
The Epigenetics of Psychosis: A Structured Review with Representative Loci


Abstract
1. Introduction
2. MCHR1
2.1. Background for Environmental Impacts on the MCH System and Findings Relevant to Psychosis
2.2. Genetic Linkage and Differential Methylation of MCHR1 Identified for Schizophrenia and Bipolar Disorder with Psychosis


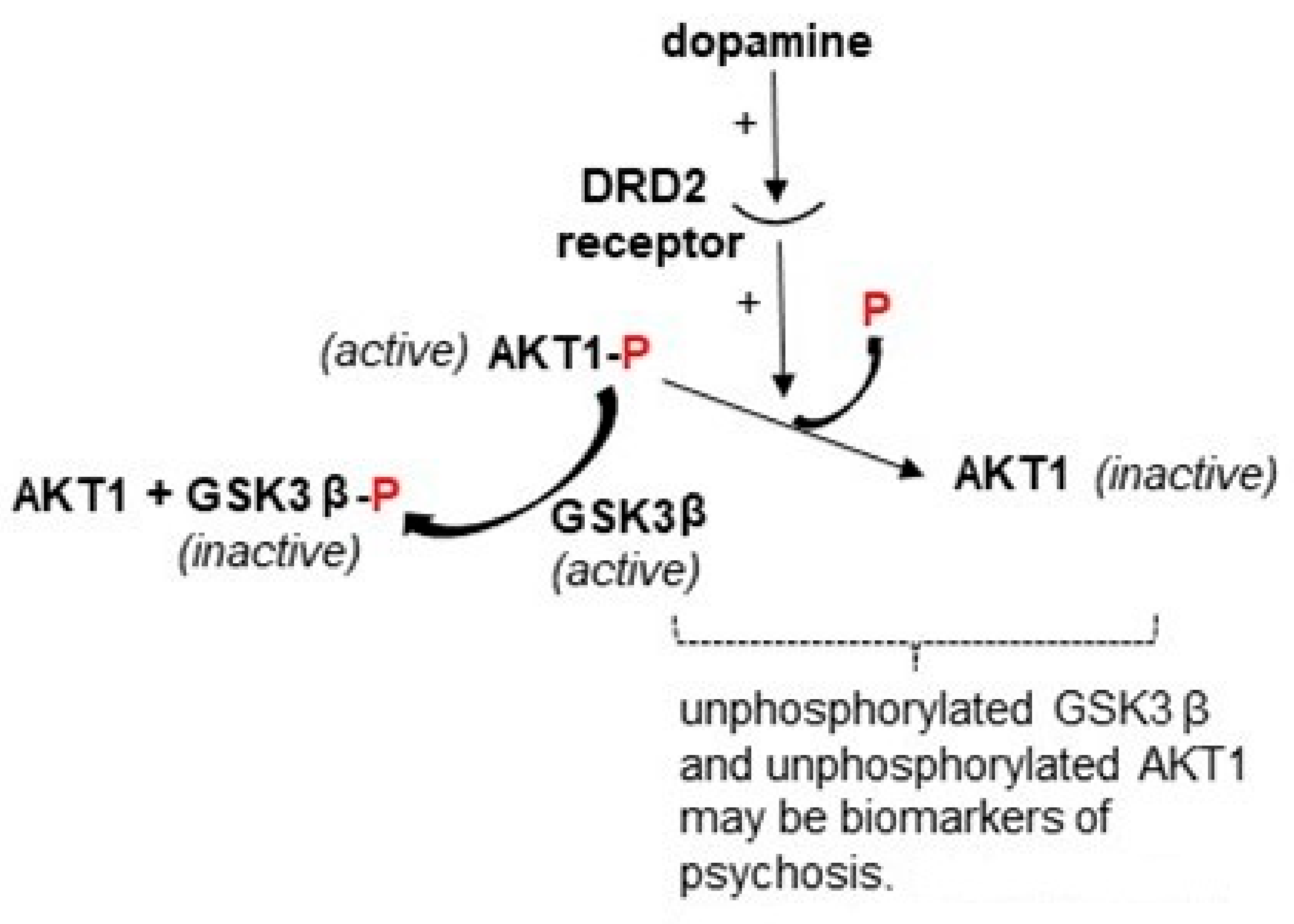
3. AKT1
3.1. Background on Cannabis-Induced Psychosis and the AKT1 Gene as a Model System for This Environmental Effect
3.2. Genetic Linkage of AKT1 to Psychotic Disorders, Gene-Environment Interaction with Cannabis Use and Potentially Relevant Methylation of AKT1 Identified

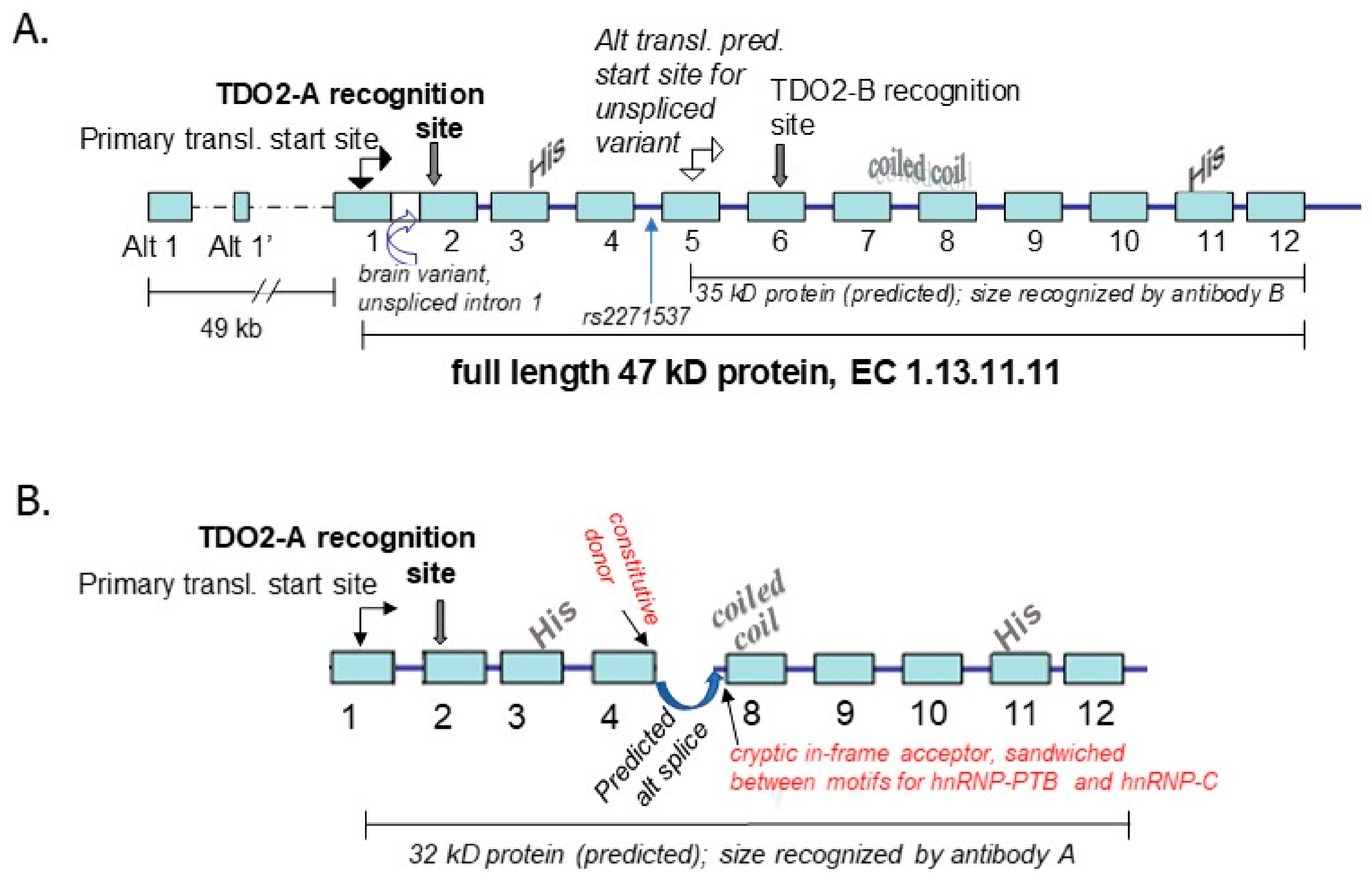
4. TDO2
4.1. Background on Relevance of the Kynurenine Pathway to Psychosis and the Response of the TDO2 Gene to Relevant Environmental Stimuli

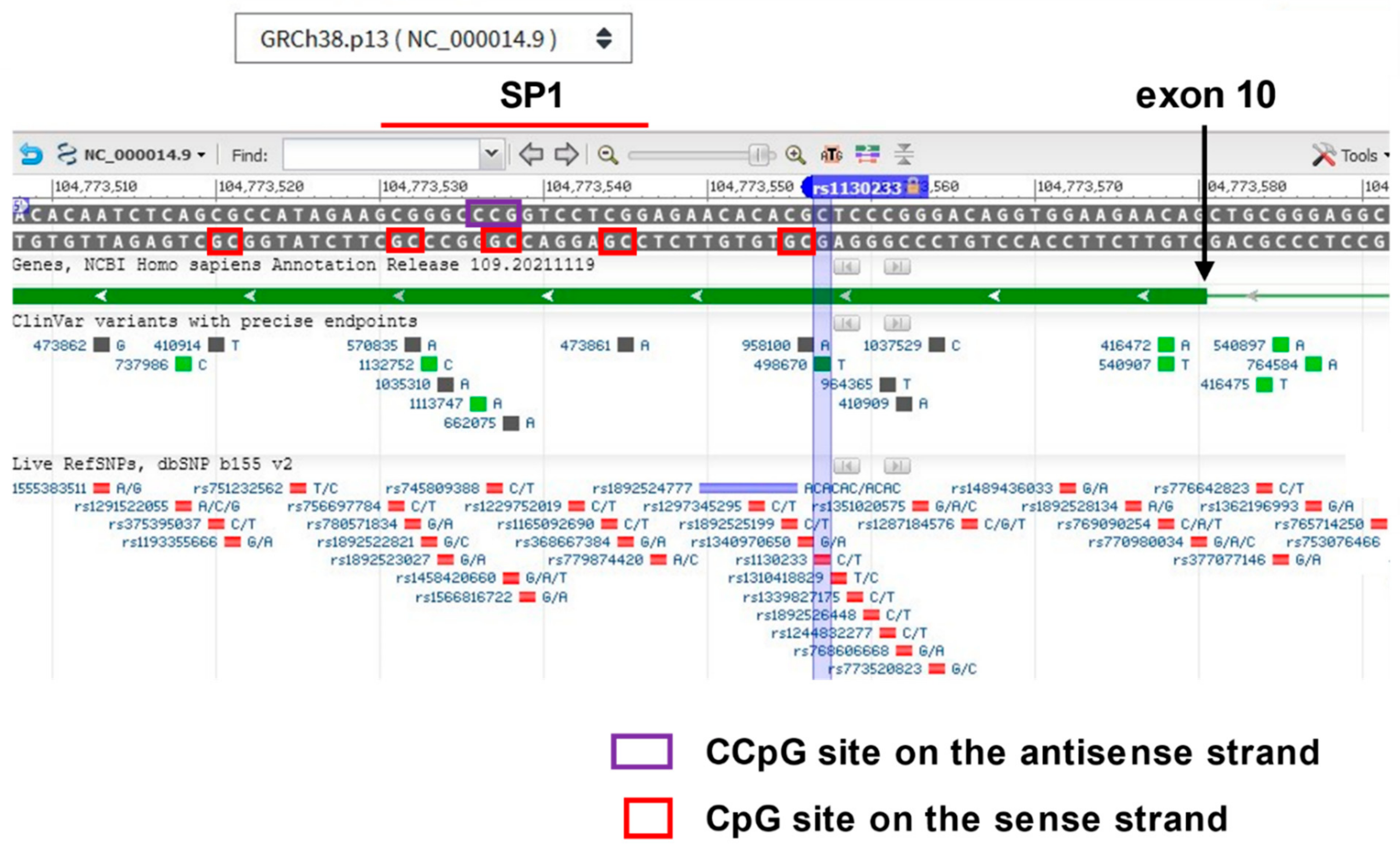
4.2. Genetic Association Study Results for TDO2 in Schizophrenia and Bipolar Disorder with Psychosis, Key Regulatory Sites within the Gene, and Potentially Relevant Methylation Induced by Stress

5. Discussion
6. Acronyms, Their Description and Any Associated Websites Used in This Review
Funding
Conflicts of Interest
References
- Gottesman, I.I.; Shields, J. A Polygenic Theory of Schizophrenia. Proc. Natl. Acad. Sci. USA 1967, 58, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, P.B.; Pedersen, C.B.; Westergaard, T.; Wohlfahrt, J.; Ewald, H.; Mors, O.; Andersen, P.K.; Melbye, M. Effects of Family History and Place and Season of Birth on the Risk of Schizophrenia. N. Engl. J. Med. 1999, 340, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Boydell, J.; Dean, K.; Dutta, R.; Giouroukou, E.; Fearon, P.; Murray, R. A Comparison of Symptoms and Family History in Schizophrenia with and without Prior Cannabis Use: Implications for the Concept of Cannabis Psychosis. Schizophr. Res. 2007, 93, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Ruhrmann, S.; Schultze-Lutter, F.; Salokangas, R.K.R.; Heinimaa, M.; Linszen, D.; Dingemans, P.; Birchwood, M.; Patterson, P.; Juckel, G.; Heinz, A.; et al. Prediction of Psychosis in Adolescents and Young Adults at High Risk: Results from the Prospective European Prediction of Psychosis Study. Arch. Gen. Psychiatry 2010, 67, 241–251. [Google Scholar] [CrossRef]
- Giordano, G.N.; Ohlsson, H.; Sundquist, K.; Sundquist, J.; Kendler, K.S. The Association between Cannabis Abuse and Subsequent Schizophrenia: A Swedish National Co-Relative Control Study. Psychol. Med. 2015, 45, 407–414. [Google Scholar] [CrossRef]
- Calafato, M.S.; Thygesen, J.H.; Ranlund, S.; Zartaloudi, E.; Cahn, W.; Crespo-Facorro, B.; Díez-Revuelta, Á.; Di Forti, M.; Genetic Risk and Outcome of Psychosis (GROUP) consortium; Hall, M.H.; et al. Use of Schizophrenia and Bipolar Disorder Polygenic Risk Scores to Identify Psychotic Disorders. Br. J. Psychiatry 2018, 213, 535–541. [Google Scholar] [CrossRef]
- Walser, J.C.; Furano, A.V. The Mutational Spectrum of Non-CpG DNA Varies with CpG Content. Genome. Res. 2010, 20, 875–882. [Google Scholar] [CrossRef]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute Exercise Remodels Promoter Methylation in Human Skeletal Muscle. Cell. Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef]
- Almouzni, G.; Cedar, H. Maintenance of Epigenetic Information. Cold Spring Harb. Perspect. Biol. 2016, 8, a019372. [Google Scholar] [CrossRef]
- Bohacek, J.; Mansuy, I.M. A Guide to Designing Germline-Dependent Epigenetic Inheritance Experiments in Mammals. Nat. Methods 2017, 14, 243–249. [Google Scholar] [CrossRef]
- Radford, E.J. Exploring the Extent and Scope of Epigenetic Inheritance. Nat. Rev. Endocrinol. 2018, 14, 345–355. [Google Scholar] [CrossRef]
- Zhang, S.; Li, B.; Du, K.; Liang, T.; Dai, M.; Huang, W.; Zhang, H.; Ling, Y.; Zhang, H. Epigenetically Modified N6-Methyladenine Inhibits DNA Replication by Human DNA Polymerase Iota. Biochimie 2020, 168, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Schuebel, K.; Gitik, M.; Domschke, K.; Goldman, D. Making Sense of Epigenetics. Int. J. Neuropsychopharmacol. 2016, 19, pyw058. [Google Scholar] [CrossRef]
- Lewis, C.A.; Crayle, J.; Zhou, S.; Swanstrom, R.; Wolfenden, R. Cytosine Deamination and the Precipitous Decline of Spontaneous Mutation during Earth’s History. Proc. Natl. Acad. Sci. USA 2016, 113, 8194–8199. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y. TET-Mediated Active DNA Demethylation: Mechanism, Function and Beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA Methylation of the First Exon Is Tightly Linked to Transcriptional Silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Relton, C.L. Statistical and Integrative System-Level Analysis of DNA Methylation Data. Nat. Rev. Genet. 2018, 19, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gao, Y.; Wu, Q.; Cheng, A.S.L.; Yip, K.Y. New Guidelines for DNA Methylome Studies Regarding 5-Hydroxymethylcytosine for Understanding Transcriptional Regulation. Genome Res. 2019, 29, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Lewis, S.; Carroll, T.; Menon, S.; Nicholson, A.; Manasterski, P.J.; Winton, D.J.; Buczacki, S.J.A.; Murrell, A. 5-Hydroxymethylcytosine and Gene Activity in Mouse Intestinal Differentiation. Sci. Rep. 2020, 10, 546. [Google Scholar] [CrossRef] [PubMed]
- Montano, C.; Taub, M.A.; Jaffe, A.; Briem, E.; Feinberg, J.I.; Trygvadottir, R.; Idrizi, A.; Runarsson, A.; Berndsen, B.; Gur, R.C.; et al. Association of DNA Methylation Differences With Schizophrenia in an Epigenome-Wide Association Study. JAMA Psychiatry 2016, 73, 506–514. [Google Scholar] [CrossRef]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-Wide Isoform-Level Dysregulation in ASD, Schizophrenia, and Bipolar Disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef] [PubMed]
- Kebir, O.; Chaumette, B.; Rivollier, F.; Miozzo, F.; Lemieux Perreault, L.P.; Barhdadi, A.; Provost, S.; Plaze, M.; Bourgin, J.; ICAAR team; et al. Methylomic Changes during Conversion to Psychosis. Mol. Psychiatry 2017, 22, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Tomassi, S.; Tosato, S. Epigenetics and Gene Expression Profile in First-Episode Psychosis: The Role of Childhood Trauma. Neurosci. Biobehav. Rev. 2017, 83, 226–237. [Google Scholar] [CrossRef]
- Hannon, E.; Dempster, E.L.; Mansell, G.; Burrage, J.; Bass, N.; Bohlken, M.M.; Corvin, A.; Curtis, C.J.; Dempster, D.; Di Forti, M.; et al. DNA Methylation Meta-Analysis Reveals Cellular Alterations in Psychosis and Markers of Treatment-Resistant Schizophrenia. eLife 2021, 10, e58430. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.I.; Ball, J.N. Evidence for a Dual Pituitary Control of Teleost Melanophores. Gen. Comp. Endocrinol. 1975, 25, 147–152. [Google Scholar] [CrossRef]
- Kishida, M.; Baker, B.I.; Bird, D.J. Localisation and Identification of Melanocyte-Stimulating Hormones in the Fish Brain. Gen. Comp. Endocrinol. 1988, 71, 229–242. [Google Scholar] [CrossRef]
- Castrucci, A.M.; Hadley, M.E.; Lebl, M.; Zechel, C.; Hruby, V.J. Melanocyte Stimulating Hormone and Melanin Concentration Hormone May Be Structurally and Evolutionarily Related. Regul. Pept. 1989, 24, 27–35. [Google Scholar] [CrossRef]
