagnostics; Grody, W.W., Nakamura, R.M., Strom, C.M., Kiechle, F.L., Eds.; Academic Press: San Diego, CA, USA, 2010; pp. 215–223. [Google Scholar]
- Gunter, J.; Kruithof-de Julio, M.; Zoni, E. Editorial: “Personalized Medicine for Urological Cancers: Targeting Cancer Metabolism”. Front. Oncol. 2022, 12, 617. [Google Scholar] [CrossRef]
- Du, R.; Wang, X.; Ma, L.; Larcher, L.M.; Tang, H.; Zhou, H.; Chen, C.; Wang, T. Adverse reactions of targeted therapy in cancer patients: A retrospective study of hospital medical data in China. BMC Cancer 2021, 21, 206. [Google Scholar] [CrossRef]
- Spaans, J.N.; Goss, G.D. Drug resistance to molecular targeted therapy and its consequences for treatment decisions in non-small-cell lung cancer. Front. Oncol. 2014, 4, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavilá, J.; Serra, V.; Prat, A.; Paré, L.; et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Braña, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di Tomaso, E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Invest. N. Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef]
- Krop, I.E.; Mayer, I.A.; Ganju, V.; Dickler, M.; Johnston, S.; Morales, S.; Yardley, D.A.; Melichar, B.; Forero-Torres, A.; Lee, S.C.; et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Wilhoit, T.; Patrick, J.M.; May, M.B. Alpelisib: A Novel Therapy for Patients With PIK3CA-Mutated Metastatic Breast Cancer. J. Adv. Pract. Oncol. 2020, 11, 768–775. [Google Scholar]
- Verret, B.; Cortes, J.; Bachelot, T.; Andre, F.; Arnedos, M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann. Oncol. 2019, 30 (Suppl. 10), x12–x20. [Google Scholar] [CrossRef]
- Liang, C.; Yu, X.; Xiong, N.; Zhang, Z.; Sun, Z.; Dong, Y. Pictilisib Enhances the Antitumor Effect of Doxorubicin and Prevents Tumor-Mediated Bone Destruction by Blockade of PI3K/AKT Pathway. Front. Oncol. 2021, 10, 615146. [Google Scholar] [CrossRef]
- Mensah, F.A.; Blaize, J.-P.; Bryan, L.J. Spotlight on copanlisib and its potential in the treatment of relapsed/refractory follicular lymphoma: Evidence to date. Onco Targets 2018, 11, 4817–4827. [Google Scholar] [CrossRef] [Green Version]
- Vangapandu, H.V.; Jain, N.; Gandhi, V. Duvelisib: A phosphoinositide-3 kinase δ/γ inhibitor for chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2017, 26, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Cheah, C.Y.; Fowler, N.H. Idelalisib in the management of lymphoma. Blood 2016, 128, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
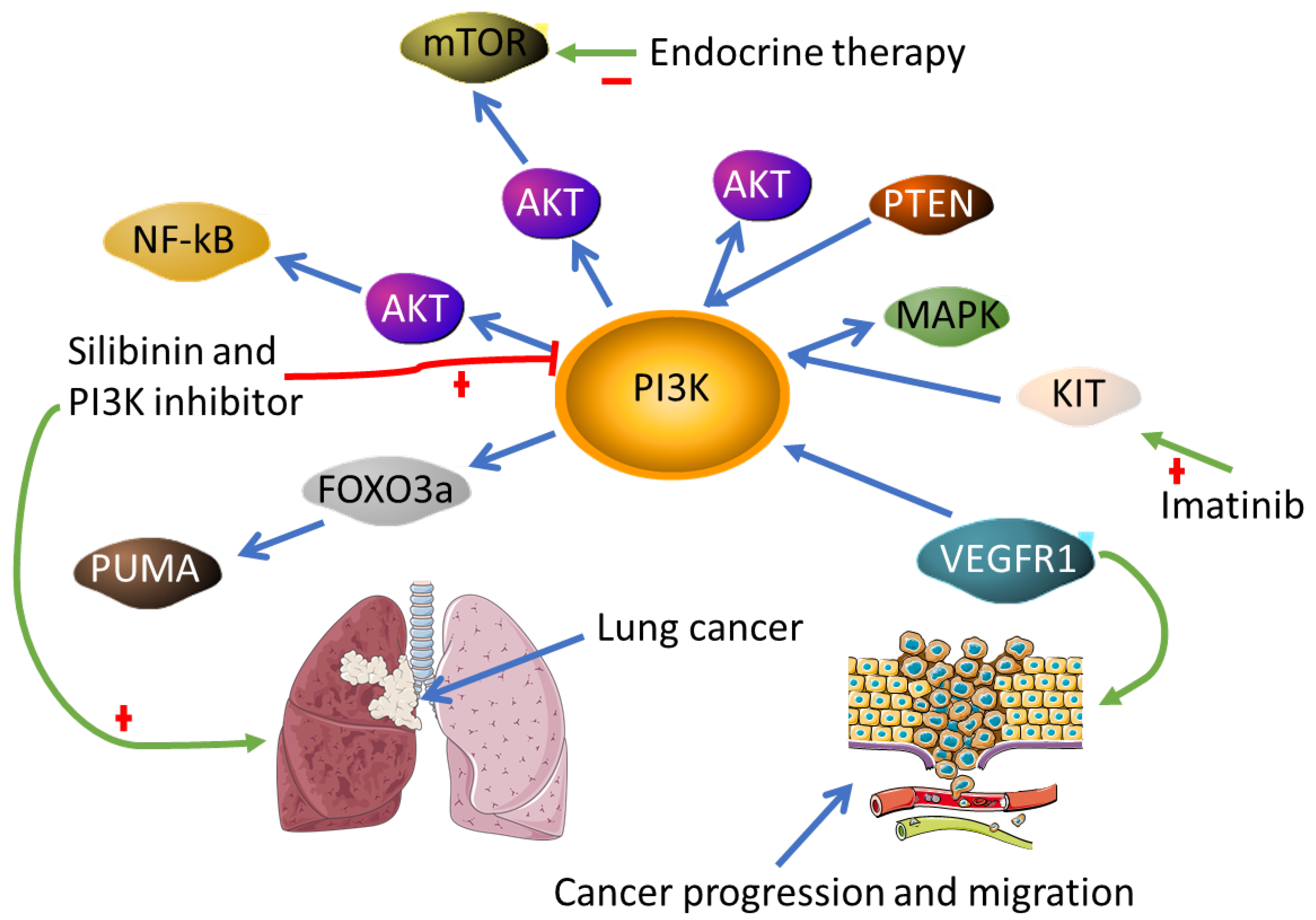
- Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Yi, J. Oxymatrine induces nasopharyngeal cancer cell death through inhibition of PI3K/AKT and