- Nahon, J.L.; Presse, F.; Bittencourt, J.C.; Sawchenko, P.E.; Vale, W. The Rat Melanin-Concentrating Hormone Messenger Ribonucleic Acid Encodes Multiple Putative Neuropeptides Coexpressed in the Dorsolateral Hypothalamus. Endocrinology 1989, 125, 2056–2065. [Google Scholar] [CrossRef]
- Mercer, J.G.; Moar, K.M.; Ross, A.W.; Hoggard, N.; Morgan, P.J. Photoperiod Regulates Arcuate Nucleus POMC, AGRP, and Leptin Receptor MRNA in Siberian Hamster Hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R271–R281. [Google Scholar] [CrossRef]
- Clarke, I.J.; Rao, A.; Chilliard, Y.; Delavaud, C.; Lincoln, G.A. Photoperiod Effects on Gene Expression for Hypothalamic Appetite-Regulating Peptides and Food Intake in the Ram. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R101–R115. [Google Scholar] [CrossRef][Green Version]
- Pereira-da-Silva, M.; Torsoni, M.A.; Nourani, H.V.; Augusto, V.D.; Souza, C.T.; Gasparetti, A.L.; Carvalheira, J.B.; Ventrucci, G.; Marcondes, M.C.C.G.; Cruz-Neto, A.P.; et al. Hypothalamic Melanin-Concentrating Hormone Is Induced by Cold Exposure and Participates in the Control of Energy Expenditure in Rats. Endocrinology 2003, 144, 4831–4840. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nahon, J.-L. The Melanocortins and Melanin-Concentrating Hormone in the Central Regulation of Feeding Behavior and Energy Homeostasis. C. R. Biol. 2006, 329, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Presse, F.; Conductier, G.; Rovere, C.; Nahon, J.-L. The Melanin-Concentrating Hormone Receptors: Neuronal and Non-Neuronal Functions. Int. J. Obes. Suppl. 2014, 4, S31–S36. [Google Scholar] [CrossRef] [PubMed]
- Anukulkitch, C.; Rao, A.; Dunshea, F.R.; Clarke, I.J. A Test of the Lipostat Theory in a Seasonal (Ovine) Model under Natural Conditions Reveals a Close Relationship between Adiposity and Melanin Concentrating Hormone Expression. Domest. Anim. Endocrinol. 2009, 36, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Henningsen, J.B.; Scheele, C. Brown Adipose Tissue: A Metabolic Regulator in a Hypothalamic Cross Talk? Annu. Rev. Physiol. 2021, 83, 279–301. [Google Scholar] [CrossRef]
- Balber, T.; Benčurová, K.; Kiefer, F.W.; Kulterer, O.C.; Klebermass, E.-M.; Egger, G.; Tran, L.; Wagner, K.-H.; Viernstein, H.; Pallitsch, K.; et al. In Vitro Radiopharmaceutical Evidence for MCHR1 Binding Sites in Murine Brown Adipocytes. Front Endocrinol. 2019, 10, 324. [Google Scholar] [CrossRef]
- Izawa, S.; Yoneshiro, T.; Kondoh, K.; Nakagiri, S.; Okamatsu-Ogura, Y.; Terao, A.; Minokoshi, Y.; Yamanaka, A.; Kimura, K. Melanin-Concentrating Hormone-Producing Neurons in the Hypothalamus Regulate Brown Adipose Tissue and Thus Contribute to Energy Expenditure. J. Physiol. 2021, 600, 815–827. [Google Scholar] [CrossRef]
- Glick, M.; Segal-Lieberman, G.; Cohen, R.; Kronfeld-Schor, N. Chronic MCH Infusion Causes a Decrease in Energy Expenditure and Body Temperature, and an Increase in Serum IGF-1 Levels in Mice. Endocrine 2009, 36, 479–485. [Google Scholar] [CrossRef]
- Arnold, V.K.; Rosenthal, T.L.; Dupont, R.T.; Hilliard, D. Redundant Clothing: A Readily Observable Marker for Schizophrenia in the Psychiatric Emergency Room Population. J. Behav. Exp. Psychiatry 1993, 24, 45–47. [Google Scholar] [CrossRef]
- Chong, T.W.H.; Castle, D.J. Layer upon Layer: Thermoregulation in Schizophrenia. Schizophr. Res. 2004, 69, 149–157. [Google Scholar] [CrossRef]
- Shiloh, R.; Weizman, A.; Stryjer, R.; Kahan, N.; Waitman, D.-A. Altered Thermoregulation in Ambulatory Schizophrenia Patients: A Naturalistic Study. World J. Biol. Psychiatry 2009, 10, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Mahintamani, T.; Ram, D.; Mitra, S. Inside the Mind of Poor Tom—A Multidimensional Approach to Determine Causes for Redundant Clothing in Patients with Schizophrenia. Psychiatry Res. 2015, 227, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Altschuler, E. Shakespeare Knew the Layered Clothing Sign of Schizophrenia. BMJ 1999, 319, 520. [Google Scholar] [CrossRef][Green Version]
- Komagata, N.; Latifi, B.; Rusterholz, T.; Bassetti, C.L.A.; Adamantidis, A.; Schmidt, M.H. Dynamic REM Sleep Modulation by Ambient Temperature and the Critical Role of the Melanin-Concentrating Hormone System. Curr. Biol. 2019, 29, 1976–1987.e4. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, S. Sleep Disturbances in Patients with Schizophrenia : Impact and Effect of Antipsychotics. CNS Drugs 2008, 22, 939–962. [Google Scholar] [CrossRef]
- Diniz, G.B.; Bittencourt, J.C. The Melanin-Concentrating Hormone as an Integrative Peptide Driving Motivated Behaviors. Front. Syst. Neurosci. 2017, 11, 32. [Google Scholar] [CrossRef]
- Berbari, N.F.; Johnson, A.D.; Lewis, J.S.; Askwith, C.C.; Mykytyn, K. Identification of Ciliary Localization Sequences within the Third Intracellular Loop of G Protein-Coupled Receptors. Mol. Biol. Cell 2008, 19, 1540–1547. [Google Scholar] [CrossRef]
- Diniz, G.B.; Battagello, D.S.; Klein, M.O.; Bono, B.S.M.; Ferreira, J.G.P.; Motta-Teixeira, L.C.; Duarte, J.C.G.; Presse, F.; Nahon, J.-L.; Adamantidis, A.; et al. Ciliary Melanin-Concentrating Hormone Receptor 1 (MCHR1) Is Widely Distributed in the Murine CNS in a Sex-Independent Manner. J. Neurosci. Res. 2020, 98, 2045–2071. [Google Scholar] [CrossRef]
- Williams, S.M.; Goldman-Rakic, P.S. Widespread Origin of the Primate Mesofrontal Dopamine System. Cereb. Cortex 1998, 8, 321–345. [Google Scholar] [CrossRef]
- Miller, C.L.; Hruby, V.J.; Matsunaga, T.O.; Bickford, P.C. Alpha-MSH and MCH Are Functional Antagonists in a CNS Auditory Gating Paradigm. Peptides 1993, 14, 431–440. [Google Scholar] [CrossRef]
- O’Hare, A.; Walsh, D.; Torrey, F. Seasonality of Schizophrenic Births in Ireland. Br. J. Psychiatry 1980, 137, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.; Welham, J.; Chant, D.; Torrey, E.F.; McGrath, J. A Systematic Review and Meta-Analysis of Northern Hemisphere Season of Birth Studies in Schizophrenia. Schizophr. Bull. 2003, 29, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L. Evidence for Phenotypic Plasticity in Response to Photic Cues and the Connection with Genes of Risk in Schizophrenia. Front. Behav. Neurosci. 2013, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Chant, D.C.; Welham, J.L.; McGrath, J.J. The Incidence and Prevalence of Schizophrenia Varies with Latitude. Acta Psychiatr. Scand. 2006, 114, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Ludewig, S.; Ludewig, K.; Geyer, M.A.; Hell, D.; Vollenweider, F.X. Prepulse Inhibition Deficits in Patients with Panic Disorder. Depress. Anxiety 2002, 15, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Borowsky, B.; Durkin, M.M.; Ogozalek, K.; Marzabadi, M.R.; DeLeon, J.; Lagu, B.; Heurich, R.; Lichtblau, H.; Shaposhnik, Z.; Daniewska, I.; et al. Antidepressant, Anxiolytic and Anorectic Effects of a Melanin-Concentrating Hormone-1 Receptor Antagonist. Nat. Med. 2002, 8, 825–830. [Google Scholar] [CrossRef]
- Chaki, S.; Shimazaki, T.; Nishiguchi, M.; Funakoshi, T.; Iijima, M.; Ito, A.; Kanuma, K.; Sekiguchi, Y. Antidepressant/Anxiolytic Potential and Adverse Effect Liabilities of Melanin-Concentrating Hormone Receptor 1 Antagonists in Animal Models. Pharm. Biochem. Behav. 2015, 135, 154–168. [Google Scholar] [CrossRef]