NF-κB pathways. Mol. Med. Rep. 2017, 16, 9701–9706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagne, A.; Melkamu, T.; Schutten, M.M.; Qian, X.; Upadhyaya, P.; Luo, X.; Kassie, F. Enhanced inhibition of lung adenocarcinoma by combinatorial treatment with indole-3-carbinol and silibinin in A/J mice. Carcinogenesis 2011, 32, 561–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharm. 2021, 12, 628690. [Google Scholar] [CrossRef] [PubMed]
- Millis, S.Z.; Ikeda, S.; Reddy, S.; Gatalica, Z.; Kurzrock, R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors. JAMA Oncol. 2016, 2, 1565–1573. [Google Scholar] [CrossRef] [Green Version]
- Weddell, J.C.; Chen, S.; Imoukhuede, P.I. VEGFR1 promotes cell migration and proliferation through PLCγ and PI3K pathways. NPJ Syst. Biol. Appl. 2017, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Hou, Q.; Yan, W.; Luo, J.; Chen, D.; Liu, Z.; He, S.; Ding, X. PIK3CA is critical for the proliferation, invasiveness, and drug resistance of human tongue carcinoma cells. Oncol Res. 2011, 19, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhu, J.; Zhong, Y.; Geng, R.; Ji, Y.; Guan, Q.; Hong, C.; Wei, Y.; Min, N.; Qi, A.; et al. PIK3CA mutation confers resistance to chemotherapy in triple-negative breast cancer by inhibiting apoptosis and activating the PI3K/AKT/mTOR signaling pathway. Ann. Transl. Med. 2021, 9, 410. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Ma, S.; Che, K.; Pobbati, A.V.; Rubin, B.P. Inhibition of PI3K and MAPK pathways along with KIT inhibitors as a strategy to overcome drug resistance in gastrointestinal stromal tumors. PLoS ONE 2021, 16, e0252689. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; You, H.; Yang, Y.; Wei, L.; Zhang, X.; Yao, L.; Fan, D.; Yu, Q. Distinctive regulation and function of PI 3K/Akt and MAPKs in doxorubicin-induced apoptosis of human lung adenocarcinoma cells. J. Cell Biochem. 2004, 91, 621–632. [Google Scholar] [CrossRef]
- Yan, J.; Yang, S.; Tian, H.; Zhang, Y.; Zhao, H. Copanlisib promotes growth inhibition and apoptosis by modulating the AKT/FoxO3a/PUMA axis in colorectal cancer. Cell Death Dis. 2020, 11, 943. [Google Scholar] [CrossRef]
- Mezynski, M.J.; Farrelly, A.M.; Cremona, M.; Carr, A.; Morgan, C.; Workman, J.; Armstrong, P.; McAuley, J.; Madden, S.; Fay, J.; et al. Targeting the PI3K and MAPK pathways to improve response to HER2-targeted therapies in HER2-positive gastric cancer. J. Transl Med. 2021, 19, 184. [Google Scholar] [CrossRef]
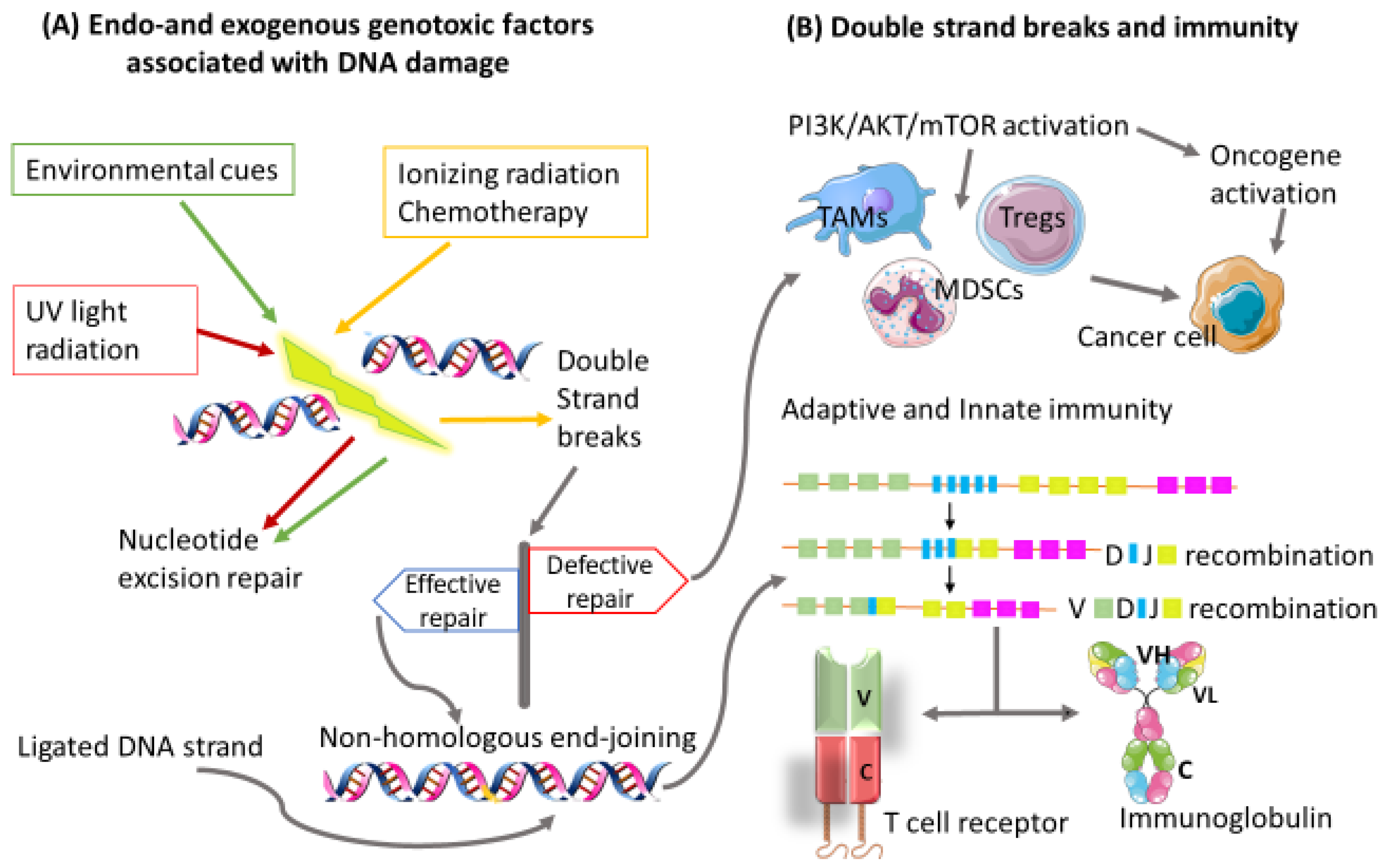
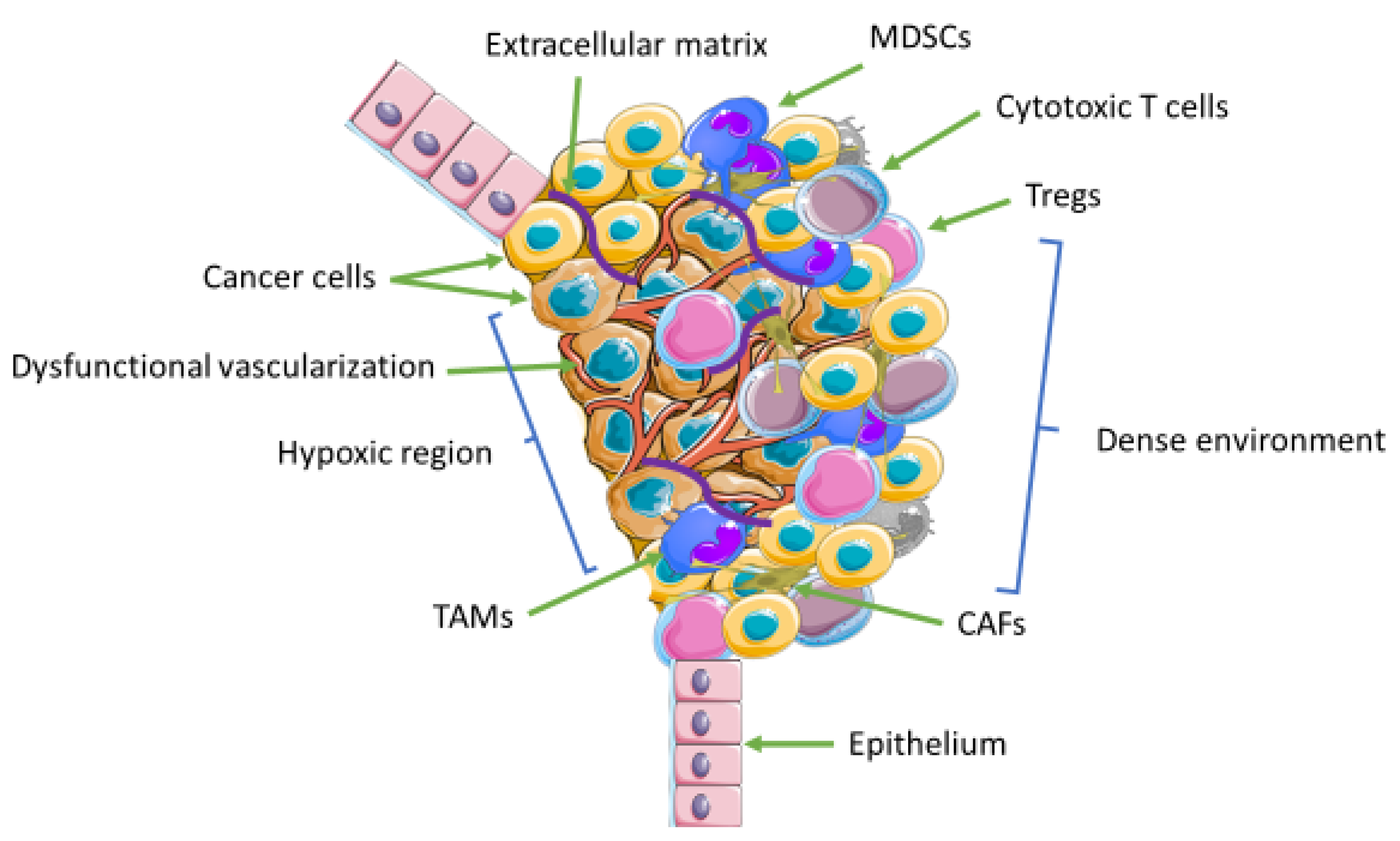
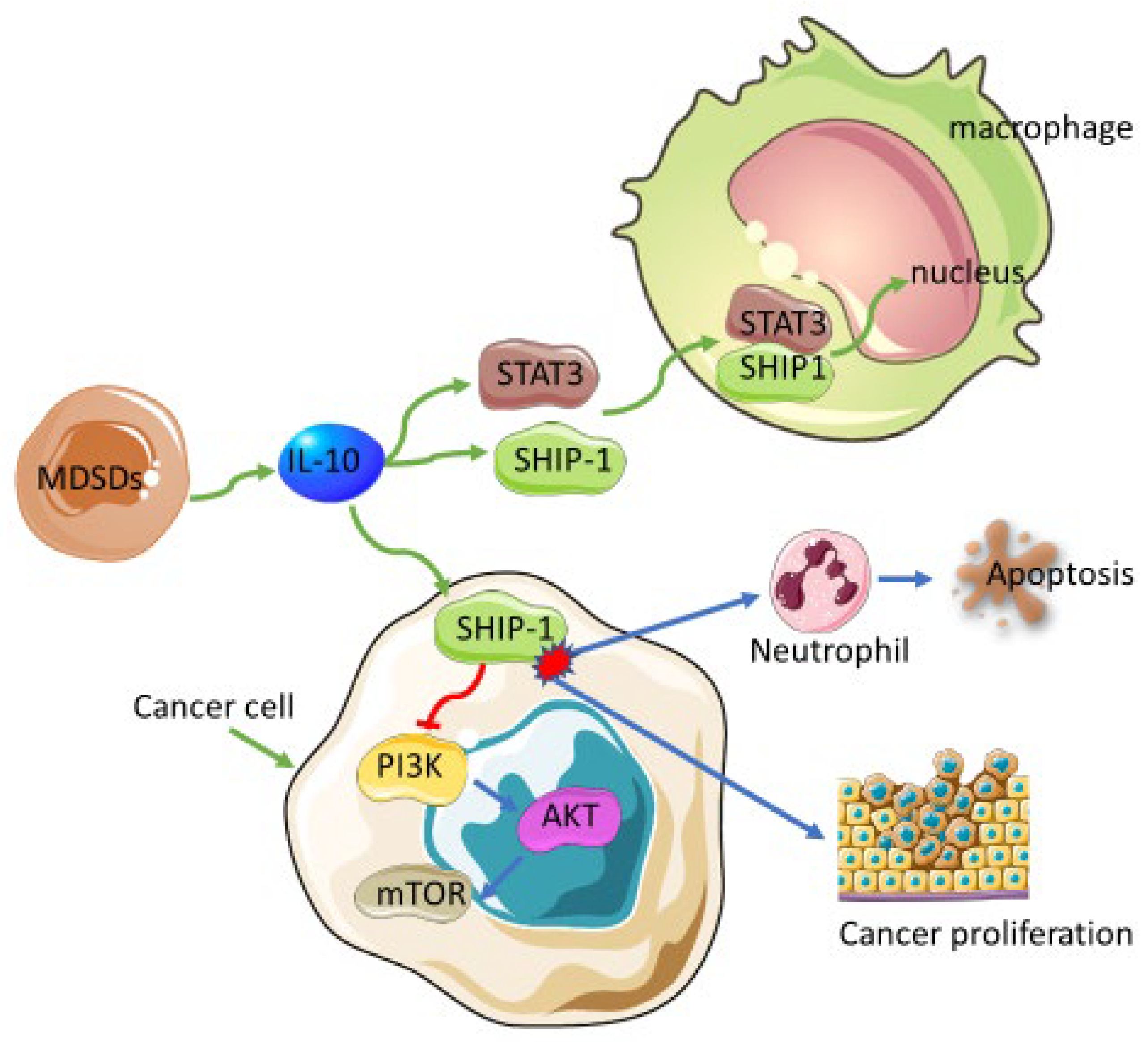
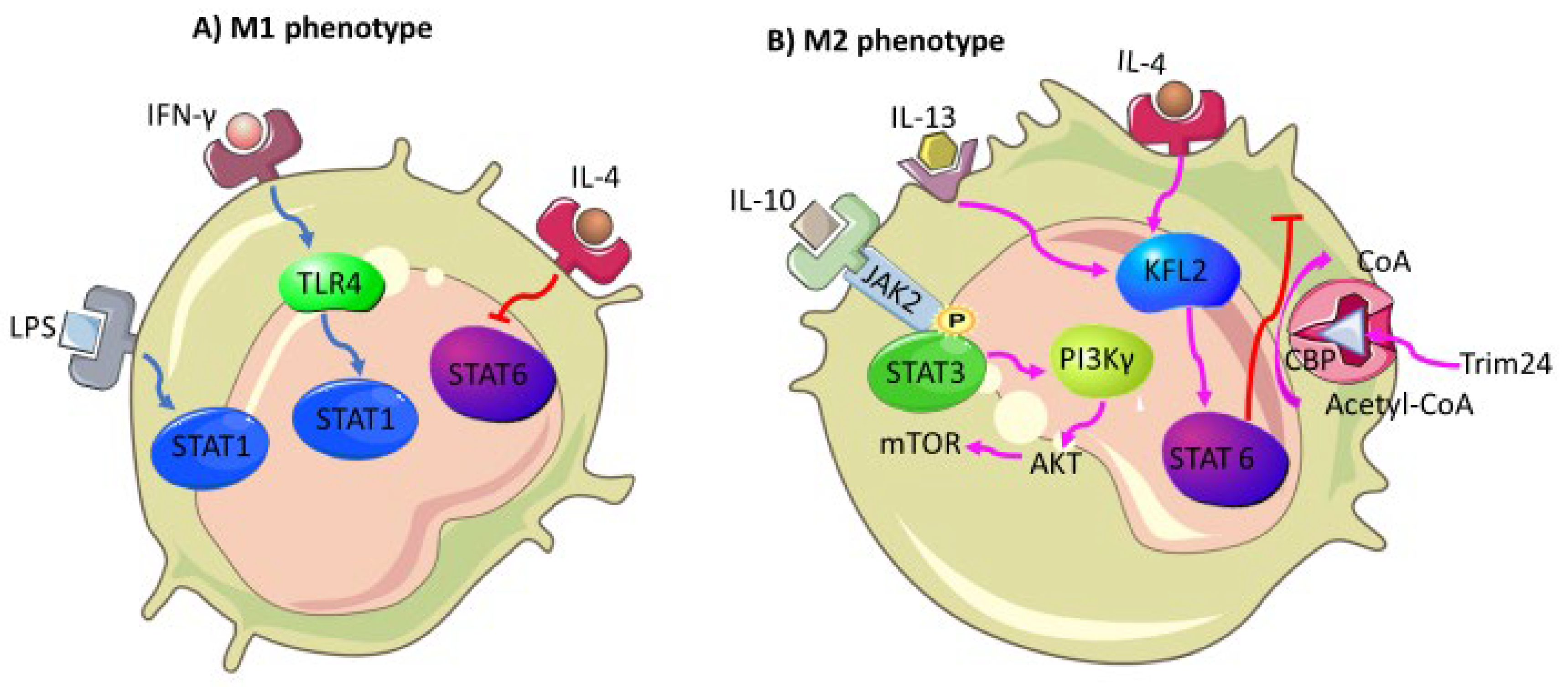
 -Inhibition.
-Inhibition.
-Inhibition.