- Smith, D.G.; Davis, R.J.; Rorick-Kehn, L.; Morin, M.; Witkin, J.M.; McKinzie, D.L.; Nomikos, G.G.; Gehlert, D.R. Melanin-Concentrating Hormone-1 Receptor Modulates Neuroendocrine, Behavioral, and Corticolimbic Neurochemical Stress Responses in Mice. Neuropsychopharmacology 2006, 31, 1135–1145. [Google Scholar] [CrossRef]
- Roy, M.; David, N.; Cueva, M.; Giorgetti, M. A Study of the Involvement of Melanin-Concentrating Hormone Receptor 1 (MCHR1) in Murine Models of Depression. Biol. Psychiatry 2007, 61, 174–180. [Google Scholar] [CrossRef]
- Vawter, M.P.; Schulmann, A.; Alhassen, L.; Alhassen, W.; Hamzeh, A.R.; Sakr, J.; Pauluk, L.; Yoshimura, R.; Wang, X.; Dai, Q.; et al. Melanin Concentrating Hormone Signaling Deficits in Schizophrenia: Association With Memory and Social Impairments and Abnormal Sensorimotor Gating. Int. J. Neuropsychopharmacol. 2020, 23, 53–65. [Google Scholar] [CrossRef]
- Oranje, B.; Geyer, M.A.; Bocker, K.B.E.; Leon Kenemans, J.; Verbaten, M.N. Prepulse Inhibition and P50 Suppression: Commonalities and Dissociations. Psychiatry Res. 2006, 143, 147–158. [Google Scholar] [CrossRef]
- Severinsen, J.E.; Als, T.D.; Binderup, H.; Kruse, T.A.; Wang, A.G.; Vang, M.; Muir, W.J.; Blackwood, D.H.R.; Mors, O.; Børglum, A.D. Association Analyses Suggest GPR24 as a Shared Susceptibility Gene for Bipolar Affective Disorder and Schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Curtin, L.; D’Andrea, W.J.; Balascio, N.L.; Shirazi, S.; Shapiro, B.; de Wet, G.A.; Bradley, R.S.; Bakke, J. Sedimentary DNA and Molecular Evidence for Early Human Occupation of the Faroe Islands. Commun. Earth. Env. 2021, 2, 1–7. [Google Scholar] [CrossRef]
- Zhu, W.G.; Srinivasan, K.; Dai, Z.; Duan, W.; Druhan, L.J.; Ding, H.; Yee, L.; Villalona-Calero, M.A.; Plass, C.; Otterson, G.A. Methylation of Adjacent CpG Sites Affects Sp1/Sp3 Binding and Activity in the P21(Cip1) Promoter. Mol. Cell Biol. 2003, 23, 4056–4065. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Murakami, P.; Ruczinski, I.; Ross, R.G.; Sinkus, M.; Sullivan, B.; Leonard, S. Two Complex Genotypes Relevant to the Kynurenine Pathway and Melanotropin Function Show Association with Schizophrenia and Bipolar Disorder. Schizophr. Res. 2009, 113, 259–267. [Google Scholar] [CrossRef]
- Stepanow, S.; Reichwald, K.; Huse, K.; Gausmann, U.; Nebel, A.; Rosenstiel, P.; Wabitsch, M.; Fischer-Posovszky, P.; Platzer, M. Allele-Specific, Age-Dependent and BMI-Associated DNA Methylation of Human MCHR1. PLoS ONE 2011, 6, e17711. [Google Scholar] [CrossRef]
- Demontis, D.; Nyegaard, M.; Christensen, J.H.; Severinsen, J.; Hedemand, A.; Hansen, T.; Werge, T.; Mors, O.; Børglum, A.D. The Gene Encoding the Melanin-Concentrating Hormone Receptor 1 Is Associated with Schizophrenia in a Danish Case-Control Sample. Psychiatr. Genet. 2012, 22, 62–69. [Google Scholar] [CrossRef]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological Insights from 108 Schizophrenia-Associated Genetic Loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef]
- Mullins, N.; Forstner, A.J.; O’Connell, K.S.; Coombes, B.; Coleman, J.R.I.; Qiao, Z.; Als, T.D.; Bigdeli, T.B.; Børte, S.; Bryois, J.; et al. Genome-Wide Association Study of More than 40,000 Bipolar Disorder Cases Provides New Insights into the Underlying Biology. Nat. Genet. 2021, 53, 817–829. [Google Scholar] [CrossRef]
- Dempster, E.L.; Pidsley, R.; Schalkwyk, L.C.; Owens, S.; Georgiades, A.; Kane, F.; Kalidindi, S.; Picchioni, M.; Kravariti, E.; Toulopoulou, T.; et al. Disease-Associated Epigenetic Changes in Monozygotic Twins Discordant for Schizophrenia and Bipolar Disorder. Hum. Mol. Genet. 2011, 20, 4786–4796. [Google Scholar] [CrossRef]
- Liu, J.; Siyahhan Julnes, P.; Chen, J.; Ehrlich, S.; Walton, E.; Calhoun, V.D. The Association of DNA Methylation and Brain Volume in Healthy Individuals and Schizophrenia Patients. Schizophr. Res. 2015, 169, 447–452. [Google Scholar] [CrossRef]
- Gao, L. Promoter CpG Island Hypermethylation in the Development of Cutaneous Melanoma. J. Natl. Cancer Inst. 2006, 98, 472–482. [Google Scholar]
- Lohr, J.B.; Flynn, K. Smoking and Schizophrenia. Schizophr. Res. 1992, 8, 93–102. [Google Scholar] [CrossRef]
- de Leon, J.; Diaz, F.J. A Meta-Analysis of Worldwide Studies Demonstrates an Association between Schizophrenia and Tobacco Smoking Behaviors. Schizophr. Res. 2005, 76, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Yang, Z.; Wong, A.; Pipinikas, C.P.; Jiao, Y.; Jones, A.; Anjum, S.; Hardy, R.; Salvesen, H.B.; Thirlwell, C.; et al. Correlation of Smoking-Associated DNA Methylation Changes in Buccal Cells With DNA Methylation Changes in Epithelial Cancer. JAMA Oncol. 2015, 1, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Lev Maor, G.; Yearim, A.; Ast, G. The Alternative Role of DNA Methylation in Splicing Regulation. Trends. Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Shayevitch, R.; Askayo, D.; Keydar, I.; Ast, G. The Importance of DNA Methylation of Exons on Alternative Splicing. RNA 2018, 24, 1351–1362. [Google Scholar] [CrossRef]
- Lunnon, K.; Hannon, E.; Smith, R.G.; Dempster, E.; Wong, C.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Kepa, A.; Schalkwyk, L.; et al. Variation in 5-Hydroxymethylcytosine across Human Cortex and Cerebellum. Genome Biol. 2016, 17, 27. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Kothari, S.; Hasse, S.; Kauser, S.; Lindsey, N.J.; Gibbons, N.C.J.; Hibberts, N.; Wood, J.M. In Situ and in Vitro Evidence for DCoH/HNF-1 Alpha Transcription of Tyrosinase in Human Skin Melanocytes. Biochem. Biophys. Res. Commun. 2003, 301, 610–616. [Google Scholar] [CrossRef]
- Xanthopoulos, K.G.; Prezioso, V.R.; Chen, W.S.; Sladek, F.M.; Cortese, R.; Darnell, J.E. The Different Tissue Transcription Patterns of Genes for HNF-1, C/EBP, HNF-3, and HNF-4, Protein Factors That Govern Liver-Specific Transcription. Proc. Natl. Acad. Sci. USA 1991, 88, 3807–3811. [Google Scholar] [CrossRef]
- Saito, Y.; Tetsuka, M.; Yue, L.; Kawamura, Y.; Maruyama, K. Functional Role of N-Linked Glycosylation on the Rat Melanin-Concentrating Hormone Receptor 1. FEBS Lett. 2003, 533, 29–34. [Google Scholar] [CrossRef]
- Tetsuka, M.; Saito, Y.; Imai, K.; Doi, H.; Maruyama, K. The Basic Residues in the Membrane-Proximal C-Terminal Tail of the Rat Melanin-Concentrating Hormone Receptor 1 Are Required for Receptor Function. Endocrinology 2004, 145, 3712–3723. [Google Scholar] [CrossRef] [PubMed]
- Belbasis, L.; Köhler, C.A.; Stefanis, N.; Stubbs, B.; van Os, J.; Vieta, E.; Seeman, M.V.; Arango, C.; Carvalho, A.F.; Evangelou, E. Risk Factors and Peripheral Biomarkers for Schizophrenia Spectrum Disorders: An Umbrella Review of Meta-Analyses. Acta Psychiatr. Scand. 2018, 137, 88–97. [Google Scholar] [CrossRef]
- Di Forti, M.; Marconi, A.; Carra, E.; Fraietta, S.; Trotta, A.; Bonomo, M.; Bianconi, F.; Gardner-Sood, P.; O’Connor, J.; Russo, M.; et al. Proportion of Patients in South London with First-Episode Psychosis Attributable to Use of High Potency Cannabis: A Case-Control Study. Lancet Psychiatry 2015, 2, 233–238. [Google Scholar] [CrossRef]