-Inhibition.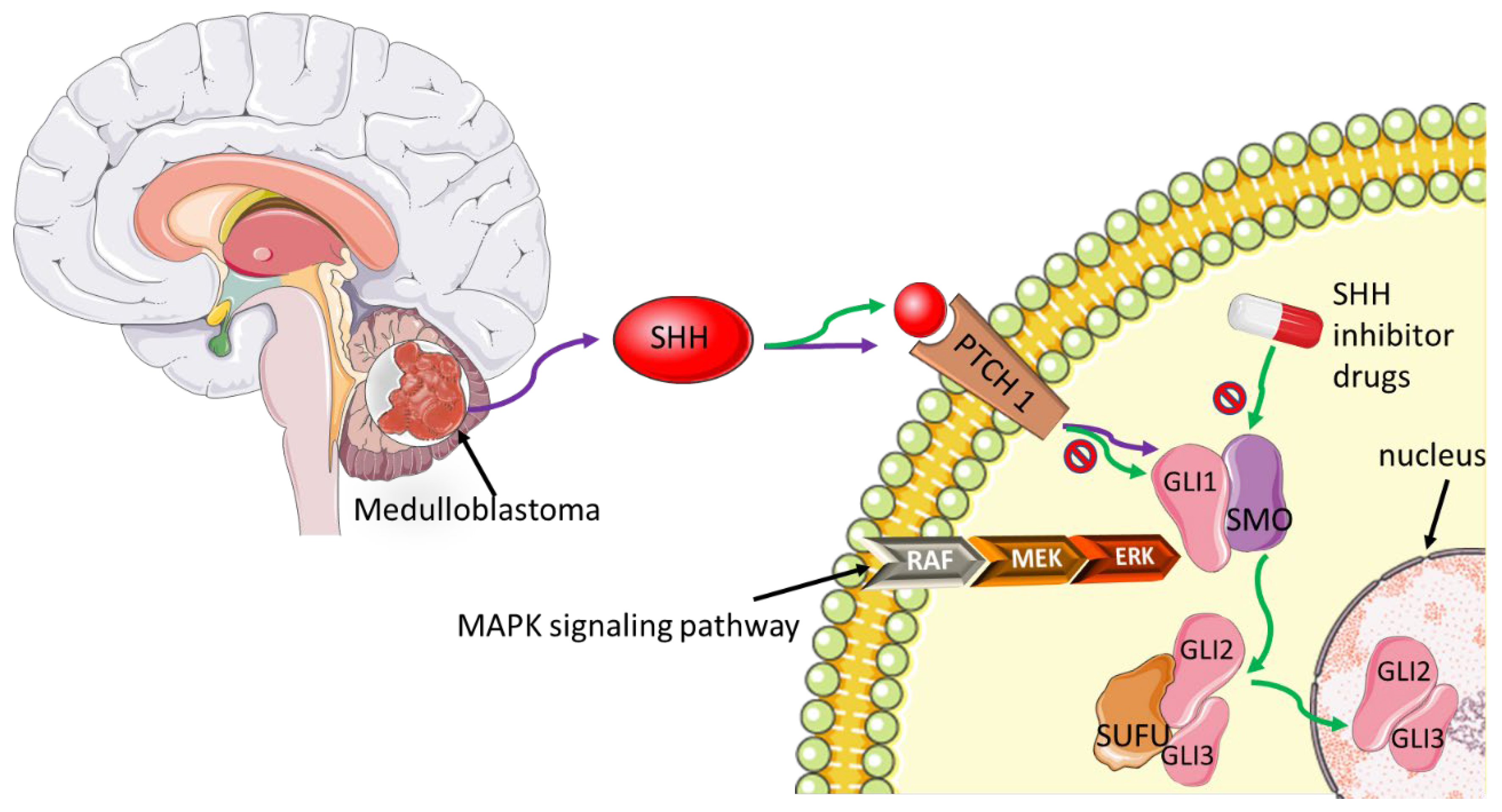

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PI3K Inhibitor | Mode of Action | Cancer Type | References |
|---|---|---|---|
| Alpelisib | PIK3CA/PI3K-δ isoform | Hormone receptor +/HER2-Breast Cancer | [131,132] |
| Copanlisib | PI3Kβ, PI3Kγ, PI3K-α & PI3K-δ isoforms | Follicular Lymphoma | [134] |
| Duvelisib | PI3K-δ and PI3K γ isoforms | Chronic Lymphocytic Leukemia | [135] |
| Idelalisib | PI3Kδ | Chronic Lymphocytic Leukemia | [136] |
| Buparlisib | PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ | Metastatic triple-negative breast and colorectal cancers | [128,129] |
| Pictilisib | PI3Kα and PI3Kδ | Advanced breast cancer and cancer of the bone | [130,133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Setlai, B.P.; Hull, R.; Bida, M.; Durandt, C.; Mulaudzi, T.V.; Chatziioannou, A.; Dlamini, Z. Immunosuppressive Signaling Pathways as Targeted Cancer Therapies. Biomedicines 2022, 10, 682. https://doi.org/10.3390/biomedicines10030682
Setlai BP, Hull R, Bida M, Durandt C, Mulaudzi TV, Chatziioannou A, Dlamini Z. Immunosuppressive Signaling Pathways as Targeted Cancer Therapies. Biomedicines. 2022; 10(3):682. https://doi.org/10.3390/biomedicines10030682
Chicago/Turabian StyleSetlai, Botle Precious, Rodney Hull, Meshack Bida, Chrisna Durandt, Thanyani Victor Mulaudzi, Aristotelis Chatziioannou, and Zodwa Dlamini. 2022. "Immunosuppressive Signaling Pathways as Targeted Cancer Therapies" Biomedicines 10, no. 3: 682. https://doi.org/10.3390/biomedicines10030682
APA StyleSetlai, B. P., Hull, R., Bida, M., Durandt, C., Mulaudzi, T. V., Chatziioannou, A., & Dlamini, Z. (2022). Immunosuppressive Signaling Pathways as Targeted Cancer Therapies. Biomedicines, 10(3), 682. https://doi.org/10.3390/biomedicines10030682