- Marconi, A.; Di Forti, M.; Lewis, C.M.; Murray, R.M.; Vassos, E. Meta-Analysis of the Association Between the Level of Cannabis Use and Risk of Psychosis. Schizophr. Bull. 2016, 42, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.C.; Perry, E.; MacDougall, L.; Ammerman, Y.; Cooper, T.; Wu, Y.-T.; Braley, G.; Gueorguieva, R.; Krystal, J.H. The Psychotomimetic Effects of Intravenous Delta-9-Tetrahydrocannabinol in Healthy Individuals: Implications for Psychosis. Neuropsychopharmacology 2004, 29, 1558–1572. [Google Scholar] [CrossRef]
- Morrison, P.D.; Nottage, J.; Stone, J.M.; Bhattacharyya, S.; Tunstall, N.; Brenneisen, R.; Holt, D.; Wilson, D.; Sumich, A.; McGuire, P.; et al. Disruption of Frontal θ Coherence by Δ9-Tetrahydrocannabinol Is Associated with Positive Psychotic Symptoms. Neuropsychopharmacology 2011, 36, 827–836. [Google Scholar] [CrossRef]
- Freeman, D.; Dunn, G.; Murray, R.M.; Evans, N.; Lister, R.; Antley, A.; Slater, M.; Godlewska, B.; Cornish, R.; Williams, J.; et al. How Cannabis Causes Paranoia: Using the Intravenous Administration of ∆9-Tetrahydrocannabinol (THC) to Identify Key Cognitive Mechanisms Leading to Paranoia. Schizophr. Bull. 2015, 41, 391–399. [Google Scholar] [CrossRef]
- Hjorthøj, C.; Posselt, C.M.; Nordentoft, M. Development Over Time of the Population-Attributable Risk Fraction for Cannabis Use Disorder in Schizophrenia in Denmark. JAMA Psychiatry 2021, 78, 1013–1019. [Google Scholar] [CrossRef]
- Skosnik, P.D.; Hajós, M.; Cortes-Briones, J.A.; Edwards, C.R.; Pittman, B.P.; Hoffmann, W.E.; Sewell, A.R.; D’Souza, D.C.; Ranganathan, M. Cannabinoid Receptor-Mediated Disruption of Sensory Gating and Neural Oscillations: A Translational Study in Rats and Humans. Neuropharmacology 2018, 135, 412–423. [Google Scholar] [CrossRef]
- Hajós, M.; Hoffmann, W.E.; Kocsis, B. Activation of Cannabinoid-1 Receptors Disrupts Sensory Gating and Neuronal Oscillation: Relevance to Schizophrenia. Biol. Psychiatry 2008, 63, 1075–1083. [Google Scholar] [CrossRef]
- Zachariou, M.; Dissanayake, D.W.N.; Coombes, S.; Owen, M.R.; Mason, R. Sensory Gating and Its Modulation by Cannabinoids: Electrophysiological, Computational and Mathematical Analysis. Cogn. Neurodyn. 2008, 2, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Atagun, M.I.; Drukker, M.; Hall, M.H.; Altun, I.K.; Tatli, S.Z.; Guloksuz, S.; van Os, J.; van Amelsvoort, T. Meta-Analysis of Auditory P50 Sensory Gating in Schizophrenia and Bipolar Disorder. Psychiatry Res. Neuroimaging 2020, 300, 111078. [Google Scholar] [CrossRef]
- Bloomfield, M.A.P.; Ashok, A.H.; Volkow, N.D.; Howes, O.D. The Effects of Δ9-Tetrahydrocannabinol on the Dopamine System. Nature 2016, 539, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Targeting the Dopamine D2 Receptor in Schizophrenia. Expert. Opin. Targets 2006, 10, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Gururajan, A.; Manning, E.E.; Klug, M.; van den Buuse, M. Drugs of Abuse and Increased Risk of Psychosis Development. Aust. N. Z. J. Psychiatry 2012, 46, 1120–1135. [Google Scholar] [CrossRef] [PubMed]
- Niemi-Pynttäri, J.A.; Sund, R.; Putkonen, H.; Vorma, H.; Wahlbeck, K.; Pirkola, S.P. Substance-Induced Psychoses Converting into Schizophrenia: A Register-Based Study of 18,478 Finnish Inpatient Cases. J. Clin. Psychiatry 2013, 74, 20155. [Google Scholar] [CrossRef]
- Ham, S.; Kim, T.K.; Chung, S.; Im, H.-I. Drug Abuse and Psychosis: New Insights into Drug-Induced Psychosis. Exp. Neurobiol. 2017, 26, 11–24. [Google Scholar] [CrossRef]
- Weidenauer, A.; Bauer, M.; Sauerzopf, U.; Bartova, L.; Nics, L.; Pfaff, S.; Philippe, C.; Berroterán-Infante, N.; Pichler, V.; Meyer, B.M.; et al. On the Relationship of First-Episode Psychosis to the Amphetamine-Sensitized State: A Dopamine D2/3 Receptor Agonist Radioligand Study. Transl. Psychiatry 2020, 10, 2. [Google Scholar] [CrossRef]
- Beaulieu, J.-M.; Sotnikova, T.D.; Marion, S.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. An Akt/Beta-Arrestin 2/PP2A Signaling Complex Mediates Dopaminergic Neurotransmission and Behavior. Cell 2005, 122, 261–273. [Google Scholar] [CrossRef]
- Emamian, E.S. AKT/GSK3 Signaling Pathway and Schizophrenia. Front. Mol. Neurosci. 2012, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Seillier, A.; Martinez, A.A.; Giuffrida, A. Differential Effects of Δ9-Tetrahydrocannabinol Dosing on Correlates of Schizophrenia in the Sub-Chronic PCP Rat Model. PLoS ONE 2020, 15, e0230238. [Google Scholar] [CrossRef]
- Emamian, E.S.; Hall, D.; Birnbaum, M.J.; Karayiorgou, M.; Gogos, J.A. Convergent Evidence for Impaired AKT1-GSK3beta Signaling in Schizophrenia. Nat. Genet. 2004, 36, 131–137. [Google Scholar] [CrossRef]
- Thiselton, D.L.; Vladimirov, V.I.; Kuo, P.-H.; McClay, J.; Wormley, B.; Fanous, A.; O’Neill, F.A.; Walsh, D.; Van den Oord, E.J.C.G.; Kendler, K.S.; et al. AKT1 Is Associated with Schizophrenia across Multiple Symptom Dimensions in the Irish Study of High Density Schizophrenia Families. Biol. Psychiatry 2008, 63, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P.N.; Ota, V.K.; Gouvea, E.S.; Pedrini, M.; Santoro, M.L.; Talarico, F.; Spindola, L.M.; Carvalho, C.M.; Noto, C.; Xavier, G.; et al. Accessing Gene Expression in Treatment-Resistant Schizophrenia. Mol. Neurobiol. 2018, 55, 7000–7008. [Google Scholar] [CrossRef]
- Nohesara, S.; Ghadirivasfi, M.; Barati, M.; Ghasemzadeh, M.-R.; Narimani, S.; Mousavi-Behbahani, Z.; Joghataei, M.; Soleimani, M.; Taban, M.; Mehrabi, S.; et al. Methamphetamine-Induced Psychosis Is Associated with DNA Hypomethylation and Increased Expression of AKT1 and Key Dopaminergic Genes. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Karege, F.; Perroud, N.; Schürhoff, F.; Méary, A.; Marillier, G.; Burkhardt, S.; Ballmann, E.; Fernandez, R.; Jamain, S.; Leboyer, M.; et al. A. Association of AKT1 Gene Variants and Protein Expression in Both Schizophrenia and Bipolar Disorder. Genes Brain Behav. 2010, 9, 503–511. [Google Scholar] [CrossRef]
- Chaumette, B.; Kebir, O.; Pouch, J.; Ducos, B.; Selimi, F.; ICAAR study group; Gaillard, R.; Krebs, M.-O. Longitudinal Analyses of Blood Transcriptome during Conversion to Psychosis. Schizophr. Bull. 2019, 45, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-Y.; Nicodemus, K.K.; Chen, Q.; Li, Z.; Brooke, J.K.; Honea, R.; Kolachana, B.S.; Straub, R.E.; Meyer-Lindenberg, A.; Sei, Y.; et al. Genetic Variation in AKT1 Is Linked to Dopamine-Associated Prefrontal Cortical Structure and Function in Humans. J. Clin. Investig. 2008, 118, 2200–2208. [Google Scholar] [CrossRef]
- Sanders, A.R.; Duan, J.; Levinson, D.F.; Shi, J.; He, D.; Hou, C.; Burrell, G.J.; Rice, J.P.; Nertney, D.A.; Olincy, A.; et al. No Significant Association of 14 Candidate Genes with Schizophrenia in a Large European Ancestry Sample: Implications for Psychiatric Genetics. Am. J. Psychiatry 2008, 165, 497–506. [Google Scholar] [CrossRef]
- Di Forti, M.; Iyegbe, C.; Sallis, H.; Kolliakou, A.; Falcone, M.A.; Paparelli, A.; Sirianni, M.; La Cascia, C.; Stilo, S.A.; Marques, T.R.; et al. Confirmation That the AKT1 (Rs2494732) Genotype Influences the Risk of Psychosis in Cannabis Users. Biol. Psychiatry 2012, 72, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Iyegbe, C.; Atakan, Z.; Martin-Santos, R.; Crippa, J.A.; Xu, X.; Williams, S.; Brammer, M.; Rubia, K.; Prata, D.; et al. Protein Kinase B (AKT1) Genotype Mediates Sensitivity to Cannabis-Induced Impairments in Psychomotor Control. Psychol. Med. 2014, 44, 3315–3328. [Google Scholar] [CrossRef]
- Slatkin, M. Linkage Disequilibrium—Understanding the Evolutionary Past and Mapping the Medical Future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M. Evolutionary Pressures in the Spread and Persistence of Infectious Agents in Vertebrate Populations. Parasitology 1995, 111, S15–S31. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, M.; Iyegbe, C.; Powell, J.; Blasi, G.; Bertolino, A.; Murray, R.M.; Di Forti, M. Interaction between DRD2 and AKT1 Genetic Variations on Risk of Psychosis in Cannabis Users: A Case-Control Study. NPJ Schizophr. 2015, 1, 15025. [Google Scholar] [CrossRef] [PubMed]
- Oetjens, M.T.; Brown-Gentry, K.; Goodloe, R.; Dilks, H.H.; Crawford, D.C. Population Stratification in the Context of Diverse Epidemiologic Surveys Sans Genome-Wide Data. Front. Genet. 2016, 7, 76. [Google Scholar] [CrossRef]
- Blest-Hopley, G.; Colizzi, M.; Prata, D.; Giampietro, V.; Brammer, M.; McGuire, P.; Bhattacharyya, S. Epigenetic Mediation of AKT1 Rs1130233′s Effect on Delta-9-Tetrahydrocannabinol-Induced Medial Temporal Function during Fear Processing. Brain Sci. 2021, 11, 1240. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, K.; Ngo, V.; Liu, C.; Fan, S.; Whitaker, J.W.; Chen, Y.; Ai, R.; Chen, Z.; Wang, J.; et al. Identification of DNA Motifs That Regulate DNA Methylation. Nucleic. Acids Res. 2019, 47, 6753–6768. [Google Scholar] [CrossRef]
- Clark, S.J.; Harrison, J.; Molloy, P.L. Sp1 Binding Is Inhibited by (m)Cp(m)CpG Methylation. Gene 1997, 195, 67–71. [Google Scholar] [CrossRef]
- Mancini, D.N.; Singh, S.M.; Archer, T.K.; Rodenhiser, D.I. Site-Specific DNA Methylation in the Neurofibromatosis (NF1) Promoter Interferes with Binding of CREB and SP1 Transcription Factors. Oncogene 1999, 18, 4108–4119. [Google Scholar] [CrossRef]
- Camchong, J.; Lim, K.O.; Kumra, S. Adverse Effects of Cannabis on Adolescent Brain Development: A Longitudinal Study. Cereb. Cortex 2017, 27, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
- Guida, F.; Sandanger, T.M.; Castagné, R.; Campanella, G.; Polidoro, S.; Palli, D.; Krogh, V.; Tumino, R.; Sacerdote, C.; Panico, S.; et al. Dynamics of Smoking-Induced Genome-Wide Methylation Changes with Time since Smoking Cessation. Hum. Mol. Genet. 2015, 24, 2349–2359. [Google Scholar] [CrossRef]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S.; et al. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef]
- Stueve, T.R.; Li, W.-Q.; Shi, J.; Marconett, C.N.; Zhang, T.; Yang, C.; Mullen, D.; Yan, C.; Wheeler, W.; Hua, X.; et al. Epigenome-Wide Analysis of DNA Methylation in Lung Tissue Shows Concordance with Blood Studies and Identifies Tobacco Smoke-Inducible Enhancers. Hum. Mol. Genet. 2017, 26, 3014–3027. [Google Scholar] [CrossRef] [PubMed]
- Ringh, M.V.; Hagemann-Jensen, M.; Needhamsen, M.; Kular, L.; Breeze, C.E.; Sjöholm, L.K.; Slavec, L.; Kullberg, S.; Wahlström, J.; Grunewald, J.; et al. Tobacco Smoking Induces Changes in True DNA Methylation, Hydroxymethylation and Gene Expression in Bronchoalveolar Lavage Cells. EBioMedicine 2019, 46, 290–304. [Google Scholar] [CrossRef]
- Davis, J.A.; James, J.R.; Siegel, S.J.; Gould, T.J. Withdrawal from Chronic Nicotine Administration Impairs Contextual Fear Conditioning in C57BL/6 Mice. J. Neurosci. 2005, 25, 8708–8713. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, M.G.; Gould, T.J. Nicotine Modulation of Fear Memories and Anxiety: Implications for Learning and Anxiety Disorders. Biochem. Pharm. 2015, 97, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Bryans, M.; Lucas, J.; Wilkie, N.; Burrow, K.; Clark, A.; Lang, J. Binding of Transcription Factor Sp1 to Exon-1 of C-Myc Is Altered in Burkitts-Lymphoma. Int. J. Oncol. 1992, 1, 175–180. [Google Scholar] [CrossRef]
- Lin, C.J.; Tam, R.C. Transcriptional Regulation of CD28 Expression by CD28GR, a Novel Promoter Element Located in Exon 1 of the CD28 Gene. J. Immunol. 2001, 166, 6134–6143. [Google Scholar] [CrossRef] [PubMed]
- Pierce, R.A.; Moore, C.H.; Arikan, M.C. Positive Transcriptional Regulatory Element Located within Exon 1 of Elastin Gene. Am. J. Physiol. Lung. Cell Mol. Physiol. 2006, 291, L391–L399. [Google Scholar] [CrossRef][Green Version]
- Caiafa, P.; Zampieri, M. DNA Methylation and Chromatin Structure: The Puzzling CpG Islands. J. Cell. Biochem. 2005, 94, 257–265. [Google Scholar] [CrossRef]
- Cho-Chung, Y.S.; Pitot, H.C. Feedback Control of Rat Liver Tryptophan Pyrrolase. I. End Product Inhibition of Trytophan Pyrrolase Activity. J. Biol. Chem. 1967, 242, 1192–1198. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan. Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Varo, N.; Alegre, E.; Díaz, A.; Melero, I. Immunosuppression Routed via the Kynurenine Pathway: A Biochemical and Pathophysiologic Approach. Adv. Clin. Chem. 2008, 45, 155–197. [Google Scholar] [CrossRef]
- Orhan, F. Deciphering the Interplay between Peripheral and Central Cytokine-and Kynurenine Pathways: Importance for the Pathophysiology of Schizophrenia; Karolinska Institutet: Stockholm, Sweden, 2018. [Google Scholar]
- van Crevel, R.; Ottenhoff, T.H.M.; van der Meer, J.W.M. Innate Immunity to Mycobacterium Tuberculosis. Clin. Microbiol. Rev. 2002, 15, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, E.R.; Rebhun, S.; Eckel, M. Characterization of an Indoleamine 2,3-Dioxygenase Induced by Gamma-Interferon in Cultured Human Fibroblasts. J. Interferon. Res. 1986, 6, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Behm, C.; Blufstein, A.; Gahn, J.; Kubin, B.; Moritz, A.; Rausch-Fan, X.; Andrukhov, O. Continuing Effect of Cytokines and Toll-Like Receptor Agonists on Indoleamine-2,3-Dioxygenase-1 in Human Periodontal Ligament Stem/Stromal Cells. Cells 2020, 9, 2696. [Google Scholar] [CrossRef] [PubMed]
- Hankes, L.V.; Leklem, J.E.; Brown, R.R.; Mekel, R.C. Tryptophan Metabolism in Patients with Pellagra: Problem of Vitamin B 6 Enzyme Activity and Feedback Control of Tryptophan Pyrrolase Enzyme. Am. J. Clin. Nutr. 1971, 24, 730–739. [Google Scholar] [CrossRef]
- Spivak, J.L.; Jackson, D.L. Pellagra: An Analysis of 18 Patients and a Review of the Literature. Johns Hopkins Med. J. 1977, 140, 295–309. [Google Scholar]
- Rapoport, M.I.; Beisel, W.R. Studies of Tryptophan Metabolism in Experimental Animals and Man during Infectious Illness. Am. J. Clin. Nutr. 1971, 24, 807–814. [Google Scholar] [CrossRef]
- Zerez, C.R.; Roth, E.F.; Schulman, S.; Tanaka, K.R. Increased Nicotinamide Adenine Dinucleotide Content and Synthesis in Plasmodium Falciparum-Infected Human Erythrocytes. Blood 1990, 75, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.; Ho, M.; Acharya, G.; Tiwari, M.; Gallati, H. Interleukin-6, Gamma Interferon, and Tumor Necrosis Factor Receptors in Typhoid Fever Related to Outcome of Antimicrobial Therapy. Antimicrob. Agents Chemother. 1993, 37, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Nagineni, C.N.; Pardhasaradhi, K.; Martins, M.C.; Detrick, B.; Hooks, J.J. Mechanisms of Interferon-Induced Inhibition of Toxoplasma Gondii Replication in Human Retinal Pigment Epithelial Cells. Infect. Immun. 1996, 64, 4188–4196. [Google Scholar] [CrossRef]
- Reinhard, J.F. Altered Tryptophan Metabolism in Mice with Herpes Simplex Virus Encephalitis: Increases in Spinal Cord Quinolinic Acid. Neurochem. Res. 1998, 23, 661–665. [Google Scholar] [CrossRef]
- Sanni, L.A.; Thomas, S.R.; Tattam, B.N.; Moore, D.E.; Chaudhri, G.; Stocker, R.; Hunt, N.H. Dramatic Changes in Oxidative Tryptophan Metabolism along the Kynurenine Pathway in Experimental Cerebral and Noncerebral Malaria. Am. J. Pathol. 1998, 152, 611–619. [Google Scholar] [PubMed]
- Bodaghi, B.; Goureau, O.; Zipeto, D.; Laurent, L.; Virelizier, J.L.; Michelson, S. Role of IFN-Gamma-Induced Indoleamine 2,3 Dioxygenase and Inducible Nitric Oxide Synthase in the Replication of Human Cytomegalovirus in Retinal Pigment Epithelial Cells. J. Immunol. 1999, 162, 957–964. [Google Scholar]
- Yap, G.S.; Sher, A. Effector Cells of Both Nonhemopoietic and Hemopoietic Origin Are Required for Interferon (IFN)-Gamma- and Tumor Necrosis Factor (TNF)-Alpha-Dependent Host Resistance to the Intracellular Pathogen, Toxoplasma Gondii. J. Exp. Med. 1999, 189, 1083–1092. [Google Scholar] [CrossRef]
- Grant, R.S.; Naif, H.; Thuruthyil, S.J.; Nasr, N.; Littlejohn, T.; Takikawa, O.; Kapoor, V. Induction of Indoleamine 2,3-Dioxygenase in Primary Human Macrophages by HIV-1. Redox Rep. 2000, 5, 105–107. [Google Scholar] [CrossRef]
- Däubener, W.; Spors, B.; Hucke, C.; Adam, R.; Stins, M.; Kim, K.S.; Schroten, H. Restriction of Toxoplasma Gondii Growth in Human Brain Microvascular Endothelial Cells by Activation of Indoleamine 2,3-Dioxygenase. Infect. Immun. 2001, 69, 6527–6531. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, K.; Riley, E.M. Innate Immune Response to Malaria: Rapid Induction of IFN-Gamma from Human NK Cells by Live Plasmodium Falciparum-Infected Erythrocytes. J. Immunol. 2002, 169, 2956–2963. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the Mammalian Brain: When Physiology Meets Pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Tishler, P.V.; Woodward, B.; O’Connor, J.; Holbrook, D.A.; Seidman, L.J.; Hallett, M.; Knighton, D.J. High Prevalence of Intermittent Acute Porphyria in a Psychiatric Patient Population. Am. J. Psychiatry 1985, 142, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Cederlöf, M.; Bergen, S.E.; Larsson, H.; Landén, M.; Lichtenstein, P. Acute Intermittent Porphyria: Comorbidity and Shared Familial Risks with Schizophrenia and Bipolar Disorder in Sweden. Br. J. Psychiatry 2015, 207, 556–557. [Google Scholar] [CrossRef]
- Wetterberg, L.; Yuwiler, A.; Geller, E. Tryptophan Oxygenase Changes Following δ-Aminolevulinic Acid Administration in the Rat. Life Sci. 1969, 8, 1047–1049. [Google Scholar] [CrossRef]
- Ren, S.; Correia, M.A. Heme: A Regulator of Rat Hepatic Tryptophan 2,3-Dioxygenase? Arch. Biochem. Biophys. 2000, 377, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.A.; Cetas, J.S. Steroid Psychosis: A Review for Neurosurgeons. J. Neurooncol. 2012, 109, 439–447. [Google Scholar] [CrossRef]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.Q.; Medoff, D.; Tamminga, C.A.; Roberts, R.C. Increased Cortical Kynurenate Content in Schizophrenia. Biol. Psychiatry 2001, 50, 521–530. [Google Scholar] [CrossRef]
- Erhardt, S.; Blennow, K.; Nordin, C.; Skogh, E.; Lindström, L.H.; Engberg, G. Kynurenic Acid Levels Are Elevated in the Cerebrospinal Fluid of Patients with Schizophrenia. Neurosci. Lett. 2001, 313, 96–98. [Google Scholar] [CrossRef]
- Miller, C.L.; Llenos, I.C.; Dulay, J.R.; Weis, S. Upregulation of the Initiating Step of the Kynurenine Pathway in Postmortem Anterior Cingulate Cortex from Individuals with Schizophrenia and Bipolar Disorder. Brain Res. 2006, 1073, 25–37. [Google Scholar] [CrossRef]
- Miller, C.L.; Llenos, I.C.; Cwik, M.; Walkup, J.; Weis, S. Alterations in Kynurenine Precursor and Product Levels in Schizophrenia and Bipolar Disorder. Neurochem. Int. 2008, 52, 1297–1303. [Google Scholar] [CrossRef]
- Linderholm, K.R.; Skogh, E.; Olsson, S.K.; Dahl, M.-L.; Holtze, M.; Engberg, G.; Samuelsson, M.; Erhardt, S. Increased Levels of Kynurenine and Kynurenic Acid in the CSF of Patients With Schizophrenia. Schizophr. Bull. 2012, 38, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Llenos, I.C.; Dulay, J.R.; Barillo, M.M.; Yolken, R.H.; Weis, S. Expression of the Kynurenine Pathway Enzyme Tryptophan 2,3-Dioxygenase Is Increased in the Frontal Cortex of Individuals with Schizophrenia. Neurobiol. Dis. 2004, 15, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.K.; Müller, A.; Heseler, K.; Woite, C.; Spekker, K.; MacKenzie, C.R.; Däubener, W. Antimicrobial and Immunoregulatory Properties of Human Tryptophan 2,3-Dioxygenase. Eur. J. Immunol. 2009, 39, 2755–2764. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An Endogenous Tumour-Promoting Ligand of the Human Aryl Hydrocarbon Receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kindler, J.; Lim, C.K.; Weickert, C.S.; Boerrigter, D.; Galletly, C.; Liu, D.; Jacobs, K.R.; Balzan, R.; Bruggemann, J.; O’Donnell, M.; et al. Dysregulation of Kynurenine Metabolism Is Related to Proinflammatory Cytokines, Attention, and Prefrontal Cortex Volume in Schizophrenia. Mol. Psychiatry 2020, 25, 2860–2872. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.A.; Pankratz, V.S.; Bostwick, J.M. The Lifetime Risk of Suicide in Schizophrenia: A Reexamination. Arch. Gen. Psychiatry 2005, 62, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Randall, J.R.; Walld, R.; Finlayson, G.; Sareen, J.; Martens, P.J.; Bolton, J.M. Acute Risk of Suicide and Suicide Attempts Associated with Recent Diagnosis of Mental Disorders: A Population-Based, Propensity Score-Matched Analysis. Can. J. Psychiatry 2014, 59, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, S.; Lim, C.K.; Linderholm, K.R.; Janelidze, S.; Lindqvist, D.; Samuelsson, M.; Lundberg, K.; Postolache, T.T.; Träskman-Bendz, L.; Guillemin, G.J.; et al. Connecting Inflammation with Glutamate Agonism in Suicidality. Neuropsychopharmacology 2013, 38, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Brundin, L.; Sellgren, C.M.; Lim, C.K.; Grit, J.; Pålsson, E.; Landén, M.; Samuelsson, M.; Lundgren, K.; Brundin, P.; Fuchs, D.; et al. An Enzyme in the Kynurenine Pathway That Governs Vulnerability to Suicidal Behavior by Regulating Excitotoxicity and Neuroinflammation. Transl. Psychiatry 2016, 6, e865. [Google Scholar] [CrossRef]
- Birner, A.; Platzer, M.; Bengesser, S.A.; Dalkner, N.; Fellendorf, F.T.; Queissner, R.; Pilz, R.; Rauch, P.; Maget, A.; Hamm, C.; et al. Increased Breakdown of Kynurenine towards Its Neurotoxic Branch in Bipolar Disorder. PLoS ONE 2017, 12, e0172699. [Google Scholar] [CrossRef]
- Messaoud, A.; Mensi, R.; Douki, W.; Neffati, F.; Najjar, M.F.; Gobbi, G.; Valtorta, F.; Gaha, L.; Comai, S. Reduced Peripheral Availability of Tryptophan and Increased Activation of the Kynurenine Pathway and Cortisol Correlate with Major Depression and Suicide. World J. Biol. Psychiatry 2019, 20, 703–711. [Google Scholar] [CrossRef]
- O’Callaghan, E.; Gibson, T.; Colohan, H.A.; Walshe, D.; Buckley, P.; Larkin, C.; Waddington, J.L. Season of birth in schizophrenia. Evidence for confinement of an excess of winter births to patients without a family history of mental disorder. Br. J. Psychiatry 1991, 158, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.R.; Keohan, J.T.; Reppert, S.M. Definition of a Prenatal Sensitive Period for Maternal-Fetal Communication of Day Length. Am. J. Physiol. 1987, 253, E701–E704. [Google Scholar] [CrossRef]
- Elliott, J.A.; Goldman, B.D. Reception of Photoperiodic Information by Fetal Siberian Hamsters: Role of the Mother’s Pineal Gland. J. Exp. Zool. 1989, 252, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Pyter, L.M.; Nelson, R.J. Enduring Effects of Photoperiod on Affective Behaviors in Siberian Hamsters (Phodopus Sungorus). Behav. Neurosci. 2006, 120, 125–134. [Google Scholar] [CrossRef]
- Walton, J.C.; Weil, Z.M.; Nelson, R.J. Influence of Photoperiod on Hormones, Behavior, and Immune Function. Front Neuroendocr. 2011, 32, 303–319. [Google Scholar] [CrossRef]
- Levitan, R.D.; Sqapi, M.; Atkinson, L.; Murphy, K.; Levitt, A.; Bocking, A.; Post, M.; Knight, J.A.; Matthews, S.G. Seasonality of Plasma Tryptophan and Kynurenine in Pregnant Mothers with a History of Seasonal Affective Disorder: Vulnerability or Adaptation? World J. Biol. Psychiatry 2020, 21, 529–538. [Google Scholar] [CrossRef]
- Aleman, A.; Kahn, R.S.; Selten, J.-P. Sex Differences in the Risk of Schizophrenia: Evidence from Meta-Analysis. Arch. Gen. Psychiatry 2003, 60, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Riecher-Rössler, A. Psychotic Disorders and Menopause: The Untold Story. Menopausal. Transit. 2009, 175, 115–126. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Modulation of Tryptophan and Serotonin Metabolism as a Biochemical Basis of the Behavioral Effects of Use and Withdrawal of Androgenic-Anabolic Steroids and Other Image- and Performance-Enhancing Agents. Int. J. Tryptophan. Res. 2018, 11, 1178646917753422. [Google Scholar] [CrossRef]
- Vacher, S.; Castagnet, P.; Chemlali, W.; Lallemand, F.; Meseure, D.; Pocard, M.; Bieche, I.; Perrot-Applanat, M. High AHR Expression in Breast Tumors Correlates with Expression of Genes from Several Signaling Pathways Namely Inflammation and Endogenous Tryptophan Metabolism. PLoS ONE 2018, 13, e0190619. [Google Scholar] [CrossRef]
- Vassos, E.; Sham, P.; Kempton, M.; Trotta, A.; Stilo, S.A.; Gayer-Anderson, C.; Di Forti, M.; Lewis, C.M.; Murray, R.M.; Morgan, C. The Maudsley Environmental Risk Score for Psychosis. Psychol. Med. 2020, 50, 2213–2220. [Google Scholar] [CrossRef]
- Phillips, L.J.; McGorry, P.D.; Garner, B.; Thompson, K.N.; Pantelis, C.; Wood, S.J.; Berger, G. Stress, the Hippocampus and the Hypothalamic-Pituitary-Adrenal Axis: Implications for the Development of Psychotic Disorders. Aust. N. Z. J. Psychiatry 2006, 40, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, D.B.; Miller, B.J. Meta-Analysis of Blood Cortisol Levels in Individuals with First-Episode Psychosis. Psychoneuroendocrinology 2019, 104, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P.; Koh, C.-S.; Froehlich, U.; Lapointe, E.; Couture, S.; Inkel, L.; Bramard, A.; Paquet, E.R.; Watier, V.; Durand, M.; et al. Multiple and Specific MRNA Processing Targets for the Major Human HnRNP Proteins. Mol. Cell Biol. 2008, 28, 6033–6043. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Ballester, A.; Forouhar, F.; Kim, S.-M.; Lew, S.; Wang, Y.; Karkashon, S.; Seetharaman, J.; Batabyal, D.; Chiang, B.-Y.; Hussain, M.; et al. Molecular Basis for Catalysis and Substrate-Mediated Cellular Stabilization of Human Tryptophan 2,3-Dioxygenase. Sci. Rep. 2016, 6, 35169. [Google Scholar] [CrossRef]
- Dick, R.; Murray, B.P.; Reid, M.J.; Correia, M.A. Structure–Function Relationships of Rat Hepatic Tryptophan 2,3-Dioxygenase: Identification of the Putative Heme-Ligating Histidine Residues. Arch. Biochem. Biophys. 2001, 392, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Bush, N.R.; Edgar, R.D.; Park, M.; MacIsaac, J.L.; McEwen, L.M.; Adler, N.E.; Essex, M.J.; Kobor, M.S.; Boyce, W.T. The biological embedding of early-life socioeconomic status and family adversity in children’s genome-wide DNA methylation. Epigenomics 2018, 10, 1445–1461. Available online: https://www.futuremedicine.com/doi/pdfplus/10.2217/epi-2018-0042 (accessed on 1 November 2021). [CrossRef]
- Wicks, S.; Hjern, A.; Gunnell, D.; Lewis, G.; Dalman, C. Social adversity in childhood and the risk of developing psychosis: A national cohort study. Am. J. Psychiatry 2005, 162, 1652–1657. [Google Scholar] [CrossRef]
- Werner, S.; Malaspina, D.; Rabinowitz, J. Socioeconomic Status at Birth Is Associated with Risk of Schizophrenia: Population-Based Multilevel Study. Schizophr. Bull. 2007, 33, 1373–1378. [Google Scholar] [CrossRef]
- Hakulinen, C.; Webb, R.T.; Pedersen, C.B.; Agerbo, E.; Mok, P.L.H. Association Between Parental Income During Childhood and Risk of Schizophrenia Later in Life. JAMA Psychiatry 2020, 77, 17–24. [Google Scholar] [CrossRef]
- Arseneault, L.; Cannon, M.; Fisher, H.L.; Polanczyk, G.; Moffitt, T.E.; Caspi, A. Childhood Trauma and Children’s Emerging Psychotic Symptoms: A Genetically Sensitive Longitudinal Cohort Study. Am. J. Psychiatry 2011, 168, 65–72. [Google Scholar] [CrossRef]
- Soichot, M.; Vaast, A.; Vignau, J.; Guillemin, G.J.; Lhermitte, M.; Broly, F.; Allorge, D. Characterization of Functional Polymorphisms and Glucocorticoid-Responsive Elements in the Promoter of TDO2, a Candidate Gene for Ethanol-Induced Behavioural Disorders. Alcohol Alcohol. 2013, 48, 415–425. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jin, J.; Lian, T.; Gu, C.; Yu, K.; Gao, Y.Q.; Su, X.-D. The Effects of Cytosine Methylation on General Transcription Factors. Sci. Rep. 2016, 6, 29119. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356, e2239. [Google Scholar] [CrossRef]
- Grøntved, L.; John, S.; Baek, S.; Liu, Y.; Buckley, J.R.; Vinson, C.; Aguilera, G.; Hager, G.L. C/EBP Maintains Chromatin Accessibility in Liver and Facilitates Glucocorticoid Receptor Recruitment to Steroid Response Elements. EMBO J. 2013, 32, 1568–1583. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Prentzell, M.T.; Mohapatra, S.R.; Sahm, F.; Zhao, Z.; Grummt, I.; Wick, W.; Opitz, C.A.; Platten, M.; Green, E.W. Constitutive Expression of the Immunosuppressive Tryptophan Dioxygenase TDO2 in Glioblastoma Is Driven by the Transcription Factor C/EBPβ. Front. Immunol. 2020, 11, 657. [Google Scholar] [CrossRef]
- Li, J.; Bidlingmaier, M.; Petru, R.; Pedrosa Gil, F.; Loerbroks, A.; Angerer, P. Impact of Shift Work on the Diurnal Cortisol Rhythm: A One-Year Longitudinal Study in Junior Physicians. J. Occup. Med. Toxicol. 2018, 13, 23. [Google Scholar] [CrossRef]
- Clarkson-Townsend, D.A.; Everson, T.M.; Deyssenroth, M.A.; Burt, A.A.; Hermetz, K.E.; Hao, K.; Chen, J.; Marsit, C.J. Maternal Circadian Disruption Is Associated with Variation in Placental DNA Methylation. PLoS ONE 2019, 14, e0215745. [Google Scholar] [CrossRef]
- Mostafizar, M.; Cortes-Pérez, C.; Snow, W.; Djordjevic, J.; Adlimoghaddam, A.; Albensi, B.C. Challenges with Methods for Detecting and Studying the Transcription Factor Nuclear Factor Kappa B (NF-ΚB) in the Central Nervous System. Cells 2021, 10, 1335. [Google Scholar] [CrossRef]
- Héberlé, É.; Bardet, A.F. Sensitivity of Transcription Factors to DNA Methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Kesanakurti, D.; Chetty, C.; Rajasekhar Maddirela, D.; Gujrati, M.; Rao, J.S. Essential Role of Cooperative NF-ΚB and Stat3 Recruitment to ICAM-1 Intronic Consensus Elements in the Regulation of Radiation-Induced Invasion and Migration in Glioma. Oncogene 2013, 32, 5144–5155. [Google Scholar] [CrossRef] [PubMed]
- Sikdar, S.; Joehanes, R.; Joubert, B.R.; Xu, C.-J.; Vives-Usano, M.; Rezwan, F.I.; Felix, J.F.; Ward, J.M.; Guan, W.; Richmond, R.C.; et al. Comparison of Smoking-Related DNA Methylation between Newborns from Prenatal Exposure and Adults from Personal Smoking. Epigenomics 2019, 11, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Wigner, P.; Synowiec, E.; Jóźwiak, P.; Czarny, P.; Bijak, M.; Białek, K.; Szemraj, J.; Gruca, P.; Papp, M.; Śliwiński, T. The Effect of Chronic Mild Stress and Venlafaxine on the Expression and Methylation Levels of Genes Involved in the Tryptophan Catabolites Pathway in the Blood and Brain Structures of Rats. J. Mol. Neurosci. 2020, 70, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.A.; Churchill, L.; Rehman, A.; Ellis, G.; Mémet, S.; Israël, A.; Krueger, J.M. Sleep Deprivation Increases the Activation of Nuclear Factor Kappa B in Lateral Hypothalamic Cells. Brain Res. 2004, 1004, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.-L.; Nie, J.; Ku, J.; Dougherty, U.; West-Szymanski, D.C.; Collin, F.; Ellison, C.K.; Sieh, L.; Ning, Y.; Deng, Z.; et al. A Human Tissue Map of 5-Hydroxymethylcytosines Exhibits Tissue Specificity through Gene and Enhancer Modulation. Nat. Commun. 2020, 11, 6161. [Google Scholar] [CrossRef]
- Quigley, H.; MacCabe, J.H. The Relationship between Nicotine and Psychosis. Adv. Psychopharmacol. 2019, 9, 2045125319859969. [Google Scholar] [CrossRef]
- Dalack, G.W.; Meador-Woodruff, J.H. Smoking, Smoking Withdrawal and Schizophrenia: Case Reports and a Review of the Literature. Schizophr. Res. 1996, 22, 133–141. [Google Scholar] [CrossRef]
- Zammit, S.; Allebeck, P.; Dalman, C.; Lundberg, I.; Hemmingsson, T.; Lewis, G. Investigating the association between cigarette smoking and schizophrenia in a cohort study. Am. J. Psychiatry 2003, 160, 2216–2221. [Google Scholar] [CrossRef]
- Adler, L.E.; Hoffer, L.J.; Griffith, J.; Waldo, M.C.; Freedman, R. Normalization by Nicotine of Deficient Auditory Sensory Gating in the Relatives of Schizophrenics. Biol. Psychiatry 1992, 32, 607–616. [Google Scholar] [CrossRef]
- Olincy, A.; Freedman, R. Nicotinic Mechanisms in the Treatment of Psychotic Disorders: A Focus on the A7 Nicotinic Receptor. Handb. Exp. Pharm. 2012, 213, 211–232. [Google Scholar] [CrossRef]
- Poddar, M.K.; Ghosh, J.J. Effect of Cannabis Extract, 9 -Tetrahydrocannabinol and Lysergic Acid Diethylamide on Rat Liver Enzymes. Biochem. Pharm. 1972, 21, 3301–3303. [Google Scholar] [CrossRef]
- Wund, M.A.; Baker, J.A.; Clancy, B.; Golub, J.L.; Foster, S.A. A Test of the “Flexible Stem” Model of Evolution: Ancestral Plasticity, Genetic Accommodation, and Morphological Divergence in the Threespine Stickleback Radiation. Am. Nat. 2008, 172, 449–462. [Google Scholar] [CrossRef]
- Tebbich, S.; Sterelny, K.; Teschke, I. The Tale of the Finch: Adaptive Radiation and Behavioural Flexibility. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Muschick, M.; Barluenga, M.; Salzburger, W.; Meyer, A. Adaptive Phenotypic Plasticity in the Midas Cichlid Fish Pharyngeal Jaw and Its Relevance in Adaptive Radiation. BMC Evol. Biol. 2011, 11, 116. [Google Scholar] [CrossRef]
- Kaushik, M.; Mahendru, S.; Kumar, M.; Chaudhary, S.; Kukreti, S. Genomic Databases and Softwares: In Pursuit of Biological Relevance through Bioinformatics. Adv. Tech. Biol. Med. 2016, 4, 1–6. [Google Scholar] [CrossRef]
- Whitaker, D.T.; Ostrander, E.A. Hair of the Dog: Identification of a Cis-Regulatory Module Predicted to Influence Canine Coat Composition. Genes 2019, 10, E323. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Important environmental cue reviewed | Temperature, changes with season and latitude |
| Direction of effect | Cold temperature stimulates expression of peptide agonist; MCHR1 controls thermo-regulation to lower body temperature and conserve BAT |
| Risk of schizophrenia (scz) consistent with known gene function? | Yes:
|
| Strength of genetic association | Moderate: 4 positive studies; and 1 positive when part of a complex genotype; contributes to PRS; the alleles conferring risk depend on the population |
| Epigenetic data of relevance? | Yes:
|
| Consistency of gene expression data | One study: gene expression decreased in scz, but may be confounded by smoking based on methylation pattern identified in smokers |
| Important environmental cue reviewed | Cannabis use |
| Direction of effect | Cannabis use predicted to inactivate AKT1 thru increased dopaminergic signaling, though animal data yields mixed results |
| Risk of schizophrenia (scz) consistent with known gene function? | Yes: schizophrenia is associated with increased dopaminergic tone, which should inactivate AKT1 via DRD2 agonists; drugs that are antagonists for DRD2 exert an antipsychotic effect |
| Strength of genetic association | Weak as a single locus: studies showing association as well as many showing lack of association; however, contributes to PRS |
| Epigenetic data of relevance? | Yes:
|
| Consistency of gene expression data | Mixed results for scz and related disorders Expression: Some studies show decreased gene expression, some no change and some increased expression; variability could be attributable to the effect of smoking seen in methylation data |
| Important environmental cue reviewed | Stress |
| Direction of effect | Stress is predicted to increase expression of gene via the established effect of glucocorticoids to stimulate TDO2 mRNA expression |
| Risk of schizophrenia (scz) consistent with known gene function? | Yes:
|
| Strength of genetic association | Weak as a single locus: one study showing association only as part of complex genotype; however, contributes to PRS |
| Epigenetic data of relevance? | Yes:
|
| Consistency of gene expression data | Consistent:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, C.L. The Epigenetics of Psychosis: A Structured Review with Representative Loci. Biomedicines 2022, 10, 561. https://doi.org/10.3390/biomedicines10030561
Miller CL. The Epigenetics of Psychosis: A Structured Review with Representative Loci. Biomedicines. 2022; 10(3):561. https://doi.org/10.3390/biomedicines10030561
Chicago/Turabian StyleMiller, Christine L. 2022. "The Epigenetics of Psychosis: A Structured Review with Representative Loci" Biomedicines 10, no. 3: 561. https://doi.org/10.3390/biomedicines10030561
APA StyleMiller, C. L. (2022). The Epigenetics of Psychosis: A Structured Review with Representative Loci. Biomedicines, 10(3), 561. https://doi.org/10.3390/biomedicines10030561